
Abstract
N-Methyl-d-aspartate (NMDA) receptors are key mediators of fast excitatory synaptic transmission within the mammalian central nervous system (CNS) and play vital roles in learning, memory, and synaptic development. Overactivation of NMDA receptors (NMDARs) at glutamatergic synapses often results in excitotoxicity, which can cause neuronal injury and death, and is associated with many neurological disorders such as ischemic stroke, epilepsy, neuropathic pain, traumatic brain and spinal injuries; ocular disorders such as glaucoma; as well as neurodegenerative diseases, including amyotrophic lateral sclerosis, Parkinson’s, Huntington’s, and Alzheimer’s diseases. Numerous NMDAR antagonists have been developed that have been effective in reducing cell damage and death in both in vitro and in vivo experimental models of neurological disease, such as ischemia and traumatic brain injury. Unfortunately, clinical use of these NMDAR antagonists has been limited by intolerable side effects, likely due to the necessity of NMDARs in normal brain function. Intracellular signaling pathways that couple to NMDAR activation can promote either cell survival or cell death, depending on NMDAR subunit composition and/or subcellular localization. Ideal therapeutic strategies targeting NMDAR-mediated neurotoxicity should selectively block pro-death signaling, while sparing pro-survival signals. Neuroprotectants targeting downstream effectors of NMDAR-mediated cell death, rather than NMDARs directly, would maintain normal NMDAR function and minimize adverse effects commonly associated with traditional NMDAR antagonists in the treatment of neurological disorders.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
In the mammalian central nervous system (CNS), the principal excitatory neurotransmitter is glutamate. This neurotransmitter acts on two classes of receptors: ionotropic glutamate receptors that form a central ion channel pore and mediate fast excitatory synaptic transmission and metabotropic glutamate receptors, which are G protein-coupled receptors that play neuromodulatory roles within the CNS (Traynelis et al. 2010). Three types of ionotropic glutamate receptors have been identified and classified based on electrophysiological and pharmacological properties: N-Methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), and kainate, each being named for their preferred pharmacologic agonist (Nakanishi 1992; Hollmann and Heinemann 1994). Activation of ionotropic glutamate receptors leads to the opening of their associated ion channel, which, depending on receptor type, is permeable to sodium (Na+), potassium (K+), and calcium (Ca2+) ions. While AMPA and kainate receptors have modest Ca2+ permeability, NMDA receptors (NMDARs) are highly Ca2+-permeable (MacDermott et al. 1986; McBain and Mayer 1994; Dingledine et al. 1999), and it is this Ca2+ influx that is thought to play a major role in both the physiological as well as pathophysiological functions of the NMDAR (Choi 1988a, b; Tymianski et al. 1993; Ghosh and Greenberg 1995).
The term “excitotoxicity,” first coined by Olney (1969), refers to the overactivation of glutamate receptors due to excessive synaptic release of glutamate. Initial studies on glutamate toxicity were done by Lucas and Newhouse (1957), who showed that systemic injections of l-glutamate into mice destroy the inner neural layers of the retina. These observations of neuronal death following excessive glutamate administration were later extended to the brain (Olney 1969; Olney and de Gubareff 1978) and spinal cord (Regan and Choi 1991). Excitotoxicity is now considered an important mechanism involved in many acute CNS insults, such as ischemia and traumatic brain injury (TBI) as well as in chronic neurodegenerative diseases, such as Parkinson’s (PD), Huntington’s (HD), and Alzheimer’s (AD) diseases (Lipton and Rosenberg 1994; Arundine and Tymianski 2004; Lau and Tymianski 2010).
Glutamate-mediated neurotoxicity arises from excessive Ca2+ influx through NMDARs (Berdichevsky et al. 1983; Choi 1987, 1988a, b; Sattler and Tymianski 2000). Ca2+ overload can trigger many detrimental cellular processes, including activation of calcium-dependent proteases, mitochondrial dysfunction, and death signals (Pivovarova and Andrews 2010; Szydlowska and Tymianski 2010). Additionally, excessive cellular Ca2+ can induce the production of harmful reactive oxygen species. For example, nitric oxide (NO), synthesized by the Ca2+-dependent enzyme neuronal nitric oxide synthase (nNOS), can react with the superoxide anion (O2 −) to form the highly reactive species peroxynitrite, which can cause damage to proteins, lipids, and DNA (Radi et al. 1991a, b; Aarts et al. 2003b). NMDARs are key mediators of excitotoxicity in the CNS and have thus been the targets of many neuroprotective strategies.
2 NMDA Receptors and Their Antagonists
Structurally, NMDARs exist as heterotetramers, typically consisting of two obligatory NR1 subunits and two regulatory NR2 subunits and less commonly including NR3 subunits (Das et al. 1998; Chatterton et al. 2002; Paoletti and Neyton 2007; Cavara and Hollmann 2008; Pachernegg et al. 2012). Four subtypes of NR2 (NR2A-D) can assemble with the NR1 subunit, most commonly NR2A or NR2B in the forebrain. These receptors are typically diheteromeric in nature; that is, they are comprised of either NR1/NR2A or NR1/NR2B subunits. Some NMDARs can also incorporate two different NR2 subunits (Chazot et al. 1994; Chazot and Stephenson 1997); however, few studies have addressed the functional implications of these receptors.
The type of NR2 subunit determines the biophysical and pharmacological properties of NMDARs; furthermore, these subunits govern distinct protein-protein interactions and downstream signaling pathways, by way of the structural diversity of their carboxyl (C)-terminus (Hardingham and Bading 2003; Cull-Candy and Leszkiewicz 2004; Prybylowski and Wenthold 2004; Kohr 2006; Ryan et al. 2008). Alternatively spliced isoforms also exist for some subunits, such as NR1, creating additional functional diversity (Sugihara et al. 1992; Zukin and Bennett 1995). At resting membrane potential, the channel pore of the NMDAR is blocked by magnesium (Mg2+) which prevents ion flow through the channel. Upon membrane depolarization, Mg2+ is expelled, allowing for the passage of ions through the channel. Thus, NMDAR activation requires not only the presynaptic release of glutamate but also the depolarization of the postsynaptic membrane. NMDARs also require simultaneous binding of glutamate and glycine (or d-serine) for activation (Johnson and Ascher 1987; Kleckner and Dingledine 1988; Lerma et al. 1990). The glutamate-binding site is located on the NR2 subunits, while the NR1 subunits provide the glycine-binding site (Furukawa and Gouaux 2003; Furukawa et al. 2005).
Evidence from preclinical research has demonstrated a critical role for NMDAR-mediated neurotoxicity in cellular and animal models of neurological disorders, such as ischemic stroke and TBI (Choi 1988a, b; Tymianski et al. 1993; Arundine et al. 2003). NMDA receptor antagonists targeting the glutamate-binding site, the glycine-binding site, or the channel pore, as well as NR2B subunit-selective antagonists, all showed neuroprotection in preclinical studies of excitotoxicity-related CNS diseases (Danysz and Parsons 2002; Wang and Shuaib 2005). Unfortunately, all clinical trials for neuroprotection in acute disorders including stroke and TBI using NMDAR antagonists, such as Selfotel, Aptiganel, and Gavestinel, have failed. This was largely due either to adverse effects or to limited clinical efficacy (Wood and Hawkinson 1997; Morris et al. 1999; Gladstone et al. 2002; Ikonomidou and Turski 2002; Hoyte et al. 2004; Muir 2006; Kalia et al. 2008; Lau and Tymianski 2010). In addition to undesirable side effects, a short therapeutic window may be a limiting factor with the use of NMDAR antagonists as neuroprotectants in stroke and TBI (Ikonomidou and Turski 2002; Roesler et al. 2003).
In the case of more chronic disorders, an anti-excitotoxic approach using NMDAR antagonists may have a role. For example, memantine is the only NMDAR antagonist currently approved for the treatment of moderate to severe Alzheimer’s disease (AD) in most of Europe, the USA, and Canada, either as a monotherapy or in combination with cholinesterase inhibitors (Thomas and Grossberg 2009). Memantine is a moderate affinity, uncompetitive NMDAR antagonist with strong voltage-dependency and fast kinetics, which allow rapid binding to and quick dissociation from NMDARs. These properties allow memantine to be well tolerated and have few side effects, unlike other NMDAR channel blockers (Danysz and Parsons 2012); however, there is no clear benefit to date for the use of memantine in milder stages of AD and only a small beneficial effect on cognition in vascular dementia (Parsons et al. 1999; McShane et al. 2006; Rammes et al. 2008; Herrmann et al. 2011). Memantine has been studied in several other neurological diseases, both at the preclinical and clinical levels (Stieg et al. 1999; Rao et al. 2001; Volbracht et al. 2006; Rojas et al. 2008; Thomas and Grossberg 2009; Milnerwood et al. 2010), and may have clinical benefit in some disorders, including PD (Aarsland et al. 2009), HD (Beister et al. 2004; Ondo et al. 2007) and HIV-associated dementia (Schifitto et al. 2007; Zhao et al. 2010). Further work is required to establish the full potential of memantine in these diseases.
3 Dual Roles of NMDARs in Health and Disease: Implications for Neuroprotectants
3.1 Importance of NMDAR Subcellular Localization
The critical roles that NMDARs play in normal brain function may help explain why clinical treatment of stroke and brain trauma with NMDAR antagonists has failed. An ideal therapeutic approach to antagonize NMDAR overactivity in neurological disorders would include blocking downstream excitotoxic signaling without directly blocking NMDARs or inhibiting normal excitatory neurotransmission. Recently, dual roles for the NMDAR in both neuronal death and survival have been increasingly appreciated. These depend, at least in part, on the subcellular NMDAR pool that is activated (Hardingham and Bading 2010). At the neuronal plasma membrane, NMDARs are localized to both the synapse and to extrasynaptic regions, and these two receptor populations may have different roles in physiological and pathophysiological processes. Although NMDARs at both locations can mediate excitotoxicity (Sattler et al. 2000; Liu et al. 2007), studies in cultured cortical and hippocampal neurons showed that stimulation of synaptic NMDARs promotes cell survival pathways, whereas extrasynaptic NMDAR activation is more strongly associated with neuronal death (Vanhoutte and Bading 2003; Soriano and Hardingham 2007; Leveille et al. 2008; Hardingham and Bading 2010). This dual role of NMDARs in cell survival and death stems from distinct intracellular signaling pathways that are coupled to synaptic and extrasynaptic NMDARs. Synaptically localized NMDARs promote cell survival by suppressing apoptotic gene expression (Leveille et al. 2010) and activating pro-survival factors such as cyclic AMP response element-binding protein (CREB), a transcription factor that induces expression of genes such as brain-derived neurotrophic factor (BDNF) that is important for survival (Hardingham et al. 2002). In contrast, extrasynaptic NMDARs can mediate cell death by attenuating the CREB pathway, blocking BDNF expression, and promoting mitochondrial dysfunction and cell death (Hardingham et al. 2002; Gouix et al. 2009). Recent work has demonstrated that, similar to cortical neurons, striatal medium-sized spiny neurons also show increased and decreased CREB signaling after stimulation of synaptic and extrasynaptic NMDARs, respectively (Kaufman et al. 2012).
Alterations in synaptic and extrasynaptic NMDAR localization, leading to an imbalance between pro-survival and proapoptotic signaling, have been associated with several neurological disorders (Gladding and Raymond 2011; Sanz-Clemente et al. 2012). For example, enhanced activity of extrasynaptically localized NMDARs was observed in cerebral ischemia (Tu et al. 2010) and Huntington’s disease (HD; Okamoto et al. 2009; Milnerwood et al. 2010). In the striatum of transgenic HD mice, increased NR2B-containing extrasynaptic NMDAR expression, increased current, and associated reductions in nuclear CREB activation were found. Furthermore, this reduction in CREB activity, along with associated motor learning deficits, was reversed by treatment of HD mice with memantine, which was shown to preferentially block extrasynaptic NMDARs (Leveille et al. 2008; Okamoto et al. 2009; Xia et al. 2010). The selective block of extrasynaptic NMDARs by memantine may contribute to its clinical tolerability; that is, memantine may spare physiological synaptic transmission needed for normal brain function but selectively antagonize extrasynaptic NMDARs that are excessively activated under pathological conditions.
3.2 Importance of NMDAR NR2 Subunit Subtype
Seemingly contrary to the idea that neuronal survival and death pathways are mediated by synaptic and extrasynaptic NMDARs, respectively, the well-characterized synapse-specific protein PSD95 (postsynaptic density-95) is required for NMDAR-mediated excitotoxic neuronal death (Sattler et al. 1999; Aarts et al. 2002; Zhou et al. 2010). This apparent paradox may be explained by the temporally and spatially regulated expression of different NMDAR NR2 subunits (Traynelis et al. 2010). In adult forebrain neurons, NR2A-containing NMDARs are preferentially localized to synapses, while NR2B-containing NMDARs are primarily expressed at extrasynaptic sites (Tovar and Westbrook 1999; Liu et al. 2007; Groc et al. 2009). In addition, NR2A-containing NMDARs are thought to mediate cell survival signals, whereas NR2B-containing NMDARs are associated with cell death pathways, in both in vitro and in vivo models of stroke and TBI (DeRidder et al. 2006; Zhou and Baudry 2006; Liu et al. 2007; Chen et al. 2008; Terasaki et al. 2010). However, this distribution profile of NMDAR NR2 subunits is not absolute. NR2A-containing receptors can be found at extrasynaptic sites; likewise, NR2B-containing NMDARs are expressed in the postsynaptic membrane, where they associate with other postsynaptic density proteins, such as PSD95 (Groc et al. 2006; Thomas et al. 2006; Harris and Pettit 2007). Therefore, neuronal death pathways may be activated by either synaptic or extrasynaptic NR2B-containing NMDARs, while activation of either synaptic or extrasynaptic NR2A-containing NMDARs promotes neuronal survival (Liu et al. 2007).
NMDARs are localized in the cell membrane, including that of synapses, by an array of scaffolding proteins. Among these is the abundant PSD95 protein, discussed in greater detail in sections below. In synapses, PSD95 binds the C-termini of NMDAR NR2 subunits and links NMDARs to other proteins found in the multiprotein complex (MPC) with which NMDARs associate. In doing so, PSD95 links NMDARs to signaling proteins within the MPC that mediate excitotoxic neuronal death (Sattler et al. 1999; Aarts et al. 2002; Lai and Wang 2010; Zhou et al. 2010). The linkage between NMDARs and downstream toxic signaling pathways may be strongest with NR2B-containing NMDARs. Lending support to this idea is a recent study (Martel et al. 2012) that used chimeric constructs of NMDAR NR2A and NR2B subunits with reciprocal exchanges of their C-terminal domains to demonstrate that the NR2B C-terminal domain is important for NMDAR-mediated toxicity, regardless of receptor location. As discussed above in the case of PSD95, the precise mechanisms by which NMDAR subunit composition determines functional outcome of the receptor may lie in the specific intracellular signaling proteins that couple to NMDARs, most often via their C-terminal tails. Ideally, a therapeutic strategy for excitotoxicity-related neurological disorders would selectively target only those NMDARs or their downstream signaling components associated with neuronal death, while sparing the cell survival-promoting NMDAR population.
4 NMDAR Signaling at the Postsynaptic Density
At the postsynaptic membrane of central excitatory synapses, ionotropic glutamate receptors are localized into specialized MPCs in an electron-dense region termed the postsynaptic density (PSD) (Kennedy 1997; Sheng 2001). NMDARs are abundantly expressed in association with the PSD (Moon et al. 1994), which is composed of both membranous and cytoplasmic proteins (Ziff 1997; Okabe 2007). Scaffolding proteins, which are major components of the MPC, associate with each other and other MPC proteins via highly specific and often unique protein-protein interactions. Such interactions govern cell-to-cell adhesion, regulation of receptor clustering, and modulation of receptor function (Feng and Zhang 2009; Verpelli et al. 2012). PSD95 (also known as SAP90), the first PSD scaffolding protein to be identified (Cho et al. 1992), is a member of a larger group of scaffolding proteins known as membrane-associated guanylate kinase (MAGUK) proteins (Gardoni 2008; Zheng et al. 2011). MAGUKs contain modular protein interaction domains, such as SH3 (Src homology 3), GK (guanylate kinase), and PDZ (Postsynaptic density-95/Discs large homolog/Zona occludens-1) domains (Sheng and Sala 2001; Gardoni 2008; Zheng et al. 2011).
Many surface receptors, such as the NMDAR, bind to MAGUK PDZ domains via their C-terminal PDZ-binding domains. For example, through its first two PDZ domains, PSD95 can bind NMDAR NR2 subunits at their C-terminus PDZ-binding motif (ESDV) (Kornau et al. 1995; Niethammer et al. 1996). PSD95 connects NMDARs to intracellular signaling proteins, such as neuronal nitric oxide synthase (nNOS). PSD95 also binds directly to nNOS via a PDZ-PDZ interaction that involves the second PDZ domain of PSD95 (Brenman et al. 1996; Craven and Bredt 1998; Christopherson et al. 1999; Tezuka et al. 1999). By physically bringing together the NMDAR and nNOS, PSD95 allows Ca2+ that permeates through NMDARs to preferentially induce the activation of nNOS and thus couples NMDAR activity to the production of nitric oxide (NO), a signaling molecule that mediates NMDAR-dependent excitotoxicity (Dawson et al. 1991, 1993). Disrupting the NMDAR-PSD95-nNOS complex in cultured cortical neurons using antisense oligonucleotides against PSD95 selectively attenuated NMDAR-mediated excitotoxicity, without affecting NMDAR expression or function (Sattler et al. 1999).
5 NMDAR NR2B Signaling: Downstream Effectors of Excitotoxicity as Therapeutic Targets in Neurological Diseases
5.1 NR2B-PSD95-nNOS Signaling Complex
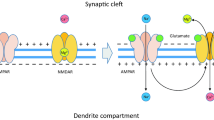
Because suppression of PSD95 using antisense oligonucleotides is impractical in the treatment of excitotoxicity-related disorders in humans, an alternative strategy for disrupting the NR2B-PSD95-nNOS signaling complex is to use drug compounds that block protein interactions within this complex. One such drug compound is NR2B9c (Aarts et al. 2002). The NR2B9c peptide, comprising the nine C-terminal residues of NR2B (KLSSIESDV), can be rendered cell permeant by fusion to the 11 residue protein transduction domain of the human immunodeficiency virus type 1 (HIV-1) Tat protein (Schwarze et al. 1999). This produces a 20-amino acid peptide termed Tat-NR2B9c. This peptide permeates through cell membranes and disrupts the intracellular interaction of NMDARs with PSD95 (See Fig. 1). This effectively dissociates NMDARs from downstream neurotoxic signaling. However, unlike NMDAR antagonists, this occurs without blocking synaptic activity or calcium influx. This approach is feasible in vivo: when applied either before, 1 or 3 h after an insult, Tat-NR2B9c protected cultured neurons from excitotoxicity and reduced transient middle cerebral artery occlusion (MCAO)-mediated ischemic brain damage in rats (Aarts et al. 2002; Sun et al. 2008).

The NMDAR-PSD95-nNOS signaling complex. (a) By binding to both the NMDAR and nNOS, PSD95 brings nNOS in close proximity to the NMDAR and allows Ca2+ entering the neuron through the NMDAR to preferentially induce the activation of nNOS, leading to cell death. (b) The Tat-NR2B9c peptide disrupts the NMDAR-PSD95-nNOS complex, dissociating NMDARs from downstream neurotoxic signaling, without blocking synaptic activity or calcium influx
A proteomic and biochemical analysis of the interactions of Tat-NR2B9c with most or all PDZs in the human genome revealed that the disruption of the NMDAR-PSD95-nNOS complex by Tat-NR2B9c was highly specific. Because the peptide targets the first and second PDZ domains of PSD95, where both the NMDAR and nNOS bind, Tat-NR2B9c inhibited the interaction between both PSD95 and NR2 subunits as well as between PSD95 and nNOS (Cui et al. 2007). Importantly, Tat-NR2B9c administered 3 h after both permanent and transient focal ischemia reduced infarct volumes in rats and improved long-term neurobehavior, including sensorimotor function, emotionality, and cognition (Sun et al. 2008), raising the possibility for clinical usefulness in a wide therapeutic window. Further downstream, the consequences of administering Tat-NR2B9c are to ameliorate excitotoxic neuronal loss in vitro and ischemic cortical damage in vivo by impairing pro-death p38 signaling, without affecting NMDAR-mediated pro-survival pathways involving CREB or Akt (Soriano et al. 2008; Martel et al. 2009). Other studies have proposed that compounds other than Tat-NR2B9c that target the NMDAR-PSD95 interaction may also have neuroprotective effects. Disruption of the PSD95-nNOS interaction using a small molecule inhibitor (ZL006) is suggested to be neuroprotective in ischemia (Zhou et al. 2010). In addition, the use of a dimeric inhibitor, Tat-NPEG4(IETDV)2 (Tat-N-dimer), which binds the tandem PDZ1-2 domain of PSD95, reduced infarct volume in mice subjected to cerebral ischemia (Bach et al. 2012).
Recently, Tat-NR2B9c was used for the treatment of stroke in cynomolgus macaques, higher-order gyrencephalic nonhuman primates, which bear anatomical and behavioral similarities to humans (Cook et al. 2012). Animals treated with Tat-NR2B9c up to 3 h after MCAO showed significant reduction in infarct volume, as measured by magnetic resonance imaging and histology. In addition, Tat-NR2B9c-treated macaques showed improved neurobehavioral outcomes assessed using the nonhuman primate stroke scale (NHPSS). This neuroprotection preserved cellular functionality, as gauged by the capacity for gene transcription in ischemic brain tissue. The 3-h time frame for neuroprotection with Tat-NR2B9c is significant as it suggests a broader clinical applicability. Tat-NR2B9c has already entered clinical trials; the ENACT (Evaluating Neuroprotection in Aneurysm Coiling Therapy) Phase 2 clinical trial (ClinicalTrials.gov number, NCT00728182) to determine the safety and efficacy of Tat-NR2B9c in reducing embolic strokes in patients undergoing endovascular repair of brain aneurysms was recently successfully completed (Hill et al. 2012). The encouraging results from this trial warrant further investigation of Tat-NR2B9c in the clinical treatment of stroke.
Since the first study (Aarts et al. 2002) utilizing blocking peptides against PSD95 in stroke, subsequent work using similar strategies has been carried out to investigate other excitotoxicity-mediated neurological diseases, such as epilepsy, AD, and neuropathic pain. Administration of Tat-NR2B9c in rats 3 h after the termination of status epilepticus reduced cell loss in regions of the hippocampus (Dykstra et al. 2009). Perturbing the NR2B-PSD95 association with Tat-NR2B9c ameliorated Aβ-mediated toxicity in vitro and was sufficient to prevent lethality and memory deficits in an AD mouse model (Ittner et al. 2010). The importance of the NR2B9c-PSD95-nNOS signaling complex in neuropathic pain has been demonstrated using a variety of genetic, biochemical, and proteomic techniques. These include the use of antisense oligonucleotides against PSD95 (Tao et al. 2001, 2003), small molecule inhibitors that block the interaction between PSD95 and nNOS (Florio et al. 2009), or Tat-fusion peptides comprising regions of PSD95 (Tao et al. 2008), nNOS (Florio et al. 2009), or the NMDAR NR2B subunit (D’Mello et al. 2011).
5.2 NR2B-ND2-Src Signaling Complex
NMDAR activity is governed in part by the balance between phosphorylation and dephosphorylation (Salter and Kalia 2004). Tyrosine phosphorylation upregulates the activity of NMDARs (Wang and Salter 1994; Zheng et al. 1998), and the nonreceptor tyrosine kinase Src (Ohnishi et al. 2011) can mediate this upregulation. Within the NMDAR complex, Src, along with the phosphotyrosine phosphatase STEP (striatal enriched tyrosine phosphatase), act in opposition to phosphorylate or dephosphorylate the NMDAR, respectively. STEP61, the membrane-associated STEP isoform (Sharma et al. 1995; Bult et al. 1996), suppresses NMDAR activity by opposing the actions of Src (Pelkey et al. 2002). Src-mediated enhanced phosphorylation of NMDARs is associated with synaptic plasticity (Rostas et al. 1996; Salter 1998) as well as pathophysiological conditions such as ischemia (Cheung et al. 2003), HD (Song et al. 2003), epilepsy (Sanna et al. 2000; Moussa et al. 2001), and neuropathic pain (Salter and Pitcher 2012). Inhibition of Src activity may attenuate brain injury in ischemia (Jiang et al. 2008; Liang et al. 2009), supporting a role for Src in excitotoxic processes.
Src is anchored to the NMDAR via the adaptor protein NADH dehydrogenase 2 (ND2) (Gingrich et al. 2004). ND2 brings Src in close proximity to the NMDAR and allows Src to upregulate NMDAR function through phosphorylation of NR2 subunits. Disruption of the NMDAR-ND2-Src signaling complex has been shown to suppress pain behaviors. A 10-amino acid peptide, Src40-49, comprising the region of Src needed to bind ND2, was able to inhibit the binding of Src to ND2, thus releasing Src from the NMDAR complex and inhibiting Src-mediated enhancement of NMDAR activity (Liu et al. 2008). Importantly, uncoupling of Src from the NMDAR complex with the membrane-permeable Src40-49Tat fusion peptide suppressed pain hypersensitivity induced by inflammation and peripheral nerve injury (see Fig. 2). Thus, by regulating the function of NMDARs through phosphorylation and dephosphorylation, Src and STEP play vital roles in both physiological as well as pathophysiological glutamatergic neurotransmission. Furthermore, both Src and STEP themselves are subject to regulation. For example, differential regulation of STEP61 may play an important role in mediating the dual roles of the NMDAR in cell survival and death (Xu et al. 2009). Synaptic NMDAR activation promotes the ubiquitination and degradation of STEP61, concomitant with ERK1/2 activation and cell survival. In contrast, extrasynaptic NMDAR stimulation leads to calpain-mediated proteolysis of STEP61, activation of p38, and cell death. Aberrant regulation of both Src and STEP may therefore be involved in NMDAR-mediated pathophysiological conditions (Hossain et al. 2012; Salter and Pitcher 2012). Although targeting STEP or the Src:ND2 interaction has not been tested in ischemia, it is another example of a protein-protein interaction that may be targeted therapeutically in disorders that depend on NMDAR overactivity.

The NMDAR-ND2-Src signaling complex. (a) Src is anchored within the NMDAR complex via the adaptor protein NADH dehydrogenase 2 (ND2). By bringing Src in close proximity to the NMDAR, ND2 allows Src to phosphorylate the receptor and enhance NMDAR activity, resulting in hypersensitivity to pain. (b) Using a Src40–49Tat peptide to block the interaction between Src and ND2 dissociates Src from ND2, thereby inhibiting Src-mediated upregulation of NMDAR activity and suppressing pain behaviors (Figure adapted from Liu et al. 2008)
5.3 Other Signaling Pathways Implicated in NMDAR-Mediated Neurotoxicity
Other signaling pathways downstream of NMDARs have been identified to play roles in excitotoxic signaling. For example, during cerebral ischemia, death-associated protein kinase 1 (DAPK1) is recruited to extrasynaptic NMDAR NR2B subunits in the cortex of mice (Tu et al. 2010). DAPK1 directly binds to NR2B, phosphorylates it, and enhances NMDAR channel conductance, leading to increased excitotoxic signaling. Peptides that inhibit the interaction between DAPK1 and the NMDAR NR2B subunit reduced brain infarction and improved neurological function in mice. DAPK1 itself is subject to regulation by other signaling molecules, such as ERK1/2, which may affect NMDAR activity (Liu et al. 2012). Another signaling protein involved in cell death downstream from the NMDAR is PTEN (phosphatase and tensin homolog deleted on chromosome 10). PTEN is recruited selectively to NMDAR NR2B subunits (Ning et al. 2004), potentiates neurotoxic NMDAR activity, and inhibits the PI3K (phosphatidylinositol 3-kinase) survival pathway, thus contributing to excitotoxic neuronal death in stroke. PTEN-induced kinase 1 (PINK1) in a ubiquitous kinase thought to function in cell survival-promoting pathways (Wilhelmus et al. 2012). Overactivation of NR2B-containing NMDARs may contribute to ischemic cell death by suppressing PINK1-dependent survival signaling (Shan et al. 2009). Thus, drug compounds that are able to increase PINK1 levels and activity may prove to be neuroprotective in NMDAR-mediated neurotoxicity.
Activation of NMDARs has been linked to the modulation of several transcription factors that can mediate either pro-survival or pro-death pathways (Hardingham et al. 2002; Zou and Crews 2006; Lai et al. 2011), and these may be useful targets for therapeutic intervention. One example is the transcription factor SREBP-1 (sterol regulatory element-binding protein-1) (Taghibiglou et al. 2009), which typically controls lipid biosynthesis genes but whose activation is essential in NMDAR-mediated excitotoxic neuronal death in both in vitro and in vivo models of stroke. Therapies aimed at pro-death transcription factors downstream of NMDAR-mediated excitotoxic pathways may offer some novel therapeutic targets in the treatment of neurological diseases. Since activation of these transcription factors is often delayed, relative to NMDAR activation, neuroprotective strategies targeting these proteins may offer a wider therapeutic window if such pathways participate in the cell injury process.
Cell death pathways also exist that may occur in parallel with and converge upon NMDAR-mediated excitotoxic signaling mechanisms (Tymianski 2011). TRPM7 and TRPM2 are members of the transient receptor potential (TRP) channel superfamily (Nilius and Owsianik 2011) that are widely expressed and function in diverse cellular processes, including death, survival, and proliferation. Both TRPM2 and TRPM7 are nonselective cation channels that are activated by oxidative stress and are thought to play significant roles in ischemic cell death (Aarts et al. 2003a; MacDonald et al. 2006; Sun et al. 2009; Xie et al. 2010). TRPM2 and TRPM7 may represent additional Ca2+ influx pathways, other than NMDARs, in mediating neurotoxicity in ischemia. Neurotoxic signals such as reactive oxygen species downstream of NMDARs may in turn activate TRPM2 and TRPM7 channels to form a positive feedback loop that perpetuates ischemic cell death.
6 Conclusion
NMDARs have important physiological roles mediating synaptic plasticity and neurodevelopment. However, overactivation of NMDARs leads to excitotoxicity, common to many neurological diseases such as ischemic stroke, epilepsy, neuropathic pain, traumatic brain and spinal injuries; ocular disorders such as glaucoma; as well as neurodegenerative diseases, including amyotrophic lateral sclerosis, Parkinson’s, Huntington’s, and Alzheimer’s diseases. Antagonists that directly inhibit NMDARs have limited use clinically, possibly due to intolerable side effects at anti-excitotoxic doses. Intracellular signaling pathways that couple to NMDAR activation can promote either cell survival or cell death, depending on NMDAR subunit composition and/or subcellular localization. Extrasynaptic NMDARs, and especially those receptors containing NR2B subunits, engage in cell death pathways upon excessive glutamate stimulation. Conversely, activation of synaptically localized NMDARs, and especially those receptors containing NR2A subunits, may result in neuroprotection. Therapeutic strategies targeting NMDAR-mediated neurotoxicity should preferentially block pro-death signaling while sparing pro-survival signals. Since neuroprotectants that selectively target the molecular components involved in pro-death signaling downstream of the NMDAR do not directly inhibit NMDARs themselves, they may be better tolerated than traditional NMDAR antagonists.
The last decade of scientific research has shed light on the molecular mechanisms involved in NMDAR-mediated excitotoxicity and has revealed additional targets for neuroprotective strategies in the treatment of neurological disorders; however, many questions remain unanswered. First, in addition to NR1/NR2A and NR1/NR2B diheteromeric NMDARs, triheteromeric receptors comprising NR1/NR2A/N2B are also believed to exist. The roles of these triheteromeric receptors, in both health and disease, have yet to be elucidated. Second, several signaling molecules downstream of NMDARs, such as nNOS, DAPK1, and PTEN, play roles in excitotoxic signaling to promote cell damage and death. Whether and how these signaling molecules cross talk with one another is not known. Third, in conditions such as ischemia, excessive Ca2+ influx through NMDARs leads to the formation of reactive oxygen species, which can activate other plasma membrane channels, such as TRPM2 and TRPM7 channels; these channels thus represent additional therapeutic targets. Lastly, a major part of our understanding about NMDAR-mediated cell death pathways comes from models of cerebral ischemia. Future work will need to determine whether these pathways also play a role in other neurological diseases that involve excitotoxic mechanisms and whether preclinical neuroprotective therapies can be translated to clinical use.
References
Aarsland, D., Ballard, C., Walker, Z., Bostrom, F., Alves, G., Kossakowski, K., Leroi, I., Pozo-Rodriguez, F., Minthon, L., & Londos, E. (2009). Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: A double-blind, placebo-controlled, multicentre trial. Lancet Neurology, 8, 613–618.
Aarts, M., Liu, Y., Liu, L., Besshoh, S., Arundine, M., Gurd, J. W., Wang, Y. T., Salter, M. W., & Tymianski, M. (2002). Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science, 298, 846–850.
Aarts, M., Iihara, K., Wei, W. L., Xiong, Z. G., Arundine, M., Cerwinski, W., MacDonald, J. F., & Tymianski, M. (2003a). A key role for TRPM7 channels in anoxic neuronal death. Cell, 115, 863–877.
Aarts, M. M., Arundine, M., & Tymianski, M. (2003b). Novel concepts in excitotoxic neurodegeneration after stroke. Expert Reviews in Molecular Medicine, 5, 1–22.
Arundine, M., & Tymianski, M. (2004). Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cellular and Molecular Life Sciences, 61, 657–668.
Arundine, M., Chopra, G. K., Wrong, A., Lei, S., Aarts, M. M., MacDonald, J. F., & Tymianski, M. (2003). Enhanced vulnerability to NMDA toxicity in sublethal traumatic neuronal injury in vitro. Journal of Neurotrauma, 20, 1377–1395.
Bach, A., Clausen, B. H., Moller, M., Vestergaard, B., Chi, C. N., Round, A., Sorensen, P. L., Nissen, K. B., Kastrup, J. S., Gajhede, M., Jemth, P., Kristensen, A. S., Lundstrom, P., Lambertsen, K. L., & Stromgaard, K. (2012). A high-affinity, dimeric inhibitor of PSD-95 bivalently interacts with PDZ1-2 and protects against ischemic brain damage. Proceedings of the National Academy of Sciences of the United States of America, 109, 3317–3322.
Beister, A., Kraus, P., Kuhn, W., Dose, M., Weindl, A., & Gerlach, M. (2004). The N-methyl-d-aspartate antagonist memantine retards progression of Huntington’s disease. Journal of Neural Transmission. Supplementum, 68, 117–122.
Berdichevsky, E., Riveros, N., Sanchez-Armass, S., & Orrego, F. (1983). Kainate, N-methylaspartate and other excitatory amino acids increase calcium influx into rat brain cortex cells in vitro. Neuroscience Letters, 36, 75–80.
Brenman, J. E., Chao, D. S., Gee, S. H., McGee, A. W., Craven, S. E., Santillano, D. R., Wu, Z., Huang, F., Xia, H., Peters, M. F., Froehner, S. C., & Bredt, D. S. (1996). Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell, 84, 757–767.
Bult, A., Zhao, F., Dirkx, R., Jr., Sharma, E., Lukacsi, E., Solimena, M., Naegele, J. R., & Lombroso, P. J. (1996). STEP61: A member of the family of brain-enriched PTPs is localized to the endoplasmic reticulum. The Journal of Neuroscience, 16, 7821–7831.
Cavara, N. A., & Hollmann, M. (2008). Shuffling the deck anew: How NR3 tweaks NMDA receptor function. Molecular Neurobiology, 38, 16–26.
Chatterton, J. E., Awobuluyi, M., Premkumar, L. S., Takahashi, H., Talantova, M., Shin, Y., Cui, J., Tu, S., Sevarino, K. A., Nakanishi, N., Tong, G., Lipton, S. A., & Zhang, D. (2002). Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature, 415, 793–798.
Chazot, P. L., & Stephenson, F. A. (1997). Molecular dissection of native mammalian forebrain NMDA receptors containing the NR1 C2 exon: Direct demonstration of NMDA receptors comprising NR1, NR2A, and NR2B subunits within the same complex. Journal of Neurochemistry, 69, 2138–2144.
Chazot, P. L., Coleman, S. K., Cik, M., & Stephenson, F. A. (1994). Molecular characterization of N-methyl-d-aspartate receptors expressed in mammalian cells yields evidence for the coexistence of three subunit types within a discrete receptor molecule. The Journal of Biological Chemistry, 269, 24403–24409.
Chen, M., Lu, T. J., Chen, X. J., Zhou, Y., Chen, Q., Feng, X. Y., Xu, L., Duan, W. H., & Xiong, Z. Q. (2008). Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke, 39, 3042–3048.
Cheung, H. H., Teves, L., Wallace, M. C., & Gurd, J. W. (2003). Inhibition of protein kinase C reduces ischemia-induced tyrosine phosphorylation of the N-methyl-d-aspartate receptor. Journal of Neurochemistry, 86, 1441–1449.
Cho, K. O., Hunt, C. A., & Kennedy, M. B. (1992). The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron, 9, 929–942.
Choi, D. W. (1987). Ionic dependence of glutamate neurotoxicity. The Journal of Neuroscience, 7, 369–379.
Choi, D. W. (1988a). Glutamate neurotoxicity and diseases of the nervous system. Neuron, 1, 623–634.
Choi, D. W. (1988b). Calcium-mediated neurotoxicity: Relationship to specific channel types and role in ischemic damage. Trends in Neurosciences, 11, 465–469.
Christopherson, K. S., Hillier, B. J., Lim, W. A., & Bredt, D. S. (1999). PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. The Journal of Biological Chemistry, 274, 27467–27473.
Cook, D. J., Teves, L., & Tymianski, M. (2012). Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature, 483, 213–217.
Craven, S. E., & Bredt, D. S. (1998). PDZ proteins organize synaptic signaling pathways. Cell, 93, 495–498.
Cui, H., Hayashi, A., Sun, H. S., Belmares, M. P., Cobey, C., Phan, T., Schweizer, J., Salter, M. W., Wang, Y. T., Tasker, R. A., Garman, D., Rabinowitz, J., Lu, P. S., & Tymianski, M. (2007). PDZ protein interactions underlying NMDA receptor-mediated excitotoxicity and neuroprotection by PSD-95 inhibitors. The Journal of Neuroscience, 27, 9901–9915.
Cull-Candy, S. G. & Leszkiewicz, D. N. (2004). Role of distinct NMDA receptor subtypes at central synapses. Science STKE, 2004, re16.
Danysz, W., & Parsons, C. G. (2002). Neuroprotective potential of ionotropic glutamate receptor antagonists. Neurotoxicity Research, 4, 119–126.
Danysz, W., & Parsons, C. G. (2012). Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine – Searching for the connections. British Journal of Pharmacology, 167, 324–352.
Das, S., Sasaki, Y. F., Rothe, T., Premkumar, L. S., Takasu, M., Crandall, J. E., Dikkes, P., Conner, D. A., Rayudu, P. V., Cheung, W., Chen, H. S. V., Lipton, S. A., & Nakanishi, N. (1998). Increased NMDA current and spine density in mice lacking the NMDA receptor subunit NR3A. Nature, 393, 377–381.
Dawson, V. L., Dawson, T. M., London, E. D., Bredt, D. S., & Snyder, S. H. (1991). Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proceedings of the National Academy of Sciences of the United States of America, 88, 6368–6371.
Dawson, V. L., Dawson, T. M., Bartley, D. A., Uhl, G. R., & Snyder, S. H. (1993). Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. The Journal of Neuroscience, 13, 2651–2661.
DeRidder, M. N., Simon, M. J., Siman, R., Auberson, Y. P., Raghupathi, R., & Meaney, D. F. (2006). Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiology of Disease, 22, 165–176.
Dingledine, R., Borges, K., Bowie, D., & Traynelis, S. F. (1999). The glutamate receptor ion channels. Pharmacological Reviews, 51, 7–61.
D’Mello, R., Marchand, F., Pezet, S., McMahon, S. B., & Dickenson, A. H. (2011). Perturbing PSD-95 interactions with NR2B-subtype receptors attenuates spinal nociceptive plasticity and neuropathic pain. Molecular Therapy, 19, 1780–1792.
Dykstra, C. M., Ratnam, M., & Gurd, J. W. (2009). Neuroprotection after status epilepticus by targeting protein interactions with postsynaptic density protein 95. Journal of Neuropathology and Experimental Neurology, 68, 823–831.
Feng, W., & Zhang, M. (2009). Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nature Reviews Neuroscience, 10, 87–99.
Florio, S. K., Loh, C., Huang, S. M., Iwamaye, A. E., Kitto, K. F., Fowler, K. W., Treiberg, J. A., Hayflick, J. S., Walker, J. M., Fairbanks, C. A., & Lai, Y. (2009). Disruption of nNOS-PSD95 protein-protein interaction inhibits acute thermal hyperalgesia and chronic mechanical allodynia in rodents. British Journal of Pharmacology, 158, 494–506.
Furukawa, H., & Gouaux, E. (2003). Mechanisms of activation, inhibition and specificity: Crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO Journal, 22, 2873–2885.
Furukawa, H., Singh, S. K., Mancusso, R., & Gouaux, E. (2005). Subunit arrangement and function in NMDA receptors. Nature, 438, 185–192.
Gardoni, F. (2008). MAGUK proteins: New targets for pharmacological intervention in the glutamatergic synapse. European Journal of Pharmacology, 585, 147–152.
Ghosh, A., & Greenberg, M. E. (1995). Calcium signaling in neurons: Molecular mechanisms and cellular consequences. Science, 268, 239–247.
Gingrich, J. R., Pelkey, K. A., Fam, S. R., Huang, Y., Petralia, R. S., Wenthold, R. J., & Salter, M. W. (2004). Unique domain anchoring of Src to synaptic NMDA receptors via the mitochondrial protein NADH dehydrogenase subunit 2. Proceedings of the National Academy of Sciences of the United States of America, 101, 6237–6242.
Gladding, C. M., & Raymond, L. A. (2011). Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Molecular and Cellular Neurosciences, 48, 308–320.
Gladstone, D. J., Black, S. E., & Hakim, A. M. (2002). Toward wisdom from failure: Lessons from neuroprotective stroke trials and new therapeutic directions. Stroke, 33, 2123–2136.
Gouix, E., Leveille, F., Nicole, O., Melon, C., Had-Aissouni, L., & Buisson, A. (2009). Reverse glial glutamate uptake triggers neuronal cell death through extrasynaptic NMDA receptor activation. Molecular and Cellular Neurosciences, 40, 463–473.
Groc, L., Bard, L., & Choquet, D. (2009). Surface trafficking of N-methyl-d-aspartate receptors: Physiological and pathological perspectives. Neuroscience, 158, 4–18.
Groc, L., Heine, M., Cousins, S. L., Stephenson, F. A., Lounis, B., Cognet, L., & Choquet, D. (2006). NMDA receptor surface mobility depends on NR2A-2B subunits. Proceedings of the National Academy of Sciences of the United States of America, 103, 18769–18774.
Hardingham, G. E., & Bading, H. (2003). The Yin and Yang of NMDA receptor signaling. Trends in Neurosciences, 26, 81–89.
Hardingham, G. E., & Bading, H. (2010). Synaptic versus extrasynaptic NMDA receptor signaling: Implications for neurodegenerative disorders. Nature Reviews Neuroscience, 11, 682–696.
Hardingham, G. E., Fukunaga, Y., & Bading, H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nature Neuroscience, 5, 405–414.
Harris, A. Z., & Pettit, D. L. (2007). Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. The Journal of Physiology, 584, 509–519.
Herrmann, N., Li, A., & Lanctot, K. (2011). Memantine in dementia: A review of the current evidence. Expert Opinion on Pharmacotherapy, 12, 787–800.
Hill, M. D. M., Martin, R. H., Mikulis, D., Wong, J. H., Silver, F. L., terBrugge, K. G., Milot, G., Clarke, W. M., MacDonald, R. L., Kelly, M. E., Boulton, M., Fleetwood, I., MacDougall, C., Gunnarsson, T., Chow, M., Lum, C., Dodd, R., Poublanc, J., Krings, T., Demchuk, A. M., Goyal, M., Anderson, R., Bishop, J., Garman, D., & Tymianski, M. (2012). Safety and efficacy of NA-1 for neuroprotection in iatrogenic stroke after endovascular aneurysm repair: A randomized controlled trial. Lancet Neurology, 12, 70225–70229.
Hollmann, M., & Heinemann, S. (1994). Cloned glutamate receptors. Annual Review of Neuroscience, 17, 31–108.
Hossain, M. I., Kamaruddin, M. A., & Cheng, H. C. (2012). Aberrant regulation and function of Src family tyrosine kinases: Their potential contributions to glutamate-induced neurotoxicity. Clinical and Experimental Pharmacology and Physiology, 39, 684–691.
Hoyte, L., Barber, P. A., Buchan, A. M., & Hill, M. D. (2004). The rise and fall of NMDA antagonists for ischemic stroke. Current Molecular Medicine, 4, 131–136.
Ikonomidou, C., & Turski, L. (2002). Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurology, 1, 383–386.
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., Wolfing, H., Chieng, B. C., Christie, M. J., Napier, I. A., Eckert, A., Staufenbiel, M., Hardeman, E., & Gotz, J. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell, 142, 387–397.
Jiang, X., Mu, D., Biran, V., Faustino, J., Chang, S., Rincon, C. M., Sheldon, A., & Ferriero, D. M. (2008). Activated Src kinases interact with the N-methyl-d-aspartate receptor after neonatal brain ischemia. Annals of Neurology, 63, 632–641.
Johnson, J. W., & Ascher, P. (1987). Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature, 325, 529–531.
Kalia, L. V., Kalia, S. K., & Salter, M. W. (2008). NMDA receptors in clinical neurology: Excitatory times ahead. Lancet Neurology, 7, 742–755.
Kaufman, A. M., Milnerwood, A. J., Sepers, M. D., Coquinco, A., She, K., Wang, L., Lee, H., Craig, A. M., Cynader, M., & Raymond, L. A. (2012). Opposing roles of synaptic and extrasynaptic NMDA receptor signaling in cocultured striatal and cortical neurons. The Journal of Neuroscience, 32, 3992–4003.
Kennedy, M. B. (1997). The postsynaptic density at glutamatergic synapses. Trends in Neurosciences, 20, 264–268.
Kleckner, N. W., & Dingledine, R. (1988). Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science, 241, 835–837.
Kohr, G. (2006). NMDA receptor function: Subunit composition versus spatial distribution. Cell and Tissue Research, 326, 439–446.
Kornau, H. C., Schenker, L. T., Kennedy, M. B., & Seeburg, P. H. (1995). Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science, 269, 1737–1740.
Lai, T. W., & Wang, Y. T. (2010). Fashioning drugs for stroke. Nature Medicine, 16, 1376–1378.
Lai, T. W., Shyu, W. C., & Wang, Y. T. (2011). Stroke intervention pathways: NMDA receptors and beyond. Trends in Molecular Medicine, 17, 266–275.
Lau, A., & Tymianski, M. (2010). Glutamate receptors, neurotoxicity and neurodegeneration. Pflügers Archiv, 460, 525–542.
Lerma, J., Zukin, R. S., & Bennett, M. V. (1990). Glycine decreases desensitization of N-methyl-d-aspartate (NMDA) receptors expressed in Xenopus oocytes and is required for NMDA responses. Proceedings of the National Academy of Sciences of the United States of America, 87, 2354–2358.
Leveille, F., El Gaamouch, F., Gouix, E., Lecocq, M., Lobner, D., Nicole, O., & Buisson, A. (2008). Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. The FASEB Journal, 22, 4258–4271.
Leveille, F., Papadia, S., Fricker, M., Bell, K. F., Soriano, F. X., Martel, M. A., Puddifoot, C., Habel, M., Wyllie, D. J., Ikonomidou, C., Tolkovsky, A. M., & Hardingham, G. E. (2010). Suppression of the intrinsic apoptosis pathway by synaptic activity. The Journal of Neuroscience, 30, 2623–2635.
Liang, S., Pong, D., Gonzales, C., Chen, Y., Ling, H. P., Mark, R. J., Boschelli, F., Boschelli, D. H., Ye, F., Barrios Sosa, A. C., Mansour, T. S., Frost, P., Wood, A., Pangalos, M. N., & Zaleska, M. M. (2009). Neuroprotective profile of novel Src kinase inhibitors in rodent models of cerebral ischemia. The Journal of Pharmacology and Experimental Therapeutics, 331, 827–835.
Lipton, S. A., & Rosenberg, P. A. (1994). Excitatory amino acids as a final common pathway for neurologic disorders. The New England Journal of Medicine, 330, 613–622.
Liu, Y., Wong, T. P., Aarts, M., Rooyakkers, A., Liu, L., Lai, T. W., Wu, D. C., Lu, J., Tymianski, M., Craig, A. M., & Wang, Y. T. (2007). NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. The Journal of Neuroscience, 27, 2846–2857.
Liu, X. J., Gingrich, J. R., Vargas-Caballero, M., Dong, Y. N., Sengar, A., Beggs, S., Wang, S. H., Ding, H. K., Frankland, P. W., & Salter, M. W. (2008). Treatment of inflammatory and neuropathic pain by uncoupling Src from the NMDA receptor complex. Nature Medicine, 14, 1325–1332.
Liu, S. B., Zhang, N., Guo, Y. Y., Zhao, R., Shi, T. Y., Feng, S. F., Wang, S. Q., Yang, Q., Li, X. Q., Wu, Y. M., Ma, L., Hou, Y., Xiong, L. Z., Zhang, W., & Zhao, M. G. (2012). G-protein-coupled receptor 30 mediates rapid neuroprotective effects of estrogen via depression of NR2B-containing NMDA receptors. The Journal of Neuroscience, 32, 4887–4900.
Lucas, D. R., & Newhouse, J. P. (1957). The toxic effect of sodium l-glutamate on the inner layers of the retina. AMA Archives of Ophthalmology, 58, 193–201.
MacDermott, A. B., Mayer, M. L., Westbrook, G. L., Smith, S. J., & Barker, J. L. (1986). NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature, 321, 519–522.
MacDonald, J. F., Xiong, Z. G., & Jackson, M. F. (2006). Paradox of Ca2+ signaling, cell death and stroke. Trends in Neurosciences, 29, 75–81.
Martel, M. A., Soriano, F. X., Baxter, P., Rickman, C., Duncan, R., Wyllie, D. J., & Hardingham, G. E. (2009). Inhibiting pro-death NMDA receptor signaling dependent on the NR2 PDZ ligand may not affect synaptic function or synaptic NMDA receptor signaling to gene expression. Channels (Austin, Tex.), 3, 12–15.
Martel, M. A., Ryan, T. J., Bell, K. F. S., Fowler, J. H., McMahon, A., Al-Mubarak, B., Komiyama, N. H., Horsburgh, K., Kind, P. C., Grant, S. G. N., & Wyllie, D. J. A. (2012). The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron, 74, 543–556.
McBain, C. J., & Mayer, M. L. (1994). N-methyl-d-aspartic acid receptor structure and function. Physiological Reviews, 74, 723–760.
McShane, R., Areosa, S. A., & Minakaran, N. (2006). Memantine for dementia. Cochrane Database of Systematic Reviews, 2, 1–42.
Milnerwood, A. J., Gladding, C. M., Pouladi, M. A., Kaufman, A. M., Hines, R. M., Boyd, J. D., Ko, R. W. Y., Vasuta, O. C., Graham, R. K., Hayden, M. R., Murphy, T. H., & Raymond, L. A. (2010). Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron, 65, 178–190.
Moon, I. S., Apperson, M. L., & Kennedy, M. B. (1994). The major tyrosine-phosphorylated protein in the postsynaptic density fraction is N-methyl-d-aspartate receptor subunit 2B. Proceedings of the National Academy of Sciences of the United States of America, 91, 3954–3958.
Morris, G. F., Bullock, R., Marshall, S. B., Marmarou, A., Maas, A., & Marshall, L. F. (1999). Failure of the competitive N-Methyl-d-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two Phase III clinical trials. The Selfotel Investigators. Journal of Neurosurgery, 91, 737–743.
Moussa, R. C., Ikeda-Douglas, C. J., Thakur, V., Milgram, N. W., & Gurd, J. W. (2001). Seizure activity results in increased tyrosine phosphorylation of the N-methyl-d-aspartate receptor in the hippocampus. Brain Research. Molecular Brain Research, 95, 36–47.
Muir, K. W. (2006). Glutamate-based therapeutic approaches: clinical trials with NMDA antagonists. Current Opinion in Pharmacology, 6, 53–60.
Nakanishi, S. (1992). Molecular diversity of glutamate receptors and implications for brain function. Science, 258, 597–603.
Niethammer, M., Kim, E., & Sheng, M. (1996). Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. The Journal of Neuroscience, 16, 2157–2163.
Nilius, B., & Owsianik, G. (2011). The transient receptor potential family of ion channels. Genome Biology, 12, 218–228.
Ning, K., Pei, L., Liao, M., Liu, B., Zhang, Y., Jiang, W., Mielke, J. G., Li, L., Chen, Y., El-Hayek, Y. H., Fehlings, M. G., Zhang, X., Liu, F., Eubanks, J., & Qan, Q. (2004). Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. The Journal of Neuroscience, 24, 4052–4060.
Ohnishi, H., Murata, Y., Okazawa, H., & Matozaki, T. (2011). Src family kinases: modulators of neurotransmitter receptor function and behavior. Trends in Neurosciences, 34, 629–637.
Okabe, S. (2007). Molecular anatomy of the postsynaptic density. Molecular and Cellular Neurosciences, 34, 503–518.
Okamoto, S., Pouladi, M. A., Talantova, M., Yao, D., Xia, P., Ehrnhoefer, D. E., Zaidi, R., Clemente, A., Kaul, M., Graham, R. K., Zhang, D., Chen, H. S. V., Tong, G., Hayden, M. R., & Lipton, S. A. (2009). Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nature Medicine, 15, 1407–1413.
Olney, J. W. (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science, 164, 719–721.
Olney, J. W., & de Gubareff, T. (1978). The fate of synaptic receptors in the kainate-lesioned striatum. Brain Research, 140, 340–343.
Ondo, W. G., Mejia, N. I., & Hunter, C. B. (2007). A pilot study of the clinical efficacy and safety of memantine for Huntington’s disease. Parkinsonism & Related Disorders, 13, 453–454.
Pachernegg, S., Strutz-Seebohm, N., & Hollmann, M. (2012). GluN3 subunit-containing NMDA receptors: Not just one-trick ponies. Trends in Neurosciences, 35, 240–249.
Paoletti, P., & Neyton, J. (2007). NMDA receptor subunits: function and pharmacology. Current Opinion in Pharmacology, 7, 39–47.
Parsons, C. G., Danysz, W., & Quack, G. (1999). Memantine is a clinically well tolerated N-methyl-d-aspartate (NMDA) receptor antagonist–a review of preclinical data. Neuropharmacology, 38, 735–767.
Pelkey, K. A., Askalan, R., Paul, S., Kalia, L. V., Nguyen, T. H., Pitcher, G. M., Salter, M. W., & Lombroso, P. J. (2002). Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron, 34, 127–138.
Pivovarova, N. B., & Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. The FEBS Journal, 277, 3622–3636.
Prybylowski, K., & Wenthold, R. J. (2004). N-Methyl-d-aspartate receptors: subunit assembly and trafficking to the synapse. The Journal of Biological Chemistry, 279, 9673–9676.
Radi, R., Beckman, J. S., Bush, K. M., & Freeman, B. A. (1991a). Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. The Journal of Biological Chemistry, 266, 4244–4250.
Radi, R., Beckman, J. S., Bush, K. M., & Freeman, B. A. (1991b). Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Archives of Biochemistry and Biophysics, 288, 481–487.
Rammes, G., Danysz, W., & Parsons, C. G. (2008). Pharmacodynamics of memantine: An update. Current Neuropharmacology, 6, 55–78.
Rao, V. L., Dogan, A., Todd, K. G., Bowen, K. K., & Dempsey, R. J. (2001). Neuroprotection by memantine, a non-competitive NMDA receptor antagonist after traumatic brain injury in rats. Brain Research, 911, 96–100.
Regan, R. F., & Choi, D. W. (1991). Glutamate neurotoxicity in spinal cord cell culture. Neuroscience, 43, 585–591.
Roesler, R., Quevedo, J., & Schroder, N. (2003). Is it time to conclude that NMDA antagonists have failed? Lancet Neurology, 2, 13.
Rojas, J. C., Saavedra, J. A., & Gonzalez-Lima, F. (2008). Neuroprotective effects of memantine in a mouse model of retinal degeneration induced by rotenone. Brain Research, 1215, 208–217.
Rostas, J. A., Brent, V. A., Voss, K., Errington, M. L., Bliss, T. V., & Gurd, J. W. (1996). Enhanced tyrosine phosphorylation of the 2B subunit of the N-methyl-d-aspartate receptor in long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America, 93, 10452–10456.
Ryan, T. J., Emes, R. D., Grant, S. G., & Komiyama, N. H. (2008). Evolution of NMDA receptor cytoplasmic interaction domains: Implications for organisation of synaptic signaling complexes. BMC Neuroscience, 9, 6.
Salter, M. W. (1998). Src, N-methyl-d-aspartate (NMDA) receptors, and synaptic plasticity. Biochemical Pharmacology, 56, 789–798.
Salter, M. W., & Kalia, L. V. (2004). Src kinases: A hub for NMDA receptor regulation. Nature Reviews Neuroscience, 5, 317–328.
Salter, M. W., & Pitcher, G. M. (2012). Dysregulated Src upregulation of NMDA receptor activity: A common link in chronic pain and schizophrenia. The FEBS Journal, 279, 2–11.
Sanna, P. P., Berton, F., Cammalleri, M., Tallent, M. K., Siggins, G. R., Bloom, F. E., & Francesconi, W. (2000). A role for Src kinase in spontaneous epileptiform activity in the CA3 region of the hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 97, 8653–8657.
Sanz-Clemente, A., Nicoll, R. A., & Roche, K. W. (2012). Diversity in NMDA receptor composition: Many regulators, many consequences. Neuroscientist, 19, 62–75.
Sattler, R., & Tymianski, M. (2000). Molecular mechanisms of calcium-dependent excitotoxicity. Journal of Molecular Medicine (Berlin), 78, 3–13.
Sattler, R., Xiong, Z., Lu, W. Y., Hafner, M., MacDonald, J. F., & Tymianski, M. (1999). Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science, 284, 1845–1848.
Sattler, R., Xiong, Z., Lu, W. Y., MacDonald, J. F., & Tymianski, M. (2000). Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. The Journal of Neuroscience, 20, 22–33.
Schifitto, G., Navia, B. A., Yiannoutsos, C. T., Marra, C. M., Chang, L., Ernst, T., Jarvik, J. G., Miller, E. N., Ginger, E. J., Ellis, R. J., Kolson, D. L., Simpson, D., Nath, A., Berger, J., Shriver, S. L., Millar, L. L., Colquhoun, D., Lenkinski, R., Gonzalez, R. G., Lipton, S. A., & Adult AIDS Clinical Trial Group (ACTG) 301, 700 Teams, HIV MRS Consortium. (2007). Memantine and HIV-associated cognitive impairment: A neuropsychological and proton magnetic resonance spectroscopy study. AIDS, 21, 1877–1886.
Schwarze, S. R., Ho, A., Vocero-Akbani, A., & Dowdy, S. F. (1999). In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science, 285, 1569–1572.
Shan, Y., Liu, B., Li, L., Chang, N., Li, L., Wang, H., Wang, D., Feng, H., Cheung, C., Liao, M., Cui, T., Sugita, S., & Wan, Q. (2009). Regulation of PINK1 by NR2B-containing NMDA receptors in ischemic neuronal injury. Journal of Neurochemistry, 111, 1149–1160.
Sharma, E., Zhao, F., Bult, A., & Lombroso, P. J. (1995). Identification of two alternatively spliced transcripts of STEP: A subfamily of brain-enriched protein tyrosine phosphatases. Brain Research. Molecular Brain Research, 32, 87–93.
Sheng, M. (2001). Molecular organization of the postsynaptic specialization. Proceedings of the National Academy of Sciences of the United States of America, 98, 7058–7061.
Sheng, M., & Sala, C. (2001). PDZ domains and the organization of supramolecular complexes. Annual Review of Neuroscience, 24, 1–29.
Song, C., Zhang, Y., Parsons, C. G., & Liu, Y. F. (2003). Expression of polyglutamine-expanded huntingtin induces tyrosine phosphorylation of N-methyl-d-aspartate receptors. The Journal of Biological Chemistry, 278, 33364–33369.
Soriano, F. X., & Hardingham, G. E. (2007). Compartmentalized NMDA receptor signaling to survival and death. The Journal of Physiology, 584, 381–387.
Soriano, F. X., Martel, M. A., Papadia, S., Vaslin, A., Baxter, P., Rickman, C., Forder, J., Tymianski, M., Duncan, R., Aarts, M., Clarke, P. G. H., Wyllie, D. J. A., & Hardingham, G. E. (2008). Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. The Journal of Neuroscience, 28, 10696–10710.
Stieg, P. E., Sathi, S., Warach, S., Le, D. A., & Lipton, S. A. (1999). Neuroprotection by the NMDA receptor-associated open-channel blocker memantine in a photothrombotic model of cerebral focal ischemia in neonatal rat. European Journal of Pharmacology, 375, 115–120.
Sugihara, H., Moriyoshi, K., Ishii, T., Masu, M., & Nakanishi, S. (1992). Structures and properties of seven isoforms of the NMDA receptor generated by alternative splicing. Biochemical and Biophysical Research Communications, 185, 826–832.
Sun, H. S., Doucette, T. A., Liu, Y., Fang, Y., Teves, L., Aarts, M., Ryan, C. L., Bernard, P. B., Lau, A., Forder, J. P., Salter, M. W., Wang, Y. T., Tasker, A., & Tymianski, M. (2008). Effectiveness of PSD95 inhibitors in permanent and transient focal ischemia in the rat. Stroke, 39, 2544–2553.
Sun, H. S., Jackson, M. F., Martin, L. J., Jansen, K., Teves, L., Cui, H., Kiyonaka, S., Mori, Y., Jones, M., Forder, J. P., Golde, T. E., Orser, B. A., MacDonald, J. F., & Tymianski, M. (2009). Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nature Neuroscience, 12, 1300–1307.
Szydlowska, K., & Tymianski, M. (2010). Calcium, ischemia and excitotoxicity. Cell Calcium, 47, 122–129.
Taghibiglou, C., Martin, H. G., Lai, T. W., Cho, T., Prasad, S., Kojic, L., Lu, J., Liu, Y., Lo, E., Zhang, S., Wu, J. Z. Z., Li, Y. P., Wen, Y. H., Imm, J. H., Cynader, M. S., & Wang, Y. T. (2009). Role of NMDA receptor-dependent activation of SREBP1 in excitotoxic and ischemic neuronal injuries. Nature Medicine, 15, 1399–1406.
Tao, F., Tao, Y. X., Gonzalez, J. A., Fang, M., Mao, P., & Johns, R. A. (2001). Knockdown of PSD-95/SAP90 delays the development of neuropathic pain in rats. Neuroreport, 12, 3251–3255.
Tao, F., Tao, Y. X., Mao, P., & Johns, R. A. (2003). Role of postsynaptic density protein-95 in the maintenance of peripheral nerve injury-induced neuropathic pain in rats. Neuroscience, 117, 731–739.
Tao, F., Su, Q., & Johns, R. A. (2008). Cell-permeable peptide Tat-PSD-95 PDZ2 inhibits chronic inflammatory pain behaviors in mice. Molecular Therapy, 16, 1776–1782.
Terasaki, Y., Sasaki, T., Yagita, Y., Okazaki, S., Sugiyama, Y., Oyama, N., Omura-Matsuoka, E., Sakoda, S., & Kitagawa, K. (2010). Activation of NR2A receptors induces ischemic tolerance through CREB signaling. Journal of Cerebral Blood Flow and Metabolism, 30, 1441–1449.
Tezuka, T., Umemori, H., Akiyama, T., Nakanishi, S., & Yamamoto, T. (1999). PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-d-aspartate receptor subunit NR2A. Proceedings of the National Academy of Sciences of the United States of America, 96, 435–440.
Thomas, S. J., & Grossberg, G. T. (2009). Memantine: A review of studies into its safety and efficacy in treating Alzheimer’s disease and other dementias. Clinical Interventions in Aging, 4, 367–377.
Thomas, C. G., Miller, A. J., & Westbrook, G. L. (2006). Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. Journal of Neurophysiology, 95, 1727–1734.
Tovar, K. R., & Westbrook, G. L. (1999). The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. The Journal of Neuroscience, 19, 4180–4188.
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., Hansen, K. B., Yuan, H., Myers, S. J., & Dingledine, R. (2010). Glutamate receptor ion channels: Structure, regulation, and function. Pharmacological Reviews, 62, 405–496.
Tu, W., Xu, X., Peng, L., Zhong, X., Zhang, W., Soundarapandian, M. M., Balel, C., Wang, M., Jia, N., Zhang, W., Lew, F., Chan, S. L., Chen, Y., & Lu, Y. (2010). DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell, 140, 222–234.
Tymianski, M. (2011). Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nature Neuroscience, 14, 1369–1373.
Tymianski, M., Charlton, M. P., Carlen, P. L., & Tator, C. H. (1993). Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. The Journal of Neuroscience, 13, 2085–2104.
Vanhoutte, P., & Bading, H. (2003). Opposing roles of synaptic and extrasynaptic NMDA receptors in neuronal calcium signaling and BDNF gene regulation. Current Opinion in Neurobiology, 13, 366–371.
Verpelli, C., Schmeisser, M. J., Sala, C., & Boeckers, T. M. (2012). Scaffold proteins at the postsynaptic density. Advances in Experimental Medicine and Biology, 970, 29–61.
Volbracht, C., van Beek, J., Zhu, C., Blomgren, K., & Leist, M. (2006). Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. The European Journal of Neuroscience, 23, 2611–2622.
Wang, Y. T., & Salter, M. W. (1994). Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature, 369, 233–235.
Wang, C. X., & Shuaib, A. (2005). NMDA/NR2B selective antagonists in the treatment of ischemic brain injury. Current Drug Targets. CNS and Neurological Disorders, 4, 143–151.
Wilhelmus, M. M., Nijland, P. G., Drukarch, B., de Vries, H. E., & van Horssen, J. (2012). Involvement and interplay of Parkin, PINK1, and DJ1 in neurodegeneration and neuroinflammatory disorders. Free Radical Biology & Medicine, 53, 983–992.
Wood, P. L., & Hawkinson, J. E. (1997). N-methyl-d-aspartate antagonists for stroke and head trauma. Expert Opinion on Investigational Drugs, 6, 389–397.
Xia, P., Chen, H. S., Zhang, D., & Lipton, S. A. (2010). Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. The Journal of Neuroscience, 30, 11246–11250.
Xie, Y. F., MacDonald, J. F., & Jackson, M. F. (2010). TRPM2, calcium and neurodegenerative diseases. International Journal of Physiology, Pathophysiology and Pharmacology, 2, 95–103.
Xu, J., Kurup, P., Zhang, Y., Goebel-Goody, S. M., Wu, P. H., Hawasli, A. H., Baum, M. L., Bibb, J. A., & Lombroso, P. J. (2009). Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. The Journal of Neuroscience, 29, 9330–9343.
Zhao, Y., Navia, B. A., Marra, C. M., Singer, E. J., Chang, L., Berger, J., Ellis, R. J., Kolson, D. L., Simpson, D., Miller, E. N., Lipton, S. A., Evans, S. R., Schifitto, G., & The Adult AIDS Clinical Trial Group (ACTG) 301 Team. (2010). Memantine for AIDS dementia complex: Open-label report of ACTG 301. HIV Clinical Trials, 11, 59–67.
Zheng, F., Gingrich, M. B., Treynelis, S. F., & Conn, P. J. (1998). Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nature Neuroscience, 1, 185–191.
Zheng, C. Y., Seabold, G. K., Horak, M., & Petralia, R. S. (2011). MAGUKs, synaptic development, and synaptic plasticity. The Neuroscientist, 17, 493–512.
Zhou, M., & Baudry, M. (2006). Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. The Journal of Neuroscience, 26, 2956–2963.
Zhou, L., Li, F., Xu, H. B., Luo, C. X., Wu, H. Y., Zhu, M. M., Lu, W., Ji, X., Zhou, Q. G., & Zhu, D. Y. (2010). Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nature Medicine, 16, 1439–1443.
Ziff, E. B. (1997). Enlightening the postsynaptic density. Neuron, 19, 1163–1174.
Zou, J., & Crews, F. (2006). CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cellular and Molecular Neurobiology, 26, 385–405.
Zukin, R. S., & Bennett, M. V. (1995). Alternatively spliced isoforms of the NMDARI receptor subunit. Trends in Neurosciences, 18, 306–313.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this entry
Cite this entry
Vetiska, S.M., Tymianski, M. (2014). Neuroprotectants Targeting NMDA Receptor Signaling. In: Kostrzewa, R. (eds) Handbook of Neurotoxicity. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5836-4_168
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5836-4_168
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5835-7
Online ISBN: 978-1-4614-5836-4
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences