
Abstract
The present study was carried out to ascertain the impact of replacing the sulfonate group of TAU with thiosulfonate, as present in thiotaurine (TTAU), on the protective actions of TAU against hepatocellular damage and biochemical alterations related to oxidative stress and glutathione redox cycling, synthesis, and utilization caused by a high dose of acetaminophen (APAP). To this end, male Sprague-Dawley rats, 225–250 g, were intraperitoneally treated with a 2.4 mmol/kg dose of TAU (or TTAU), followed 30 min later by 800 mg/kg of APAP. A reference group received 2.4 mmol/kg of N-acetylcysteine (NAC) prior to APAP. Naive rats served as controls. The animals were sacrificed 6 h after receiving APAP and their blood and livers were collected. Plasma and liver homogenates were analyzed for indices of cell damage (plasma transaminases, plasma lactate dehydrogenase), oxidative stress (malondialdehyde = MDA, reduced glutathione = GSH, glutathione disulfide = GSSG, catalase, glutathione peroxidase, superoxide dismutase), glutathione cycling (glutathione reductase), utilization (glutathione S-transferase), and synthesis (γ-glutamylcysteine synthetase) activities. APAP increased MDA formation and lowered the GSH/GSSG ratio and all enzyme activities, especially those of antioxidant enzymes. In general, TTAU was equipotent with NAC and more potent than TAU in protecting the liver. Taken into account the results of a previous study comparing the actions of TAU and hypotaurine (HTAU), the sulfinate analog of TAU, it appears that the sulfinate and thiosulfonate analogs are somewhat more effective than the parent sulfonate TAU in counteracting APAP-induced hepatic alterations in the liver and plasma.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Glutathione Reductase
- Mercapturic Acid
- Glutathione Disulfide
- Cytochrome P450 Enzyme System
- APAP Overdose
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Acetaminophen (N-acetyl-p-aminophenol, APAP) is a synthetic centrally acting compound widely used for its analgesic and antipyretic effects in infants, children, and adults. At therapeutic doses this over-the-counter medication is well tolerated, but in overdoses it is one of the most common causes of both intentional and unintentional poisoning and toxicity, often culminating in hepatic failure and even death (Chung et al. 2009; Lee 2007).
The toxicity of APAP is closely linked to its hepatic metabolism, which may involve the depletion of protein and glutathione thiol groups, the elevation of the cytoplasmic Ca2+ (Moore et al. 1985), and the development of oxidative/nitrosative stress (Jaeschke et al. 2003; van de Straat et al. 1987).
After the ingestion of a standard dose of APAP, up to 90% of this phenolic compound enters into hepatic conjugation at the meta-position to yield innocuous glucuronide (∼55%) and sulfate (35%) conjugates, with only a small fraction (about 5–10%) undergoing metabolism to the toxic electrophile N-acetyl-para-benzoquinoimine (NAPQI) by hepatic cytochrome P450 isoforms, mainly CYP 2E1, CYP 1A2, and CYP 3A2 (Bessems and Vermeulen 2001; Patten et al. 1993; Smilkstein et al. 1988). At low doses of APAP, NAPQI is conjugated with glutathione and subsequently excreted in the urine as the mercapturic acid and cysteine derivatives (Patten et al. 1993). At high doses of APAP, however, the normal conjugating pathways for this compound become rapidly saturated and a greater proportion of the drug is available for conversion to NAPQI, which then depletes the hepatic stores of glutathione. As a result, NAPQI is left unopposed to effect cell injury by covalently binding to hepatic proteins, primarily though not exclusively, to cysteine residues to generate stable 3-(cystein-S-yl) APAP adducts (Nelson and Bruschi 2003). While this mechanism may account for APAP-related cell injury, the fraction of the dose of APAP entering into covalent binding to hepatic proteins is small. Hence, additional mechanisms have been postulated to account for the bulk of liver injury by APAP. One such proposal considers lipid peroxidation (LPO) as a mechanism of cell death based on the increased formation of reactive oxygen (ROS) and nitrogen (RNS) species observed in hepatocytes undergoing necrotic changes (Hinson et al. 2010). In this case, the binding of NAPQI to mitochondrial proteins of the respiratory chain will decrease mitochondrial respiration, oxidative phosphorylation, and ATP formation, and the flow of electrons will be diverted towards oxygen to generate superoxide anion (Jaeschke et al. 2003). In turn, superoxide anion arising from mitochondrial stress will propagate and amplify the liver injury upon reacting with nitric oxide to form peroxynitrite, a powerful oxidant and nitrating agent with the ability to modify cellular macromolecules and to aggravate mitochondrial dysfunction and ATP depletion (Jaeschke et al. 2003). Furthermore, mitochondria oxidative stress will alter calcium homeostasis and calcium-controlled cellular processes and will stimulate signaling pathways for the activation of transduction responses ending in mitochondrial permeability transition and the loss of membrane potential, events that further contribute to centrilobular hepatic necrosis and acute liver failure (Hinson et al. 2010).
In parallel with its depleting action of the intrahepatic glutathione, APAP is also capable of exerting varying effects on the levels of glutathione disulfide and of lowering the activities of antioxidant enzymes (catalase, glutathione peroxidase, superoxide dismutase) as well as of enzymes participating in glutathione redox cycling (glutathione reductase), utilization (glutathione S-transferase), and synthesis (γ-glutamylcysteine synthetase) (Acharya and Lau-Cam 2010).
In general, the treatment of APAP poisoning has been directed at inhibiting its activation by the cytochrome P450 enzyme system or at restoring hepatic glutathione reserves to sustain conjugation with glutathione. Since protection of the liver against APAP toxicity by decreasing the formation of NAPQI through inhibition of the cytochrome P450 enzyme system with cysteamine, the descarboxy analog of l-cysteine, was not high enough (Miller and Jollow 1986), most antidotal approaches for APAP poisoning have been targeted at restoring the levels of intracellular glutathione. In spite of the numerous attempts to develop prodrugs of l-cysteine for GSH synthesis, only N-acetylcysteine (NAC) has received recognition as a first-line treatment for APAP poisoning. In addition to reversing APAP-induced depletion of glutathione and insuring the excretion of both APAP and NAPQ in the bile as glutathione conjugates (Lauterburg et al. 1983), NAC is also an effective antioxidant (Cotter et al. 2007; Ozaras et al. 2003; Sathish et al. 2011; Victor et al. 2003).
In a previous study we compared the effects of NAC with those of the sulfonic (-SO3H) and sulfinic (-SO2H) analogs of 2-aminoethane, namely, taurine (TAU) and hypotaurine (HYTAU), as protection against APAP-mediated LPO, changes in glutathione redox state, and declines in the activities of enzymes involved in glutathione redox cycling, transfer, and synthesis in the rat (Acharya and Lau-Cam 2010). The rationale for testing these sulfur-containing compounds as protection against APAP hepatotoxicity stemmed from previous recognition that TAU was capable of attenuating LPO, apoptosis, necrosis, and DNA fragmentation in hepatocytes from rats treated with a toxic dose of APAP (Waters et al. 2001) and that TAU and HYTAU effectively lowered LPO, the fall of the GSH/GSSG ratio, and the loss of antioxidant enzyme activities in erythrocytes from diabetic rats (Gossai and Lau-Cam 2009). Since these results pointed to a determining role for the sulfur-containing functionality in the potency differences noted in erythrocytes between TAU and HYTAU, the present study was undertaken to further validate this assumption by comparing the actions of TAU in APAP-related hepatotoxicity against those of thiotaurine (TTAU), the thiosulfonate (-SO2-SH) analog thiotaurine which has also been found to possess antioxidant actions (Egawa et al. 1999; Yoshiyuki 1998; Yoshiyuki and Yoshiki 2000).To better define the degree of protective action of TAU and TTAU in APAP-induced hepatotoxicity, their activities were compared against those of NAC.
2 Methods
2.1 Chemicals
The chemicals used in the present study were obtained from commercial sources in the USA. NAC, APAP, and chemicals used in the preparation of the biological samples and in the biochemical assays were purchased from Sigma-Aldrich, St. Louis, MO. TTAU was from Wako Chemicals USA, Inc., Richmond, VA.
2.2 Animals, Treatments, and Sample Collections
Male Sprague-Dawley rats, weighing 200–250 g, were obtained from Taconic, Germantown, New York, USA. The study received the approval of the Institutional Animal Care and Use Committee of St. John’s University, Jamaica, New York, and the animals were cared in accordance with guidelines established by the United States Department of Agriculture. The experimental groups consisted of six rats each and they were used in the nonfasted state. The treatment solutions were prepared just prior to an experiment. The APAP solution was made in warm 50% polyethylene glycol (PEG) 400 and allowed to cool to ambient temperature before its administration. The test compounds (NAC, TAU, TTAU) were dissolved in distilled water. A treatment compound was administered as a single, 2.4 mmol/kg/2 mL, dose 30 min before a hepatotoxic, 800 mg/kg/2 mL, dose of APAP. Animals serving as controls only received 50% PEG 400 in a volume equal to 2 mL. All treatments were carried out by the intraperitoneal route. At 6 h after the administration of APAP or 50% PEG 400, the animals were sacrificed by decapitation and their blood collected in heparinized tubes. Immediately thereafter the livers were removed using the freeze-clamp technique of Wollenberger et al. (1960). From each blood sample, a portion was set aside for the assay of reduced (GSH) and disulfide (GSSG) glutathione, and the remaining portion was centrifuged at 3,000 rpm (500 ´g) and 4°C for 10 min to obtain the corresponding plasma fraction. For each liver, a 500 mg portion, kept cold on an ice bath, was mixed in a 1:20 (w/v) ratio with Tris buffer pH 7.0 containing 1 mg of phenylmethylsulfonyl fluoride and homogenized using a handheld electric blender. The resulting suspension was centrifuged at 12,000 rpm (8000 ´ g) and 4°C for 30 min to isolate the supernatant, which was kept on ice until needed.
2.3 Assay of Liver Malondialdehyde
Malondialdehyde (MDA) was determined as thiobarbituric acid-reactive substances (TBARS) by the endpoint assay method of Buege and Aust (1978). The amount of TBARS in the sample was derived from a calibration curve of MDA prepared from serial dilutions of a stock solution of 1,1,3,3-tetraethoxypropane which had been treated in the same manner as the sample preparation, and the results were reported as nM of MDA/mg of tissue.
2.4 Assay of the Plasma and Hepatic Levels of GSH and GSSG
The concentration of GSH in plasma and liver samples was measured by the method of Hissin and Hilf (1976), after reaction with ortho-phthalaldehyde to form a highly fluorescent product. The concentration of GSSG was measured in another aliquot of the same sample, following removal of any preexisting GSH upon reaction with N-ethylmaleimide. The concentrations of GSH and GSSG in the sample preparation were determined by reference to standard curves of these compounds prepared on the day of the assay and were reported either as nM/mL of plasma or as μM/g of tissue.
2.5 Assay of Plasma and Liver Antioxidant Enzymes
The catalase (CAT) activity was measured by the spectrophotometric method of Aebi (1984), the glutathione peroxidase (GPX) activity was measured as described by Flohé and Günzler (1984), and the activity of CuZn superoxide dismutase (SOD) was measured using the spectrophotometric method of Misra (1985). These activities were expressed as U/min/mg of protein.
2.6 Assay of the Plasma and Liver g -Glutamylcysteine Synthetase (GCS), Glutathione S-Transferase (GST) and Glutathione Reductase (GR) Activities
The GCS activity was measured according to the method of Zhou and Freed (2005), the GST activity was measured as described by Habig et al. (1974), and the GR activity was measured by the spectrophotometric method of Wheeler et al. (1990). The results were expressed in U/min/mg of protein.
2.7 Statistical Analysis of the Data
The experimental results are reported as mean ± SEM for n = 6. They were analyzed for statistical significance using unpaired Student’s t-test and a commercial computer software (JMP 7, JMP® Statistical Discovery Software, Cary, NC 27513) followed by one-way analysis of variance and Newman-Keuls post hoc test. Intergroup differences were considered to be statistically significant at p ≤ 0.05.
3 Results and Discussion
The protective effects of TAU and TTAU against APAP-induced hepatotoxicity were assessed by measuring biochemical parameters consonant with cellular injury and oxidative stress. To more accurately define their respective potencies, they were further compared with an equidose (2.4 mmol/kg) of NAC, the current antidote of choice for APAP overdoses.
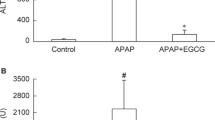
The degree of injury to the hepatocytes by a high dose of APAP was investigated by measuring the plasma ALT, AST, and LDH, three abundant intrahepatic enzymes whose release into the circulation is taken as evidence of liver injury (Duong and Loh 2006). As shown in Table 20.1, the plasma activities of all three enzymes were significantly elevated (p < 0.001), with the values decreasing in the order LDH (+292%) > AST (+135%) > ALT (+64%) probably in direct proportion to their intracellular abundance and location (Moss et al. 1986). TAU and TTAU were highly protective and about equipotent, with the increases amounting to only ∼104% (p < 0.001), ∼37% (p < 0.01), and ∼33% (p < 0.01) of corresponding control values, respectively. NAC was more potent than either TAU or TTAU since the enzyme activities of LDH, AST, and ALT were only 60% (p < 0.001), 28% (p < 0.01), and 21% (p < 0.05), respectively, above control values.

The effects of NAC, TAU, and TTAU on liver and plasma MDA levels of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at *p < 0.05 and ***p < 0.001 and from APAP at †† p < 0.01 and ††† p<0.001. Values are shown as mean ± SEM for n = 6
Hepatic LPO is a common consequence of acute APAP intoxication which is typically manifested in rodents by increased ethane inhalation, increased hepatic MDA, and GSH depletion (Wendel 1983). The detection of MDA accumulation is a rather sensitive indicator of hepatotoxicity since it is increased before the appearance of necrosis (Nakae et al. 1990). The results presented in Fig. 20.1 indicate that APAP markedly raised the MDA level in the plasma (by >200%) and liver (by >130%). Both TAU and TTAU were able to attenuate these increases, with TTAU appearing more potent (+48%, p < 0.01, and +14%, respectively) than TAU (+91%, p < 0.001, and +22%, p < 0.05, respectively). On the other hand, NAC was more potent than either TAU or TTAU especially in the plasma (32%, p < 0.01, and 16%, p < 0.05, respectively).

The effects of NAC, TAU, and TTAU on liver and plasma GSH levels of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at *p < 0.05, **p < 0.01, and ***p < 0.001 and from APAP at †† p<0.01 and ††† p<0.001. Values are shown as mean ± SEM for n = 6
Experiments in rodents have demonstrated that an acute high dose of APAP can lower the hepatic GSH levels as early as 15 min after its administration (Lores Arnaiz et al. 1995; Lauterburg et al. 1983). From the results presented in Fig. 20.2, it is apparent that APAP depleted GSH both in the liver (by 76%, p < 0.001) and plasma (by 39%, p < 0.01), changes that were much less pronounced in the presence of TTAU (+5% and −11%, respectively). By comparison, protection was less in the presence of TAU (−20%, p < 0.01, and −46%, p < 0.001, respectively) and about equal following a treatment with NAC (−10% and −2%, respectively) relative to TTAU.

The effects of NAC, TAU, and TTAU on liver and plasma GSSG levels of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at *p < 0.05 and **p < 0.01 and from APAP at † p<0.05 and †† p<0.01. Values are shown as mean ± SEM for n = 6
The loss of GSH upon the administration of APAP has occurred concomitantly with a proportional decrease in arterial plasma GSH concentration but not with a corresponding increase in GSSG, findings that are consistent with the hypothesis that the liver is a major source of plasma GSH (Adams et al. 1983). In the present study, APAP (800 mg/kg) lowered the GSSG levels in both plasma (−25%, p < 0.01) and liver (−47%, p < 0.001) (Fig. 20.3), effects that were counteracted by TAU (−19% and −22%, p < 0.05, respectively) and TTAU (−15%, p < 0.05, and +66%, p < 0.001) relative to control values. NAC, on the other hand, was about equipotent with TTAU (−14% and +46%, p < 0.001, respectively). In parallel with a decrease in hepatic GSH and GSSG, there was also a corresponding decrease in the GSH/GSSG ratio both in the plasma (−19%, p < 0.05) and liver (−54%, p < 0.001) (Fig. 20.4). These changes were attenuated by TAU (−23% and −29%, respectively, both at p < 0.05) and reversed by TTAU (+10% and 66%, p < 0.001, respectively) and NAC (+4 and +46%, p < 0.001, respectively).

The effects of NAC, TAU, and TTAU on the liver and plasma GSH/GSSG ratios of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at *p < 0.05, **p < 0.01, and ***p < 0.001 and from APAP at † p<0.05, †† p<0.01, and ††† p < 0.001. Values are shown as mean ± SEM for n = 6

The effects of NAC, TAU, and TTAU on the activities of liver and plasma CAT of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at *p < 0.05, **p < 0.01, and ***p < 0.001 and from APAP at †† p<0.01 and ††† p < 0.001. Values are shown as mean ± SEM for n = 6
At high doses APAP can make the liver more vulnerable to the deleterious effects of oxidative stress through its inhibitory interaction with antioxidant enzymes known to interact and destroy specific ROS (Matés 2000). In agreement with the results reported by other investigators in rats (Sabina et al. 2009) and mice (Olaleye and Rocha 2008), this work verified that the activity of CAT and SOD was decreased by a high dose of APAP both in the plasma and liver. This situation will certainly add to the susceptibility of liver cells to oxidative stress inasmuch as each of these enzymes is involved in ROS detoxification (hydrogen peroxide by CAT, hydrogen peroxide and other peroxides by GPx, superoxide anion by SOD). As seen in Figs. 20.5 and 20.6, the plasma activities of these enzymes were reduced by 56% and 29%, respectively, relative to control values (p ≤ 0.01) and by 41% and 58%, respectively (p ≤ 0.01), in the liver. All the pretreatment compounds were able to prevent the losses in antioxidant enzyme activities to a significant extent in the plasma (CAT by only 1–11%, SOD by ∼11%) and liver (CAT 7–15%, SOD by 26–35%, p < 0.01), with TAU providing a nonsignificantly greater protection than either TTAU or NAC. While the activity of CAT may protect against LPO by preventing iron-catalyzed generation of ROS, an increase in SOD activity may curtail the accumulation of MDA in parallel with liver necrosis (Nakae et al. 1990).

The effects of NAC, TAU, and TTAU on the activities of liver and plasma SOD of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at **p < 0.01 and ***p < 0.001 and from APAP at † p<0.01 and ††† p < 0.001. Values are shown as mean ± SEM for n = 6

The effects of NAC, TAU, and TTAU on the liver and plasma activities of GPx of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at ***p < 0.001 and from APAP at †† p<0.01 and ††† p < 0.001. Values are shown as mean ± SEM for n = 6
In vitro studies in which isolated rat hepatocytes were exposed to an oxidant in the presence and absence of inhibitors of GPx and GR have suggested that the GPx/GR system can protect hepatocytes from damage by oxidative stress and can decrease cell susceptibility to APAP toxicity in response to oxidative stress initiated by ROS, particularly by peroxides normally handled by the GPx/GR system (Adamson and Harman 1989). Overdoses of APAP may negatively affect the activity of GPx by lowering the availability of its cofactor GSH, by promoting the formation of ROS capable of oxidizing critical thiol groups, and through arylation by NAPQI (Lores Arnaiz et al. 1995; Tirmenstein and Nelson 1990). On the other hand, GR, an enzyme that converts GSSG back to GSH, may be inhibited by APAP-GSH conjugate (Roušar et al. 2010). Not surprisingly, in the present study the activities of GPX and GR were significantly decreased by APAP in the plasma (−57% and −34%, p < 0.01, respectively) and liver (−69%, p < 0.001, and −23%, p < 0.05, respectively) (Figs. 20.7 and 20.8). A pretreatment with a sulfur-containing compound ameliorated these losses to rather similar extents in both the plasma (36–43% for GPx, p < 0.01; 3–22% for GR) and liver (40–52%, p ≤ 0.01, for GPX; ≤12% for GR).

The effects of NAC, TAU, and TTAU on the activities of liver and plasma GR of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at *p < 0.05 and **p < 0.01 and from APAP at † p<0.05 and †† p<0.01. Values are shown as mean ± SEM for n = 6

The effects of NAC, TAU, and TTAU on the liver and plasma activities of GCS of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at *p < 0.05 and from APAP at † p<0.05 and †† p<0.01. Values are shown as mean ± SEM for n = 6
γ-Glutamylcysteine synthetase (γ-GCS) catalyzes the rate-limiting step in de novo synthesis of GSH in liver cells and, consequently, plays a major role in the antioxidant capacity of these cells. The protective role of this enzyme was demonstrated by knocking down the heavy chain mRNA of γ-GCS in a rat model to induce a protracted GSH depletion and to potentiate APAP toxicity when compared with normal rats (Akai et al. 2007). For this reason, individuals who are heterozygous for γ-GCS deficiency may have a limited capacity for detoxifying NAPQI through conjugation with GSH (Spielberg 1985). Relative to control values, APAP drastically lowered the plasma and hepatic activity of γ-GCS by 61% and 70% (p < 0.001), respectively (Fig. 20.9). A pretreatment with TTAU virtually reversed the effects (≤3% decrease) and one with TAU was markedly protective (−19%, p < 0.05, and −8%, respectively). NAC was equipotent with TTAU.
GSTs are a group of cytosolic and membrane-associated isoenzymes with the ability to catalyze the nucleophilic addition of the thiol of reduced glutathione to a variety of electrophiles (Hayes and Strange 1995; Rushmore and Pickett 1993). At low doses of APAP, GST is responsible for the bulk of the detoxification of NAPQI through conjugation with GSH (Henderson et al. 2000; Ketterer et al. 1983). However, a protective role for GST in APAP overdoses has been questioned after experiments showing that mice nulled for GST became resistant to the hepatotoxicity of APAP and that wild and nulled animals showed no difference in APAP metabolism and the same degree of APAP-reactive metabolites binding to cellular proteins, thus indicating that GST does not contribute in vivo to the formation of GSH conjugates of APAP but instead plays an unexpected role in the toxicity of this compound (Henderson et al. 2000).
Under the present experimental conditions, the plasma activity of GST was found to be reduced by APAP significantly (p < 0.001) in the plasma (by 70%) and liver (by 61%) when compared to control values (Fig. 20.10). These decreases were more than halved in the plasma (28–31% decreases, p < 0.01) and nearly halved (34–42% decreases, p < 0.01) in the liver by a pretreatment with TAU, TTAU, or INS compared to controls. These results contrast with those reported by Polaniak et al. (2011) in rats chronically treated with a daily 2.4 g/kg intraesophageal dose of APAP for periods up to 12 weeks and who found the activities of GST and GR to be elevated while that of GPx was decreased. The existence of a wide variability in the type of effect exerted by APAP on GSH-related enzymes is exemplified by the results of two studies conducted in rodents. In one study conducted in rats, treatment with a single 300 mg/kg dose of APAP lowered the activities of GPx, GR, and GST to below control values; in the other, the treatment of mice with a single 90–150 mg/kg dose of APAP enhanced the activity of GST in the serum but had the opposite effect on that associated with liver microsomes and homogenate (Wang and Peng 1993).

The effects of NAC, TAU, and TTAU on the activities of liver and plasma GST of rats treated with a hepatotoxic (800 mg/kg i.p.) dose of APAP. Differences were significant from control at **p < 0.01 and ***p < 0.001 and from APAP at ††† p < 0.001. Values are shown as mean ± SEM for n = 6
4 Conclusion
In short, this work has verified that the protective actions of the TAU molecule are maintained upon its conversion to the thiosulfonate analog TTAU. Even though the pattern of protective actions derived from these 2-aminoethane derivatives is identical and comparable to that expressed by NAC, TTAU appears to be more potent than TAU in terms of preserving the glutathione redox status of the cell. From the present results and those gathered in an early study with TAU and HTAU, it is apparent that the protection rendered by these sulfur-containing compounds is highly dependent on the sulfur-containing functionality and that their protective effect against APAP hepatotoxicity is the result of an antioxidant action that translates into reduced LPO, preservation of antioxidant defenses, and maintenance of enzymatic mechanisms needed for GSH redox cycling, synthesis, and utilization. When ranked according to their relative protective potencies, TTAU was more akin to NAC and somewhat more effective than TAU.
Overall, the sulfinate analog of 2-aminoethane HYTAU appears to be a better antioxidant than either the thiosulfonate or sulfonate analogs.
Abbreviations
- APAP:
-
Acetaminophen
- TAU:
-
Taurine
- TTAU:
-
Thiotaurine
- NAC:
-
N-acetylcysteine
- ALT:
-
Alanine transaminase
- AST:
-
Aspartate transaminase
- LDH:
-
Lactate dehydrogenase
- MDA:
-
Malondialdehyde
- GSH:
-
Reduced glutathione
- GSSG:
-
Glutathione disulfide
- CAT:
-
Catalase
- SOD:
-
Superoxide dismutase
- GPx:
-
Glutathione peroxidase
- GS:
-
γ-Glutamylcysteine synthetase
- GR:
-
Glutathione reductase
- GST:
-
Glutathione S-transferase
References
Acharya M, Lau-Cam CA (2010) Comparison of the protective actions of N-acetylcysteine, hypotaurine and taurine against acetaminophen-induced hepatotoxicity in the rat. J Biomed Sci 17(Suppl 1):S35
Adams JD, Lauterburg BH, Mitchell JR (1983) Plasma glutathione and glutathione disulfide in the rat: regulation and response to oxidative stress. J Pharmacol Exp Ther 227:749–754
Adamson GM, Harman AW (1989) A role for the glutathione peroxidase/reductase enzyme system in the protection from paracetamol toxicity in isolated mouse hepatocytes. Biochem Pharmacol 38:3323–3330
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Akai S, Hosomi H, Minami K, Tsuneyama K, Katoh M, Nakajima M, Yokoi T (2007) Knock down of γ-glutamylcysteine synthetase in rat causes acetaminophen-induced hepatotoxicity. J Biol Chem 282:23996–24003
Bessems JG, Vermeulen NP (2001) Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches. Crit Rev Toxicol 31:55–138
Buege JA, Aust SD (1978) Microsomal lipid peroxidation. Methods Enzymol 52:302–310
Cotter MA, Thomas J, Cassidy P et al (2007) Melanoma in mice stress/damage and delays onset of ultraviolet-induced N-acetylcysteine. Clin Cancer Res 13:5952–5958
Chung LJ, Tong MJ, Busuttil RW, Hiatt JR (2009) Acetaminophen hepatotoxicity and acute liver failure. J Clin Gastroenterol 43:342–349
Duong CD, Loh JY (2006) Basic review. Laboratory monitoring in oncology. J Oncol Pharm Pract 12:223–236
Egawa M, Kohno Y, Kumano Y (1999) Oxidative effects of cigarette smoke on the human skin. Int J Cosmet Sci 21:83–98
Gossai D, Lau-Cam CA (2009) The effects of taurine, taurine homologs and hypotaurine on cell and membrane antioxidative system alterations caused by type 2 diabetes in rat erythrocytes. Adv Exp Med Biol 643:359–368
Flohé L, Günzler WA (1984) Assays of glutathione peroxidase. Methods Enzymol 105:114–121
Habig WH, Pabst MJ, Jakoby WB (1974) Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem 246:7130–7139
Hayes JD, Strange RC (1995) Potential contribution of the glutathione S-transferase supergene family to resistance to oxidative stress. Free Radic Res 22:193–207
Henderson CJ, Wolf CR, Kitteringham N, Powell H, Otto D, Park BK (2000) Increased resistance to acetaminophen hepatotoxicity in mice lacking glutathione S-transferase Pi. Proc Natl Acad Sci USA 97:12741–12745
Hinson JA, Roberts DW, James LP (2010) Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol (196):369–405
Hissin PJ, Hilf R (1976) A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem 74:214–226
Jaeschke H, Knight TR, Bajt ML (2003) The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett 144:279–288
Ketterer B, Coles B, Meyer DJ (1983) The role of glutathione in detoxication. Environ Health Perspect 49:59–69
Lauterburg BH, Corcoran GB, Mitchell JR (1983) Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 71:980–991
Lee WM (2007) Acetaminophen toxicity: changing perceptions on a social/medical issue. Hepatology 46:966–970
Lores Arnaiz S, Llesuy S, Cutrín JC, Boveris A (1995) Oxidative stress by acute acetaminophen administration in mouse liver. Free Radic Biol Med 19:303–310
Matés JM (2000) Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 153:83–104
Miller MG, Jollow DJ (1986) Acetaminophen hepatotoxicity: studies on the mechanism of cysteamine protection. Toxicol Appl Pharmacol 83:115–125
Misra HP (1985) Adrenochrome assay. In: Greenwald RA (ed) CRC Handbook of Methods for Oxygen Radical Research. CRC Press, Boca Raton, FL, pp 237–241
Moore M, Thor H, Moore G, Nelson SD, Moldeus P, Orrenius S (1985) The toxicity of acetaminophen and N-acetyl-p-benzoquinone imine in isolated hepatocytes is associated with thiol depletion and increased cytosolic Ca2+. J Biol Chem 260:13035–13040
Moss DW, Henderson AR, Kachman JF (1986) Enzymes. In: Tietz NW (ed) Textbook of Clinical Chemistry, W B Saunders, Philadelphia, PA, pp 663–667
Nakae D, Yoshiji H, Yamamoto K et al (1990) Influence of timing of administration of liposome-encapsulated superoxide dismutase on its prevention of acetaminophen-induced liver cell necrosis in rats. Acta Pathol Jpn 40:568–573
Nelson SD, Bruschi SA (2003) Mechanisms of acetaminophen-induced liver disease. In: Kaplowitz N, DeLeve LD (eds) Drug-Induced Liver Disease. Marcel Decker, New York, NY, pp 287–325
Olaleye MT, Rocha BT (2008) Acetaminophen-induced liver damage in mice: effects of some medicinal plants on the oxidative defense system. Exp Toxicol Pathol 59:319–327
Ozaras R, Tahan V, Aydin S, Uzun H, Kaya S, Senturk H (2003) N-Acetylcysteine attenuates alcohol-induced oxidative stress in the rat. World J Gastroenterol 9:125–128
Patten CJ, Thomas PE, Guy RL et al (1993) Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 6:511–518
Polaniak R, Bułdak RJ, Jacheć W (2011) Long-term exposure to acetaminophen is a crucial for activity of selected antioxidative enzymes and level of lipid peroxidation process in rat liver. J Bioequiv Availab 3:182–186
Roušar T, Pařík P, Kučera O, Bartoš M, Červinková Z (2010) Glutathione reductase is inhibited by acetaminophen-glutathione conjugate in vitro. Physiol Res 59:225–232
Rushmore TH, Pickett CB (1993) Glutathione S-transferases, structure, regulation, and therapeutic implications. J Biol Chem 268:11475–11478
Sabina EP, Mathew J, RajappaRamya S et al (2009) Hepatoprotective and antioxidant potential of Spirulina fusiformis on acetaminophen-induced hepatotoxicity in mice. Int J Integr Med 6:1–5
Sathish P, Paramasivan V, Palani V, Sivanesan K (2011) N-Acetylcysteine attenuates dimethylnitrosamine induced oxidative stress in rats. Eur J Pharmacol 654:181–186
Smilkstein MJ, Knapp GL, Kulig KW et al (1988) Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose: analysis of the national multicenter study (1976–85). N Engl J Med 319:1557–1562
Spielberg SP (1985) Acetaminophen toxicity in lymphocytes heterozygous for glutathione synthetase deficiency. Can J Physiol Pharmacol 63:468–471
Tirmenstein MA, Nelson SD (1990) Acetaminophen-induced oxidation of protein thiols. Contribution of impaired thiol-metabolizing enzymes and the breakdown of adenine nucleotides. J Biol Chem 265:3059–3065
van de Straat R, de Vries J, Debets AJ, Vermeulen NP (1987) The mechanism of prevention of paracetamol-induced hepatotoxicity by 3,5-dialkyl substitution: the roles of glutathione depletion and oxidative stress. Biochem Pharmacol 36:2066–2070
Victor VM, Rocha M, De la Fuente M (2003) Regulation of macrophage function by the antioxidant N-acetylcysteine in mouse-oxidative stress by endotoxin. Int Immunopharmacol 3:97–106
Wang H, Peng RX (1993) Effects of paracetamol on glutathione S-transferase activity in mice. Zhongguo Yao Li Xue Bao 14(Suppl):S41–S44
Waters E, Wang JH, Redmond HP, Wu QD, Kay E, Bouchier-Hayes D (2001) Role of taurine in preventing acetaminophen-induced hepatic injury in the rat. Am J Physiol Gastrointest Liver Physiol 280:G1274–G1279
Wendel A (1983) Hepatic lipid peroxidation: caused by acute drug intoxication, prevented by liposomal glutathione. Int J Clin Pharmacol Res 3:443–447
Wheeler CR, Salzman JA, Elsayed NM, Omaye ST, Korte DW Jr (1990) Automated assays for superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase activity. Anal Biochem 184:193–199
Wollenberger A, Ristau O, Schoffa G (1960) Eine einfache technik der extrem schnellen abkühlung größerer gewebestücke. Pflügers Arch Gesamte Physiol 270:399–412
Yoshiyuki K (1998) New raw materials and new technologies for cosmetics. (Part I). Development and its application of “sebum antioxidant thiotaurine” for cosmetics. Fragr J 26:9–14
Yoshiyuki K, Yoshiki M (2000) Peroxidation in the skin and its prevention. Jpn J Inflamm 20:119–129
Zhou W, Freed CR (2005) DJ-1 upregulates glutathione synthesis during oxidative stress and inhibits A53T α-synuclein toxicity. J Biol Chem 280:43150–43158
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this paper
Cite this paper
Acharya, M., Lau-Cam, C.A. (2013). Comparative Evaluation of the Effects of Taurine and Thiotaurine on Alterations of the Cellular Redox Status and Activities of Antioxidant and Glutathione-Related Enzymes by Acetaminophen in the Rat. In: El Idrissi, A., L'Amoreaux, W. (eds) Taurine 8. Advances in Experimental Medicine and Biology, vol 776. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6093-0_20
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6093-0_20
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6092-3
Online ISBN: 978-1-4614-6093-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)