
Abstract
Statins are a family of lipid-lowering agents, long known to be beneficial in conditions where dyslipidemia occurs, such as atherosclerosis. Very recently, statins also have been proposed for use in neurodegenerative conditions, including Alzheimer’s disease (AD). However, it is clear that the purported effectiveness of statins in neurodegenerative disorders is not directly related to cholesterol-lowering effects of these agents but, rather, to their pleiotropic functions.
Moreover, evidence from randomized, double-blind clinical trials demonstrated that statins have only limited beneficial effects in improving cognitive function in AD patients with moderate dementia. There is also a suggestion that in nondemented elderly people, statin use can be associated with cognitive impairments. Possible mechanisms underlying these effects are discussed along with the pros and cons of the use of statins in neurodegenerative disorders.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

Keywords
1 Basic Pharmacology of Statins
Statins are a family of drugs with pleiotropic functions. To this class belong eight drugs: mevastatin, lovastatin, pravastatin, and simvastatin, which are natural compounds derived from fungal products, whereas fluvastatin, atorvastatin, rosuvastatin, and pitavastatin are distinct synthetic compounds (Shitara and Sugiyama 2006). In addition, lovastatin and simvastatin have a lactone ring in the chemical structure which increases their liposolubility, whereas the remainder statins have open acid forms (Shitara and Sugiyama 2006). These differences in structure could modify the ability of individual statins to cross the blood-brain barrier.
From a pharmacodynamic viewpoint, statins inhibit hydroxy-methyl-glutaryl-CoA (HMG-CoA) reductase, which makes statins effective for the treatment of dyslipidemias (Shitara and Sugiyama 2006). By inhibiting HMG-CoA reductase, statins block the first step in cholesterol biosynthesis, namely, the conversion of HMG-CoA into mevalonate (Shitara and Sugiyama 2006; Bersot 2011). As a result of statin administration, low-density lipoprotein (LDL) cholesterol synthesis decreases in hepatocytes that in turn leads to reduced cholesterol blood level. In addition to this effect, statins reduce triglyceride and increase HDL cholesterol plasma levels. Taken together, statins are cardiovascular agents, due to their ability to counteract hyperlipidemias that are a major cause of atherosclerosis which, in turn, is a common pathogenic mechanism for coronary artery disease, ischemic cerebrovascular disease, and peripheral vascular disease (Shitara and Sugiyama 2006; Bersot 2011). In addition to this main pharmacological effect, statins are endowed with other pleiotropic activities. Statins reduce primary cardiac risk in many types of cardiac and vascular surgeries, with greater benefits in high-risk patients (Gajendragadkar et al. 2009). Furthermore, due to the well-known immunomodulatory effect, statins prevent graft versus host disease and allogeneic transplantation (Shimabukuro-Vornhagen et al. 2009).
Although all statins share the same main mechanism of action, their pharmacokinetic profile is quite different. Statins are well absorbed by the intestine when given by oral route, even though they undergo marked first-pass effects by the liver, which reduces the systemic bioavailability (5–30 %) (Bellosta et al. 2004). With the exception of simvastatin and lovastatin, which are prodrugs and require hepatic activation, other statins are administered as β-hydroxy-acids. Upon administration, statins reach Cmax (peak plasma concentration) ranging from 10 ng/ml (lovastatin and simvastatin) to 448 ng/ml (fluvastatin), with a Tmax (time to reach Cmax) from 0.5–2 h (fluvastatin and pravastatin) to 2–4 h (atorvastatin, lovastatin, rosuvastatin, simvastatin) (Bellosta et al. 2004). In the bloodstream, statins are bound to albumin (43–99 %), which accounts for their variable half-life. Atorvastatin and rosuvastatin are the statins with the longest half-life (15–30 h and 20.8 h, respectively), whereas fluvastatin, lovastatin, pravastatin, and simvastatin have half-lives around 0.5–3 h (Bellosta et al. 2004). All statins are metabolized by the liver through the isoforms 3A4 (atorvastatin, lovastatin, and simvastatin) and 2C9 (fluvastatin and rosuvastatin) of the cytochrome P-450 (CYP) system, with the exception of pravastatin which undergoes sulfation (Bellosta et al. 2004). The primary route of elimination is fecal, and only a minor fraction of statins is eliminated via urine (Bellosta et al. 2004; Shitara and Sugiyama 2006; Bersot 2011).
2 Statins’ Toxicology
The main adverse effects of statins are hepatotoxicity and myopathy. A transient elevation of serum transaminases (up to three times the baseline value) is a common outcome of statin therapy (Bersot 2011). However, the incidence of this side effect is low and dose dependent and does not imply the contraindication of statins in individuals with concomitant liver diseases such as hepatitis C (Bersot 2011). Myalgia is often associated with statin use and is paralleled by a 10-fold increase in plasma creatine kinase (Bersot 2011). Rhabdomyolysis is quite rare, and the risk of developing this side effect is correlated with the statin dose and plasma concentration (Bersot 2011). About 30 cases of serious hepatic failure and 42 cases of death due to rhabdomyolysis associated with statin administration were reported to the FDA over the last 15 years (Bersot 2011; Law and Rudnicka 2006). Particularly serious was the occurrence of fatal rhabdomyolysis in 31 patients treated with cerivastatin in the USA; among these patients, 12 received cerivastatin and gemfibrozil, thus suggesting that the cerivastatin-induced miotoxicity was potentiated by gemfibrozil (Staffa et al. 2002; Shitara and Sugiyama 2006). Due to this severe adverse effect, Bayer voluntarily withdrew cerivastatin from the market. Rare adverse effects due to statin therapy are tendinous disorders. Tendinitis and tendon rupture were mainly associated with atorvastatin, simvastatin, and pravastatin therapy (Marie et al. 2008).
The main determinants of statin toxicity are the following.
2.1 Transporters
The organic anion-transporting polypeptide (OATP1B1) is expressed in the sinusoidal membranes of hepatocytes and is the most important influx transporter of statins (Niemi 2010; Neuvonen 2010). The activity of OATP1B1, whose encoding gene SLCO1B1 undergoes genetic variability among individuals, accounts for important differences in statin plasma levels and toxicity. Two common single nucleotide polymorphism (SNP) variants of the SLCO1B1 gene were identified: c.388A>G (p.Asn130Asp; rs2306283) and c.521T>C (p.Val174Ala; rs4149056). Individuals who carry the c.388A>G (haplotype SLCO1B1*1B) have an increased OATP1B1 activity which is associated with a 35 % reduction in pravastatin bioavailability (measured as area under the concentration-time curve, AUC) but without significant clinical outcomes (Niemi 2010). Conversely, subjects harboring the haplotypes SLCO1B1*5 (c.521T>C) and SLCO1B1*15 (c.521T>C together with c.388A>G) have a reduced activity of the OATP1B1 transporter and an increased statin plasma concentration (Niemi 2010). Both the latter haplotypes are differentially distributed in the population, with a combined allele frequency of 15–20 % in Caucasians, 10–15 % in Asians, and 2 % in sub-Saharan Africans and African Americans (Niemi 2010). Individuals with the c.521CC genotype have an increased bioavailability of simvastatin, pitavastatin, atorvastatin, pravastatin, and rosuvastatin of 221 %, 173 %, 144 %, 90 %, and 87 %, respectively, with respect to subjects carrying the c.521TT genotype (Niemi 2010; Neuvonen 2010). These genetic polymorphisms have indeed important clinical outcomes. The administration of 80 mg/day simvastatin to individuals with myocardial infarction was associated with myopathy, and the odds ratios were 4.5 per copy of the C allele and 16.9 in c.521CC as compared with the c.521TT homozygotes (Link et al. 2008; Niemi 2010). More than 60 % of these cases of myopathy were attributable to the C variant (Link et al. 2008). Similar results were obtained when patients were treated with 40 mg simvastatin/day with a relative risk of 2.6 per copy of the c.521C allele (Link et al. 2008; Niemi 2010). It is also important to emphasize the c.521T>C polymorphism could be responsible for the onset of myopathy even when other statins are administered. In order to reduce the risk of adverse effects, Caucasian adults harboring the SLCO1B1 c.521TC genotype should be treated with a half dose of simvastatin, atorvastatin, pravastatin, rosuvastatin, and pitavastatin, whereas those with the c.521CC genotype should receive a quarter dose of simvastatin, atorvastatin, and pitavastatin (Niemi 2010).
ABCG2 encodes the ATP-binding cassette G2 (ABCG2), an efflux transporter located on the apical membranes of several cell types including intestinal epithelial cells and hepatocytes. The ABCG2 gene undergoes genetic variability, and the more common SNP is the c.421C>A (p.Gln141Lys; rs 2231142) which reduces the transport function of ABCG2. Individuals who carry the c.421AA genotype have an increased AUC of simvastatin lactone (111 %) as well as atorvastatin and fluvastatin (72 %) with respect to those with the c.421CC genotype (Niemi 2010). Individuals with a c.421AA genotype have a greater risk to develop myopathy if treated with rosuvastatin, atorvastatin, and fluvastatin, even if no clinical trial definitely proved this possibility (Niemi 2010).
2.2 Drug Interactions
In order to reduce the incidence of hepatotoxicity and myopathy, statins should not be used in association with inhibitors of CYP3A4, including grapefruit juice (see below). Several cases of rhabdomyolysis were associated with the concomitant treatment of simvastatin, lovastatin, or atorvastatin with ritonavir, cyclosporine, azole antifungals, macrolides antibiotics, and calcium channel blockers (Neuvonen et al. 2006; Neuvonen 2010). Also the combination of statins and fibrates should be avoided, in particular gemfibrozil (Bersot 2011). For further information about the potential harmful effects deriving from the interaction of statins with other drugs or nutraceuticals, see below.
3 Rationale for the Use of Statins in Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by cognitive dysfunction, difficulties in performing the activities of daily living, and mood changes. At autopsy, the AD brain is characterized by the accumulation of amyloid-β peptide (Aβ) and hyperphosphorylated tau protein (Sultana and Butterfield 2010; Citron 2010; Querfurth and LaFerla 2010). Amyloid-β peptide is formed by the secretase-mediated cleavage of the amyloid precursor protein (APP). β-Secretase cleaves APP at the N-terminus, generating an extracellular soluble fragment named sAPPβ and leaves an intramembrane fragment called C99 (Mancuso et al. 2011; Querfurth and LaFerla 2010). The latter is cleaved at the C-terminus by γ-secretase and Aβ is released (Mancuso et al. 2011; Querfurth and LaFerla 2010). Once formed, Aβ aggregates as oligomers and fibrils, which form the central core of senile plaques (Sultana et al. 2009; Citron 2010; Querfurth and LaFerla 2010). Current research suggests that Aβ oligomers are more toxic than aggregates (Shankar et al. 2008; Butterfield et al. 2007; Querfurth and LaFerla 2010), and this evidence provides a plausible explanation of the reason why drugs targeted to facilitate the disaggregation of Aβ fibrils into oligomers, e.g., bapineuzumab and solanezumab, failed to improve cognitive function in AD patients (Ayrolles-Torro et al. 2011; Mancuso et al. 2011). Interestingly, the gene encoding APP is located on the chromosome 21; therefore, individuals with down syndrome (DS), who have 3 copies of this chromosome, have an increased risk of developing AD (Bush and Beail 2004). Autopsy findings in individuals with DS revealed senile plaques and neurofibrillary tangles in the brain, which are secondary to the formation and deposition of Aβ and tau protein, respectively (Gyure et al. 2001; Itoh and Yagishita 1998).
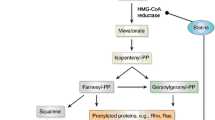
Amyloid-β peptide and tau protein synergize and exert their neurotoxic effects through several mechanisms: (a) generation of superoxide anion and nitric oxide through the activation of NADPH oxidase and inducible nitric oxide synthase (iNOS), respectively (Butterfield et al. 2007; Querfurth and LaFerla 2010), which react each other and form peroxynitrite (ONOO–); (b) mitochondrial impairment secondary to the inhibition of key enzymes involved in the respiratory chain and Krebs cycle (Querfurth and LaFerla 2010); and (c) stimulation of the ionotropic glutamate receptor NMDA and increase of Ca2+ overload thus leading to excitotoxic cell death (Querfurth and LaFerla 2010). Taking into consideration these multiple toxic pathways, statins may be particularly useful for AD. Statins can inhibit endothelial O2 −• formation by preventing the isoprenylation of p21 Rac, which is critical for the assembly of NADPH oxidase after activation of protein kinase C (Wallerath et al. 2003) (Fig. 1). In addition, the cellular superoxide clearance activity is increased given that SOD3 activity is more than doubled by simvastatin. Further, simvastatin treatment also increases the number of functionally active endothelial progenitor cells (Landmesser et al. 2005). Moreover, statins increase the expression of endothelial nitric oxide synthase (eNOS) through the inhibition of Rho isoprenylation (Laufs et al. 1998), and statins can also directly activate eNOS via posttranslational mechanisms involving activation of the PI3K/Akt pathway (Kureishi et al. 2000). Statins showed positive effects against Aβ-induced oxidative stress in mouse models of AD (Kurinami et al. 2008; Tong et al. 2009) as well as a reduction in the phosphorylation of tau protein in human cerebrospinal fluid (CSF) (Riekse et al. 2006). However, although statins’ treatment appears to provide benefits, it is difficult to determine whether the benefits are due to lower cholesterol levels or to statin pleiotropy. Atorvastatin treatment was neuroprotective against cell degeneration induced by Aβ(1-40), reducing inflammatory and oxidative responses and increasing the expression of glutamatergic transporters (Piermartiri et al. 2010). Murphy et al. showed that long-term atorvastatin did not reduce Aβ levels, despite a significant reduction in β-secretase 1 (BACE1) protein levels and activity in the brains of aged beagles (Murphy et al. 2010). Subsequently, Barone et al. found that although no changes in Aβ levels occur, long-term atorvastatin significantly reduced lipid peroxidation, protein oxidation and nitration, and increased GSH levels in the parietal cortex of treated dogs (Barone et al. 2011). This effect was cholesterol and Aβ independent and specific for brain (Barone et al. 2011). Another possible mechanism by which statins exert neuroprotective effects is the enhancement of the cell stress response. The administration of atorvastatin 80 mg/day for 14.5 months to aged beagles increased the expression of the heme oxygenase-1/biliverdin reductase-A (HO-1/BVR-A) system, a main effector of the adaptive stress response (Mancuso and Barone 2009), and this correlated with a significant reduction in both oxidative and nitrosative stress biomarkers in the parietal cortex of treated beagles that showed improved cognition (Barone et al. 2012; Butterfield et al. 2011). Conversely, side effects of long-term statin treatment include a decrease in serum and tissue CoQ10 levels, which could result in an energy metabolism impairment in heart, skeletal muscle, and liver (Martin et al. 2011; Bliznakov and Wilkins 1998). The importance of statin-induced energy metabolism impairment and its role in the development of their side effects was demonstrated also in humans. The administration of 240 mg/day CoQ10 reverted fatigue, myalgia, and peripheral neuropathy in 50 patients chronically treated with statins (Langsjoen et al. 2005).

Cholesterol-dependent and cholesterol-independent effects of statins and repercussion on the redox status of the cell. For further information, see text. Abbreviations: 3-NT 3-nitrotyrosine, BVR-A biliverdin reductase-A, CoQ10 coenzyme Q10, eNOS endothelial nitric oxide synthase, FPP farnesyl pyrophosphate, GPP geranyl pyrophosphate, GSH reduced glutathione, HMG-CoA hydroxyl-methyl-glutaryl-CoA, HNE 4-hydroxynonenals, HO-1 heme oxygenase-1, PC protein carbonyls, SOD3 extracellular superoxide dismutase
More detailed research into the pharmacology of statins is necessary, particularly the concentrations achieved in the central nervous system and the level at which they block the production of cholesterol and they modulate all the above pathways, in order to prove their beneficial effect and support the potential use of statins in neurodegenerative disorders.
4 Statins and Dementia: The Support Given by the Evidence-Based Medicine
Although the majority of the preclinical evidence shows neuroprotective effects of statins in ameliorating cognitive dysfunction, clinical data largely do not show similar benefits. In addition, some clinical studies show the opposite results depending on whether they are observational studies or randomized clinical trials (RCT).
Early cohort and case-control studies demonstrated that statins reduced the risk of developing dementia, including AD, and this protective effect was maintained over a 6-year follow-up period (Beydoun et al. 2010; Li et al. 2010; Smeeth et al. 2009; Haag et al. 2009; Horsdal et al. 2009; Rosenberg and Allard 2008; Sparks et al. 2008; Cramer et al. 2008). These findings were recently challenged by Benito-Leon et al., who demonstrated that statins did not improve cognitive performance in nondemented elderly subjects with a median age of 72 years (Benito-Leon et al. 2010). Similar results were obtained in a large cohort study, which involved more than two million participants aged 30–84 years of whom 10.7 % received statins (Hippisley-Cox and Coupland 2010). A cross-sectional research study, with a follow-up of 5 years, demonstrated that in 123 Caucasian subjects with DS (41–78 years), total cholesterol levels > 200 mg/dl positively correlated with the development of AD (hazard rate 2.59), whereas individuals taking statins had less than half the risk of developing AD with respect to nonusers (hazard rate 0.402) (Zigman et al. 2007).
In order to confirm these epidemiological studies, some RCT were performed with comparable results. The PROSPER study, which enrolled approximately 6,000 people aged 70–82 years, demonstrated that pravastatin (40 mg/day) did not have any effect on cognitive function over a follow-up of 3 years (Trompet et al. 2010; Shepherd et al. 1999). The LEADe study tested the hypothesis that atorvastatin (80 mg/day) over 72 weeks delayed cognitive decline in patients with mild-to-moderate AD. The results of this study did not support any significant positive effect of atorvastatin on cognitive or global function in patients receiving the statin compared to those with placebo (Feldman et al. 2010). In August 2011, Sano et al. published the results of an RCT of simvastatin (20 mg/day for 6 weeks and 40 mg/day for the remainder 18 months) in participants with mild-to-moderate AD that also demonstrated the lack of efficacy (Sano et al. 2011). On the other hand, the ADCLT trial demonstrated that atorvastatin (80 mg/day) for 1 year exhibited a significant positive effect on cognitive performance after 6 months of therapy compared with placebo. However, this beneficial effect was selective for individuals who matched restricted criteria, such as a higher MMSE score at baseline, total cholesterol levels higher than 200 mg/dl, and the presence of an apolipoprotein E-4 allele (APOE-4) (Sparks et al. 2006a, b). When all the RCT study results are combined, a meta-analysis suggests that there is insufficient evidence to recommend statins for the treatment of dementia and AD (McGuinness et al. 2010). This statement matches the guidelines of the British Association for Psychopharmacology who also do not recommend statins for the prevention or treatment of AD (O'Brien and Burns 2010). However, in a pilot clinical trial, a modest improvement in cognition was detected in individuals with mild cognitive impairment (MCI), arguably the earliest form of AD, treated with statins (Sparks et al. 2010).
5 Unresolved Issues
In order to understand the reasons for the inconsistent beneficial effects of statins in aged or demented individuals, several issues need to be addressed and carefully evaluated.
Ability to Cross the Blood-Brain Barrier. As mentioned earlier, the presence of a lactone ring in the chemical structures of simvastatin and lovastatin makes these latter very liposoluble, whereas the open acids atorvastatin, fluvastatin, and pitavastatin have an intermediate liposolubility, and pravastatin has the lowest one (Shitara and Sugiyama 2006). Such different degree of liposolubility might suggest the use of lipophilic statins in demented patients to increase the bioavailability in the brain. However, both the LEADe and CLASP trials failed to demonstrate a beneficial effect of lipophilic atorvastatin and simvastatin on cognitive function in AD patients, similar to the results obtained in the PROSPER study involving the lipophobic pravastatin. Thus, the different degrees of liposolubility are unlikely to be a key determinant of the limited effectiveness of statins in these clinical trials.
Age. Epidemiological data demonstrate that the incidence of AD increases with age and doubles every 5 years after 65 years of age with 1,275 new cases/1,00,000 persons/year (Querfurth and LaFerla 2010). In the Western hemisphere, the prevalence of AD was calculated at approximately 1% in subjects aged 60–64 but increases to between 33 % and 50 % in people aged 85 or older (Mayeux 2010). Much of the epidemiological and clinical studies designed to examine the role of statins in AD enrolled included individuals aged 65–84 years. Although the clinical studies and the meta-analysis discussed above did not support an overall beneficial effect for statins in dementia and AD, it is noteworthy to mention that those studies in which statins had a major effect on cognitive functions included individuals aged 68–74 years (Simons et al. 2002; Li et al. 2010; Haag et al. 2009). Consequently, it is possible that 68–74 years of age should be considered an optimal age for statin efficacy in preventing dementia. On the other hand, both the LEADe and PROSPER studies, which recruited individuals within the same range of age, failed to demonstrate any beneficial effect of atorvastatin and pravastatin in people with AD. Subjects aged 80 or older also did not have any beneficial effects from statins (Li et al. 2010).
Cholesterol Blood Levels Before and After Treatment with Statins. An important question to address when studying the HMG-CoA reductase-independent effects of statins is the concomitant reduction of LDL cholesterol plasma levels. This issue is quite important when the pleiotropic effects of statins are studied in the nervous system because cholesterol is a main component of cell membranes, in particular myelin (Saher et al. 2005), and if cholesterol blood levels fall due to uncontrolled therapy with lipid lowering agents, nervous function would also decrease. The majority of AD patients recruited for clinical trials had serum LDL cholesterol at baseline around 131–147 mg/dL, which was significantly reduced by 50–54 % after the administration of atorvastatin (80 mg for 52 weeks) or simvastatin (40 mg for 26 weeks) (McGuinness et al. 2009; McGuinness et al. 2010). These values of LDL cholesterol, before and after statin treatment, are acceptable and do not imply any possible adverse effects. However, although there is a marked effect of statins on LDL cholesterol plasma levels, no beneficial effects on cognitive function are observed in normocholesterolemic patients (McGuinness et al. 2009; McGuinness et al. 2010). Indeed, even in hypercholesterolemic patients, the administration of statins did not have any disease-modifying effect on AD (Hoglund et al. 2004; Riekse et al. 2006). Evans et al. showed that in AD patients heterozygous for APOE-4 allele or carriers of PS1 mutations, the administration of simvastatin or atorvastatin only slightly reduced the concentration of CSF cholesterol at 6–7 months followed by a peak at 2 years and a return to baseline levels after 3 years (Evans et al. 2009). This finding suggests that despite changes in plasma cholesterol levels, statins cause only minimal changes in brain cholesterol, and, therefore, the effect on cognitive functions may be independent of systemic cholesterol metabolism.
Interaction with Xenobiotics. Patients with AD, as well as other types of dementia, usually take additional drugs for other age-related disorders or comorbidities. As mentioned above, all statins, with the exception of pravastatin, are metabolized by CYP3A4 or CYP2C9, and their plasma levels could be reduced or increased with concomitant administration of drugs that induce or inhibit these CYP isoforms. Donepezil and galantamine, two acetylcholinesterase inhibitors provided to AD patients, are metabolized by CYP3A4 (Jann et al. 2002) and, therefore, could compete with statins. As a consequence of this competition, statin plasma levels could increase as well as the risk of side effects. Also, AD patients may also be supplemented with nutraceuticals including curcumin, green tea extracts, or grapefruit juice as these natural substances are widely considered to be free radical scavengers and therefore neuroprotective. Unfortunately, these natural substances are inhibitors of CYP3A4 and, therefore, increase plasma concentrations of statins (Kiani and Imam 2007; Stump et al. 2006; Hare and Elliott 2003). Interestingly, it was reported that the concomitant administration of simvastatin and consumption of grapefruit or green tea causes rhabdomyolysis (Dreier and Endres 2004; Werba et al. 2008).
Taking into consideration the results from evidence-based medicine, it is possible to suggest that select individuals with AD may benefit more from statins: aged 65–74 years, carrier of an ApoE-4 allele (Sparks et al. 2006a) but without the haplotype SLCO1B1*5 or SLCO1B1*15 or SLCO1B1*1B/*1B (Niemi 2010), normocholesterolemic and sparing user of drugs which inhibit CYP3A4.
6 Conclusion
Although preclinical data suggest beneficial effects of statins in the treatment of dementia and AD, results derived from epidemiological studies and RCT are contradictory. Several critical issues may need to be considered (1) the genetic profile of individuals with regard to specific genes involved in statin absorption, (2) the rate of inhibition of cholesterol synthesis in normocholesterolemic patients, (3) age of the individual, and (4) concomitant administration of drugs or nutraceuticals. Thus, future RCT using statins may consider from recruiting AD patients aged 65–75 years, which is an age range that appears to benefit from statin therapy, with primary endpoint, the analysis of cognitive function, and a significant longer follow-up. Another suggestion is to study the effect of statins in individuals with MCI in order to understand whether or not the neuroprotective effect of statins could block or slow the transition to AD. This recommendation could be especially important since preclinical studies with atorvastatin decreased oxidative stress in aged dogs, and this benefit was correlated with levels of Aβ(1–42), which is same sequence as that of humans (Barone et al. 2011). Oxidative stress is strongly associated with amnestic MCI and AD (Perluigi et al. 2009; Sultana et al. 2010; Sultana et al. 2009), and Aβ is hypothesized to contribute to this oxidative stress (Butterfield et al. 2001).
In conclusion, the current clinical evidence is not strong enough to support the widespread use of statins to treat dementia and AD. However, it is critical that researchers and clinicians in the near future investigate whether or not statin therapy should be restricted to selected populations of demented individuals with the best chance of efficacy derived from evidence-based medicine is recommended.
References
Ayrolles-Torro, A., Imberdis, T., Torrent, J., Toupet, K., Baskakov, I. V., Poncet-Montange, G., Gregoire, C., Roquet-Baneres, F., Lehmann, S., Rognan, D., Pugniere, M., Verdier, J. M., & Perrier, V. (2011). Oligomeric-induced activity by thienyl pyrimidine compounds traps prion infectivity. Journal of Neuroscience, 31(42), 14882–14892. doi:31/42/14882 [pii] 10.1523/JNEUROSCI.0547-11.2011.
Barone, E., Cenini, G., Di Domenico, F., Martin, S., Sultana, R., Mancuso, C., Murphy, M. P., Head, E., & Butterfield, D. A. (2011). Long-term high-dose atorvastatin decreases brain oxidative and nitrosative stress in a preclinical model of Alzheimer disease: A novel mechanism of action. Pharmacological Research, 63(3), 172–180. doi:S1043-6618(10)00232-X [pii] 10.1016/j.phrs.2010.12.007.
Barone, E., Mancuso, C., Di Domenico, F., Sultana, R., Murphy, M. P., Head, E., & Butterfield, D. A. (2012). Biliverdin reductase-A: A novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. Journal of Neurochemistry, 120(1), 135–146. doi:10.1111/j.1471-4159.2011.07538.x.
Bellosta, S., Paoletti, R., & Corsini, A. (2004). Safety of statins: Focus on clinical pharmacokinetics and drug interactions. Circulation, 109(23 Suppl 1), III50–III57. doi:10.1161/01.CIR.0000131519.15067.1f 109/23_suppl_1/III-50 [pii].
Benito-Leon, J., Louis, E. D., Vega, S., & Bermejo-Pareja, F. (2010). Statins and cognitive functioning in the elderly: A population-based study. Journal of Alzheimer's Disease, 21(1), 95–102. doi:T60L52283451G644 [pii] 10.3233/JAD-2010-100180.
Bersot, T. P. (2011). Drug therapy for hypercholesterolemia and dyslipidemia. In L. B. Laurence (Ed.), Goodman and Gilman’s, The pharmacological basis of therapeutics (12th ed., pp. 877–908). New York: McGraw-Hill.
Beydoun, M. A., Beason-Held, L. L, Kitner-Triolo, M. H., Beydoun, H. A., Ferrucci, L., Resnick, S. M., Zonderman, A. B. (2010). Statins and serum cholesterol's associations with incident dementia and mild cognitive impairment. Journal of Epidemiology and Community Health. doi:jech.2009.100826 [pii]. 10.1136/jech.2009.100826.
Bliznakov, E. G., & Wilkins, D. J. (1998). Biochemical and clinical consequences of inhibiting coenzyme Q(10) biosynthesis by lipid-lowering HMG-CoA reductase inhibitors (statins): A critical overview. Advances in Therapy, 15(4), 218–228.
Bush, A., & Beail, N. (2004). Risk factors for dementia in people with down syndrome: issues in assessment and diagnosis. American Journal of Mental Retardation, 109(2), 83–97. doi:10.1352/0895-8017(2004)109<83:RFFDIP>2.0.CO;2.
Butterfield, D. A., Drake, J., Pocernich, C., & Castegna, A. (2001). Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends in Molecular Medicine, 7(12), 548–554. doi:S1471-4914(01)02173-6 [pii].
Butterfield, D. A., Reed, T., Newman, S. F., & Sultana, R. (2007). Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radical Biology & Medicine, 43(5), 658–677. doi:S0891-5849(07)00392-9 [pii] 10.1016/j.freeradbiomed.2007.05.037.
Butterfield, D. A., Barone, E., Di Domenico, F., Cenini, G., Sultana, R., Murphy, M. P., Mancuso, C., Head, E. (2011). Atorvastatin treatment in a dog preclinical model of Alzheimer’s disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. International Journal of Neuropsychopharmacol:1–7. doi:S1461145711001118 [pii]. 10.1017/S1461145711001118.
Citron, M. (2010). Alzheimer’s disease: Strategies for disease modification. Nature Reviews Drug Discovery, 9(5), 387–398. doi:nrd2896 [pii]. 10.1038/nrd2896.
Cramer, C., Haan, M. N., Galea, S., Langa, K. M., & Kalbfleisch, J. D. (2008). Use of statins and incidence of dementia and cognitive impairment without dementia in a cohort study. Neurology, 71(5), 344–350. doi:71/5/344 [pii]. 10.1212/01.wnl.0000319647.15752.7b.
Dreier, J. P., & Endres, M. (2004). Statin-associated rhabdomyolysis triggered by grapefruit consumption. Neurology, 62(4), 670.
Evans, B. A., Evans, J. E., Baker, S. P., Kane, K., Swearer, J., Hinerfeld, D., Caselli, R., Rogaeva, E., St George-Hyslop, P., Moonis, M., & Pollen, D. A. (2009). Long-term statin therapy and CSF cholesterol levels: implications for Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders, 27(6), 519–524. doi:000221835 [pii]. 10.1159/000221835.
Feldman, H. H., Doody, R. S., Kivipelto, M., Sparks, D. L., Waters, D. D., Jones, R. W., Schwam, E., Schindler, R., Hey-Hadavi, J., DeMicco, D. A., & Breazna, A. (2010). Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology, 74(12), 956–964. doi:WNL.0b013e3181d6476a [pii]. 10.1212/WNL.0b013e3181d6476a.
Gajendragadkar, P. R., Cooper, D. G., Walsh, S. R., Tang, T. Y., Boyle, J. R., & Hayes, P. D. (2009). Novel uses for statins in surgical patients. International Journal of Surgery, 7(4), 285–290. doi:S1743-9191(09)00066-1 [pii]. 10.1016/j.ijsu.2009.04.016.
Gyure, K. A., Durham, R., Stewart, W. F., Smialek, J. E., & Troncoso, J. C. (2001). Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Archives of Pathology & Laboratory Medicine, 125(4), 489–492. doi:10.1043/0003-9985(2001)125<0489:IAAPDO>2.0.CO;2.
Haag, M. D., Hofman, A., Koudstaal, P. J., Stricker, B. H., & Breteler, M. M. (2009). Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity The Rotterdam Study. Journal of Neurology, Neurosurgery and Psychiatry, 80(1), 13–17. doi:jnnp.2008.150433 [pii]. 10.1136/jnnp.2008.150433.
Hare, J. T., & Elliott, D. P. (2003). Grapefruit juice and potential drug interactions. The Consultant Pharmacist, 18(5), 466–472.
Hippisley-Cox, J., & Coupland, C. (2010). Unintended effects of statins in men and women in England and Wales: Population based cohort study using the QResearch database. BMJ, 340, c2197.
Hoglund, K., Wiklund, O., Vanderstichele, H., Eikenberg, O., Vanmechelen, E., & Blennow, K. (2004). Plasma levels of beta-amyloid(1-40), beta-amyloid(1-42), and total beta-amyloid remain unaffected in adult patients with hypercholesterolemia after treatment with statins. Archives of Neurology, 61(3), 333–337. doi:10.1001/archneur.61.3.333. 61/3/333 [pii].
Horsdal, H. T., Olesen, A. V., Gasse, C., Sorensen, H. T., Green, R. C., & Johnsen, S. P. (2009). Use of statins and risk of hospitalization with dementia: A Danish population-based case-control study. Alzheimer Disease and Associated Disorders, 23(1), 18–22. doi:10.1097/WAD.0b013e318180f55b.
Itoh, Y., & Yagishita, S. (1998). Scanning electron microscopical study of neurofibrillary tangles in a presenile patient with Down’s syndrome. Acta Neuropathologica, 96(2), 179–184.
Jann, M. W., Shirley, K. L., & Small, G. W. (2002). Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clinical Pharmacokinetics, 41(10), 719–739. doi:411003 [pii].
Kiani, J., & Imam, S. Z. (2007). Medicinal importance of grapefruit juice and its interaction with various drugs. Nutrition Journal, 6, 33. doi:1475-2891-6-33 [pii]. 10.1186/1475-2891-6-33.
Kureishi, Y., Luo, Z., Shiojima, I., Bialik, A., Fulton, D., Lefer, D. J., Sessa, W. C., & Walsh, K. (2000). The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nature Medicine, 6(9), 1004–1010. doi:10.1038/79510.
Kurinami, H., Sato, N., Shinohara, M., Takeuchi, D., Takeda, S., Shimamura, M., Ogihara, T., & Morishita, R. (2008). Prevention of amyloid beta-induced memory impairment by fluvastatin, associated with the decrease in amyloid beta accumulation and oxidative stress in amyloid beta injection mouse model. International Journal of Molecular Medicine, 21(5), 531–537.
Landmesser, U., Bahlmann, F., Mueller, M., Spiekermann, S., Kirchhoff, N., Schulz, S., Manes, C., Fischer, D., de Groot, K., Fliser, D., Fauler, G., Marz, W., & Drexler, H. (2005). Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation, 111(18), 2356–2363. doi:01.CIR.0000164260.82417.3F [pii]. 10.1161/01.CIR.0000164260.82417.3F.
Langsjoen, P. H., Langsjoen, J. O., Langsjoen, A. M., & Lucas, L. A. (2005). Treatment of statin adverse effects with supplemental Coenzyme Q10 and statin drug discontinuation. Biofactors, 25(1–4), 147–152.
Laufs, U., La Fata, V., Plutzky, J., & Liao, J. K. (1998). Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation, 97(12), 1129–1135.
Law, M., & Rudnicka, A. R. (2006). Statin safety: A systematic review. The American Journal of Cardiology, 97(8A), 52C–60C. doi:S0002-9149(05)02143-0 [pii]. 10.1016/j.amjcard.2005.12.010.
Li, G., Shofer, J. B., Rhew, I. C., Kukull, W. A., Peskind, E. R., McCormick, W., Bowen, J. D., Schellenberg, G. D., Crane, P. K., Breitner, J. C., & Larson, E. B. (2010). Age-varying association between statin use and incident Alzheimer’s disease. Journal of American Geriatrics Society, 58(7), 1311–1317. doi:JGS2906 [pii]. 10.1111/j.1532-5415.2010.02906.x.
Link, E., Parish, S., Armitage, J., Bowman, L., Heath, S., Matsuda, F., Gut, I., Lathrop, M., & Collins, R. (2008). SLCO1B1 variants and statin-induced myopathy–a genomewide study. The New England Journal of Medicine, 359(8), 789–799. doi:NEJMoa0801936 [pii]. 10.1056/NEJMoa0801936.
Mancuso, C., & Barone, E. (2009). The heme oxygenase/biliverdin reductase pathway in drug research and development. Current Drug Metabolism, 10(6), 579–594. doi:Abstract no. 0015 [pii].
Mancuso, C., Siciliano, R., Barone, E., Butterfield, D. A., & Preziosi, P. (2011). Pharmacologists and Alzheimer disease therapy: To boldly go where no scientist has gone before. Expert Opinion on Investigational Drugs, 20(9), 1243–1261. doi:10.1517/13543784.2011.601740.
Marie, I., Delafenetre, H., Massy, N., Thuillez, C., & Noblet, C. (2008). Tendinous disorders attributed to statins: A study on ninety-six spontaneous reports in the period 1990-2005 and review of the literature. Arthritis and Rheumatism, 59(3), 367–372. doi:10.1002/art.23309.
Martin, S. B., Cenini, G., Barone, E., Dowling, A. L., Mancuso, C., Butterfield, D. A., Murphy, M. P., & Head, E. (2011). Coenzyme Q10 and cognition in atorvastatin treated dogs. Neuroscience Letters, 501(2), 92–95. doi:S0304-3940(11)01009-3 [pii]. 10.1016/j.neulet.2011.06.054.
Mayeux, R. (2010). Clinical practice. Early Alzheimer’s disease. The New England Journal of Medicine, 362(23), 2194–2201. doi:362/23/2194 [pii]. 10.1056/NEJMcp0910236.
McGuinness, B., Craig, D., Bullock, R., Passmore, P. (2009). Statins for the prevention of dementia. Cochrane Database System Review (2):CD003160. doi:10.1002/14651858.CD003160.pub2.
McGuinness, B., O’Hare, J., Craig, D., Bullock, R., Malouf, R., Passmore, P. (2010). Statins for the treatment of dementia. Cochrane Database System Review (8):CD007514. doi:10.1002/14651858.CD007514.pub2.
Murphy, M. P., Morales, J., Beckett, T. L., Astarita, G., Piomelli, D., Weidner, A., Studzinski, C. M., Dowling, A. L., Wang, X., Levine, H., 3rd, Kryscio, R. J., Lin, Y., Barrett, E., & Head, E. (2010). Changes in cognition and amyloid-beta processing with long term cholesterol reduction using atorvastatin in aged dogs. Journal of Alzheimer's Disease, 22(1), 135–150. doi:R81617377L4X3511 [pii]. 10.3233/JAD-2010-100639.
Neuvonen, P. J. (2010). Drug interactions with HMG-CoA reductase inhibitors (statins): The importance of CYP enzymes, transporters and pharmacogenetics. Current Opinion in Investigational Drugs, 11(3), 323–332.
Neuvonen, P. J., Niemi, M., & Backman, J. T. (2006). Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clinical Pharmacology and Therapeutics, 80(6), 565–581. doi:S0009-9236(06)00360-2 [pii].10.1016/j.clpt.2006.09.003.
Niemi, M. (2010). Transporter pharmacogenetics and statin toxicity. Clinical Pharmacology and Therapeutics, 87(1), 130–133. doi:clpt2009197 [pii]. 10.1038/clpt.2009.197.
O'Brien, J. T., Burns A. (2010) Clinical practice with anti-dementia drugs: a revised (second) consensus statement from the British Association for Psychopharmacology. J Psychopharmacol. doi:0269881110387547 [pii]. 10.1177/0269881110387547.
Perluigi, M., Sultana, R., Cenini, G., Di Domenico, F., Memo, M., Pierce, W. M., Coccia, R., & Butterfield, D. A. (2009). Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer's disease: Role of lipid peroxidation in Alzheimer's disease pathogenesis. Proteomics. Clinical Applications, 3(6), 682–693. doi:10.1002/prca.200800161.
Piermartiri, T. C., Figueiredo, C. P., Rial, D., Duarte, F. S., Bezerra, S. C., Mancini, G., de Bem, A. F., Prediger, R. D., & Tasca, C. I. (2010). Atorvastatin prevents hippocampal cell death, neuroinflammation and oxidative stress following amyloid-beta(1-40) administration in mice: evidence for dissociation between cognitive deficits and neuronal damage. Experimental Neurology, 226(2), 274–284. doi:S0014-4886(10)00329-8 [pii]. 10.1016/j.expneurol.2010.08.030.
Querfurth, H. W., & LaFerla, F. M. (2010). Alzheimer’s disease. The New England Journal of Medicine, 362(4), 329–344. doi:362/4/329 [pii].
Riekse, R. G., Li, G., Petrie, E. C., Leverenz, J. B., Vavrek, D., Vuletic, S., Albers, J. J., Montine, T. J., Lee, V. M., Lee, M., Seubert, P., Galasko, D., Schellenberg, G. D., Hazzard, W. R., & Peskind, E. R. (2006). Effect of statins on Alzheimer's disease biomarkers in cerebrospinal fluid. Journal of Alzheimer's Disease, 10(4), 399–406.
Rosenberg, H., & Allard, D. (2008). Women and statin use: A women’s health advocacy perspective. Scandinavian Cardiovascular Journal, 42(4), 268–273. doi:791850044 [pii]. 10.1080/14017430801993180.
Saher, G., Brugger, B., Lappe-Siefke, C., Mobius, W., Tozawa, R., Wehr, M. C., Wieland, F., Ishibashi, S., & Nave, K. A. (2005). High cholesterol level is essential for myelin membrane growth. Nature Neuroscience, 8(4), 468–475. doi:nn1426 [pii]. 10.1038/nn1426.
Sano, M., Bell, K. L., Galasko, D., Galvin, J. E., Thomas, R. G., van Dyck, C. H., & Aisen, P. S. (2011). A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology, 77(6), 556–563. doi:WNL.0b013e318228bf11 [pii]. 10.1212/WNL.0b013e318228bf11.
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., Brett, F. M., Farrell, M. A., Rowan, M. J., Lemere, C. A., Regan, C. M., Walsh, D. M., Sabatini, B. L., & Selkoe, D. J. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature Medicine, 14(8), 837–842. doi:nm1782 [pii]. 10.1038/nm1782.
Shepherd, J., Blauw, G. J., Murphy, M. B., Cobbe, S. M., Bollen, E. L., Buckley, B. M., Ford, I., Jukema, J. W., Hyland, M., Gaw, A., Lagaay, A. M., Perry, I. J., Macfarlane, P. W., Meinders, A. E., Sweeney, B. J., Packard, C. J., Westendorp, R. G., Twomey, C., & Stott, D. J. (1999). The design of a prospective study of Pravastatin in the Elderly at Risk (PROSPER). PROSPER Study Group. PROspective Study of Pravastatin in the Elderly at Risk. The American Journal of Cardiology, 84(10), 1192–1197. doi:S0002914999005330 [pii].
Shimabukuro-Vornhagen, A., Glossmann, J., Liebig, T., Scheid, C., & von Bergwelt-Baildon, M. (2009). The use of statins in hematopoietic stem cell transplantation. Current Stem Cell Research & Therapy, 4(4), 260–265. doi:ABSTRACT # 12.
Shitara, Y., & Sugiyama, Y. (2006). Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacology and Therapeutics, 112(1), 71–105. doi:S0163-7258(06)00040-4 [pii]. 10.1016/j.pharmthera.2006.03.003.
Simons, M., Schwarzler, F., Lutjohann, D., von Bergmann, K., Beyreuther, K., Dichgans, J., Wormstall, H., Hartmann, T., & Schulz, J. B. (2002). Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: A 26-week randomized, placebo-controlled, double-blind trial. Annals of Neurology, 52(3), 346–350. doi:10.1002/ana.10292.
Smeeth, L., Douglas, I., Hall, A. J., Hubbard, R., & Evans, S. (2009). Effect of statins on a wide range of health outcomes: A cohort study validated by comparison with randomized trials. British Journal of Clinical Pharmacology, 67(1), 99–109. doi:BCP3308 [pii]. 10.1111/j.1365-2125.2008.03308.x.
Sparks, D. L., Connor, D. J., Sabbagh, M. N., Petersen, R. B., Lopez, J., & Browne, P. (2006a). Circulating cholesterol levels, apolipoprotein E genotype and dementia severity influence the benefit of atorvastatin treatment in Alzheimer’s disease: Results of the Alzheimer’s Disease Cholesterol-Lowering Treatment (ADCLT) trial. Acta Neurologica Scandinavica. Supplementum, 185, 3–7. doi:ANE690 [pii]. 10.1111/j.1600-0404.2006.00690.x.
Sparks, D. L., Sabbagh, M., Connor, D., Soares, H., Lopez, J., Stankovic, G., Johnson-Traver, S., Ziolkowski, C., & Browne, P. (2006b). Statin therapy in Alzheimer’s disease. Acta Neurologica Scandinavica. Supplementum, 185, 78–86. doi:ANE689 [pii]. 10.1111/j.1600-0404.2006.00689.x.
Sparks, D. L., Kryscio, R. J., Sabbagh, M. N., Connor, D. J., Sparks, L. M., & Liebsack, C. (2008). Reduced risk of incident AD with elective statin use in a clinical trial cohort. Current Alzheimer Research, 5(4), 416–421.
Sparks, D. L., Kryscio, R. J., Connor, D. J., Sabbagh, M. N., Sparks, L. M., Lin, Y., & Liebsack, C. (2010). Cholesterol and cognitive performance in normal controls and the influence of elective statin use after conversion to mild cognitive impairment: results in a clinical trial cohort. Neurodegenerative Diseases, 7(1–3), 183–186. doi:000295660 [pii] 10.1159/000295660.
Staffa, J. A., Chang, J., & Green, L. (2002). Cerivastatin and reports of fatal rhabdomyolysis. The New England Journal of Medicine, 346(7), 539–540. doi:10.1056/NEJM200202143460720. 346/7/539-a [pii].
Stump, A. L., Mayo, T., & Blum, A. (2006). Management of grapefruit-drug interactions. American Family Physician, 74(4), 605–608.
Sultana, R., & Butterfield, D. A. (2010). Role of oxidative stress in the progression of Alzheimer’s disease. Journal of Alzheimer's Disease, 19(1), 341–353. doi:0U88727833343HV6 [pii]. 10.3233/JAD-2010-1222.
Sultana, R., Perluigi, M., & Butterfield, D. A. (2009). Oxidatively modified proteins in Alzheimer's disease (AD), mild cognitive impairment and animal models of AD: Role of Abeta in pathogenesis. Acta Neuropathologica, 118(1), 131–150. doi:10.1007/s00401-009-0517-0.
Sultana, R., Perluigi, M., Newman, S. F., Pierce, W. M., Cini, C., Coccia, R., & Butterfield, D. A. (2010). Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer’s disease. Antioxidants & Redox Signaling, 12(3), 327–336. doi:10.1089/ars.2009.2810.
Tong, X. K., Nicolakakis, N., Fernandes, P., Ongali, B., Brouillette, J., Quirion, R., & Hamel, E. (2009). Simvastatin improves cerebrovascular function and counters soluble amyloid-beta, inflammation and oxidative stress in aged APP mice. Neurobiology of Disease, 35(3), 406–414. doi:DOI 10.1016/j.nbd.2009.06.003.
Trompet, S., van Vliet, P., de Craen, A. J., Jolles, J., Buckley, B. M., Murphy, M. B., Ford, I., Macfarlane, P. W., Sattar, N., Packard, C. J., Stott, D. J., Shepherd, J., Bollen, E. L., Blauw, G. J., Jukema, J. W., & Westendorp, R. G. (2010). Pravastatin and cognitive function in the elderly. Results of the PROSPER study. Journal of Neurology, 257(1), 85–90. doi:10.1007/s00415-009-5271-7.
Wallerath, T., Poleo, D., Li, H., & Forstermann, U. (2003). Red wine increases the expression of human endothelial nitric oxide synthase: a mechanism that may contribute to its beneficial cardiovascular effects. Journal of the American College of Cardiology, 41(3), 471–478. doi:S0735109702028267 [pii].
Werba, J. P., Giroli, M., Cavalca, V., Nava, M. C., Tremoli, E., & Dal Bo, L. (2008). The effect of green tea on simvastatin tolerability. Annals of Internal Medicine, 149(4), 286–287. doi:149/4/286 [pii].
Zigman, W. B., Schupf, N., Jenkins, E. C., Urv, T. K., Tycko, B., & Silverman, W. (2007). Cholesterol level, statin use and Alzheimer’s disease in adults with Down syndrome. Neuroscience Letters, 416(3), 279–284. doi:S0304-3940(07)00176-0 [pii] 10.1016/j.neulet.2007.02.023.
Acknowledgments
The authors are grateful to Dr. Marina Mele for expert graphical assistance. This work was in the frame of a grant of the Italian Ministry of Education, University and Research PRIN 2009 to CM and was supported in part by a NIH grant to D.A.B. [AG-05119] and by an Alzheimer’s Association grant to EH (IIRG 035673).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this entry
Cite this entry
Mancuso, C., Head, E., Barone, E., Perluigi, M., Preziosi, P., Butterfield, D.A. (2014). Potential Therapeutic Effects of Statins in Alzheimer’s Disease. In: Kostrzewa, R. (eds) Handbook of Neurotoxicity. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5836-4_171
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5836-4_171
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5835-7
Online ISBN: 978-1-4614-5836-4
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences