
Abstract
The pathogenic role of the Epstein-Barr virus (EBV) has been suggested because it is consistently found in some lymphoproliferative disorders. More than 90% of the population is exposed to the virus, usually during childhood, and after initial exposure, the virus survives for the lifetime of individuals as an episome, and expresses a limited set of proteins depending on the immune status of the host. The variable expression of viral proteins is known as latency pattern with minimal expression of viral products in immunocompetent hosts, but expression of numerous viral products in the immunocompromised host. Thus, the more virus products that are expressed, the more targets for an immune reaction. Since it is acknowledged that the neoplastic process including lymphoproliferative disorders result from a multistep chain of events, multiple pathways are affected, and in particular in EBV-positive lymphoproliferations, EBV products interact with abnormally activated pathways in the context of the immune response of the host. In this review we analyze the structure of the virus and its interaction in the immunocompetent as well as in the immunocompromised host, and discuss the possible roles of EBV products in various lymphoproliferative disorders. Finally, we discuss the potential role of expressed EBV viral proteins and activation of pathogenic pathways in the setting of specific diagnostic categories with the identification of potential molecular targets.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Hematopoietic Stem Cell Transplant
- Lymphoproliferative Disorder
- Burkitt Lymphoma
- Primary Effusion Lymphoma
- Posttransplant Lymphoproliferative Disorder
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Epstein-Barr Virus Structure, Expressed Genes, and Pathogenesis
Epstein-Barr virus (EBV) or human herpes virus 4 is a ubiquitous virus that infects more than 90% of the human population and usually establishes a persistent latent infection in the host. Primary infection occurs via salivary contact in infancy and is usually asymptomatic, however when primary infection occurs in adolescents or young adults it may manifest as infectious mononucleosis. After primary infection, the virus usually remains in an asymptomatic latent state within resting memory B-cells for the lifetime of the host. Cytotoxic T-cells, both CD8+ and CD4+, and natural killer (NK) cells are in charge to recognize and eliminate the virus-infected cells [1]. When infected B-cells evade or suppress the T-cell immune response, or the host’s cellular immune system fails to control EBV-induced B-cell proliferations, the infected B-cells can transform into neoplastic cells and eventually manifest as a lymphoproliferative disorder. EBV-associated lymphoproliferative disorders encompass a heterogeneous group of disorders, including chronic active EBV infection, B-cell lymphoma, T/NK-cell lymphoma or leukemia, classical Hodgkin lymphoma, as well as immunodeficiency and posttransplant lymphoproliferative disorders.
EBV infection occurs through the binding of the major viral envelope glycoprotein gp350 to the CD21 receptor on the surface of B cells and through the binding of glycoprotein gp42 to human leukocyte antigen (HLA) as a coreceptor [2]. The viral genome is linear and becomes circular by fusion of either end of the genome, and the virus persists within cells as an episome. EBV virus expresses genes in two distinct programs known as the lytic cycle and the latent cycle [3, 4]. During the lytic cycle, there is production of infectious virus and expression of viral genes that encode proteins involved in DNA replication and in assembly of viral particles. In contrast, during the latent cycle only a limited number of viral genes are expressed, and these include six nuclear proteins, referred to as EBV nuclear antigens (EBNA 1, 2, 3A, 3B, 3C, and EBNA-LP), and three latent membrane proteins (LMP 1, 2A and 2B), which are associated with transforming activity [5]. There are two nontranslated RNA molecules known as the EBV-encoded RNAs designated as EBER1 and EBER2. In addition, there are EBV microRNAs (miRNAs), which are encoded in two regions: BHRF1 (Bam HI fragment H rightward open reading frame 1) and BART (Bam HI-A region rightward transcript). miRNAs are the only known functional products of the BARTs transcripts [6]. BHRF1 miRNAs have been found highly expressed in immortalized lymphoblastoid cell lines (LCL), whereas BARTs miRNAs have been found in all EBV-infected cell lines as well as in biopsies of lymphoma cases [6].
There are four types of latent infection, designated as latency 0, I, II, and III, that reflect the extent of viral protein expression allowed by the host in vivo. Latency 0 refers to infection of cells where most viral genes are not expressed and therefore cells are not recognized or targeted by EBV-specific T lymphocytes; this stage is also called “in vivo latency” [1, 7]. EBV resides in memory B cells, which constitute a long-term reservoir for EBV. Thus, in the absence of production of viral proteins or antigens, there is no EBV-specific T cells neither is there an immune response against EBV. Activation of EBV-infected memory B cells can lead to their differentiation into plasma cells, a process that might switch-on the lytic cycle of EBV and produce viral particles [8]. Three latency patterns are associated with different types of lymphoid proliferations (Table 1). In latency type I there is selective expression of EBNA-1 and LMP-2A, and this pattern is found typically in Burkitt lymphoma (BL). In latency type II, there is expression of EBNA-1, LMP-1, LMP-2A, and LMP-2B, and this pattern is found in classical Hodgkin lymphoma (HL), primary effusion lymphoma, angioimmmunoblastic T-cell lymphoma (AITL), peripheral T-cell lymphoma (PTCL) and NK/T cell lymphoma or leukemia. In latency type III there is expression of all nine latent cycle EBV antigens, and this pattern is typically found in posttransplant lymphoproliferative disorders (PTLD) and in AIDS-related lymphoma. Because there are more viral proteins expressed in latency type II and III infections, respective EBV-associated lymphoproliferations are more immunogenic than diseases associated with a latency type I.
EBV-Related Lymphoproliferative Disorders
Burkitt Lymphoma
Burkitt lymphoma (BL) is a highly aggressive B-cell lymphoma that was initially described in children around equatorial Africa [9]. There are three clinical variants of BL: endemic, sporadic, and human immunodeficiency virus (HIV)-associated. The EBV genome is present in almost all cases of endemic BL and there is strong epidemiological link with endemic malaria. In sporadic BL, EBV is identified in 15–30% of cases, while it is more common in HIV-associated BL.
The neoplastic cells of BL express B-cell lineage and B-cell germinal center cell markers CD10 and Bcl6. At the molecular level, the neoplastic cells undergo somatic hypermutation (SHM) and class-switch recombination (CSR) supporting the notion that they derive from germinal center cells. It is suspected that SHM and CSR predispose GC cells to chromosome translocations or mutations in non-Ig genes [10]. Virtually all BL cases carry one of three characteristic chromosomal translocations between the MYC gene in chromosome 8 and one of the immunoglobulin genes on chromosomes 2, 14, or 22 [1]. The role of MYC deregulation as the key factor in the pathogenesis of BL is compelling [11–13]. Most cases of BL show DNA breakpoints in rearranged VJ regions or in S regions of the immunoglobulin heavy chain (IgH) loci, thus it is generally accepted that the chromosomal translocations are mediated by aberrant SHM or CSR, which require the intervention of DNA-modifying enzymes known as activation-induced deaminase (AID). Most MYC/Ig breakpoints in EBV-positive endemic BL appear to originate from aberrant SHM. On the other hand, the translocations in sporadic cases mostly involve the Ig switch regions of the IgH locus [14]. EBV may be accounted for the difference in MYC breakpoints between EBV-positive BL and EBV-negative BL [15].
Genes downstream of MYC regulate cell cycle progression, cell growth, apoptosis, and senescence. Deregulated expression of MYC induces p53 response and triggers apoptosis [16, 17].
EBV has a latency type I pattern, with expression of EBNA1 and LMP2A. Since EBNA1 is the only viral gene product in all latency patterns, it is suspected that EBNA1 promotes lymphomagenesis in EBV+ processes. Although Kang et al. [18] suggested that EBNA1 is limited to maintenance of the viral genome, other researchers showed that EBNA1 may activate the catalytic subunit p91 of the NADPH oxidase2 (NOX2) at the transcriptional level. Thus EBNA1 generates reactive oxygen species that may contribute to DNA damage and genomic instability [19]. Furthermore, other researchers reported that EBNA1 binds to the deubiquitinating enzyme HAUSP/USP7 and together sequester p53, contributing to p53 degradation [20].
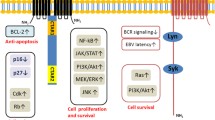
LMP2A expression was recently confirmed in endemic BL using a sensitive RT-PCR assay [13, 21]. LMP2A contains a 119 amino terminal cytoplasmic domain that includes eight tyrosine residues, two of which form an immunoreceptor tyrosine-based activation motif (ITAM) [22]. Experimental data using EBV LCLs in vitro and LMP2A-transgenic mice indicate that the cytoplasmic tail of LMP2A mimic signals used by the B-cell receptor (BCR) and promotes B-cell development. In effect, LMP2A constitutively phosphorylates Lyn and Syk, with Lyn binding to tyrosine 112 and Syk binding to the ITAM motif of LMP2A. Additional studies using LMP2A-transgenic mice demonstrate that LMP2A constitutively phosphorylates and activates many of the proteins induced by BCR, such as Lyn, Syk, BLNK, BTK, Ras, P13K, NF-κB, and MAP kinases [23–26]. LMP2A also increases the levels of anti-apoptotic Bcl family members and protect B cells from apoptosis [13].
Chemotherapy is the standard of care for immunocompetent patients with EBV-associated BL; however it is expected that targeting EBV therapy may contribute to better outcomes, or the use of EBV vaccines may decrease the incidence of endemic BL.
Diffuse Large B-Cell Lymphoma of the Elderly
EBV-positive diffuse large B-cell lymphoma (EBV+ DLBCL) of the elderly, also known as age-related EBV+ lymphoproliferative disorder, accounts for 8–10% of DLBCL cases [27]. Neoplastic cells are B-lymphocytes that lack germinal center cell markers CD10 and Bcl6, consistent with post GC B-cells. The neoplastic cells most often display an EBV type III latency, positive for EBV protein products including LMP1 and EBNA-2, although some cases express latency II, and lack EBNA expression [28, 29] The variable presence of EBNA2 expression is attributed to the variable degrees of immune surveillance in aging individuals.
EBV infection and waning immunity that is part of the aging process, where a decrease in T-cell response occurs, naturally appear to be the main driving mechanisms [30]. Decrease in T-cell function leads to EBV reactivation that manifests with the expression of proteins such as LMP1 that leads to upregulation of anti-apoptotic proteins Bcl-2, MCL-1, and A20 [31, 32].
The prognosis of EBV-positive DLBCL of the elderly is worse than EBV-negative DLBCL, partially compounded by a high median age of patients between 70 and 75 years, who are often unable to tolerate aggressive therapeutic regimens, thus an optimal regimen has not been established for EBV-positive DLBCL of the elderly. The development of adoptive immunotherapy with cytotoxic T cells (CTLs) directed against EBV latency antigens has the potential of improving the outcome of this group of patients.
Classical Hodgkin Lymphoma
Classical Hodgkin lymphoma (CHL) is clinically distinct from non-Hodgkin lymphoma and histologically is characterized by an exuberant inflammatory background and only rare or few neoplastic cells. The neoplastic cells are large mononuclear or multinucleated known as Hodgkin Reed-Sternberg (HRS) cells. CHL comprises four histological subtypes [33] and the prevalence of EBV varies with the histological subtype. The prevalence is highest (∼75%) in mixed cellularity HL, and lowest (10∼40%) in nodular sclerosis HL [34]. It is also notorious that the prevalence of EBV in CHL varies with epidemiologic factors; EBV infection is more prevalent in developing countries and affects mostly childhood, and older adult age groups (age >50 years).
The neoplastic cells of CHL are monoclonal B cells at the germinal center stage of differentiation [35–37]. Analysis of the Ig variable (IgV) gene regions show evidence of somatic hypermutation, revealing a germinal center (GC) or post-GC origin [38].
EBV is believed to play a causal role in the pathogenesis of CHL. EBV is detected in HRS cells and the virus is clonal, indicating that infection occurred prior to neoplastic transformation [39, 40]. In EBV-positive CHL, the HRS cells demonstrate latency type II pattern, and express EBNA1, LMP1 and LMP2.
LMP1 is considered the major transforming protein of EBV. It has an integral membrane protein comprising 386 amino acids and consists of a short amino (N)-terminal cytoplasmic stretch, 6 trans-membrane (6TM) domains, and a long carboxyl (C)-terminal cytoplasmic region with no significant extracellular domain [41]. The 6TM domains regulate their own synthesis and degradation via the unfolded protein response (UPR) and autophagy. The carboxy terminal domain induces proliferation and survival of EBV-infected B cells in vitro and in vivo [42, 43]. LMP1 mimics CD40 and can substitute for the signaling of CD40 in B cells [41, 42, 44].
LMP1 activates the signaling pathways of nuclear factor-κB (NF-κB), activated protein 1 (AP1), and signal transducer and activator of transcription (STAT). Aberrant activation of NF-κB plays a determinant role in cell transformation, while tumor promotion is mediated by its anti-apoptotic functions. Evidence shows that NF-κB activation by LMP1 is critical for B cell transformation in vitro and in vivo [45, 46]. LMP1 can induce most of the phenotypic changes of neoplastic cells, including expression of surface antigens CD21, CD23, CD30, CD40, CD44, and Fas as well as cell adhesion molecules ICAM1, LFA1, and LFA3. LMP1 also upregulates expression of the anti-apoptotic proteins Bcl-2, A20, Bfl, and Mcl1 and stimulates production of cytokines interleukin (IL)-6 and IL-8 [47, 48]. HRS cells express constitutively CD30 that is a trans-membrane protein which belongs to the tumor necrosis factor (TNF) receptor family [49]. When stimulated by CD30 ligand, CD30 interacts with TNF receptor-associated factors TRAF2 and TRAF5, mediating signal transduction that leads to the activation of the NF-κB pathway. Another potential role of LMP1 is the downregulation of CD99. Loss of CD99 has been associated with generation of B cells with CHL immunophenotype.
LMP2A mimics the BCR and competes with BCR to bind tyrosine kinases, thereby modulating the activity of these tyrosine kinases. LMP2B is not essential for EBV-induced B-cell transformation in vitro [50].
Mechanisms and cell interactions of HRS in CHL are complex. In EBV+ CHL, most of the reactive T lymphocytes have a regulatory T-cell phenotype, and LMP1 may mediate their attraction through IL-10 secretion [51]. The presence of regulatory cells around HRS cells causes a profoundly immunosuppressive microenvironment, contributing to evade or suppress immune T-cell responses. Thus, the use of EBV-specific T-cells to deliver immunostimulatory cytokines may counteract immunoregulatory T-cells and contribute to eliminate HRS cells. Similarly, HRS cells positive for LMP1 and LMP2A can be targeted with adoptive transfer of EBV-CTL (cytotoxic T cells) [52]. Clinical data showed that LMP2-specific CTLs augmented T-cell responses, migrated to tumor deposits, and caused regression of tumors in a subset of patients with CHL [53]. LMP1 is another potential target for CHL immunotherapy. LMP1 effect or function has been targeted directly, using single-chain antibodies or antisense RNA approaches, and indirectly, by the genetic or pharmacological interception of its downstream effects on NF-κB [54].
NK/T-Cell Lymphoproliferative Disorders
EBV-related NK/T-cell lymphoproliferative disorders (LPD) include aggressive NK-cell leukemia, EBV-positive T-cell lymphoproliferative disorders of childhood, angioimmunoblastic T-cell lymphoma (AITL), extranodal NK/T-cell lymphoma, nasal type, and peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS).
EBV can infect CD4+ and CD8+ peripheral blood T-cells as well as NK-cells in a minority of patients with infectious mononucleosis. The mechanisms of access of EBV into T cells and NK cells in vivo are speculative. It has been shown that NK cells activated by EBV acquire CD21 by synaptic transfer from CD21+ B cells, and these ectopic receptors allow EBV binding to NK-cells. It is possible that NK cells in close contact with EBV-infected B cells may acquire EBV infection directly and then expand clonally [55].
Most EBV-associated T-cell lymphomas are thought to arise from chronic active EBV infection (CAEBV). CAEBV is considered as a progressive EBV infection from infected B-cells. Evidence suggests that CAEBV of NK/T-cells develops into NK/T cell lymphoma [56].
In EBV-positive T-cell LPD of childhood, there is a monoclonal proliferation of CD4+ or CD8+ cells with viral gene expression of EBNA1, LMP1 and LMP2, consistent with a latency type II pattern. In extranodal NK/T-cell lymphoma, nasal type, disease usually presents in the nasal or upper aerodigestive tract and most patients are of Asian origin.
The pathogenesis of EBV in NK/T-cell lymphoma may be similar to that of CHL, since both have a latency type II pattern of EBV infection. In this model, LMP1 mimics and activates NF-κB pathway, as previously discussed. Yang et al. [57] reported that increased IL-9 levels induced by the EBERs possess anti-apoptotic effects and promote T-cell proliferation and transformation. Further research is needed to determine how EBV infects NK/T cells and the pathogenic role of EBV.
Since LMP1 expression in EBV-associated NK/T cell lymphomas activates the NF-κB pathway, targeted therapies have been applied to inhibit NF-κB activation. Several targets that inhibit the NF-κB pathway have been identified [58]. For example, bortezomib, a proteasome inhibitor, leads to increased levels of I-κB kinase and inhibits activation of NF-κB. Dehydroxymethylepoxyquinomicin, another inhibitor of NF-κB, induces apoptosis of EBV-transformed B cells [48].
Immunodeficiency-Related Lymphoproliferative Disorders
Congenital Immunodeficiency-Related Lymphoproliferation
Congenital immunodeficiency also called primary immunodeficiency is present at the time of birth, and may occur as a result of defects in B- or T-lymphocytes, or both. X-linked lymphoproliferative disease (XLP) is an inherited syndrome characterized by extreme sensitivity to EBV infection that leads to severe infectious mononucleosis, acquired hypogammaglobulinemia, and/or malignant lymphoma [59]. The defective gene in XLP has been identified as src homology 2 domain protein 1A (SH2D1A) also known as signaling lymphocytic activation molecule (SLAM)-associated protein (SAP) gene [60]. SAP is a key regulator of normal immune function in T cells, NK cells, and in certain B-cell lines. Evidence both from knockout mice and from XLP patients show that SAP deficiency has multiple immunologic effects. These include significantly impaired Th2-like CD4+ T-cell responses, reflected as poor IL-10 production in in vitro assays of T-dependent B cell responses that associate with in vivo defective Ig class switching, affinity maturation of antibody responses, and memory B cell development [61]. Both functional cytotoxic T-cell defects and abnormal cytokine production as a result of SAP deficiency may explain the failure to control EBV infection and predisposition to B-cell lymphoma.
For some patients with primary immunodeficiency, there is promise with hematopoietic stem cell transplant (HSCT).
Lymphoproliferative Disorders Associated with Human Immunodeficiency Virus and Acquired Immunodeficiency Syndrome
There is an increased frequency of EBV infection in human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome (AIDS)-associated lymphomas. AIDS patients have 10–20 times more EBV-infected B-cells than healthy counterparts and the risk of NHL is 60–200 times higher than persons noninfected with HIV [1]. EBV is detected in up to 60% of all HIV-related lymphomas, including nearly 100% of primary CNS lymphomas, 80% of DLBCL with immunoblastic features, 30–50% of BL, 70% of primary effusion lymphomas, and nearly 100% of CHL [62, 63]. The increased risk for EBV-related lymphoma among HIV-infected individuals appears related to multiple factors, including duration and degree of immunosuppression, induction of cytokines leading to B-cell proliferation, and coinfection with HHV8. HIV infection can act as chronic stimuli for the B-cell system, characterized by marked hyperplasia of germinal centers, which greatly increases the chances of MYC translocations. HIV also induces abnormal immune response and probably increases the number of EBV-infected B-cells that are at risk of being recruited into the germinal center reaction [48]. There is constitutive expression of LMP1 that supports an EBV-driven proliferation.
The use of highly active antiretroviral therapy (HAART) has resulted in a fall in the incidence of opportunistic infections and AIDS-related malignancies, including lymphoma. However, EBV-associated lymphoma still is one of the most frequent causes of death in HIV-infected patients. Therefore, there is a need in identifying viral targets and evaluate the potential benefit of molecular-targeted chemotherapy.
Iatrogenic Immunodeficiency-Associated Lymphoproliferative Disorders
The WHO classification includes the category of “Other iatrogenic immunodeficiency-associated lymphoproliferative disorders” that are lymphoid proliferations or lymphomas that arise in patients treated with immunosuppressive drugs for autoimmune diseases or conditions other than in the transplant setting. The most common subtype of lymphoma is DLBCL. The better-known agent is methotrexate (MTX), which is commonly used in patients with rheumatoid arthritis. MTX directly reactivates EBV with subsequent release of infectious virions [64]. Since patients with rheumatoid arthritis have impaired T-cell responses to EBV products [65], therapy with MTX results in higher EBV loads in their blood and immunodeficient conditions that predispose to EBV-driven lymphoma.
Posttransplant Lymphoproliferative Disorders
Posttransplant lymphoproliferative disorders (PTLD) constitute a heterogeneous group of lymphoproliferations that occur in the setting of allogeneic transplantation of solid organs (SOT) or hematopoietic stem cells (HSCT). In patients with PTLD, the incidence of EBV ranges from 73 to 100% [5]. Morphologically, PTLD can be subdivided into monomorphic, polymorphic, plasmacytic, or HL-like variants. Most PTLD are of B-cell origin, and 10–15% are of T-cell origin [66, 67].
Patients with PTLD have impaired anti-EBV cellular immunity because of iatrogenic immunosuppression, resulting in EBV-induced transformation of B-cells. In addition, the immunomodulation used to prevent graft-versus-host disease (GVHD) remove T-cells nonspecifically from the graft and increases the risk of PLTD [68].
The pathogenic mechanisms of EBV in PTLD are presumably similar to those in CHL. Because approximately 50% cases of PTLD are derived from aberrant GC B cells that lack a functional BCR, HRS cells escape apoptosis through alternative survival signals. It is considered that LMP1 and LMP2A replace survival signals induced by activated CD40 and BCR receptors and activate NF-κB signaling pathway, inducing proliferation of neoplastic cells. As already mentioned, the decreased cytotoxic T-cell surveillance also increases the susceptibility to EBV.
In the absence of effective T-cell surveillance, EBV+ lymphomas in immunodeficient individuals usually express a latency type III pattern. All the EBNA and LMP viral proteins are expressed together with various noncoding small RNAs (EBERs and miRNAs). EBNA3 family of proteins (EBNA3A, 3B, and 3C) are nuclear phosphoproteins that act as transcriptional regulators. Only EBNA3A and EBNA3B have been shown to be essential for B-cell transformation. A number of functional domains have been characterized for these proteins, including transactivation, repression, and nuclear localization domains, but their roles have not been elucidated. It has been noted that conserved regions of EBNA3A, 3B, 3C are capable of binding to RBP, while EBNA3A and 3C can bind to the ATPase/Helicase DP103 [69]. Krauer et al. [70] showed that EBNA-3 disrupt the DNA damage and replication at the G2/M checkpoint. EBNA3A and EBNA3C together have been found to interfere with the proapoptotic protein Bim [71]. EBNA3C appears to also have repressor functions potentially mediated via its interaction with a histone deacetylase [72]. In addition, studies have shown that EBNA3C is an immortalizing oncogene capable of cooperating with (Ha)-ras in cotransformation assays and is capable of overriding Rb-mediated pathways [73].
In latency III, EBNA2 acts as a transcription factor to induce expression of the viral LMP genes and many cell genes. EBNA2 interacts with a sequence-specific DNA-binding protein, Jκ-recombination-binding protein (RBP-Jκ) [3], to transcriptionally activate cellular genes such as CD21 and CD23 and key viral genes LMP1 and LMP2A [74, 75]. In addition, EBNA2 can modify chromatin structure through recruitment of SWI/SNF. EBNA-LP interacts with EBNA2 and is required for the efficient outgrowth of virus-transformed B cells in vitro. The transcriptional activation mediated by EBNA2/EBNA-LP is modulated by the EBNA3 family of proteins, repressing transactivation.
Therapeutic Targets of EBV
The association of EBV with various lymphoproliferative disorders suggests that EBV plays a pathogenic role. Various pathways linked with tumorigenesis are activated in these processes, and plausible mechanisms involving viral products have been identified. Thus, the identification of the molecular mechanisms associated with EBV tumor promotion and progression may contribute to identify molecular targets for immune attack, small molecules or interfering RNA. Other therapeutic options include adoptive immunotherapy, antiviral therapy, and therapies against EBV-driven signaling or transfer of antigens.
Adoptive transfer of EBV-specific cytotoxic T-cells (CTLs) has been proven effective in treating posttransplant EBV-associated lymphomas. The research in this field has moved quickly from animal experiments to the bedside [75]. EBV-specific CTL have been successfully infused in patients subjected to hematopoietic stem cell transplant (HSCT) and solid organ transplant (SOT), both with therapeutic and prophylactic purposes. In a study that included 108 HSCT recipients and 21 SOT recipients, patients received virus-specific CTL as a preemptive approach. No events of lymphoproliferation were reported at follow-up, except for one patient who received an infusion lacking a well-defined EBV-specific component [76]. The success of CTL therapy in PTLD has not been reproduced in CHL and BL probably due to cell mechanisms that evade EBV-specific immune responses. For example, HRS cells create a microenvironment that suppresses EBV-specific T-cell responses.
EBV-related lymphoproliferations have different latency patterns that reflect the interaction between the immune response of the host and the virus. Thus, the virus expresses as many proteins as the immune system of the host allows. As a rule, the most immunosuppressed the host is, the more viral particles are produced, and thus EBV+ neoplasms express more viral antigens. Of the viral antigens, EBNA1, LMP1, LMP2 are the targets more frequently challenged using CTL therapy. In particular, cytotoxic LMP1 or EBNA1-specific CD4+ T cells, have proven effective not only in vitro against LCLs and infected NK/T cells, but also against naturally expressed targets in NK/T cell lymphoma and CAEBV [77, 78]. These therapies need further refinement in generating such in vitro and in demonstrating their efficacy in vivo.
Antiviral therapy is another therapeutic approach. A phase I/II trial of arginine butyrate and ganciclovir in 15 patients with refractory EBV+ lymphoid malignancies was well tolerated, achieving good antitumor response in ten patients [79]. Cidofovir, another antiviral drug, downregulated LMP1 expression and decrease BCL-2 levels in lymphoma cells.
Anti-CD30 monoclonal antibody, which inhibits growth of CD30-expressing tumor cells has been used in patients with refractory CHL, independent of EBV status, achieving success [80]. Another study showed that the combination of MTX and irradiation significantly induced apoptosis and growth inhibition in two EBV-expressing NK/T-cell lines, via downregulation of NF-κB signaling. The NF-κB inhibition highlighted an efficacious therapeutic approach for patients with nasal T/NK-cell lymphoma and other EBV-related lymphomas.
Further experience and better delivery of molecular therapies may provide safe and efficacious therapeutic benefits for EBV-related lymphoproliferative disorders, which coupled with other therapies that target simultaneously other mechanisms of oncogenesis may contribute to better management of these disorders.
References
Roschewski M, Wilson WH (2012) EBV-associated lymphomas in adults. Best Pract Res Clin Haematol 25:75–89
Nemerow GR, Mold C, Schwend VK, Tollefson V, Cooper NR (1987) Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/c3d receptor of B cells: sequence homology of gp350 and c3 complement fragment c3d. J Virol 61:1416–1420
Kieff E, Rickinson A (2001) Epstein-Barr virus. In: Knipe D, Howley P (eds) Fields virology, 4th edn. Lippincott, Philadelpia, PA, pp 2511–2573
Kieff E, Richardson AB (2007) Epstein-Barr virus and its replication. In: Knipe DM, Howley PM (eds) Fields virology, 5th edn. Lippincott, Williams and Wilkins, Philadelphia, PA, pp 2603–2654
Castillo JJ, Beltran BE, Miranda RN, Paydas S, Winer ES, Butera JN (2011) Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist 16:87–96
Qiu J, Cosmopoulos K, Pegtel M et al (2011) A novel persistence associated EBV miRNA expression profile is disrupted in neoplasia. PLoS Pathog 7:e1002193
Bornkamm GW (2009) Epstein-Barr virus and its role in the pathogenesis of Burkitt’s lymphoma: an unresolved issue. Semin Cancer Biol 19:351–365
Cader FZ, Kearns P, Young L, Murray P, Vockerodt M (2010) The contribution of the Epstein-Barr virus to the pathogenesis of childhood lymphomas. Cancer Treat Rev 36:348–353
Epstein MA, Achong BG, Barr YM (1964) Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1:702–703
Kuppers R (2005) Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer 5:251–262
Scheller H, Tobollik S, Kutzera A et al (2010) c-Myc overexpression promotes a germinal center-like program in Burkitt’s lymphoma. Oncogene 29:888–897
Allday MJ (2009) How does Epstein-Barr virus (EBV) complement the activation of Myc in the pathogenesis of Burkitt’s lymphoma? Semin Cancer Biol 19:366–376
Bieging KT, Swanson-Mungerson M, Amick AC, Longnecker R (2010) Epstein-Barr virus in Burkitt’s lymphoma: a role for latent membrane protein 2A. Cell Cycle 9:901–908
Guikema JE, de Boer C, Haralambieva E et al (2006) IGH switch breakpoints in Burkitt lymphoma: exclusive involvement of noncanonical class switch recombination. Genes Chromosomes Cancer 45:808–819
Bellan C, Lazzi S, Hummel M et al (2005) Immunoglobulin gene analysis reveals 2 distinct cells of origin for EBV-positive and EBV-negative Burkitt lymphomas. Blood 106:1031–1036
Preudhomme C, Dervite I, Wattel E et al (1995) Clinical significance of p53 mutations in newly diagnosed Burkitt’s lymphoma and acute lymphoblastic leukemia: a report of 48 cases. J Clin Oncol 13:812–820
Farrell PJ, Allan GJ, Shanahan F, Vousden KH, Crook T (1991) p53 is frequently mutated in Burkitt’s lymphoma cell lines. EMBO J 10:2879–2887
Kang MS, Hung SC, Kieff E (2001) Epstein-Barr virus nuclear antigen 1 activates transcription from episomal but not integrated DNA and does not alter lymphocyte growth. Proc Natl Acad Sci U S A 98:15233–15238
Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG (2009) The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci U S A 106:2313–2318
Saridakis V, Sheng Y, Sarkari F et al (2005) Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol Cell 18:25–36
Bell AI, Groves K, Kelly GL et al (2006) Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt’s lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J Gen Virol 87:2885–2890
Fruehling S, Swart R, Dolwick KM, Kremmer E, Longnecker R (1998) Tyrosine 112 of latent membrane protein 2A is essential for protein tyrosine kinase loading and regulation of Epstein-Barr virus latency. J Virol 72:7796–7806
Swanson-Mungerson MA, Caldwell RG, Bultema R, Longnecker R (2005) Epstein-Barr virus LMP2A alters in vivo and in vitro models of B-cell anergy, but not deletion, in response to autoantigen. J Virol 79:7355–7362
Portis T, Longnecker R (2004) Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene 23:8619–8628
Merchant M, Longnecker R (2001) LMP2A survival and developmental signals are transmitted through Btk-dependent and Btk-independent pathways. Virology 291:46–54
Engels N, Merchant M, Pappu R, Chan AC, Longnecker R, Wienands J (2001) Epstein-Barr virus latent membrane protein 2A (LMP2A) employs the SLP-65 signaling module. J Exp Med 194:255–264
Wong HH, Wang J (2009) Epstein-Barr virus positive diffuse large B-cell lymphoma of the elderly. Leuk Lymphoma 50:335–340
Asano N, Yamamoto K, Tamaru J et al (2009) Age-related Epstein-Barr virus (EBV)-associated B-cell lymphoproliferative disorders: comparison with EBV-positive classic Hodgkin lymphoma in elderly patients. Blood 113:2629–2636
Oyama T, Ichimura K, Suzuki R et al (2003) Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol 27:16–26
Lages CS, Suffia I, Velilla PA et al (2008) Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J Immunol 181:1835–1848
Park S, Lee J, Ko YH et al (2007) The impact of Epstein-Barr virus status on clinical outcome in diffuse large B-cell lymphoma. Blood 110:972–978
Kapatai G, Murray P (2007) Contribution of the Epstein Barr virus to the molecular pathogenesis of Hodgkin lymphoma. J Clin Pathol 60:1342–1349
Stein H, Delsol G, Pileri SA, Weiss LM, Poppema S, Jaffe ES (2008) Classic Hodgkin lymphoma, introduction. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds) WHO classification of tumors of haematopoietic and lymphoid tissues, 4th edn. IARC, Lyon, p 326
Hummel M, Anagnostopoulos I, Dallenbach F, Korbjuhn P, Dimmler C, Stein H (1992) EBV infection patterns in Hodgkin’s disease and normal lymphoid tissue: expression and cellular localization of EBV gene products. Br J Haematol 82:689–694
Marafioti T, Hummel M, Foss HD et al (2000) Hodgkin and reed-sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 95:1443–1450
Kuppers R, Hajadi M, Plank L et al (1996) Molecular Ig gene analysis reveals that monocytoid B cells lymphoma is a malignancy of mature B cells carrying somatically mutated V region genes and suggests that rearrangement of the kappa-deleting element(resulting in deletion of the Ig kappa enhancers) abolishes somatic hypermutation in the human. Eur J Immunol 26:1794–1800
Kanzler H, Kuppers R, Hansmann ML et al (1996) Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the out growth of a dominant tumor clone derived from(crippled) germinal center B cells. J Exp Med 184:1495–1505
Kuppers R (2002) Molecular biology of Hodgkin’s lymphoma. Adv Cancer Res 84:277–312
Farrell K, Jarrett RF (2011) The molecular pathogenesis of Hodgkin lymphoma. Histopathology 58:15–25
Gulley ML, Eagan PA, Quintanilla-Martinez L et al (1994) Epstein-Barr virus DNA is abundant and monoclonal in the Reed-Sternberg cells of Hodgkin’s disease: association with mixed cellularity subtype and Hispanic American ethnicity. Blood 83:1595–1602
Lam N, Sugden B (2003) CD40 and its viral mimic, LMP1: similar means to different ends. Cell Signal 15:9–16
Graham JP, Arcipowski KM, Bishop GA (2010) Differential B-lymphocyte regulation by CD40 and its viral mimic, latent membrane protein 1. Immunol Rev 237:226–248
Rastelli J, Homig-Holzel C, Seagal J et al (2008) LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood 111:1448–1455
Pratt ZL, Zhang J, Sugden B (2012) The latent membrane protein 1 (LMP1) oncogene of Epstein-Barr virus can simultaneously induce and inhibit apoptosis in B cells. J Virol 86:4380–4393
He Z, Xin B, Yang X, Chan C, Cao L (2000) Nuclear factor-kappaB activation is involved in LMP1-mediated transformation and tumorigenesis of rat-1 fibroblasts. Cancer Res 60:1845–1848
Cahir-McFarland ED, Carter K, Rosenwald A et al (2004) Role of NF-kappa B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J Virol 78:4108–4119
Lee IS, Kim SH, Song HG, Park SH (2003) The molecular basis for the generation of Hodgkin and Reed-Sternberg cells in Hodgkin’s lymphoma. Into J Hematology 77:330–335
Cohen JI, Bollard CM, Khanna R, Pittaluga S (2008) Current understanding of the role of Epstein-Barr virus in lymphomagenesis and therapeutic approaches to EBV-associated lymphomas. Leuk Lymphoma 49(Suppl 1):27–34
Aizawa S, Nakano H, Ishida T et al (1997) Tumor necrosis factor receptor-associated factor (TRAF) 5 and TRAF2 are involved in CD30-mediated NFkappaB activation. J Biol Chem 272:2042–2045
Longnecker R (2000) Epstein-Barr virus latency: LMP2, a regulator or means for Epstein-Barr virus persistence? Adv Cancer Res 79:175–200
Marshall NA, Culligan DJ, Tighe J, Johnston PW, Barker RN, Vickers MA (2007) The relationships between Epstein-Barr virus latent membrane protein 1 and regulatory T cells in Hodgkin’s lymphoma. Exp Hematology 35:596–604
Carbone A, Gloghini A, Dotti G (2008) EBV-associated lymphoproliferative disorders: classification and treatment. Oncologist 13:577–585
Bollard CM, Aguilar L, Straathof KC et al (2004) Cytotoxic T lymphocyte therapy for Epstein-Barr virus+ Hodgkin’s disease. J Exp Med 200:1623–1633
Young LS, Rickinson AB (2004) Epstein-Barr virus: 40 years on. Nat Rev Cancer 4:757–768
Kimura H, Ito Y, Kawabe S et al (2012) EBV-associated T/NK-cell lymphoproliferative diseases in nonimmunocompromised hosts: prospective analysis of 108 cases. Blood 119:673–686
Cohen JI, Kimura H, Nakamura S, Ko YH, Jaffe ES (2009) Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8–9 September 2008. Ann Oncol 20:1472–1482
Yang L, Aozasa K, Oshimi K, Takada K (2004) Epstein-Barr virus (EBV)-encoded RNA promotes growth of EBV-infected T cells through interleukin-9 induction. Cancer Res 64:5332–5337
Kim A, Lee JE, Jang WS et al (2012) A combination of methotrexate and irradiation promotes cell death in NK/T-cell lymphoma cells via down-regulation of NF-kappaB signaling. Leuk Res 36:350–357
Sharifi R, Sinclair JC, Gilmour KC et al (2004) SAP mediates specific cytotoxic T-cell functions in X-linked lymphoproliferative disease. Blood 103:3821–3827
Booth C, Gilmour KC, Veys P et al (2011) X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood 117:53–62
Hislop AD, Taylor GS, Sauce D, Rickinson AB (2007) Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol 25:587–617
Raphael M, Said J, Borisch B, Cesarman E, Harris NL (2008) Lymphomas associated with HIV infection. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds) WHO classification of tumors of haematopoietic and lymphoid tissues, 4th edn. IARC, Lyon, pp 340–342
Bibas M, Antinori A (2009) EBV and HIV-related lymphoma. Mediterr J Hematology Infect Dis 1:e2009032
Feng WH, Cohen JI, Fischer S et al (2004) Reactivation of latent Epstein-Barr virus by methotrexate: a potential contributor to methotrexate-associated lymphomas. J Natl Cancer Inst 96:1691–1702
Balandraud N, Roudier J, Roudier C (2005) What are the links between Epstein-Barr virus, lymphoma, and tumor necrosis factor antagonism in rheumatoid arthritis? Semin Arthritis Rheum 34:31–33
Draoua HY, Tsao L, Mancini DM, Addonizio LJ, Bhagat G, Alobeid B (2004) T-cell post-transplantation lymphoproliferative disorders after cardiac transplantation: a single institutional experience. Br J Haematol 127:429–432
Hochberg D, Middeldorp JM, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA (2004) Demonstration of the Burkitt’s lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc Natl Acad Sci U S A 101:239–244
Mautner J, Bornkamm GW (2012) The role of virus-specific CD4+ T cells in the control of Epstein-Barr virus infection. Eur J Cell Biol 91:31–35
Robertson ES, Lin J, Kieff E (1996) The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(kappa). J Virol 70(5):3068–3074
Krauer KG, Burgess A, Buck M, Flanagan J, Sculley TB, Gabrielli B (2004) The EBNA-3 gene family proteins disrupt the G2/M checkpoint. Oncogene 23:1342–1353
Anderton E, Yee J, Smith P, Crook T, White RE, Allday MJ (2008) Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 27:421–433
Radkov SA, Touitou R, Brehm A et al (1999) Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J Virol 73:5688–5697
Parker GA, Touitou R, Allday MJ (2000) Epstein-Barr virus EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear division divorced from cytokinesis. Oncogene 19:700–709
Sjoblom A, Yang WW, Palmqvist L, Jansson A, Rymo L (1998) An ATF/CRE element mediates both EBNA2-dependent and EBNA2-independent activation of the Epstein-Barr virus LMP1 gene promoter. J Virol 72:1365–1376
Lucchesi W, Brady G, Dittrich-Breiholz O, Kracht M, Russ R, Farrell PJ (2008) Differential gene regulation by Epstein-Barr virus type 1 and type 2 EBNA2. J Virol 82:7456–7466
Gustafsson A, Levitsky V, Zou JZ et al (2000) Epstein-Barr virus (EBV) load in bone marrow transplant recipients at risk to develop posttransplant lymphoproliferative disease: prophylactic infusion of EBV-specific cytotoxic T cells. Blood 95:807–814
Demachi-Okamura A, Ito Y, Akatsuka Y et al (2008) Epstein-Barr virus nuclear antigen 1-specific CD4(+) T cells directly kill Epstein-Barr virus-carrying natural killer and T cells. Cancer Sci 99:1633–1642
Kobayashi H, Nagato T, Takahara N et al (2008) Induction of EBV-latent membrane protein 1-specific MHC class II-restricted T-cell responses against natural killer lymphoma cells. Cancer Res 68:901–908
Perrine SR, Hermine O, Small T et al (2007) A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 109:2571–2578
Ansell SM, Horwitz SM, Engert A et al (2007) Phase I/II study of an anti-CD30 monoclonal antibody (MDX-060) in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. J Clin Oncol 25:2764–2769
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Rao, H., Miranda, R.N. (2013). Epstein-Barr Virus Lymphomagenesis and Therapeutic Targets. In: Quesenberry, P., Castillo, J. (eds) Non-Hodgkin Lymphoma. Cancer Drug Discovery and Development. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5851-7_4
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5851-7_4
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5850-0
Online ISBN: 978-1-4614-5851-7
eBook Packages: MedicineMedicine (R0)