
Abstract
Multiple in vitro and in vivo studies have shown that the signal transducer and activator of transcription 3 (STAT3) protein regulates key mechanisms in cardiac physiology (exercise, pregnancy) and pathophysiology (pressure overload, ischemia/reperfusion, myocardial infarction (MI), myocarditis, and cardiotoxic agents). STAT3 is activated in various cardiac cell types including cardiomyocytes, endothelial cells, fibroblasts, and cardiac progenitor cells by a multitude of factors including cytokines, growth factors, neurohormones, mechanical load, and ischemia. It acts as a signaling molecule, a transcription factor, and a mitochondrial protein involved in energy production, and it controls autocrine and paracrine pathways. While the majority of data imply rather beneficial roles of STAT3 in the heart, newer studies implicate that this is mainly the case when the expression and activation of STAT3 is precisely regulated. In contrast, continuous uncontrolled activation of STAT3 in cardiomyocytes seems to promote adverse cardiac remodeling processes especially after MI. Here, we provide an overview on STAT3 signaling and summarize the current understanding of the role of STAT3 for cardiac inflammation, metabolism, remodeling, and regeneration based on experimental and clinical studies. Finally, we highlight the consequences of targeting STAT3 for future therapeutic approaches in the setting of cardiac remodeling.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- STAT3
- Cardiac remodeling
- gp130–STAT3 signaling
- Inflammation
- Metabolism
- Angiogenesis
- Hypertrophy
- Sarcomere structure
1 Introduction
The adult heart experiences numerous insults of physiological (aging, pregnancy, exercise) and pathophysiological [ischemia/reperfusion, myocardial infarction (MI), pressure overload, infections, and cardiotoxic agents] nature, which can induce alterations in the cardiac homeostasis and architecture. These alterations may be adaptive and protective to a certain stress situation or can be maladaptive, thereby inducing, driving, and contributing to heart failure [1].
Recent findings in experimental in vitro and in vivo models implicate important intermediate signal-transduction pathways in the coordination of remodeling processes following cardiac stress stimuli [2]. It is a key issue for the clinical arena to identify both detrimental as well as protective pathways and to transfer this knowledge into clinical practice. In this regard, the STAT3 signaling system has been shown to critically impact on changes in cardiac inflammation, vasculature, extracellular matrix (ECM) composition, energetics and metabolism, and cardiomyocyte survival and architecture [3–13].
As a transcription factor, STAT3 participates in the regulation of numerous genes involved in cell survival, hypertrophic growth, proliferation, angiogenesis and neovascularization, development and regeneration, and anti-oxidative pathways [3, 4, 6–11, 13, 14]. Furthermore, STAT3 has been reported to modulate ECM response to mechanical overload and to influence inflammatory processes [4, 6–8, 10]. While many of these effects of STAT3 are achieved by its own direct transcriptional activity, recent findings identified the novel function of STAT3 to directly interact with mitochondrial function [12], which adds to the complexity of STAT3-mediated regulatory processes.
Altogether, recent studies support the conclusion that STAT3 functions as part of an integrated signaling network in the heart. Here, we provide an overview of established STAT3-related biological mechanisms in cardiac physiology and pathophysiology, we illustrate its particular role in directing cardiac cellular processes that modulate ventricular remodeling, and finally we challenge the question whether STAT3 signaling conducts beneficial or detrimental effects with respect to cardiac function and heart failure.
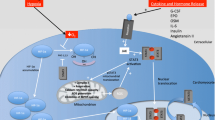
2 Canonical gp130-STAT3 Signaling, a Central Regulatory Circuit in Cardiac Physiology and Pathophysiology
Interleukin (IL)-6-type cytokines such as IL-6, IL-11, leukemia inhibitory factor (LIF), oncostatin M (OSM), ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-1), and cardiotrophin-like cytokine (CLC) are a family of helix bundle cytokines that play a key role in cellular and tissue homeostasis [15, 16]. They are involved in inflammatory and immunologic processes as well as in hematopoiesis, liver and neuronal regeneration, embryonic development, and cardiovascular physiology [16–18].
All IL-6-type cytokines interact with the glycoprotein-130 (gp130) receptor, and upon ligand binding, the homodimerization of gp130 (upon binding of IL-6 and IL-11) or heterodimerization of gp130 with LIF receptor (following binding of LIF, CNTF, CLC, OSM, and CT-1) or OSM receptor (for OSM), respectively, occurs [19–21] leading to the activation of three major downstream pathways: the Janus kinase (JAK)-STAT, the SH2 domain-containing cytoplasmic protein tyrosine phosphatase (SHP2)–extracellular signal-regulated kinase (ERK), and the phosphatidylinositol-3-kinase (PI3K)–protein kinase B (PKB, AKT) pathways [4, 18, 22–26]. The induction of the gp130-JAK-STAT pathway through IL-6-type cytokines leads mainly to activation of STAT3 and to a lesser extent to STAT1 activation [17]. Following dimerization of the gp130 receptor complex, JAK1, JAK2, JAK3, and tyrosine kinase 2 (Tyk2) constitutively connected to the intracytoplasmic membrane-proximal regions of the receptor subunits are catalytically activated and themselves transphosphorylate tyrosine residues in the gp130 receptor intracellular domain [27, 28]. This allows the recruitment of STAT proteins to the gp130 receptor complex via recognition of its phosphotyrosines by the STAT Src homology 2 domains (SH2) resulting in binding of monomeric STAT to the Y-X-X-Q motives and in phosphorylation of STAT proteins at a single carboxy-terminal tyrosine residue at amino acid position 705 (Y705) [29]. The phosphorylated STAT proteins then detach from their receptor and undergo homo- or heterodimerization through reciprocal interactions between the SH2 domain of one monomer and the phosphorylated tyrosine residue of the other [30]. Dimeric STAT proteins translocate in an importin-α-dependent manner to the nucleus, bind to specific DNA elements in the promoters of immediate early target genes, and activate specific gene expression programs.
Ligand-dependent STAT activation is a tightly controlled transient process, lasting for minutes up to several hours, during which nuclear STAT proteins are eventually inactivated by tyrosine dephosphorylation leading to their nuclear export [31]. Additional regulatory mechanisms are known to negatively control STAT activity, that is, gp130 receptors are rapidly internalized upon binding of their ligands and are degraded by the ubiquitin–proteasome pathway [32]. Furthermore, SHP-1/2 can interact with the intracytoplasmic portion of cytokine receptors and dephosphorylate JAK proteins, thereby lowering their activity [33, 34]. In addition, the suppressor of cytokine signaling (SOCS) family represents a specific negative regulatory feedback element of STAT signaling [35], since, for example, SOCS3 is a direct transcriptional target of STAT3. SOCS1, SOCS3, and SHP-1/2 interact with the kinase domain of various JAK proteins or the cytoplasmic phosphotyrosine residue (phospho-Y759 in humans, phospho-Y757 in mice) of the gp130 receptor resulting in the inhibition of STAT protein phosphorylation, thereby leading to termination of JAK-STAT signaling [36–38]. Additional negative regulators consisting of the group of protein inhibitors of activated STAT (PIAS-1, PIAS-3) inhibit the binding of phosphorylated STAT1 and STAT3 dimers to DNA, thereby reducing the transcriptional activation of STAT target genes. Finally, the protein tyrosine phosphatase receptor T (PTPRT) specifically dephosphorylates the Y705 residue of STAT3 and thereby regulates target gene expression but also cellular localization of STAT3 [39]. Importantly, the activation state of STAT3 seems to play a major role whether IL-6-gp130 signaling acts in a more anti-inflammatory or a more proinflammatory manner.
Not only is gp130-STAT3 signaling tightly controlled, but also activation of the other gp130-triggered signaling pathways is regulated to avoid unlimited cytokine signaling from the gp130 receptor with possible detrimental effects. For example, SHP2 limits gp130-SHP2-ERK signaling [18, 40, 41]. In addition, SOCS3 competes with SHP2 for binding to phosphorylated Y757 of the gp130 receptor, thereby limiting gp130-SHP2-ERK signaling and STAT activation [18, 40, 41]. Thus, simultaneous activation of SHP2-ERK and STAT maintains a balanced gp130 signaling, which appears crucial for beneficial responses mediated by this receptor system [40, 42]. Indeed, mice with a systemic deletion of all STAT-binding sites at the C terminus of the gp130 receptor, gp130 ΔSTAT, are characterized by impaired induction of the STAT1 and STAT3 activation and sustained activity of the SHP2-ERK pathway, which is associated with impaired intestinal wound healing and immune response and reduced life spans [18, 40, 41]. In contrast, gp130 Y757F mice harboring a phenylalanine substitution at Y757 (Y757F) abolish binding of SHP2 and SOCS3 leading to a prolonged and continuous activation of STAT1 and STAT3 in response to IL-6 and IL-11 without activation of ERK or AKT signaling. As a consequence, gp130 Y757F mice spontaneously develop gastric adenomas, splenomegaly, and an exaggerated immune response [18, 40, 41]. Moreover, mice with a cardiomyocyte-specific Y757F mutation (αMHC-Cre tg/-; gp130 fl/Y757F) display a high inflammatory reaction and LV rupture rate with increased mortality after MI [10].
Albeit an unrestricted high activation of gp130-STAT3 signaling seems to be detrimental in many organ systems including the heart, activation of STAT3 is absolutely required for many protective mechanisms in the heart including compensatory hypertrophy, cardiomyocyte protection, and wound healing after MI [4, 24, 43, 44].
In conclusion, it seems that a well-controlled and balanced activation of IL-6-type cytokine gp130-STAT3 signaling is beneficial for most organs including the heart, but sustained and prolonged gp130-STAT3 appears to be detrimental and is responsible for enhanced inflammation and high mortality, for example, after MI [10].
3 STAT3 in Cardiac Inflammation
STAT3 was initially described as a DNA-binding protein that recognizes the IL-6-responsive element in the promoter of the α2-macroglobulin gene whose gene product belongs to the acute-phase response of the liver [45, 46]. Further evidences for STAT3 being a mediator of inflammation include the discovery of overlapping DNA-binding sites for STAT3 and the proinflammatory nuclear factor-κB (NF-κB) in the promoter of acute-phase proteins and the finding that it is activated by most proinflammatory agents, such as IL-6-type cytokines [47, 48]. Experiments with IL-6-knockout and cardiac-specific transgenic mice reveal its role in mediating cardiac inflammation after burn injury in combination with sepsis [49].
To ensure resolution of inflammation, the infiltration of inflammatory cells and the expression of proinflammatory cytokines in response to injury are balanced by the production of the anti-inflammatory cytokine IL-10 [50]. In an ischemia/reperfusion model, IL-10 is shown to be induced in the myocardium with concomitant downregulation of IL-6 mRNA expression [51, 52]. Furthermore, IL-10 suppresses the inflammatory response by inhibiting the expression of several cytokines such as IL-1, IL-6, IL-12, and TNF-α in activated macrophages and dendritic cells and mediates its effects indirectly through activation of STAT3 [52–54]. Hereafter, the induced STAT3 activates the expression of other genes, which in turn are required for a selective control of the transcription of inflammatory genes [53]. One of those intermediate genes might be B cell lymphoma (Bcl-3), whose gene product blocks TNF expression after IL-10-mediated STAT3 activation in human macrophages [54].
Interestingly, infiltrating CD5-positive T cells have been identified as a predominant source of reperfusion-dependent IL-10 production, which express the cytokine upon induction by IL-6 [52, 55]. Another proinflammatory cytokine, tumor necrosis-α (TNF-α), is as well able to induce IL-10 synthesis by monocytes [55].
3.1 STAT3 and Inflammation Following Myocardial Ischemia
STAT3 is rapidly and transiently activated in response to myocardial ischemia per se or by reperfusion following ischemia. Mice harboring a cardiomyocyte-restricted deletion of STAT3 (αMHC-Cre tg/-; STAT3 fl/fl: STAT3-KO) display increased cardiac injury and lower survival rates after MI mainly caused by adverse remodeling suggesting that STAT3 is needed for protective mechanisms after ischemia [7]. Likewise, mice lacking the gp130 receptor and therefore having reduced STAT3 activation in the acute phase after infarction show increased mortality rates [10]. However, not only lack of STAT3 activation but also sustained uncontrolled cardiac activation of STAT3 after MI as it is present in mice carrying the mutation of gp130 Y757F only in cardiomyocytes (αMHC-Cre tg/-; gp130 fl/Y757F mice) leads to lower survival rates post-MI. The high mortality under unrestricted gp130-STAT3 activation derives mainly from enhanced and prolonged ventricular inflammation and high rupture rates [10]. In search for adverse downstream effectors of STAT3 signaling in infarcted αMHC-Cre tg/-; gp130 fl/Y757F left ventricles (LVs), the mannose-binding lectin (MBL)/lectin complement activation has been identified as a mediator of inflammation by hyperactivated STAT3 whose relevance is supported by the finding that attenuation of complement activation by the C3 antagonist cobra venom factor (CVF) reduces cardiac inflammation and improves cardiac function and survival in infarcted αMHC-Cre tg/-; gp130 fl/Y757F mice [10]. In addition, a murine SOCS3-knockout model, in which STAT3 is continuously activated because the negative feedback loop of the gp130-STAT3 signaling in cardiomyocytes is abolished, develops lethal cardiac arrhythmias and heart failure in response to pressure overload, supporting the hypothesis that too much of STAT3 activation seems to be harmful for the heart [56]. However, it seems that STAT3 not simply promotes inflammation, it can also act as a mediator of anti-inflammatory effects since it has been shown that in ischemia/reperfusion injury, the cardioprotective effects and the attenuation of myocardial inflammation by recombinant IL-10 given were lost if STAT3 activation was inhibited [57]. Likewise, intravenous administration of exogenous IL-11 evokes activation of STAT3 in cardiomyocytes in vivo and attenuates cardiac fibrosis and suppresses the expression of proinflammatory cytokines like IL-6 and TNF-α in a STAT3-dependent manner supporting the anti-inflammatory properties of activated STAT3 [58]. Here it is important to note that a majority of cytokine expression in the ischemic heart seems to derive from non-myocyte cells [59, 60].
Taken together, STAT3 plays an important role for protective mechanisms in the ischemic heart and after ischemia/reperfusion; however, the genetic and pharmacological dissection of gp130-mediated signaling indicates that only a well-controlled and balanced STAT3 activation is protective in the ischemic heart. Moreover, it seems specifically important in which cell type STAT3 is activated under ischemic condition, a feature that may explain the somehow contrary roles of STAT3 with regard to cardiac inflammation under ischemic conditions.
3.2 STAT3 and Myocarditis
It seems that STAT3 acts as a protective factor in viral myocarditis since cardiac-specific overexpression of SOCS3 increases the susceptibility to viral infection of the myocardium [61]. Further evidences for STAT3 regulating cardiac inflammation in responses to pathogens derive from STAT3-KO mice, which display a significantly higher TNF-α secretion and cardiac apoptosis rate in response to lipopolysaccharide (LPS)-induced inflammation [11].
With regard to cardiac inflammation, the question whether STAT3 is a friend or a foe cannot be answered in general. STAT3-mediated responses depend on the abundance of the particular cytokines, which activate the gp130-STAT3 signaling cascade, and on the cell type where these cytokines and the receptor system are expressed and activated in order to be either pro- or anti-inflammatory. Furthermore, the activation status of STAT3 is of major importance for the behaviors as a friend or a foe, since too much STAT3 activation after MI or cardiac deletion of STAT3 during LPS-mediated inflammation could be detrimental. Thus, the key issue seems to be a balanced and well-controlled activation of STAT3 in all cells affected by inflammatory processes in the heart.
4 STAT3 in Angiogenesis and Extracellular Matrix Composition
One of the earliest evidence of a potential role of STAT3 in angiogenesis was derived from observations showing that granulocyte-macrophage colony-stimulating factor (GM-CSF) via activated STAT3 contributes to vessel formation [62].
However, genetic analysis in mice with an endothelial-specific deficiency of STAT3 (Tie2-Cre tg/-; STAT3 fl/fl), where STAT3 is depleted at E8 in angioblasts, showed that these mice survive into adulthood without major vascular defects implicating that endothelial STAT3 is optional for vessel formation during development [63].
While it seems that STAT3 is not directly required for vessel formation by endothelial cells, it has been shown that STAT3 acts as a transcription factor for angiogenic mediators such as vascular endothelial growth factor (VEGF) and as a suppressor of angiostatic genes [64]. The mechanisms of vascular formation in the postnatal heart involve both auto- and paracrine circuits where specifically secreted paracrine factors from cardiomyocytes seem to play a pivotal role for cardiac angiogenesis [65]. Indeed, it has been shown that STAT3 participates in the upregulation of VEGF and VE-cadherin expression in cardiomyocytes [64, 66], a feature that is in line with the observation that STAT3 acts as a paracrine mediator in cardiomyocytes for vascular growth as, for example, demonstrated in mice with cardiomyocyte-specific overexpression of STAT3 (αMHC-STAT3tg) that displays increased myocardial capillary density [64, 67].
In contrast to αMHC-STAT3tg mice, the STAT3-KO (αMHC-Cre tg/-; STAT3 fl/fl) in cardiomyocytes leads to a rarification of cardiac capillaries, an enhanced fibrosis, and the development of an age-dependent heart failure suggesting a crucial role of STAT3 in controlling paracrine mechanisms involved in postnatal angiogenesis [7, 11]. Surprisingly, STAT3-KO mice show no differences in the expression of VEGF or VEGF receptors. However, they display an increase in the expression of antiangiogenic factors, which are able to inhibit endogenous VEGF activity or release such as connective tissue growth factor (CTGF) and thrombospondin-1 (TSP-1) [7, 68, 69]. Other potent antiangiogenic factors are as well upregulated like tissue inhibitor of metalloproteinase-1 (TIMP-1), tenascin C (TNC), and plasminogen activator-1 (PAI-1), which act by promoting apoptosis of endothelial cells or alter ECM composition [7, 8, 70, 71]. In this regard, supernatants of STAT3-KO cardiomyocytes are able to repress the proliferation of endothelial cells [7]. Additionally, TIMP-1 is well known to enhance fibrosis by inhibition of matrix-degrading enzymes such as matrix metalloproteinases (MMPs) [72]. This regulatory alteration may explain the age-dependent increase in interstitial fibrosis and enhanced deposition of collagens of STAT3-KO mice, indicating impaired ECM homeostasis [7, 11]. Thus, deterioration of angiogenesis in the aging myocardium of STAT3-KO mice is caused at least in part by altered structural and regulatory components of the matrix scaffold of the vascular bed and by enhanced collagen accumulation [7, 73]. Additionally, disturbance of ECM homeostasis may cause insufficient migration of angiogenic cells such as endothelial cell precursors or blood-derived primitive stem cells to the side of injured myocardium and thereby reduces the neoangiogenic capacity [74].
Moreover, STAT3 is involved in angiogenesis under pathophysiological conditions like MI. In this regard, activated STAT3 induces angiogenesis in the ischemic preconditioned infarcted myocardium [75]. In a STAT3-dependent manner, granulocyte colony-stimulating factor (G-CSF) is involved in cardiac remodeling after MI by increasing the number of endothelial cells in the border zone of the infarcted myocardium. This observation is diminished in dominant-negative STAT3 transgenic mice, suggesting that the enhanced vascularization is mediated STAT3-dependently in cardiomyocytes [76].
Furthermore, in αMHC-Cre tg/-; gp130 fl/Y757F with a continuous activation of STAT3 in cardiomyocytes, the prolonged activation of STAT3 contributes to a higher capillary density in the nonischemic myocardium in the subacute phase after MI [10]. But the increase in capillarization is not sufficient to improve the outcome after MI. In fact, the high vessel density in the border zone of infarcted αMHC-Cre tg/-; gp130 fl/Y757F mice may weaken the tissue or may promote further inflammation and thereby even contribute to adverse effects such as LV rupture and dilatation of the scar. During elimination of necrotic tissue and digestion of collagens in the border zone and later in the infarct scar, the adverse remodeling is mediated by MMPs released from inflammatory cells. In fact, in αMHC-Cre tg/-; gp130 fl/Y757F mice, MMP-1 and MMP-13 expression is upregulated, but is not counterbalanced by upregulated expression of their inhibitors such as TIMP-1 [10].
Additionally, STAT3 signaling influences the fate of cardiac stem or progenitor cells by the modulation of paracrine circuits in the heart or the direct induction of differentiation of stem cells. In this context, STAT3 activation (mediated by LIF stimulation) is able to directly induce endothelial differentiation of cardiac Sca-1+ stem cells contributing to neovascularization during cardiac remodeling [77, 78]. Moreover, cardiomyocyte-specific deficiency of STAT3 alters the cardiac microenvironment by the release of STAT3-dependent paracrine factors such as erythropoietin (EPO). The depletion of EPO within the cardiac microenvironment attenuates the endothelial differentiation of an endogenous cardiac progenitor cell population by arresting their chemokine ligand 2 (CCL2)/chemokine ligand receptor 2 (CCR2) system, which as a consequence impairs the vasculogenic regeneration of the heart [79]. Interestingly, the endothelial differentiation seems to be VEGF independent [79]. These aspects suggest a potential positive role of STAT3 signaling for the fate of cardiac stem or progenitor cells contributing to the homeostasis of the heart, especially after cardiac injury, and harbor an endogenous source for the generation of new capillaries and vessels. The formation of new vessels could contribute to the preservation of the cardiac function.
Finally, STAT3 seems to play a key role in the regulation of cardiac angiogenesis during pregnancy-induced adaptive processes such as physiological hypertrophy of the maternal heart. In fact, cardiac-restricted deletion of STAT3 leads to peripartum cardiomyopathy (PPCM) with a massive loss of cardiac microvessels as the major phenotype [9]. This pathologic decrease in blood vessels is mainly caused by unbalanced oxidative stress in cardiomyocytes that promotes the release of activated cathepsin D, which subsequently cleaves the nursing hormone prolactin in the angiostatic factor 16 kDa prolactin. It is important to note that STAT3 expression and activation is downregulated in cardiac tissue of patients with PPCM suggesting that STAT3 activation is also crucial for cardioprotection from pregnancy-related stress in humans.
Conclusively, these data suggest that STAT3 plays an important role in protecting the vasculature as well as inducing neoangiogenesis in the heart under physiological and pathophysiological conditions by controlling paracrine circuits.
The proangiogenic features of STAT3 seem to involve paracrine mechanisms in cardiomyocytes and non-myocytes including the expression and regulation of VEGF, which indicate that activation of STAT3 controls vessel growth in vivo. In addition, STAT3 seems important to suppress the expression of antiangiogenic factors such as CTGF, TSP-1, and TIMP-1 and the generation of angiostatic peptides such as 16 kDa prolactin. Especially in cardiac adaptation processes in response to physiological stress (pregnancy) or in response to injury, STAT3 is essential for the formation and preservation of blood vessels and ultimately for the maintenance of the cardiac function. However, too much proangiogenic activity induced by a continuous activation of STAT3, for example, in the infarcted heart may be detrimental since enhanced vessel formation in the border zone of the infarcted LV may weaken the tissue and promote inflammation, ventricular rupture, and scar dilatation. Therefore, a precise regulation of STAT3 expression and activity is also necessary for beneficial cardiac angiogenesis.
5 STAT3 and Heart Metabolism
It is inevitable for the cardiac muscle to consistently produce high amounts of ATP to meet the demand of energy required to maintain cardiac pump function. There is ample evidence that derangements in myocardial fuel selection and energetics occur in heart failure; however, causes and consequences of these defects remain poorly understood [80]. With its important role as a mediator of cardiac vessel growth, STAT3 is important to provide the pipes to transport energy and oxygen to the cardiac muscle. Apart from its role in angiogenesis, distinct forms of STAT3 actions have been implied in the direct modulation of substrate metabolism and energy generation. However, so far data are derived from liver and from tumor biology, while effects on the role of STAT3 on cardiac energy generation are less elaborated.
STAT3 has been shown to act as a master metabolic regulator in STAT3-dependent cancer cell lines where it enhances aerobic glycolysis and downregulates mitochondrial activity [81]. For instance, in human breast cancer cells, IL-6 enhances glycolysis by STAT3-mediated upregulation of the glycolytic enzymes hexokinase 2 (HK2) and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), a result that could be confirmed in mouse embryonic fibroblast and links inflammation to alterations in cancer metabolism [82, 83]. In liver, a link between STAT3 signaling and modulation of glycolysis has been suggested since hepatocyte-specific STAT3 deletion results in hepatic insulin resistance [84]. In the heart of a transgenic mouse model with a cardiac-specific overexpression of peroxisome proliferator-activated receptor (PPAR)-α, insulin-induced STAT3 activation measured as phosphorylation at Y705 is diminished and therefore associated with an elevated oxidation of fatty acids and downregulation of genes involved in glycolysis [85].
With regard to substrate use, there is a major difference between cancer cells which strongly rely on glucose oxidation as their preferred way of energy generation and striated muscle cells which mostly favor fatty acids as the substrate for ATP production. In this context, STAT3 was attributed to mediate leptin-induced lipid oxidation in skeletal muscle, while the effect of STAT3 on cardiac fatty acid oxidation has yet to be investigated [86].
However, it is known that with the development of heart failure, the cardiac muscle switches from fatty acid utilization to glucose oxidation as the preferred substrate for energy generation [87]. Whether this switch is protective as it adapts the heart to different energy demands or contributes to the development of heart failure is a matter of debate and not really understood. A contribution of STAT3 in the regulation of genes involved in cardiac substrate utilization has not been evaluated yet. However, a direct function of STAT3 in mitochondrial ATP production in the heart and in cardiomyocytes has recently been demonstrated [12]. In fact, evidence has been presented that STAT3 is present in mitochondria of B cells and cardiomyocytes, where it binds to complex I and II of the electron transport chain (ETC) [12 88]. This direct protein–protein interaction of STAT3 with ETC components requires phosphorylation at S727 but seems to be independent from Y705 phosphorylation or DNA binding. Deletion of STAT3 in this model leads to a drastic reduction in complex I and II activities, a lower membrane potential and ATP production, and an increase in the production of reactive oxygen species (ROS) [12]. The mitochondrial localization of STAT3 could recently be confirmed, and additionally a Tom20-mediated import of the protein was described [88]. Genetic deletion as well as pharmacologic inhibition of STAT3 phosphorylation at both sites, Y705 and S727, leads to a decreased ADP-stimulated respiration in the presence of complex I substrates [88]. No effect of STAT3 on complex II, complex IV, or uncoupled respiration was detected in cardiomyocyte mitochondria [88].
Also, an interaction between the mitochondrial protein cyclophilin D that is part of the mitochondrial permeability transition pore complex and STAT3 is reported, and STAT3 deletion or inhibition is found to increase the susceptibility of pore opening [88]. Interestingly, mice overexpressing a transgenic STAT3 protein with a mitochondrial target sequence but a mutated DNA-binding site in cardiomyocytes also display reduced complex I and II activities. However, this reduction is, unlike in the STAT3-deficient situation, not associated with augmented ROS, or decreased ATP production and cardiac function and left ventricular dimensions are comparable to wild-type mice at 1 year of age [89]. Mitochondria from these transgenic mice are protected against ischemia-induced ROS production via complex I and show diminished cytochrome c release in response to ischemic insult [89]. It therefore seems likely that mitochondrial-targeted STAT3 on top of the endogenous protein protects against ischemic damage by partially inhibiting electron flow through complex I [89].
In the aforedescribed studies, the effect of mitochondrial-located STAT3 on cellular respiration at the level of the ETC is attributed to direct interactions of STAT3 and ETC proteins. However, to generate sufficient amounts of ATP required for maximal myocardial performance, nearly equimolar amounts of STAT3 and complex I and II proteins would be necessary. Using different proteomic approaches, it was shown that the ratio of STAT3 to complex I/II is ∼10−5 [90]. From these experiments it can be concluded that direct regulation of cellular respiration by protein–protein interaction of STAT3 and respiratory chain proteins is not feasible and that the observed modulations in ATP production by STAT3 might rather be due to regulation of the expression of mitochondrial genes [90].
Taken together, STAT3 plays an important role in the heart and in cardiac cells for upstream processes like transport of nutrition and oxygen, while little is known about the role of STAT3 in cardiac fuel selection and substrate oxidation. However, strong evidence exists that STAT3 is essential to mediate ATP production in mitochondria via the ETC. However, the exact mechanisms by which STAT3 influences ATP generation especially at the level of complex I and II are controversially discussed. Additional studies are therefore necessary to decipher the contribution of disrupted metabolism in adverse structural remodeling of the myocardium in general and to determine the role of STAT3 in this context.
6 STAT3 in Hypertrophy and Sarcomere Composition
The role of the JAK-STAT pathway for the development of cardiac hypertrophy and a putative therapeutic utility still remains a matter of debate. While early studies demonstrated that under massive STAT3 overexpression (αMHC-STAT3 tg), mice develop spontaneous concentric cardiac hypertrophy [67], the role of physiological expression and activation of STAT3 for cardiac hypertrophy was until recently unclear [10]. Some studies have favored a decisive contribution of STAT3 to the development of cardiac hypertrophy, particularly in response to certain pathophysiological stimuli, such as MI and ischemia/reperfusion [7], pressure overload, and hormones [91]. In this regard, upregulation of STAT3 signaling in response to MI may contribute to an adaptive component of remodeling processes in an effort to maintain function and forward flow in the acute phase. However, prolonged and unbalanced activation of JAK-STAT3 signaling appears detrimental after MI due to enhanced inflammation and left ventricular instability with increased rupture rate as mentioned above in the αMHC-Cre tg/-; gp130 fl/Y757F mice [10].
Further evidence for the involvement of STAT3 signaling in cardiomyocyte hypertrophy is provided by in vitro experiments using the activator of gp130 receptor signaling, LIF, which induces cell growth in cardiomyocytes, a feature that is prevented by concomitant overexpression of SOCS3 [38]. However, LIF mainly induces a distinct type of cell growth that is marked by thinning of the width and elongation of the length, a phenotype that is considered as an eccentric cell growth, rather than a concentric hypertrophy. A link between STAT3 and the ubiquitin–proteasome system (UPS) was identified in a microRNA-dependent manner. In this circuit, STAT3 negatively regulates the expression of miR-199a on the transcriptional level, which in turn fine-tunes the expression of at least two ubiquitin-conjugating enzymes, the Ube2i and Ube2g, on the posttranscriptional level [5]. Under circumstances of low STAT3 expression, for example, as it occurs in the end-stage failing heart [92], the expression of miR-199a is upregulated with a subsequent impairment of the UPS. Consequently, this leads to disturbed homeostasis of certain sarcomeric proteins, such as α- and β-MHC proteins, a severe derangement of the sarcomere ultrastructure, and an eccentric type of cardiomyocyte growth—a phenotype that corresponds to the architecture of the cardiomyocyte syncytium in dilated cardiomyopathy. Moreover, the derailed UPS activity and protein turnover also result in alteration of the cardiomyocyte secretome with accumulation of the endogenous endothelial nitric oxide synthase (eNOS) inhibitor asymmetric dimethylarginine (ADMA), which in a paracrine fashion impairs endothelial function of adjacent vessels [5].
Conclusively, STAT3 regulates sarcomere composition, cardiomyocyte growth, and hypertrophy and thus determines the transition from adaptive to maladaptive remodeling depending on the extent, time point, and duration of signaling activity. While under certain conditions a transiently enhanced STAT3 activity may promote adaptive concentric hypertrophy, prolonged STAT3 activation or deficiency result in ventricular instability or eccentric ventricular dilatation, respectively. Notably the STAT3-related processes with regard to regulation of sarcomeric gene composition via microRNAs and the UPS are not restricted exclusively to the cardiomyocyte itself but may also impact via release of metabolites such as ADMA in a paracrine manner on the non-myocyte compartment especially the myocardial vasculature.
7 Conclusion
In conclusion, STAT3 is involved in multiple biological mechanisms in the developing, the adult, and the injured heart. A timely regulated STAT3 activation appears cardioprotective by positively modulating anti-oxidative defense, angiogenesis, eventually metabolism and energy production, hypertrophy, and survival, as reported in previous studies [7, 8, 11, 12, 67, 93]. However, upregulation and sustained activation of STAT3 promotes ventricular inflammation and rupture after ischemic insults resulting in high mortality. These antithetic effects clearly demonstrate that a precise regulation of STAT3 expression and activity is required for beneficial effects of STAT3 in the heart.
This double-faced nature of STAT3 may offer an explanation for discrepancies existing between experimental findings that demonstrated mainly beneficial effects of gp130-STAT3 signaling for the cardiovascular system and clinical studies which found that high serum levels of gp130 ligands, that is, IL-6, predict a poor outcome in patients after MI or with heart failure [94–96]. Based on the data summarized in this report, it could be speculated that high IL-6 serum levels in patients indicate a high and continuous activation state of STAT3 which has been shown to be detrimental especially after MI. Therefore, it should be explored whether there is a link between high IL-6 serum levels, activation state of cardiac STAT3, and poor prognosis in patients after MI. If this is the case, targeting STAT3 by small molecule inhibitors as currently tested in cancer therapy [97] might be a novel therapy option in these patients. In turn, in other scenarios such as heart failure due to myocarditis, peripartum cardiomyopathy, or heart failure due to cardiotoxic treatment strategies, therapies that enhance STAT3 activation may be beneficial and promote healing and regeneration. Taken together, in most cardiac physiological and pathophysiological situations, balanced STAT3 expression and activation is essential for cardioprotection and behaves as a friend, but an unbalanced STAT3 activation can turn a friend into a foe. Therefore, therapeutic success of targeting STAT3 in positive or negative ways will require the possibility to precisely control the activation status of STAT3.
Abbreviations
- ADMA:
-
Asymmetric dimethylarginine
- Bcl-3:
-
B cell lymphoma 3
- CCL2:
-
Chemokine ligand 2
- CCR2:
-
Chemokine ligand receptor 2
- CLC:
-
Cardiotrophin-like cytokine
- CNTF:
-
Ciliary neurotrophic factor
- CT-1:
-
Cardiotrophin 1
- CTGF:
-
Connective tissue growth factor
- CVF:
-
Cobra venom factor
- ECM:
-
Extracellular matrix
- eNOS:
-
Endothelial nitric oxide synthase
- EPO:
-
Erythropoietin
- ERK:
-
Extracellular signal-regulated kinase
- ETC:
-
Electron transport chain
- G-CSF:
-
Granulocyte colony-stimulating factor
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- gp130:
-
Glycoprotein-130
- HK2:
-
Hexokinase 2
- IL:
-
Interleukin
- JAK:
-
Janus kinase
- LIF:
-
Leukemia inhibitory factor
- LPS:
-
Lipopolysaccharide
- LV:
-
Left ventricle
- MBL:
-
Mannose-binding lectin
- MHC:
-
Myosin heavy chain
- MI:
-
Myocardial infarction
- MMP:
-
Matrix metalloproteinase
- OSM:
-
Oncostatin M
- miRNA:
-
microRNA
- NF-κB:
-
Nuclear factor-κB
- PAI-1:
-
Plasminogen activator-1
- PFKFB3:
-
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
- PKB:
-
Protein kinase B, also known as AKT
- PPAR:
-
Peroxisome proliferator-activated receptor
- PPCM:
-
Peripartum cardiomyopathy
- PIAS:
-
Protein inhibitors of activated STAT
- PTPRT:
-
Protein tyrosine phosphatase receptor T
- ROS:
-
Reactive oxygen species
- SH2:
-
Src homology 2 domain
- SHP2:
-
SH2 domain-containing cytoplasmatic protein
- SOCS:
-
Suppressor of cytokine signaling
- STAT3:
-
Signal transducer and activator of transcription 3
- STAT3-KO:
-
STAT3-knockout
- TAC:
-
Transverse aortic constriction
- TIMP-1:
-
Tissue inhibitor of metalloproteinase-1
- TNC:
-
Tenascin C
- TNF-α:
-
Tumor necrosis factor-α
- TSP-1:
-
Thrombospondin-1
- Ube:
-
Ubiquitin-conjugating enzyme
- UPS:
-
Ubiquitin–proteasome system
- VEGF:
-
Vascular endothelial growth factor
References
Forrester JS, Wyatt HL, Da Luz PL et al (1976) Functional significance of regional ischemic contraction abnormalities. Circulation 54:64–70
Dorn GW (2009) Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat Rev Cardiol 6:283–291
Boengler K, Hilfiker-Kleiner D, Drexler H et al (2008) The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther 120:172–185
Fischer P, Hilfiker-Kleiner D (2007) Survival pathways in hypertrophy and heart failure: the gp130-STAT axis. Basic Res Cardiol 102:393–411
Haghikia A, Missol-Kolka E, Tsikas D et al (2011) Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: a key role for ubiquitin-conjugating enzymes. Eur Heart J 32:1287–1297
Haghikia A, Stapel B, Hoch M, Hilfiker-Kleiner D (2011) STAT3 and cardiac remodeling. Heart Fail Rev 16:35–47
Hilfiker-Kleiner D, Hilfiker A, Fuchs M et al (2004) Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res 95:187–195
Hilfiker-Kleiner D, Hilfiker A, Drexler H (2005) Many good reasons to have STAT3 in the heart. Pharmacol Ther 107:131–137
Hilfiker-Kleiner D, Kaminski K, Podewski E et al (2007) A Cathepsin D-Cleaved 16 kDa Form of Prolactin Mediates Postpartum Cardiomyopathy. Cell 128:589–600
Hilfiker-Kleiner D, Shukla P, Klein G et al (2010) Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation 122:145–155
Jacoby JJ, Kalinowski A, Liu MG et al (2003) Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci USA 100:12929–12934
Wegrzyn J, Potla R, Chwae YJ et al (2009) Function of mitochondrial Stat3 in cellular respiration. Science 323:793–797
Luedde M, Spaich S, Hippe HJ et al (2011) Affixin (beta-parvin) promotes cardioprotective signaling via STAT3 activation. J Mol Cell Cardiol 50:919–923
Kurdi M, Booz GW (2009) JAK redux: a second look at the regulation and role of JAKs in the heart. Am J Physiol Heart Circ Physiol 297:H1545–1556
Betz UA, Bloch W, van den Broek M et al (1998) Postnatally induced inactivation of gp130 in mice results in neurological, cardiac, hematopoietic, immunological, hepatic, and pulmonary defects. J Exp Med 188:1955–1965
Keller ET, Wanagat J, Ershler WB (1996) Molecular and cellular biology of interleukin-6 and its receptor. Front Biosci 1:d340–357
Heinrich PC, Behrmann I, Muller-Newen G et al (1998) Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334:297–314
Heinrich PC, Behrmann I, Haan S et al (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 374:1–20
Olivier C, Auguste P, Chabbert M et al (2000) Identification of a gp130 cytokine receptor critical site involved in oncostatin M response. J Biol Chem 275:5648–5656
Pennica D, Wood WI, Chien KR (1996) Cardiotrophin-1: a multifunctional cytokine that signals via LIF receptor-gp 130 dependent pathways. Cytokine Growth Factor Rev 7:81–91
Plun-Favreau H, Perret D, Diveu C et al (2003) Leukemia inhibitory factor (LIF), cardiotrophin-1, and oncostatin M share structural binding determinants in the immunoglobulin-like domain of LIF receptor. J Biol Chem 278:27169–27179
Funamoto M, Hishinuma S, Fujio Y et al (2000) Isolation and characterization of the murine cardiotrophin-1 gene: expression and norepinephrine-induced transcriptional activation. J Mol Cell Cardiol 32:1275–1284
Hirano T, Nakajima K, Hibi M (1997) Signaling mechanisms through gp130: a model of the cytokine system. Cytokine Growth Factor Rev 8:241–252
Hirota H, Chen J, Betz UA et al (1999) Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 97:189–198
Kunisada K, Hirota H, Fujio Y et al (1996) Activation of JAK-STAT and MAP kinases by leukemia inhibitory factor through gp130 in cardiac myocytes. Circulation 94:2626–2632
Oh H, Fujio Y, Kunisada K et al (1998) Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70 S6 kinase phosphorylation in cardiac myocytes. J Biol Chem 273:9703–9710
Kishimoto T (1994) Signal transduction through homo- or heterodimers of gp130. Stem Cells 12(Suppl 1):37–44, discussion 44–35
Murakami M, Hibi M, Nakagawa N et al (1993) IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science 260:1808–1810
Abe K, Hirai M, Mizuno K et al (2001) The YXXQ motif in gp 130 is crucial for STAT3 phosphorylation at Ser727 through an H7-sensitive kinase pathway. Oncogene 20:3464–3474
Chen X, Vinkemeier U, Zhao Y et al (1998) Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell 93:827–839
Haspel RL, Salditt-Georgieff M, Darnell JE Jr (1996) The rapid inactivation of nuclear tyrosine phosphorylated Stat1 depends upon a protein tyrosine phosphatase. EMBO J 15:6262–6268
Heinrich PC, Bode J, Decker M et al (2001) Termination and modulation of IL-6-type cytokine signaling. Adv Exp Med Biol 495:153–160
David M, Chen HE, Goelz S et al (1995) Differential regulation of the alpha/beta interferon-stimulated Jak/Stat pathway by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol Cell Biol 15:7050–7058
Fischer P, Lehmann U, Sobota RM et al (2004) The role of the inhibitors of interleukin-6 signal transduction SHP2 and SOCS3 for desensitization of interleukin-6 signalling. Biochem J 378:449–460
Starr R, Hilton DJ (1999) Negative regulation of the JAK/STAT pathway. Bioessays 21:47–52
Lehmann U, Schmitz J, Weissenbach M et al (2003) SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem 278:661–671
Yasukawa H, Misawa H, Sakamoto H et al (1999) The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J 18:1309–1320
Yasukawa H, Hoshijima M, Gu Y et al (2001) Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J Clin Invest 108:1459–1467
Zhang X, Guo A, Yu J et al (2007) Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci USA 104:4060–4064
Ernst M, Jenkins BJ (2004) Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet 20:23–32
Fischer P, Hilfiker-Kleiner D (2008) Role of gp130-mediated signalling pathways in the heart and its impact on potential therapeutic aspects. Br J Pharmacol 153(Suppl 1):414–427
Tebbutt NC, Giraud AS, Inglese M et al (2002) Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med 8:1089–1097
Yamauchi-Takihara K, Hirota H, Kunisada K et al (1996) Roles of gp130 signaling pathways in cardiac myocytes: recent advances and implications for cardiovascular disease. J Card Fail 2(4 Suppl):S63–68
Yamauchi-Takihara K (2002) Gp130-mediated pathway and left ventricular remodeling. J Card Fail 8(6 Suppl):S374–378
Akira S, Nishio Y, Inoue M et al (1994) Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77:63–71
Zhong Z, Wen Z, Darnell JE Jr (1994) Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264:95–98
Aggarwal BB, Kunnumakkara AB, Harikumar KB et al (2009) Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci 1171:59–76
Zhang Z, Fuller GM (1997) The competitive binding of STAT3 and NF-kappa B on an overlapping DNA binding site. Biochem Biophys Res Commun 237:90–94
Zhang H, Wang HY, Bassel-Duby R et al (2007) Role of interleukin-6 in cardiac inflammation and dysfunction after burn complicated by sepsis. Am J Physiol Heart Circ Physiol 292:H2408–2416
Krishnamurthy P, Rajasingh J, Lambers E et al (2009) IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res 104:e9–18
Dewald O, Ren G, Duerr GD et al (2004) Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol 164:665–677
Frangogiannis NG, Mendoza LH, Lindsey ML et al (2000) IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol 165:2798–2808
Murray PJ (2006) STAT3-mediated anti-inflammatory signalling. Biochem Soc Trans 34:1028–1031
Williams LM, Sarma U, Willets K et al (2007) Expression of constitutively active STAT3 can replicate the cytokine-suppressive activity of interleukin-10 in human primary macrophages. J Biol Chem 282:6965–6975
Daftarian PM, Kumar A, Kryworuchko M, Diaz-Mitoma F (1996) IL-10 production is enhanced in human T cells by IL-12 and IL-6 and in monocytes by tumor necrosis factor-alpha. J Immunol 157:12–20
Yajima T, Murofushi Y, Zhou H et al (2011) Absence of SOCS3 in the cardiomyocyte increases mortality in a gp130-dependent manner accompanied by contractile dysfunction and ventricular arrhythmias. Circulation 124:2690–2701
Manukyan MC, Alvernaz CH, Poynter JA et al (2011) Interleukin-10 protects the ischemic heart from reperfusion injury via the STAT3 pathway. Surgery 150:231–239
Obana M, Maeda M, Takeda K et al (2010) Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation 121:684–691
Deten A, Volz HC, Briest W, Zimmer HG (2003) Differential cytokine expression in myocytes and non-myocytes after myocardial infarction in rats. Mol Cell Biochem 242:47–55
Yue P, Massie BM, Simpson PC, Long CS (1998) Cytokine expression increases in nonmyocytes from rats with postinfarction heart failure. Am J Physiol 275:H250–258
Yajima T, Yasukawa H, Jeon ES et al (2006) Innate defense mechanism against virus infection within the cardiac myocyte requiring gp130-STAT3 signaling. Circulation 114:2364–2373
Valdembri D, Serini G, Vacca A et al (2002) In vivo activation of JAK2/STAT-3 pathway during angiogenesis induced by GM-CSF. FASEB J 16:225–227
Kano A, Wolfgang MJ, Gao Q et al (2003) Endothelial cells require STAT3 for protection against endotoxin-induced inflammation. J Exp Med 198:1517–1525
Osugi T, Oshima Y, Fujio Y et al (2002) Cardiac-specific Activation of Signal Transducer and Activator of Transcription 3 Promotes Vascular Formation in the Heart. J Biol Chem 277:6676–6681
Dor Y, Djonov V, Abramovitch R et al (2002) Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J 21:1939–1947
Funamoto M, Fujio Y, Kunisada K et al (2000) Signal transducer and activator of transcription 3 is required for glycoprotein 130-mediated induction of vascular endothelial growth factor in cardiac myocytes. J Biol Chem 275:10561–10566
Kunisada K, Negoro S, Tone E et al (2000) Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci USA 97:315–319
Inoki I, Shiomi T, Hashimoto G et al (2002) Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J 16:219–221
Rodriguez-Manzaneque JC, Lane TF, Ortega MA et al (2001) Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci USA 98:12485–12490
Bloomston M, Shafii A, Zervos EE, Rosemurgy AS (2002) TIMP-1 overexpression in pancreatic cancer attenuates tumor growth, decreases implantation and metastasis, and inhibits angiogenesis. J Surg Res 102:39–44
Volpert OV, Zaichuk T, Zhou W et al (2002) Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat Med 8:349–357
Ahmed MS, Oie E, Vinge LE et al (2004) Connective tissue growth factor–a novel mediator of angiotensin II-stimulated cardiac fibroblast activation in heart failure in rats. J Mol Cell Cardiol 36:393–404
Edelberg JM, Reed MJ (2003) Aging and angiogenesis. Front Biosci 8:s1199–1209
Ingber DE (2002) Mechanical signaling and the cellular response to extracellular matrix in angiogenesis and cardiovascular physiology. Circ Res 91:877–887
Fukuda S, Kaga S, Sasaki H et al (2004) Angiogenic signal triggered by ischemic stress induces myocardial repair in rat during chronic infarction. J Mol Cell Cardiol 36:547–559
Harada M, Qin Y, Takano H et al (2005) G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-Stat pathway in cardiomyocytes. Nat Med 11:305–311
Mohri T, Fujio Y, Maeda M et al (2006) Leukemia inhibitory factor induces endothelial differentiation in cardiac stem cells. J Biol Chem 281:6442–6447
Mohri T, Fujio Y, Obana M et al (2009) Signals through glycoprotein 130 regulate the endothelial differentiation of cardiac stem cells. Arterioscler Thromb Vasc Biol 29:754–760
Hoch M, Fischer P, Stapel B et al (2011) Erythropoietin preserves the endothelial differentiation capacity of cardiac progenitor cells and reduces heart failure during anticancer therapies. Cell Stem Cell 9:131–143
van Bilsen M, van Nieuwenhoven FA, van der Vusse GJ (2009) Metabolic remodelling of the failing heart: beneficial or detrimental? Cardiovasc Res 81:420–428
Demaria M, Giorgi C, Lebiedzinska M et al (2010) A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY) 2:823–842
Ando M, Uehara I, Kogure K et al (2010) Interleukin 6 enhances glycolysis through expression of the glycolytic enzymes hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3. J Nihon Med Sch 77:97–105
Jiang S, Zhang LF, Zhang HW et al (2012) A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J 31:1985–1998
Inoue H, Ogawa W, Ozaki M et al (2004) Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat Med 10:168–174
Park SY, Cho YR, Finck BN et al (2005) Cardiac-specific overexpression of peroxisome proliferator-activated receptor-alpha causes insulin resistance in heart and liver. Diabetes 54:2514–2524
Akasaka Y, Tsunoda M, Ogata T et al (2010) Direct evidence for leptin-induced lipid oxidation independent of long-form leptin receptor. Biochim Biophys Acta 1801:1115–1122
Neubauer S (2007) The failing heart–an engine out of fuel. N Engl J Med 356:1140–1151
Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R (2010) Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 105:771–785
Szczepanek K, Chen Q, Derecka M et al (2011) Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem 286:29610–29620
Phillips D, Reilley MJ, Aponte AM et al (2010) Stoichiometry of STAT3 and mitochondrial proteins: Implications for the regulation of oxidative phosphorylation by protein-protein interactions. J Biol Chem 285:23532–23536
Beckles DL, Mascareno E, Siddiqui MA (2006) Inhibition of Jak2 phosphorylation attenuates pressure overload cardiac hypertrophy. Vascul Pharmacol 45:350–357
Podewski EK, Hilfiker-Kleiner D, Hilfiker A et al (2003) Alterations in Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation 107:798–802
Negoro S, Kunisada K, Fujio Y et al (2001) Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation 104:979–981
Rattazzi M, Puato M, Faggin E et al (2003) C-reactive protein and interleukin-6 in vascular disease: culprits or passive bystanders? J Hypertens 21:1787–1803
Tsutamoto T, Hisanaga T, Wada A et al (1998) Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol 31:391–398
Ohtsuka T, Hamada M, Inoue K et al (2004) Relation of circulating interleukin-6 to left ventricular remodeling in patients with reperfused anterior myocardial infarction. Clin Cardiol 27:417–420
Heimberger AB, Priebe W (2008) Small molecular inhibitors of p-STAT3: novel agents for treatment of primary and metastatic CNS cancers. Recent Patents CNS Drug Discov 3:179–188
Acknowledgment
We thank the DFG for supporting this work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Ricke-Hoch, M., Stapel, B., Gorst, I., Haghikia, A., Hilfiker-Kleiner, D. (2013). The STAT3 Pathway and Downstream Mechanisms in Cardiac Remodeling: Friend or Foe. In: Jugdutt, B., Dhalla, N. (eds) Cardiac Remodeling. Advances in Biochemistry in Health and Disease, vol 5. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5930-9_20
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5930-9_20
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5929-3
Online ISBN: 978-1-4614-5930-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)