
Abstract
The perception of Parkinson’s disease (PD) as a disease centered on dopaminergic striatonigral neurodegeneration has changed fundamentally since 1997 when the first mutation in the SNCA gene (PARK1) encoding α-synuclein was discovered (Polymeropoulos et al. 1997). This discovery formed the basis for a new description of brain pathology characterized by the presence of α-synuclein aggregates in brain cell inclusions that are the hallmarks of PD and other synucleinopathies: dementia with Lewy bodies (DLB) and multiple system atrophy (MSA). This field has been thoroughly covered by many reviews during the last decade (Gai et al. 1998; Spillantini and Goedert 2000; Huang et al. 2004; Ubhi et al. 2011). This review will briefly highlight the historical breakthroughs but focus on α-synuclein modifications, human neuropathology, biomarker potential, current animal models and the new concepts emerging after the significance of extracellular α-synuclein has gained support.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

Keywords
- Animal models
- Biomarkers
- Dementia with Lewy bodies
- Lewy bodies
- Multiple systems atrophy
- Parkinson disease
- Phosphorylation
- Truncation
- α-synuclein
1 Introduction
The perception of Parkinson’s disease (PD) as a disease centered on dopaminergic striatonigral neurodegeneration has changed fundamentally since 1997 when the first mutation in the SNCA gene (PARK1) encoding α-synuclein was discovered (Polymeropoulos et al. 1997). This discovery formed the basis for a new description of brain pathology characterized by the presence of α-synuclein aggregates in brain cell inclusions that are the hallmarks of PD and other synucleinopathies: dementia with Lewy bodies (DLB) and multiple system atrophy (MSA). This field has been thoroughly covered by many reviews during the last decade (Gai et al. 1998; Spillantini and Goedert 2000; Huang et al. 2004; Ubhi et al. 2011). This review will briefly highlight the historical breakthroughs but focus on recent developments in concepts and hypotheses.
2 Genetics
Missense mutations in the SNCA gene substituting single amino acids (A30P, E46K, and A53T) cause early-onset autosomal dominant PD and DLB (Polymeropoulos et al. 1997; Kruger et al. 1998; Zarranz et al. 2004). Additionally, genomic multiplications of the SNCA gene have been found in patients with early-onset PD and DLB (Singleton et al. 2003) demonstrating that simple overexpression and perhaps developmentally dysregulated expression of the normal protein is sufficient to initiate disease development. The discovery of these rare cases was essential because they causally tie α-synuclein to the development of a PD syndrome, leading to the widely accepted hypothesis that a gain of function in α-synuclein contributes to the pathogenesis of PD. A possible role for α-synuclein in sporadic PD is corroborated by a series of genome-wide association studies carried out on ethnically diverse populations of PD patients. The single-nucleotide polymorphisms conferring the strongest risk are in the SNCA gene (Simon-Sanchez et al. 2009). Although SNCA is the strongest risk gene, polymorphisms within this gene only increase the risk 2–3 times above the low prevalence of about 0.14 % but serve to corroborate a mechanistic link between the development of sporadic PD and α-synuclein.
3 Structure of α-Synuclein
α-Synuclein is a small 140-amino acid protein, which was first identified as associated with purified synaptic vesicles from the electric ray Torpedo californica (Maroteaux et al. 1988). It is one of three synuclein family members (α, β, and γ) identified in humans. The α-synuclein protein can be divided into two regions: (i) the N-terminal region (residues 1–95), which contains seven imperfect repeats with a highly conserved hexameric motif (KTKEGV) and the aggregation-prone non-amyloid component (NAC) sequence (residues 60–95), and (ii) the unstructured C-terminal region (96–140), which is rich in acidic residues (Ueda et al. 1993; George et al. 1995).
The heat-stable nature of human α-synuclein was already described in 1994 (Jakes et al. 1994), and it was ascribed to its natively unfolded structure that possesses an ability to form α-helical structures in apolar solvents (Weinreb et al. 1996) and upon interactions with acidic phospholipid membranes (Clayton and George 1998). When analyzed by gel filtration, the unfolded structure caused the 14.4 kDa α-synuclein peptide to elute corresponding to an approximately 55 kDa globular protein. Moreover, certain dimers appearing on denaturing SDS-polyacrylamide gels were due to misincorporation of a cysteine residue in some preparations of bacterially expressed human α-synuclein (Masuda et al. 2006). Despite its unfolded nature, purified α-synuclein has been demonstrated to exhibit dynamic long-range interactions between the C-terminus and the central NAC domain and even further toward the N-terminus (Cho et al. 2009). Moreover, it undergoes a dramatic structural change upon binding to acidic liposomes where the N-terminal repeat regions fold into two α-helices (Bussell and Eliezer 2003; Chandra et al. 2003).
α-Synuclein aggregates into amyloid fibrils by a nucleation-dependent mechanism and the disease-causing mutations as well as C-terminal truncations favor the consumption of monomers into aggregates. The transformation from a monomeric state to an amyloid state proceeds through soluble oligomeric intermediates (Wood et al. 1999). Several lines of evidence suggest that the oligomers are the most toxic species. First, the disease-causing A30P mutation accelerates the initial oligomerization of α-synuclein in vitro, whereas it retards the formation of mature filamentous aggregates (Conway et al. 2000). Second, α-synuclein oligomers bind synthetic vesicles and cause transient permeabilization representing a potential mechanism of neurotoxicity (Lashuel et al. 2002). This was corroborated when oligomeric forms of α-synuclein applied extracellularly were shown to trigger calcium entry and toxicity in a neuronal cell line (Danzer et al. 2007). Third, cytotoxicity in cellular models is usually seen without heavily aggregated α-synuclein (Xu et al. 2002; Outeiro et al. 2008; Tetzlaff et al. 2008; Kragh et al. 2009). The protofibrils formed in vitro from purified α-synuclein represent a diverse group generated from simple aggregation of monomers in buffer to induction by various agents and conditions, e.g., apolar solvents, lyophilization, and oxidative cross-linking by, e.g., metals and oxidation products from lipid peroxidation (Sharon et al. 2001; Nasstrom et al. 2011). Immunological characterization of α-synuclein oligomers has been feasible using two different antibodies: the pan-oligomeric A11 antibody recognizing soluble precursors of many types of amyloid-type fibrils (Kayed et al. 2007) and the FILA-1 antibody recognizing α-synuclein-specific epitopes shared between filaments and soluble oligomers. The FILA-1 antibody recognizes elevated levels of soluble α-synuclein aggregates in DLB brain extracts (Paleologou et al. 2009) indicating commonalities between in vitro-formed oligomers and those present in vivo. At present, the structural relation between in vitro-formed α-synuclein protofibrils and those found in cells and brain tissue is unclear and structure-specific imaging agents are in high demand.
Two recent reports challenge the dogma of α-synuclein as a natively unfolded protein and show evidence supporting native tetramers rich in α-helical structure. Both reports indicate that α-synuclein from red blood cells and recombinantly expressed in Escherichia coli with a short N-terminal extension can be purified as folded tetramers that irreversibly dissociate into monomers (Bartels et al. 2011; Wang et al. 2011). Interestingly, the tetramers are unable to aggregate and amyloid-type aggregation is only possible after dissociation into monomers. Importantly, the PD-causing missense mutations destabilize the tetramers and thus favor aggregation. These findings are controversial but potentially important as they hypothesize that a stabilization of the native tetrameric state could be a fruitful therapeutic strategy.
4 Posttranslational Modifications
α-Synuclein is subject to different posttranslational modifications. It can be phosphorylated at Ser87, Ser129, and Tyr125 (Ellis et al. 2001; Anderson et al. 2006; Paleologou et al. 2010) in human brain tissue. Though predominantly found in a non-phosphorylated state in vivo, α-synuclein is phosphorylated at Ser129 in inclusions in all α-synucleinopathies (Fujiwara et al. 2002; Anderson et al. 2006). Both Ser129-phosphorylation and ubiquitination are highly enriched in α-synuclein aggregates in diseased brain, whereas other less abundant modifications occur in both soluble and insoluble fractions.
The role of Ser129-phosphorylation in promoting toxic α-synuclein aggregation is unclear as in vitro phosphorylation of this residue has been demonstrated to prevent amyloid-type insoluble aggregation (Fujiwara et al. 2002; Paleologou et al. 2008). α-Synuclein phosphorylation at Ser129 accelerates inclusion formation and/or toxicity in cell culture models (Smith et al. 2005b; Sugeno et al. 2008; Kragh et al. 2009) and correlates with pathology in transgenic Drosophila (Chen and Feany 2005). Studies in rats expressing the phosphomimic form of α-synuclein (S129D) using adeno-associated virus (AAV) have yielded conflicting results (see section on “Animal Models”). However, S129D may not be a suitable phosphomimicking variant of α-synuclein as it was demonstrated that S129D does not reproduce the effects of phosphorylation on structural and aggregation properties of α-synuclein in vitro (Paleologou et al. 2008). Mechanistically, Ser129-P may contribute functionally by conferring novel ligand-binding properties as demonstrated using a phosphopeptide co-immunoprecipitation approach (McFarland et al. 2008). Phosphorylation of tyrosines also occurs in α-synuclein (Ellis et al. 2001) and Tyr125-phosphorylation decreases with age in human brain tissue (Chen et al. 2009). Deletion of the tyrosine residues in α-synuclein increased toxicity in a Drosophila model (Chen et al. 2009) indicating a protective role for tyrosine phosphorylation that may be lost with aging. To fully understand the consequence of α-synuclein phosphorylation, it is important to identify the involved kinase(s). Casein kinase II, GRK2/5, and polo-like kinases 2/3 have been shown to phosphorylate α-synuclein in vitro, in cell culture, Drosophila, and mice (Pronin et al. 2000; Chen and Feany 2005; Arawaka et al. 2006; Inglis et al. 2009; Mbefo et al. 2010) (for a review see Oueslati et al. 2010).
Proteolytic C-terminal truncations after residues 115, 119, 122, 133, and 135 have been demonstrated in human brain tissue (Anderson et al. 2006), and such modifications increase α-synuclein aggregation in vitro (Crowther et al. 1998). Interestingly, α-synuclein is a substrate for cytoplasmic calpains, and cleaved α-synuclein is found in PD and DLB brain (Mishizen-Eberz et al. 2005; Kim et al. 2006; Dufty et al. 2007), suggesting that calpain may generate truncated toxic species of α-synuclein.
The early focus on dopaminergic neurons in substantia nigra in PD prior to the description of the Braak hypothesis and the use of L-dopa for therapy forms the basis for a large series of experiments on dopamine effects on α-synuclein and vice versa (for a review Leong et al. 2009). Key findings are that dopamine stimulates the formation of soluble off-pathway α-synuclein oligomers in vitro that do not aggregate into fibrils (Conway et al. 2001) and the dopamine oxidation product dopaminochrome promotes the formation of spherical oligomers (Norris et al. 2005). These observations were corroborated in cellular experiments where the dopamine level was modulated by expression of tyrosine hydroxylase (TH) forms (Mazzulli et al. 2006). In addition, α-synuclein has been related to different key proteins of the dopaminergic system. α-Synuclein influences dopamine synthesis by decreasing TH and/or aromatic amino acid decarboxylase activity via changes in the phosphorylation status of these two enzymes (Perez et al. 2002; Tehranian et al. 2006; Lou et al. 2010). Mutated α-synuclein A53T leads to low levels of VMAT2 and consequently to accumulation of dopamine in neurons (Lotharius et al. 2002). α-Synuclein levels have also been implicated in regulating the activity of the dopamine transporter (DAT), although controversies exist. Hence, direct binding between transgenic hα-synuclein and DAT in cells enhanced dopamine uptake and dopamine-dependent cell death (Lee et al. 2001). However, a later study coexpressing the two proteins identified a negative effect on dopamine uptake (Wersinger and Sidhu 2003). Finally, using the opposite approach by silencing endogenously expressed α-synuclein showed a decreased sensitivity to the DAT-dependent toxin MPP+ and decreased dopamine uptake (Fountaine and Wade-Martins 2007). Hence, the definitive role of dopamine-dependent α-synuclein modifications in vivo and the effect of α-synuclein levels and modifications on dopamine metabolism are still enigmatic.
5 Cellular Metabolism
α-Synuclein is synthesized as a cytosolic protein but it can exist in various subcompartments, e.g., membrane associated or encapsulated with various posttranslational modifications. Natively unmodified α-synuclein is readily degraded by the 20S proteasome (Liu et al. 2003) and by chaperone-mediated autophagy in lysosomes in vitro (Cuervo et al. 2004). How α-synuclein is degraded in the brain is less clear, and studies using cell lines and primary neuronal cultures have yielded different results (reviewed in (Xilouri and Stefanis 2011)). Neurons in culture appear to degrade the bulk of α-synuclein by chaperone-mediated autophagy and macroautophagy (Vogiatzi et al. 2008). A recent study using transgenic mice expressing human α-synuclein or α-synuclein-eGFP analyzed the levels of α-synuclein after topical application of inhibitors of the 20S proteasome and the autophagy-lysosome pathway (Ebrahimi-Fakhari et al. 2011). Using cranial window access to the brain surface combined with live imaging and tissue extraction, it was elegantly demonstrated that the basal α-synuclein turnover is mediated by the proteasome system and this process appears even more important with aging. Upon stress mediated by increased α-synuclein expression or compromised proteasomal activity, the autophagy-lysosome pathway is activated (Ebrahimi-Fakhari et al. 2011). The importance of lysosomal dysfunction in relation to Lewy body (LB) formation has been corroborated by the presence of LB pathology in Gaucher disease that is caused by dysfunctional lysosomal hydrolases (Vitner et al. 2010). Exocytosis of vesicle-bound α-synuclein was demonstrated in 2005 (Lee et al. 2005), and direct neuron-neuron transfer of α-synuclein was later confirmed using cell transplantation (Desplats et al. 2009). This formed the important experimental frame for testing whether α-synuclein by a prion-like mechanism causes the neurodegenerative progression described by the Braak hypothesis (Braak et al. 2004) ranging from development of LBs in fetal human dopaminergic neurons transplanted into PD patients (Kordower et al. 2008; Li et al. 2008) to Lewy-like pathology ascending to the brainstem after oral toxin-based generation of α-synuclein pathology in the gut (Pan-Montojo et al. 2010) and to neuronal uptake of preformed α-synuclein aggregates in vitro (Volpicelli-Daley et al. 2011).
6 Normal Functions
The normal function of α-synuclein has been difficult to pinpoint although its effective axonal transport (Jensen et al. 1999) and selective localization in nerve terminals (Maroteaux et al. 1988) suggest a function in this structure. Although it is expressed at high levels in the brain, it is also found in other tissues, e.g., hematopoietic cells (Miller et al. 2004). Reduction of α-synuclein in cultured neurons reduced the distal pool of synaptic vesicles (Murphy et al. 2000), and knockout mice corroborated a presynaptic function as evidenced by reduced striatal dopamine content and an increased neurotransmitter release upon paired stimuli (Abeliovich et al. 2000). However, the phenotype was surprisingly mild with the most striking feature being a resistance to the toxin MPTP (Dauer et al. 2002). Although knockout of α-synuclein alone or α-synuclein and β-synuclein together has minimal effects on neuronal function (Chandra et al. 2004), triple knockout mice lacking all synuclein members demonstrate a robust phenotype with gross reduction in presynaptic size and premature death (Greten-Harrison et al. 2010). These mice display deficits in assembly of SNARE complexes that are required for fusion of synaptic vesicles to the plasma membrane. Interestingly, it was recently shown that the synucleins act as chaperones for presynaptic SNARE proteins (Burre et al. 2010).
Transgenic expression of α-synuclein has been considered a rational approach for most studies of pathological α-synuclein effects because of the dominant inheritance of PD and DLB associated to mutations in the SNCA gene. Hence, wild-type and mutated human α-synuclein has been expressed in a range of cell lines, primary brain cells, yeast cells, and organisms ranging from Caenorhabditis elegans and Drosophila to mice, rats, and monkeys with variable results. Initially the criteria for success were generation of a LB-like structure and this frustrated many researchers. End points have later changed toward neuronal degeneration and cell death because it was realized that LBs may take months to years to develop in human neurons.
7 Pathogenic Functions
Mechanisms by which abnormal accumulation of α-synuclein disrupts cellular functions have been investigated intensively in several different model systems. Yeast studies have demonstrated that α-synuclein accumulation leads to a blockade of vesicular transport from the endoplasmatic reticulum to the Golgi apparatus and hereby inhibits the viability of the cells (Cooper et al. 2006). Moreover, activation of an unfolded protein response has been demonstrated in SHSY5Y cells overexpressing α-synuclein (Sugeno et al. 2008) and in postmortem brain tissue from PD patients (Hoozemans et al. 2007). However, it became clear in the yeast study that the toxic response was dependent on the α-synuclein dose as revealed by comparing high and low expressers (Su et al. 2010). A different approach has been taken in mammalian cells that tolerate expression of α-synuclein but where cytotoxicity was induced by coexpressing proteins known to stimulate α-synuclein aggregation like p25α and synphilin-1 (Engelender et al. 1999; Kragh et al. 2009). Using this approach it was demonstrated that p25α induced cellular degeneration that could be attenuated by inhibitors of α-synuclein aggregation as well as of Ser129-phosphorylation (Kragh et al. 2009). The approach of including inhibitors of α-synuclein aggregation may be readily applicable to other models and thereby allow phenotypes caused by mere overexpression of α-synuclein to be more clearly distinguished from those forming aggregations.
It has previously been difficult to identify soluble α-synuclein aggregates in cellular models. Nevertheless, they have been detected by Western blotting in mesencephalic neurons exposed to polyunsaturated fatty acids (Sharon et al. 2003) and in a dopaminergic cell line overexpressing A53T α-synuclein (Zhou and Freed 2005). Moreover, a fluorescence lifetime imaging-based technique to probe the organization of α-synuclein within the cell has demonstrated the formation of small oligomers (Klucken et al. 2006). Toxic effects have also been demonstrated by applying α-synuclein from culture media to cells, but the toxic pathways elicited by internal versus external α-synuclein are still unclear. An elegant approach was used to test whether mature aggregates or insoluble protofibrils are responsible for toxic effects caused by overexpression of α-synuclein. In this study, α-synuclein mutants designed to favor generation of soluble protofibrils exhibited increased toxic potential compared to wild-type α-synuclein in cell and animal models (Karpinar et al. 2009).
Several lines of evidence implicate a dysfunction of the ubiquitin-proteasome system in the pathogenesis of α-synucleinopathies. Studies show that LBs contain ubiquitinated proteins (Chung et al. 2001; McNaught and Jenner 2001) and that the proteasomal activity is decreased in the substantia nigra of PD patients (McNaught and Jenner 2001; McNaught et al. 2003). Moreover, aggregated α-synuclein has been demonstrated to inhibit the activity of the proteasome in vitro (Snyder et al. 2003; Lindersson et al. 2004; Emmanouilidou et al. 2010), and this may have significant effects on proteasomal-dependent degradation of short-lived proteins.
Mitochondrial dysfunction as a consequence of α-synuclein accumulation has also been a major area of interest. Abnormalities of mitochondrial function and increased free radical-mediated damage were described in PD brain before the first PD-associated gene mutations were discovered. α-Synuclein can localize to mitochondria (Hsu et al. 2000; Tanaka et al. 2001; Smith et al. 2005a) and α-synuclein expression increases cellular sensitivity to rotenone, a mitochondrial complex I inhibitor (Orth et al. 2003; Ved et al. 2005). α-Synuclein has also been shown to reduce ATP synthesis and mitochondrial membrane potential (Kamp et al. 2010). Moreover, structural abnormalities of mitochondria have been observed in transgenic mice overexpressing mutant α-synuclein (Martin et al. 2006).
The existence of cellular α-synuclein excretion and uptake by neighboring neurons as previously described may form a path for spreading of the disease process in the nervous system. The potential of such mechanisms has been corroborated by the dramatic neuronal spread of α-synuclein pathology in transgenic mice expressing human A53T mutant α-synuclein (Luk et al. 2012). Unilateral injection of homogenates from old symptomatic M83 mice into the brains of young mice caused a rapid propagation of aggregated and hyperphosphorylated α-synuclein ranging from the olfactory bulb to the spinal cord that also traversed the corpus callosum to the contralateral hemisphere. The propagation required the recruitment of endogenous mouse α-synuclein. Surprisingly, similar pathology was induced upon injection of in vitro-formed insoluble aggregates of a fusion protein consisting of C-terminally truncated human α-synuclein (1–120) and a Myc epitope tag that lacked the S129 phosphorylation site. Hence, insoluble α-synuclein aggregates may possess “prion-like” seeding activity (Luk et al. 2012).
8 Human Pathology
PD is diagnosed by two separate abnormalities in the absence of sufficient neuropathology for an alternative neurodegenerative disorder (Dickson et al. 2009). A moderate to substantial loss of the dopaminergic neurons in the substantia nigra is required, a finding that also occurs in the majority of movement disorders, and the presence of hallmark neuronal LBs in brainstem predilection sites is also required (Fig. 1a). LBs are also the hallmark pathology of DLB with the two conditions separated mainly on clinical rather than pathological grounds, particularly at end stage (McKeith et al. 2005). PD patients have a dominant slowly progressive movement disorder with cognitive impairment often intervening very late in the disease when LBs infiltrate into the cortex (Dickson et al. 2009). DLB patients have a dominant more rapidly progressive dementia disorder and have cortical LBs often with coexisting Alzheimer’s disease (Halliday et al. 2011a). Although α-synuclein forms abnormal aggregates in all synucleinopathies, neuronal LBs only occur in PD and DLB with α-synuclein inclusions in alternate cell types (oligodendroglia) or cellular locations (axonal spheroids) in other synucleinopathies (Halliday et al. 2011a). The reason for the different locations and mechanisms of neurodegeneration involving α-synuclein remain largely unexplored.


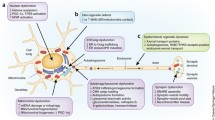
Neuropathology found in patients with α-synucleinopathies. (a) The two diagnostic pathologies required for PD are the loss of pigmented dopaminergic neurons in the midbrain substantia nigra (shown macroscopically at left) and LB formation in remaining brainstem pigmented neurons (shown at right). Macroscopic photos of transverse sections through the midbrain from a control and a patient with PD demonstrate the loss of the dark neuromelanin pigment in the ventral midbrain. LBs within remaining pigmented neurons in patients with PD can be visualized as large round intracellular inclusions observed with hematoxylin and eosin (H&E) or α-synuclein immunohistochemistry. (b) Diagrammatic representation of the Braak staging scheme for the progression of PD from the olfactory and peripheral nervous systems to invade the brain in a slow hierarchical fashion from the lower brainstem to the upper brainstem and forebrain to the cortex. (c) Photomicrographs of the typical astrocytic changes that occur in PD versus MSA. While reactive astrogliosis is typical in MSA (observed as an increase in GFAP and hypertrophy of their cell bodies compared to controls), this reaction is not typically observed in end-stage cases of PD. Rather, in PD a large number of astrocytes abnormally accumulate α-synuclein throughout their cytoplasm, a finding associated with the formation of the large round intracytoplasmic LBs in nearby neurons. (d) Photomicrographs of the microglial changes observed in PD. Amoebic, phagocytic microglia aggregate only in regions with neuronal loss like substantia nigra (right), while in areas abnormally accumulating α-synuclein, the microglia display more HLA-DR reactivity in association with an increased branching morphology (left). (e) Pathologies associated with each other in patients with the more rapidly progressing DLB and later onset PD with early dementia. The density of amyloid plaques in both the striatum (left) and cortex (middle left) is related to each other and to the severity of dementia in these patients. Lewy neurites in the hippocampus (middle right) are also related to the severity of plaque and LB pathology (at right) in such cases
In addition to PD and DLB, LBs occur in a small proportion of asymptomatic elderly, a finding which has allowed the development of a hypothesized pathological staging scheme for PD (Braak et al. 2003) and one for both PD and DLB (Beach et al. 2009). Braak hypothesis for the progression of pathology in PD states that the pathology starts peripherally in the olfactory and autonomic nervous systems and progresses through connected regions into the spinal cord and medulla oblongata (stage 1), progressing over time to the pons (stage 2) and midbrain (stage 3), to the basal ganglia (stage 4) and limbic systems (stage 5), and finally to association cortices (stage 6), largely sparing the somatosensory and motor system (Braak et al. 2003) (Fig. 1b). The alternate hypothesis for the progression of LB pathology across PD and DLB by Beach and colleagues (Beach et al. 2009) states that pathology begins in the olfactory bulb (stage 1) and then progresses in two different directions depending on clinical phenotype – for the motor phenotype (PD) LBs infiltrate the brainstem (stage 2a) and for the dementia phenotype (DLB) LBs infiltrate the limbic system (stage 2b). Finally, for both phenotypes, LBs are found in both the brainstem and limbic system (stage 3) prior to infiltrating cortical regions (stage 4) (Beach et al. 2009).
The Braak staging of PD pathology has been controversial due to discrepancies in a number of validation studies and also due to some conceptual difficulties, while the alternate staging scheme has not been independently evaluated. However, it should be noted that the majority of longitudinally followed cases with motor-dominant PD fit the Braak staging [see review (Halliday and McCann 2010)]. The most obvious difficulty with the Braak staging scheme is using asymptomatic cases with LBs to only stage the progression of PD and not DLB. The more restricted number of stages in the alternate hypothesis incorporating DLB makes this staging scheme of limited current utility. It should be noted that in ∼10 % of asymptomatic elderly, LBs occur only in cortical regions (Zaccai et al. 2008). Additionally, the clinical progression of PD relates to the severity of dopaminergic cell loss (Greffard et al. 2006). This may not be surprising if LBs mark neurons destined to die with all LB debris removed following cell death. However, if this was the case, then as the disease progresses, there should be a reduction of LB pathology as cell death is enhanced and affects more regions, rather than the concept of pathology progressing to accumulate in more and more neurons and cells still viable in the brain. Empirical data shows that even in regions thought to be heavily affected with pathology, only 4–5 % of neurons contain LBs in PD (Harding et al. 2002; Greffard et al. 2010). Furthermore, there is no widespread brain tissue loss in PD (Weintraub et al. 2011), and the pattern and severity of cell loss does not reflect the demonstrated pattern and severity of LB formation. Definitive proof of the progression of LB pathology will only occur with the development of biomarkers capable of identifying this pathology.
In addition to the cell loss and LB pathology required for diagnosis, glial changes are obvious in PD (Halliday and Stevens 2011). Astrocytes in PD disengage from neurons leaving the neuronal membranes more exposed (Knott et al. 1999). They undergo PD-specific changes in that they accumulate non-fibrillar α-synuclein and lack the typical reactive astrogliosis that usually accompanies neurodegeneration (Song et al. 2009) (Fig. 1c). In particular, there is often no obvious hypertrophy of astrocytes or upregulation of their glial fibrillary acidic protein (GFAP) or any substantial proliferation, a finding that differs significantly from reactive astrogliosis observed in MSA (Song et al. 2009) (Fig. 1c). This may suggest that any reactive trigger resolves or is removed over time in PD, possibly by cellular uptake. In contrast, the astrocytes in patients with MSA do not accumulate α-synuclein but have the typical reactivity observed in chronic neurodegenerative conditions with obvious cell hypertrophy and upregulation of GFAP (Song et al. 2009) (Fig. 1c), changes that are associated with long-lasting reorganization of tissue architecture. The accumulation of α-synuclein in astrocytes in PD is related to the severity of LB infiltration and synaptic degeneration (Braak et al. 2007), affecting more astrocytes (∼45 %; (Song et al. 2009)) compared with neurons (∼4–5 %; Harding et al. 2002; Greffard et al. 2010).
α-Synuclein in astrocytes causes an upregulation of inflammatory mediators (Klegeris et al. 2006; Lee et al. 2010) as well as neuroprotective factors (Power et al. 2002; Ishida et al. 2006; Durrenberger et al. 2009; Michael et al. 2011). The increase in α-synuclein deposition also correlates with an increase in activation of microglia (Croisier et al. 2005), and with the removal of astroglial processes from neurons, the vacated perineuronal space becomes occupied by amoeboid phagocytic microglia (Knott et al. 1999) (Fig. 1d). While activated microglia are well described as a feature of PD (see review Hirsch et al. 2012), phagocytic microglia are only found in regions of cell loss and express scavenger and high-affinity IgG receptor Fcγ (Orr et al. 2005) as well as macrophage markers (Croisier et al. 2005). These data suggest that the relatively large number of astrocytes affected by α-synuclein plays a significant role in the disease process.
PD is one of the age-associated neurodegenerative conditions and occurs often in association with other age-related neuropathologies (Compta et al. 2011). In particular, the pathology most often associated with age-related dementia is amyloid plaque accumulation (Matthews et al. 2009), which also contributes to dementia in older cases of PD (Compta et al. 2011) (Fig. 1e). It should be noted that patients with dementia and sufficient neurofibrillary tangle pathology for a diagnosis of Alzheimer’s disease are not considered to have a LB syndrome that contributes to their dementia, due to their substantially greater Alzheimer-type pathology (Dickson et al. 2009). However, amyloid plaque pathology is known to occur in nearly all patients with DLB, being much less frequent in younger PD patients, even those with dementia (Halliday et al. 2011b). This confirms in vivo PiB imaging studies in patients with PD (Gomperts et al. 2008) and suggests that, in such typical patients, the α-synuclein cortical LB pathology is sufficient for their late end-stage dementia. Therefore, there is considerable clinical heterogeneity in patients with LBs that associates with other age-related pathologies – patients with a younger onset often have a slow disease course and more pure LB pathology at end stage, while patients with an older onset are more likely to have additional age-related pathologies and a more rapid and complex clinical course, the most rapid course occurring in patients with DLB (Compta et al. 2011).
9 α-Synuclein in Human Fluids as a Biomarker for PD Progression
Aggregated α-synuclein accumulates in degenerating neurons in PD making measurements of α-synuclein levels a rational biomarker for diagnosis and prediction of disease susceptibility. α-Synuclein has been detected in blood, CSF, and saliva at low nanogram/ml levels (e.g., (Fjorback et al. 2007; Devic et al. 2011; Shi et al. 2011)). Most investigations have focused on detecting the total amount of α-synuclein but have also investigated the presence of polymers using identical monoclonal anti-α-synuclein antibodies as catching and detecting antibody (Tokuda et al. 2010). Future developments will likely aim at detecting specific α-synuclein isoforms, e.g., misfolded species as detected in brain extracts, and specific phosphorylations to allow for better stratification. For a review on α-synuclein in CSF, please see (Mollenhauer et al. 2010).
10 Modeling PD
Different organisms from flies and worms to rodents and primates have been used to model PD based on expression of wild-type or mutated human α-synuclein. In this section, we will highlight the discoveries related to mammals and refer readers to a recent review about other approaches (Pienaar et al. 2010). There are numerous human α-synuclein transgenic mice although the associated phenotypes are not always consistent probably due to the varying levels and areas of transgene expression as a consequence of the promoters used (for further reading see (Magen and Chesselet 2010; Chesselet and Richter 2011)). Disease modeling has focused on the following points: neurodegeneration in general and dopaminergic neurodegeneration in particular, basal ganglia-associated motor behavior and signs of abnormal α-synuclein folding and handling (see Table 1 for overview).
10.1 Dopaminergic Neurodegeneration
Mice lines using the dopaminergic TH promoter have been rather disappointing in modeling PD (Matsuoka et al. 2001; Rathke-Hartlieb et al. 2001) and only double-mutated α-synuclein (A30P and A53T; a condition not found in humans) has been able to induce dopaminergic failure (Richfield et al. 2002), and this was even obtained with the pan-neuronal Thy1 promoter (Ono et al. 2009). Interestingly, expressing C-terminal-truncated α-synuclein (1–120 or 1–130 α-synuclein) under the TH promoter induced dopaminergic cell death, although not progressively and related to developmental defects (Tofaris et al. 2006; Wakamatsu et al. 2007; Wakamatsu et al. 2008). Using the rat AAV model, the ability of truncated α-synuclein to promote pathological accumulation of wild-type α-synuclein was corroborated (Ulusoy et al. 2010). More widely expressing promoters like Thy1 and PDGF have also been used to model dopaminergic degeneration and subtle phenotypes have been obtained (Masliah et al. 2000; Rockenstein et al. 2002; Lam et al. 2011).
An alternative to the classic transgenic mice lines is adult transgenesis using viral vectors to induce overexpression of wild-type or mutated forms of α-synuclein in nigrostriatal neurons. This leads to progressive and selective neurodegeneration and dopaminergic loss in the basal ganglia (Kirik et al. 2002; Lo Bianco et al. 2002; Yamada et al. 2004; Maingay et al. 2006). These models have shown characteristics of α-synuclein misfolding (Lo Bianco et al. 2004; Eslamboli et al. 2007), markers of dysfunctional synapses, and neuroinflammation (Theodore et al. 2008; Chung et al. 2009; Sanchez-Guajardo et al. 2010). So far, this is the only approach that has allowed transgenesis in nonhuman primates to successfully model several of the cardinal symptoms of the disease (Eslamboli et al. 2007; Yasuda et al. 2007).
10.2 Non-dopaminergic Neurodegeneration and Braak
The Braak hypothesis posits that α-synuclein degeneration also occurs in non-dopaminergic neurons (Fig. 1b). Indeed enteric nervous system anomalies and colonic problems have been demonstrated in α-synuclein mice (Wang et al. 2008; Kuo et al. 2010). Olfactory problems being early symptoms in the disease have also been observed early in mice along with concomitant α-synuclein pathology in the olfactory bulb (Fleming et al. 2004, 2008; Wang et al. 2008). Very pronounced brainstem and spinal cord degeneration has been observed in the lines using the prion promoter (Giasson et al. 2002; Lee et al. 2002) with up to 75 % of motor neuron cell death (Gispert et al. 2003).
10.3 Pathological α-Synuclein In Vivo
Several α-synuclein transgenic mice models have shown ubiquitin-positive intracellular inclusions (Masliah et al. 2000; Kahle et al. 2001; Lee et al. 2002; Eslamboli et al. 2007; Zhou et al. 2008) and characteristics of aggregation, like resistance to proteinase K treatment (Magen and Chesselet 2010). Early dysfunctional or plastic changes have been observed in the tissue before pathological deposition of α-synuclein can be detected that appears to correlate over time with the severity of neurodegeneration (Gispert et al. 2003). The putative role of α-synuclein Ser129-phosphorylation has been seen in different models (Magen and Chesselet 2010). Phosphorylation of Ser129 is a prerequisite for dopaminergic cell death in Drosophila (Chen and Feany 2005), but this has not been corroborated in mammalian models. Three studies have been conducted comparing the neurotoxicity of expressing wild-type and pseudo-phosphorylated S129D mutant human α-synuclein. In two studies wild-type α-synuclein was more toxic than S129D, while the third reported no difference between the two α-synuclein species (Gorbatyuk et al. 2008; Azeredo da Silveira et al. 2009; McFarland et al. 2009). However, it should be kept in mind that S129D α-synuclein structurally is a poor mimic of Ser129-phosphorylated α-synuclein (Paleologou et al. 2008). Indirect approaches for increasing Ser129-phosphorylation using overexpression of phosphoprotein phosphatase 2A activity in a Thy1 transgenic mice line (Lee et al. 2011) or the α-synuclein directed kinase GRK6 using viral vectors in rats (Sato et al. 2011) report increased toxicity, but these approaches also affect the phosphorylation status of other proteins. Noteworthy, phosphorylation at Ser129 is not necessary for α-synuclein transgene-dependent cell death to occur, as demonstrated in animals overexpressing α-synuclein C-terminally truncated before the Ser129 residue (Wakamatsu et al. 2008; Ulusoy et al. 2010).
10.4 Inflammation and Microglia in the Disease
Microglia activation and neuroinflammation in PD is recapitulated in several models: changes in microglia numbers or morphology, the cytokine pattern, or the gene expression (van der Putten et al. 2000; Neumann et al. 2002; Gomez-Isla et al. 2003; Tofaris et al. 2006; Su et al. 2008; Emmer et al. 2011). The microglial activation patterns correlate with the degree of α-synuclein-dependent neuropathology in the rat viral vector PD model (Sanchez-Guajardo et al. 2010), and pro-degenerative synergism exists between the proinflammatory environment induced by local or peripheral LPS treatment and α-synuclein in transgenic models (Gao et al. 2008; Gao et al. 2011). Neuroprotection observed by both passive and active vaccination in an α-synuclein transgenic mice line demonstrates that the peripheral immune system may be harnessed therapeutically and such an approach is already in a Phase 1 trial (Masliah et al. 2005; Haggerty et al. 2011).
10.5 Interactions with Other Brain Disease-Related Genes
Regarding the possible interactions with other proteins and genes associated with PD, coexpression of A53T α-synuclein and LRRK2 (wild-type, mutated G2019S, or KD) resulted in synergistic toxicity enhancing and accelerating the α-synuclein-induced neurodegeneration (Lin et al. 2009). However, no difference in toxicity was observed in A53T α-synuclein transgenic mice when crossed with DJ1-deficient (Ramsey et al. 2010) or parkin null mice (von Coelln et al. 2006). Transgenic lines coexpressing wild-type α-synuclein and β-amyloid peptides showed more severe anomalies than single transgenics, suggesting a shared pathogenic event between both proteins (Masliah et al. 2001). In parallel, the role of α-synuclein in Huntington’s disease has also been highlighted by a decrease in neurodegenerative changes in a knock-in mouse model of Huntington’s disease with an α-synuclein knockout background (Tomas-Zapico et al. 2011).
11 Conclusion
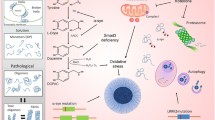
There is still considerable doubt over the species of α-synuclein that contributes most to neurotoxicity (Fig. 2). Currently there is a huge gap between data obtained through in vitro versus in vivo methods. This is highlighted by recent data that suggests the native state of α-synuclein is a folded tetramer (Bartels et al. 2011; Wang et al. 2011). If this concept proves true, it has fundamental implications for conceptualizing the potential molecular mechanisms involved in α-synucleinopathies. How Ser129-phosphorylation contributes to these structures or to pathogenesis needs to be determined, as Ser129-phosphorylation is tightly linked to human pathological lesions. To date the functional significance of Ser129-phosphorylation in cell and in vivo models has been inconclusive, findings that may be related to its structural state. Development of novel structure-specific imaging tools, like antibodies and small molecules, is likely to assist with understanding of both the structural and posttranslational changes in α-synuclein that occur in α-synucleinopathies. It is important to clarify the significance of Ser129-phosphorylation of different α-synuclein structural species to determine their role in disease initiation and progression because kinases hold great potential as drug targets.

Proposed mechanisms of neurodegeneration in PD. There are multiple potential pathways of neurodegeneration in PD. α-Synuclein aggregation and accumulation (e.g., of phosphorylated α-synuclein) can cause proteasomal, lysosomal, and mitochondrial impairment leading to neurodegeneration. In addition, α-synuclein can be secreted from neurons and taken up by microglia and astrocytes. Proinflammatory mediators such as cytokines and chemokines produced by activated microglia activate astrocytes, and the products released by activated microglia and astrocytes may exert neurotoxic effects
The novel hypothesis that the disease is spread through the uptake of extracellular α-synuclein has significant implications currently driving considerable research activity. The observation that astrocytes seem to be more prone to such uptake in PD now needs to be understood fully. Particularly, the functional significance of the early and pervasive astrocytic accumulation of α-synuclein needs to be determined. Does such behavior play a paralytic role in the neurodegeneration in PD? Regarding uptake of extracellular α-synuclein, research questions currently pursued include the following: How is α-synuclein externalized during aging and disease and in which forms? How does external α-synuclein affect cells at the cell surface and upon internalization? The answers to these questions will be important for the further development of the currently promising in vivo α-synuclein immunotherapies, as externalized α-synuclein is more amenable to such therapies. Whether there are specific α-synuclein species and/or cell types that are optimal for antibody-dependent targeting needs to be determined. Furthermore, any immunotherapy successes require replication in multiple models.
Aging is known to be the greatest risk factor for PD and LB pathology, but the mechanisms affected by aging still need to be identified. While the importance of mitochondrial function with aging needs to be considered, the specificity that mitochondrial aging precipitates only α-synucleinopathies and not other neurodegenerative conditions requires further explanation. Although speculative, Tyr125-phosphorylation may represent a α-synuclein-dependent cytoprotective mechanism associated with tetramer stability that is lost upon aging. The identification of the toxic species of α-synuclein and changes with aging will be important to determine. The identification that α-synuclein can be catabolized through autophagy and recent in vivo modeling suggests that disruption in the cross talk between proteasomal and autophagic α-synuclein catabolism may be important, particularly as the catabolism of α-synuclein changes during brain development and aging. Another significant factor may be the coexisting pathologies that accumulate with aging. Cellular defenses against misfolded proteins have been characterized in model organisms, and their role in PD needs to be clarified. Whether concentrating on α-synuclein therapeutic interventions alone is the best strategy needs to be clarified.
References
Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., Shinsky, N., Verdugo, J. M., Armanini, M., Ryan, A., Hynes, M., Phillips, H., Sulzer, D., & Rosenthal, A. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron, 25, 239–252.
Anderson, J. P., Walker, D. E., Goldstein, J. M., de Laat, R., Banducci, K., Caccavello, R. J., Barbour, R., Huang, J., Kling, K., Lee, M., Diep, L., Keim, P. S., Shen, X., Chataway, T., Schlossmacher, M. G., Seubert, P., Schenk, D., Sinha, S., Gai, W. P., & Chilcote, T. J. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. The Journal of Biological Chemistry, 281, 29739–29752.
Arawaka, S., Wada, M., Goto, S., Karube, H., Sakamoto, M., Ren, C. H., Koyama, S., Nagasawa, H., Kimura, H., Kawanami, T., Kurita, K., Tajima, K., Daimon, M., Baba, M., Kido, T., Saino, S., Goto, K., Asao, H., Kitanaka, C., Takashita, E., Hongo, S., Nakamura, T., Kayama, T., Suzuki, Y., Kobayashi, K., Katagiri, T., Kurokawa, K., Kurimura, M., Toyoshima, I., Niizato, K., Tsuchiya, K., Iwatsubo, T., Muramatsu, M., Matsumine, H., & Kato, T. (2006). The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson’s disease. The Journal of Neuroscience, 26, 9227–9238.
Azeredo da Silveira, S., Schneider, B. L., Cifuentes-Diaz, C., Sage, D., Abbas-Terki, T., Iwatsubo, T., Unser, M., & Aebischer, P. (2009). Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Human Molecular Genetics, 18, 872–887.
Bartels, T., Choi, J. G., & Selkoe, D. J. (2011). Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature, 477, 107–110.
Beach, T. G., Adler, C. H., Lue, L., Sue, L. I., Bachalakuri, J., Henry-Watson, J., Sasse, J., Boyer, S., Shirohi, S., Brooks, R., Eschbacher, J., White, C. L., 3rd, Akiyama, H., Caviness, J., Shill, H. A., Connor, D. J., Sabbagh, M. N., & Walker, D. G. (2009). Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathologica, 117, 613–634.
Braak, H., Del Tredici, K., Rub, U., de Vos, R. A., Jansen Steur, E. N., & Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging, 24, 197–211.
Braak, H., Ghebremedhin, E., Rub, U., Bratzke, H., & Del Tredici, K. (2004). Stages in the development of Parkinson’s disease-related pathology. Cell and Tissue Research, 318, 121–134.
Braak, H., Sastre, M., & Del Tredici, K. (2007). Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathologica, 114, 231–241.
Burre, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., & Sudhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science, 329, 1663–1667.
Bussell, R., Jr., & Eliezer, D. (2003). A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. Journal of Molecular Biology, 329, 763–778.
Cabin, D. E., Gispert-Sanchez, S., Murphy, D., Auburger, G., Myers, R. R., & Nussbaum, R. L. (2005). Exacerbated synucleinopathy in mice expressing A53T SNCA on a Snca null background. Neurobiology of Aging, 26, 25–35.
Chandra, S., Chen, X., Rizo, J., Jahn, R., & Sudhof, T. C. (2003). A broken alpha-helix in folded alpha-synuclein. The Journal of Biological Chemistry, 278, 15313–15318.
Chandra, S., Fornai, F., Kwon, H. B., Yazdani, U., Atasoy, D., Liu, X., Hammer, R. E., Battaglia, G., German, D. C., Castillo, P. E., & Sudhof, T. C. (2004). Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proceedings of the National Academy of Sciences of the United States of America, 101, 14966–14971.
Chen, L., & Feany, M. B. (2005). Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nature Neuroscience, 8, 657–663.
Chen, L., Periquet, M., Wang, X., Negro, A., McLean, P. J., Hyman, B. T., & Feany, M. B. (2009). Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. The Journal of Clinical Investigation, 119, 3257–3265.
Chesselet, M. F., & Richter, F. (2011). Modelling of Parkinson’s disease in mice. Lancet Neurology, 10, 1108–1118.
Cho, M. K., Nodet, G., Kim, H. Y., Jensen, M. R., Bernado, P., Fernandez, C. O., Becker, S., Blackledge, M., & Zweckstetter, M. (2009). Structural characterization of alpha-synuclein in an aggregation prone state. Protein Science: A Publication of the Protein Society, 18, 1840–1846.
Chung, K. K., Dawson, V. L., & Dawson, T. M. (2001). The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends in Neurosciences, 24, S7–S14.
Chung, C. Y., Koprich, J. B., Siddiqi, H., & Isacson, O. (2009). Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. The Journal of Neuroscience, 29, 3365–3373.
Clayton, D. F., & George, J. M. (1998). The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends in Neurosciences, 21, 249–254.
Compta, Y., Parkkinen, L., O’Sullivan, S. S., Vandrovcova, J., Holton, J. L., Collins, C., Lashley, T., Kallis, C., Williams, D. R., de Silva, R., Lees, A. J., & Revesz, T. (2011). Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain: A Journal of Neurology, 134, 1493–1505.
Conway, K. A., Lee, S. J., Rochet, J. C., Ding, T. T., Williamson, R. E., & Lansbury, P. T., Jr. (2000). Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proceedings of the National Academy of Sciences of the United States of America, 97, 571–576.
Conway, K. A., Rochet, J. C., Bieganski, R. M., & Lansbury, P. T., Jr. (2001). Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science, 294, 1346–1349.
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., Liu, K., Xu, K., Strathearn, K. E., Liu, F., Cao, S., Caldwell, K. A., Caldwell, G. A., Marsischky, G., Kolodner, R. D., Labaer, J., Rochet, J. C., Bonini, N. M., & Lindquist, S. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science, 313, 324–328.
Croisier, E., Moran, L. B., Dexter, D. T., Pearce, R. K., & Graeber, M. B. (2005). Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. Journal of Neuroinflammation, 2, 14.
Crowther, R. A., Jakes, R., Spillantini, M. G., & Goedert, M. (1998). Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Letters, 436, 309–312.
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., & Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science, 305, 1292–1295.
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., Kretzschmar, H., Hengerer, B., & Kostka, M. (2007). Different species of alpha-synuclein oligomers induce calcium influx and seeding. The Journal of Neuroscience, 27, 9220–9232.
Dauer, W., Kholodilov, N., Vila, M., Trillat, A. C., Goodchild, R., Larsen, K. E., Staal, R., Tieu, K., Schmitz, Y., Yuan, C. A., Rocha, M., Jackson-Lewis, V., Hersch, S., Sulzer, D., Przedborski, S., Burke, R. E., & Hen, R. (2002). Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proceedings of the National Academy of Sciences of the United States of America, 99, 14524–14529.
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., Spencer, B., Masliah, E., & Lee, S. J. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proceedings of the National Academy of Sciences of the United States of America, 106, 13010–13015.
Devic, I., Hwang, H., Edgar, J. S., Izutsu, K., Presland, R., Pan, C., Goodlett, D. R., Wang, Y., Armaly, J., Tumas, V., Zabetian, C. P., Leverenz, J. B., Shi, M., & Zhang, J. (2011). Salivary alpha-synuclein and DJ-1: Potential biomarkers for Parkinson’s disease. Brain: A Journal of Neurology, 134, e178.
Dickson, D. W., Braak, H., Duda, J. E., Duyckaerts, C., Gasser, T., Halliday, G. M., Hardy, J., Leverenz, J. B., Del Tredici, K., Wszolek, Z. K., & Litvan, I. (2009). Neuropathological assessment of Parkinson’s disease: Refining the diagnostic criteria. Lancet Neurology, 8, 1150–1157.
Dufty, B. M., Warner, L. R., Hou, S. T., Jiang, S. X., Gomez-Isla, T., Leenhouts, K. M., Oxford, J. T., Feany, M. B., Masliah, E., & Rohn, T. T. (2007). Calpain-cleavage of {alpha}-synuclein: Connecting proteolytic processing to disease-linked aggregation. The American Journal of Pathology, 170, 1725–1738.
Durrenberger, P. F., Filiou, M. D., Moran, L. B., Michael, G. J., Novoselov, S., Cheetham, M. E., Clark, P., Pearce, R. K., & Graeber, M. B. (2009). DnaJB6 is present in the core of Lewy bodies and is highly up-regulated in parkinsonian astrocytes. Journal of Neuroscience Research, 87, 238–245.
Ebrahimi-Fakhari, D., Cantuti-Castelvetri, I., Fan, Z., Rockenstein, E., Masliah, E., Hyman, B. T., McLean, P. J., & Unni, V. K. (2011). Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 31, 14508–14520.
Ellis, C. E., Schwartzberg, P. L., Grider, T. L., Fink, D. W., & Nussbaum, R. L. (2001). Alpha-synuclein is phosphorylated by members of the Src family of protein-tyrosine kinases. The Journal of Biological Chemistry, 276, 3879–3884.
Emmanouilidou, E., Stefanis, L., & Vekrellis, K. (2010). Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiology of Aging, 31, 953–968.
Emmer, K. L., Waxman, E. A., Covy, J. P., & Giasson, B. I. (2011). E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. The Journal of Biological Chemistry, 286, 35104–35118.
Engelender, S., Kaminsky, Z., Guo, X., Sharp, A. H., Amaravi, R. K., Kleiderlein, J. J., Margolis, R. L., Troncoso, J. C., Lanahan, A. A., Worley, P. F., Dawson, V. L., Dwson, T. M., & Ross, C. A. (1999). Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nature Genetics, 22, 110–114.
Eslamboli, A., Romero-Ramos, M., Burger, C., Bjorklund, T., Muzyczka, N., Mandel, R. J., Baker, H., Ridley, R. M., & Kirik, D. (2007). Long-term consequences of human alpha-synuclein overexpression in the primate ventral midbrain. Brain, 130, 799–815.
Fjorback, A. W., Varming, K., & Jensen, P. H. (2007). Determination of alpha-synuclein concentration in human plasma using ELISA. Scandinavian Journal of Clinical and Laboratory Investigation, 67, 431–435.
Fernagut, et al. (2007). Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. In: Fernagut, P. O., Hutson, C. B., Fleming, S. M., Tetreaut, N. A., Salcedo, J., Masliah, E., & Chesselet, M. F. Synapse, 61(12):991–1001.
Fleming, S. M., & Chesselet, M. F. (2006). Behavioral phenotypes and pharmacology in genetic mouse models of Parkinsonism. Behavioural Pharmacology, 17, 383–391.
Fleming, S. M., Salcedo, J., Fernagut, P. O., Rockenstein, E., Masliah, E., Levine, M. S., & Chesselet, M. F. (2004). Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. The Journal of Neuroscience, 24, 9434–9440.
Fleming, S. M., Tetreault, N. A., Mulligan, C. K., Hutson, C. B., Masliah, E., & Chesselet, M. F. (2008). Olfactory deficits in mice overexpressing human wildtype alpha-synuclein. The European Journal of Neuroscience, 28, 247–256.
Fountaine, T. M., & Wade-Martins, R. (2007). RNA interference-mediated knockdown of alpha-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. Journal of Neuroscience Research, 85, 351–363.
Freichel, C., Neumann, M., Ballard, T., Muller, V., Woolley, M., Ozmen, L., Borroni, E., Kretzschmar, H. A., Haass, C., Spooren, W., & Kahle, P. J. (2007). Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiology of Aging, 28, 1421–1435.
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., Shen, J., Takio, K., & Iwatsubo, T. (2002). Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biology, 4, 160–164.
Gai, W., Power, J., Blumberg, P., & Blessing, W. (1998). Multiple-system atrophy: A new α-synuclein disease? Lancet, 352, 547–548.
Gao, H. M., Kotzbauer, P. T., Uryu, K., Leight, S., Trojanowski, J. Q., & Lee, V. M. (2008). Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. The Journal of Neuroscience, 28, 7687–7698.
Gao, H. M., Zhang, F., Zhou, H., Kam, W., Wilson, B., & Hong, J. S. (2011). Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environmental Health Perspectives, 119, 807–814.
George, J., Jin, H., Woods, W., & Clayton, D. (1995). Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron, 15, 361–372.
Giasson, B. I., Duda, J. E., Quinn, S. M., Zhang, B., Trojanowski, J. Q., & Lee, V. M. (2002). Neuronal alpha-Synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron, 34, 521–533.
Gispert, S., Del Turco, D., Garrett, L., Chen, A., Bernard, D. J., Hamm-Clement, J., Korf, H. W., Deller, T., Braak, H., Auburger, G., & Nussbaum, R. L. (2003). Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence of aggregate formation. Molecular and Cellular Neurosciences, 24, 419–429.
Gomez-Isla, T., Irizarry, M. C., Mariash, A., Cheung, B., Soto, O., Schrump, S., Sondel, J., Kotilinek, L., Day, J., Schwarzschild, M. A., Cha, J. H., Newell, K., Miller, D. W., Ueda, K., Young, A. B., Hyman, B. T., & Ashe, K. H. (2003). Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30P transgenic mice. Neurobiology of Aging, 24, 245–258.
Gomperts, S. N., Rentz, D. M., Moran, E., Becker, J. A., Locascio, J. J., Klunk, W. E., Mathis, C. A., Elmaleh, D. R., Shoup, T., Fischman, A. J., Hyman, B. T., Growdon, J. H., & Johnson, K. A. (2008). Imaging amyloid deposition in Lewy body diseases. Neurology, 71, 903–910.
Gorbatyuk, O. S., Li, S., Sullivan, L. F., Chen, W., Kondrikova, G., Manfredsson, F. P., Mandel, R. J., & Muzyczka, N. (2008). The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America, 105, 763–768.
Greffard, S., Verny, M., Bonnet, A. M., Beinis, J. Y., Gallinari, C., Meaume, S., Piette, F., Hauw, J. J., & Duyckaerts, C. (2006). Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Archives of Neurology, 63, 584–588.
Greffard, S., Verny, M., Bonnet, A. M., Seilhean, D., Hauw, J. J., & Duyckaerts, C. (2010). A stable proportion of Lewy body bearing neurons in the substantia nigra suggests a model in which the Lewy body causes neuronal death. Neurobiology of Aging, 31, 99–103.
Greten-Harrison, B., Polydoro, M., Morimoto-Tomita, M., Diao, L., Williams, A. M., Nie, E. H., Makani, S., Tian, N., Castillo, P. E., Buchman, V. L., & Chandra, S. S. (2010). Alphabetagamma-synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proceedings of the National Academy of Sciences of the United States of America, 107, 19573–19578.
Haggerty, T., Credle, J., Rodriguez, O., Wills, J., Oaks, A. W., Masliah, E., & Sidhu, A. (2011). Hyperphosphorylated Tau in an alpha-synuclein-overexpressing transgenic model of Parkinson’s disease. The European Journal of Neuroscience, 33, 1598–1610.
Halliday, G. M., & McCann, H. (2010). The progression of pathology in Parkinson’s disease. Annals of the New York Academy of Sciences, 1184, 188–195.
Halliday, G. M., & Stevens, C. H. (2011). Glia: Initiators and progressors of pathology in Parkinson’s disease. Movement Disorders, 26, 6–17.
Halliday, G. M., Holton, J. L., Revesz, T., & Dickson, D. W. (2011a). Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathologica, 122, 187–204.
Halliday, G. M., Song, Y. J., & Harding, A. J. (2011b). Striatal beta-amyloid in dementia with Lewy bodies but not Parkinson’s disease. Journal of Neural Transmission, 118, 713–719.
Harding, A. J., Stimson, E., Henderson, J. M., & Halliday, G. M. (2002). Clinical correlates of selective pathology in the amygdala of patients with Parkinson’s disease. Brain: A Journal of Neurology, 125, 2431–2445.
Hirsch, E. C., Vyas, S., & Hunot, S. (2012). Neuroinflammation in Parkinson’s disease. Parkinsonism & Related Disorders, 18(Suppl 1), S210–S212.
Hoozemans, J. J., van Haastert, E. S., Eikelenboom, P., de Vos, R. A., Rozemuller, J. M., & Scheper, W. (2007). Activation of the unfolded protein response in Parkinson’s disease. Biochemical and Biophysical Research Communications, 354, 707–711.
Hsu, L. J., Sagara, Y., Arroyo, A., Rockenstein, E., Sisk, A., Mallory, M., Wong, J., Takenouchi, T., Hashimoto, M., & Masliah, E. (2000). Alpha-synuclein promotes mitochondrial deficit and oxidative stress. The American Journal of Pathology, 157, 401–410.
Huang, Y., Cheung, L., Rowe, D., & Halliday, G. (2004). Genetic contributions to Parkinson’s disease. Brain Research. Brain Research Reviews, 46, 44–70.
Inglis, K. J., Chereau, D., Brigham, E. F., Chiou, S. S., Schobel, S., Frigon, N. L., Yu, M., Caccavello, R. J., Nelson, S., Motter, R., Wright, S., Chian, D., Santiago, P., Soriano, F., Ramos, C., Powell, K., Goldstein, J. M., Babcock, M., Yednock, T., Bard, F., Basi, G. S., Sham, H., Chilcote, T. J., McConlogue, L., Griswold-Prenner, I., & Anderson, J. P. (2009). Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. The Journal of Biological Chemistry, 284, 2598–2602.
Ishida, Y., Nagai, A., Kobayashi, S., & Kim, S. U. (2006). Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: Astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. Journal of Neuropathology and Experimental Neurology, 65, 66–77.
Jakes, R., Spillantini, M. G., & Goedert, M. (1994). Identification of two distinct synucleins from human brain. FEBS Letters, 345, 27–32.
Jensen, P., Li, J.-Y., Dahlstrom, A., & Dotti, C. (1999). Axonal transport of synucleins is mediated by all rate components. Europian Journal of Neuroscience, 11, 3369–3376.
Kahle, P. J., Neumann, M., Ozmen, L., Muller, V., Odoy, S., Okamoto, N., Jacobsen, H., Iwatsubo, T., Trojanowski, J. Q., Takahashi, H., Wakabayashi, K., Bogdanovic, N., Riederer, P., Kretzschmar, H. A., & Haass, C. (2001). Selective insolubility of alpha-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. The American Journal of Pathology, 159, 2215–2225.
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., Nuscher, B., Bartels, T., Giese, A., Beyer, K., Eimer, S., Winklhofer, K. F., & Haass, C. (2010). Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. The EMBO Journal, 29, 3571–3589.
Karpinar, D. P., Balija, M. B., Kugler, S., Opazo, F., Rezaei-Ghaleh, N., Wender, N., Kim, H. Y., Taschenberger, G., Falkenburger, B. H., Heise, H., Kumar, A., Riedel, D., Fichtner, L., Voigt, A., Braus, G. H., Giller, K., Becker, S., Herzig, A., Baldus, M., Jackle, H., Eimer, S., Schulz, J. B., Griesinger, C., & Zweckstetter, M. (2009). Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. The EMBO Journal, 28, 3256–3268.
Kayed, R., Head, E., Sarsoza, F., Saing, T., Cotman, C. W., Necula, M., Margol, L., Wu, J., Breydo, L., Thompson, J. L., Rasool, S., Gurlo, T., Butler, P., & Glabe, C. G. (2007). Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Molecular Neurodegeneration, 2, 18.
Kim, H. J., Lee, D., Lee, C. H., Chung, K. C., Kim, J., & Paik, S. R. (2006). Calpain-resistant fragment(s) of alpha-synuclein regulates the synuclein-cleaving activity of 20S proteasome. Archives of Biochemistry and Biophysics, 455, 40–47.
Kirik, D., Rosenblad, C., Burger, C., Lundberg, C., Johansen, T. E., Muzyczka, N., Mandel, R. J., & Bjorklund, A. (2002). Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. The Journal of Neuroscience, 22, 2780–2791.
Klegeris, A., Giasson, B. I., Zhang, H., Maguire, J., Pelech, S., & McGeer, P. L. (2006). Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. The FASEB Journal: Official publication of the Federation of American Societies for Experimental Biology, 20, 2000–2008.
Klucken, J., Outeiro, T. F., Nguyen, P., McLean, P. J., & Hyman, B. T. (2006). Detection of novel intracellular alpha-synuclein oligomeric species by fluorescence lifetime imaging. The FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 20, 2050–2057.
Knott, C., Wilkin, G. P., & Stern, G. (1999). Astrocytes and microglia in the substantia nigra and caudate-putamen in Parkinson’s disease. Parkinsonism & Related Disorders, 5, 115–122.
Koob, A. O., Ubhi, K., Paulsson, J. F., Kelly, J., Rockenstein, E., Mante, M., Adame, A., & Masliah, E. (2010). Lovastatin ameliorates alpha-synuclein accumulation and oxidation in transgenic mouse models of alpha-synucleinopathies. Experimental Neurology, 221, 267–274.
Kordower, J. H., Chu, Y., Hauser, R. A., Freeman, T. B., & Olanow, C. W. (2008). Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nature Medicine, 14, 504–506.
Kragh, C. L., Lund, L. B., Febbraro, F., Hansen, H. D., Gai, W. P., El-Agnaf, O., Richter-Landsberg, C., & Jensen, P. H. (2009). {alpha}-synuclein aggregation and Ser-129 phosphorylation-dependent cell death in oligodendroglial cells. The Journal of Biological Chemistry, 284, 10211–10222.
Kruger, R., Kuhn, W., Muller, T., Woitalla, D., Graeber, M., Kosel, S., Przuntek, H., Epplen, J., Schols, L., & Reiss, O. (1998). Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nature Genetics, 18, 106–108.
Kuo, Y. M., Li, Z., Jiao, Y., Gaborit, N., Pani, A. K., Orrison, B. M., Bruneau, B. G., Giasson, B. I., Smeyne, R. J., Gershon, M. D., & Nussbaum, R. L. (2010). Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Human Molecular Genetics, 19, 1633–1650.
Lam, H. A., Wu, N., Cely, I., Kelly, R. L., Hean, S., Richter, F., Magen, I., Cepeda, C., Ackerson, L. C., Walwyn, W., Masliah, E., Chesselet, M. F., Levine, M. S., & Maidment, N. T. (2011). Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human alpha-synuclein. Journal of Neuroscience Research, 89, 1091–1102.
Lashuel, H. A., Petre, B. M., Wall, J., Simon, M., Nowak, R. J., Walz, T., & Lansbury, P. T., Jr. (2002). Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. Journal of Molecular Biology, 322, 1089–1102.
Lee, F. J., Liu, F., Pristupa, Z. B., & Niznik, H. B. (2001). Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 15, 916–926.
Lee, M. K., Stirling, W., Xu, Y., Xu, X., Qui, D., Mandir, A. S., Dawson, T. M., Copeland, N. G., Jenkins, N. A., & Price, D. L. (2002). Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 –> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America, 99, 8968–8973.
Lee, H. J., Patel, S., & Lee, S. J. (2005). Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. The Journal of Neuroscience, 25, 6016–6024.
Lee, H. J., Suk, J. E., Patrick, C., Bae, E. J., Cho, J. H., Rho, S., Hwang, D., Masliah, E., & Lee, S. J. (2010). Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. The Journal of Biological Chemistry, 285, 9262–9272.
Lee, K. W., Chen, W., Junn, E., Im, J. Y., Grosso, H., Sonsalla, P. K., Feng, X., Ray, N., Fernandez, J. R., Chao, Y., Masliah, E., Voronkov, M., Braithwaite, S. P., Stock, J. B., & Mouradian, M. M. (2011). Enhanced phosphatase activity attenuates alpha-synucleinopathy in a mouse model. The Journal of Neuroscience, 31, 6963–6971.
Leong, S. L., Cappai, R., Barnham, K. J., & Pham, C. L. (2009). Modulation of alpha-synuclein aggregation by dopamine: A review. Neurochemical Research, 34, 1838–1846.
Li, J. Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., Lashley, T., Quinn, N. P., Rehncrona, S., Bjorklund, A., Widner, H., Revesz, T., Lindvall, O., & Brundin, P. (2008). Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nature Medicine, 14, 501–503.
Lin, X., Parisiadou, L., Gu, X. L., Wang, L., Shim, H., Sun, L., Xie, C., Long, C. X., Yang, W. J., Ding, J., Chen, Z. Z., Gallant, P. E., Tao-Cheng, J. H., Rudow, G., Troncoso, J. C., Liu, Z., Li, Z., & Cai, H. (2009). Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron, 64, 807–827.
Lindersson, E., Beedholm, R., Hojrup, P., Moos, T., Gai, W., Hendil, K. B., & Jensen, P. H. (2004). Proteasomal inhibition by alpha-synuclein filaments and oligomers. The Journal of Biological Chemistry, 279, 12924–12934.
Liu, C. W., Corboy, M. J., DeMartino, G. N., & Thomas, P. J. (2003). Endoproteolytic activity of the proteasome. Science, 299, 408–411.
Lo Bianco, C., Ridet, J. L., Schneider, B. L., Deglon, N., & Aebischer, P. (2002). Alpha-synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, 99, 10813–10818.
Lo Bianco, C., Schneider, B. L., Bauer, M., Sajadi, A., Brice, A., Iwatsubo, T., & Aebischer, P. (2004). Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, 101, 17510–17515.
Lotharius, J., Barg, S., Wiekop, P., Lundberg, C., Raymon, H. K., & Brundin, P. (2002). Effect of mutant alpha-synuclein on dopamine homeostasis in a new human mesencephalic cell line. The Journal of Biological Chemistry, 277, 38884–38894.
Lou, H., Montoya, S. E., Alerte, T. N., Wang, J., Wu, J., Peng, X., Hong, C. S., Friedrich, E. E., Mader, S. A., Pedersen, C. J., Marcus, B. S., McCormack, A. L., Di Monte, D. A., Daubner, S. C., & Perez, R. G. (2010). Serine 129 phosphorylation reduces the ability of alpha-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. The Journal of Biological Chemistry, 285, 17648–17661.
Luk, K. C., Kehm, V. M., Zhang, B., O’ Brien, P., Trojanowski, J. Q., & Lee, V. M. (2012). Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. The Journal of Experimental Medicine, 209(5), 975–986.
Magen, I., & Chesselet, M. F. (2010). Genetic mouse models of Parkinson’s disease The state of the art. Progress in Brain Research, 184, 53–87.
Maingay, M., Romero-Ramos, M., Carta, M., & Kirik, D. (2006). Ventral tegmental area dopamine neurons are resistant to human mutant alpha-synuclein overexpression. Neurobiology of Disease, 23, 522–532.
Manning-Bog, A. B., McCormack, A. L., Purisai, M. G., Bolin, L. M., & Di Monte, D. A. (2003). Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. Journal of Neuroscience, 23, 3095–3099.
Maroteaux, L., Campanelli, J., & Scheller, R. (1988). Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 8, 2804–2815.
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., Price, D. L., & Lee, M. K. (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. The Journal of Neuroscience, 26, 41–50.
Masliah, E., Rockenstein, E., Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., Sagara, Y., Sisk, A., & Mucke, L. (2000). Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science, 287, 1265–1269.
Masliah, E., Rockenstein, E., Veinbergs, I., Sagara, Y., Mallory, M., Hashimoto, M., & Mucke, L. (2001). Beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, 98, 12245–12250.
Masliah, E., Rockenstein, E., Adame, A., Alford, M., Crews, L., Hashimoto, M., Seubert, P., Lee, M., Goldstein, J., Chilcote, T., Games, D., & Schenk, D. (2005). Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron, 46, 857–868.
Masuda, M., Dohmae, N., Nonaka, T., Oikawa, T., Hisanaga, S., Goedert, M., & Hasegawa, M. (2006). Cysteine misincorporation in bacterially expressed human alpha-synuclein. FEBS Letters, 580, 1775–1779.
Matsuoka, Y., Vila, M., Lincoln, S., McCormack, A., Picciano, M., LaFrancois, J., Yu, X., Dickson, D., Langston, W. J., McGowan, E., Farrer, M., Hardy, J., Duff, K., Przedborski, S., & Di Monte, D. A. (2001). Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiology of Disease, 8, 535–539.
Matthews, F. E., Brayne, C., Lowe, J., McKeith, I., Wharton, S. B., & Ince, P. (2009). Epidemiological pathology of dementia: Attributable-risks at death in the Medical Research Council Cognitive Function and Ageing study. PLoS Medicine, 6, e1000180.
Mazzulli, J. R., Mishizen, A. J., Giasson, B. I., Lynch, D. R., Thomas, S. A., Nakashima, A., Nagatsu, T., Ota, A., & Ischiropoulos, H. (2006). Cytosolic catechols inhibit alpha-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 26, 10068–10078.
Mbefo, M. K., Paleologou, K. E., Boucharaba, A., Oueslati, A., Schell, H., Fournier, M., Olschewski, D., Yin, G., Zweckstetter, M., Masliah, E., Kahle, P. J., Hirling, H., & Lashuel, H. A. (2010). Phosphorylation of synucleins by members of the Polo-like kinase family. The Journal of Biological Chemistry, 285, 2807–2822.
McFarland, M. A., Ellis, C. E., Markey, S. P., & Nussbaum, R. L. (2008). Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Molecular & Cellular Proteomics, 7, 2123–2137.
McFarland, N. R., Fan, Z., Xu, K., Schwarzschild, M. A., Feany, M. B., Hyman, B. T., & McLean, P. J. (2009). Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. Journal of Neuropathology and Experimental Neurology, 68, 515–524.
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., Cummings, J., Duda, J. E., Lippa, C., Perry, E. K., Aarsland, D., Arai, H., Ballard, C. G., Boeve, B., Burn, D. J., Costa, D., Del Ser, T., Dubois, B., Galasko, D., Gauthier, S., Goetz, C. G., Gomez-Tortosa, E., Halliday, G., Hansen, L. A., Hardy, J., Iwatsubo, T., Kalaria, R. N., Kaufer, D., Kenny, R. A., Korczyn, A., Kosaka, K., Lee, V. M., Lees, A., Litvan, I., Londos, E., Lopez, O. L., Minoshima, S., Mizuno, Y., Molina, J. A., Mukaetova-Ladinska, E. B., Pasquier, F., Perry, R. H., Schulz, J. B., Trojanowski, J. Q., & Yamada, M. (2005). Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology, 65, 1863–1872.
McNaught, K., & Jenner, P. (2001). Proteasomal function is impaired in substantial nigra in Parkinson’s disease. Neuroscience Letters, 297, 191–194.
McNaught, K. S., Belizaire, R., Isacson, O., Jenner, P., & Olanow, C. W. (2003). Altered proteasomal function in sporadic Parkinson’s disease. Experimental Neurology, 179, 38–46.
Michael, G. J., Esmailzadeh, S., Moran, L. B., Christian, L., Pearce, R. K., & Graeber, M. B. (2011). Up-regulation of metallothionein gene expression in Parkinsonian astrocytes. Neurogenetics, 12, 295–305.
Miller, D. W., Hague, S. M., Clarimon, J., Baptista, M., Gwinn-Hardy, K., Cookson, M. R., & Singleton, A. B. (2004). Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology, 62, 1835–1838.
Mishizen-Eberz, A. J., Norris, E. H., Giasson, B. I., Hodara, R., Ischiropoulos, H., Lee, V. M., Trojanowski, J. Q., & Lynch, D. R. (2005). Cleavage of alpha-synuclein by calpain: Potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry, 44, 7818–7829.
Mollenhauer, B., El-Agnaf, O. M., Marcus, K., Trenkwalder, C., & Schlossmacher, M. G. (2010). Quantification of alpha-synuclein in cerebrospinal fluid as a biomarker candidate: Review of the literature and considerations for future studies. Biomarkers in Medicine, 4, 683–699.
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q., & Lee, V. M. (2000). Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. The Journal of Neuroscience, 20, 3214–3220.
Nasstrom, T., Fagerqvist, T., Barbu, M., Karlsson, M., Nikolajeff, F., Kasrayan, A., Ekberg, M., Lannfelt, L., Ingelsson, M., & Bergstrom, J. (2011). The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of alpha-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radical Biology & Medicine, 50, 428–437.
Neumann, M., Kahle, P. J., Giasson, B. I., Ozmen, L., Borroni, E., Spooren, W., Muller, V., Odoy, S., Fujiwara, H., Hasegawa, M., Iwatsubo, T., Trojanowski, J. Q., Kretzschmar, H. A., & Haass, C. (2002). Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. The Journal of Clinical Investigation, 110, 1429–1439.
Nieto, M., Gil-Bea, F. J., Dalfo, E., Cuadrado, M., Cabodevilla, F., Sanchez, B., Catena, S., Sesma, T., Ribe, E., Ferrer, I., Ramirez, M. J., & Gomez-Isla, T. (2006). Increased sensitivity to MPTP in human alpha-synuclein A30P transgenic mice. Neurobiology of Aging, 27, 848–856.
Norris, E. H., Giasson, B. I., Hodara, R., Xu, S., Trojanowski, J. Q., Ischiropoulos, H., & Lee, V. M. (2005). Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. The Journal of Biological Chemistry, 280, 21212–21219.
Norris, E. H., Uryu, K., Leight, S., Giasson, B. I., Trojanowski, J. Q., & Lee, V. M. (2007). Pesticide exposure exacerbates alpha-synucleinopathy in an A53T transgenic mouse model. American Journal of Pathology, 170, 658–666.
Oksman, M., Tanila, H., & Yavich, L. (2009). Behavioural and neurochemical response of alpha-synuclein A30P transgenic mice to the effects of L-DOPA. Neuropharmacology, 56, 647–652.
Ono, K., Ikemoto, M., Kawarabayashi, T., Ikeda, M., Nishinakagawa, T., Hosokawa, M., Shoji, M., Takahashi, M., & Nakashima, M. (2009). A chemical chaperone, sodium 4-phenylbutyric acid, attenuates the pathogenic potency in human alpha-synuclein A30P+ A53T transgenic mice. Parkinsonism & Related Disorders, 15, 649–654.
Orr, C. F., Rowe, D. B., Mizuno, Y., Mori, H., & Halliday, G. M. (2005). A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain: A Journal of Neurology, 128, 2665–2674.
Orth, M., Tabrizi, S. J., Schapira, A. H., & Cooper, J. M. (2003). Alpha-synuclein expression in HEK293 cells enhances the mitochondrial sensitivity to rotenone. Neuroscience Letters, 351, 29–32.
Oueslati, A., Fournier, M., & Lashuel, H. A. (2010). Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: Implications for Parkinson’s disease pathogenesis and therapies. Progress in Brain Research, 183, 115–145.
Outeiro, T. F., Putcha, P., Tetzlaff, J. E., Spoelgen, R., Koker, M., Carvalho, F., Hyman, B. T., & McLean, P. J. (2008). Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One, 3, e1867.
Paleologou, K. E., Schmid, A. W., Rospigliosi, C. C., Kim, H. Y., Lamberto, G. R., Fredenburg, R. A., Lansbury, P. T., Jr., Fernandez, C. O., Eliezer, D., Zweckstetter, M., & Lashuel, H. A. (2008). Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. The Journal of Biological Chemistry, 283, 16895–16905.
Paleologou, K. E., Kragh, C. L., Mann, D. M., Salem, S. A., Al-Shami, R., Allsop, D., Hassan, A. H., Jensen, P. H., & El-Agnaf, O. M. (2009). Detection of elevated levels of soluble alpha-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain: A Journal of Neurology, 132, 1093–1101.
Paleologou, K. E., Oueslati, A., Shakked, G., Rospigliosi, C. C., Kim, H. Y., Lamberto, G. R., Fernandez, C. O., Schmid, A., Chegini, F., Gai, W. P., Chiappe, D., Moniatte, M., Schneider, B. L., Aebischer, P., Eliezer, D., Zweckstetter, M., Masliah, E., & Lashuel, H. A. (2010). Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 30, 3184–3198.
Pan-Montojo, F., Anichtchik, O., Dening, Y., Knels, L., Pursche, S., Jung, R., Jackson, S., Gille, G., Spillantini, M. G., Reichmann, H., & Funk, R. H. (2010). Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One, 5, e8762.
Perez, R. G., Waymire, J. C., Lin, E., Liu, J. J., Guo, F., & Zigmond, M. J. (2002). A role for alpha-synuclein in the regulation of dopamine biosynthesis. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 22, 3090–3099.
Pienaar, I. S., Gotz, J., & Feany, M. B. (2010). Parkinson’s disease: Insights from non-traditional model organisms. Progress in Neurobiology, 92, 558–571.
Polymeropoulos, M., Lavedan, C., Leroy, E., Ide, S., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenroos, E., Chandrasekharappa, S., Athanassiadou, A., Papapetropulos, T., Johnson, W., Lazzarini, A., Duvoisin, R., Di Iorio, G., Golbe, L., & Nussbaum, R. (1997). Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science, 276, 2045–2047.
Poon, H. F., Frasier, M., Shreve, N., Calabrese, V., Wolozin, B., & Butterfield, D. A. (2005). Mitochondrial associated metabolic proteins are selectively oxidized in A30P alpha-synuclein transgenic mice–a model of familial Parkinson’s disease. Neurobiology of Disease, 18, 492–498.
Power, J. H., Shannon, J. M., Blumbergs, P. C., & Gai, W. P. (2002). Nonselenium glutathione peroxidase in human brain: Elevated levels in Parkinson’s disease and dementia with lewy bodies. The American Journal of Pathology, 161, 885–894.
Pronin, A. N., Morris, A. J., Surguchov, A., & Benovic, J. L. (2000). Synucleins are a novel class of substrates for G protein-coupled receptor kinases. The Journal of Biological Chemistry, 275, 26515–26522.
Ramsey, C. P., Tsika, E., Ischiropoulos, H., & Giasson, B. I. (2010). DJ-1 deficient mice demonstrate similar vulnerability to pathogenic Ala53Thr human alpha-syn toxicity. Human Molecular Genetics, 19, 1425–1437.
Rathke-Hartlieb, S., Kahle, P. J., Neumann, M., Ozmen, L., Haid, S., Okochi, M., Haass, C., & Schulz, J. B. (2001). Sensitivity to MPTP is not increased in Parkinson’s disease-associated mutant alpha-synuclein transgenic mice. Journal of Neurochemistry, 77, 1181–1184.
Richfield, E. K., Thiruchelvam, M. J., Cory-Slechta, D. A., Wuertzer, C., Gainetdinov, R. R., Caron, M. G., Di Monte, D. A., & Federoff, H. J. (2002). Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Experimental Neurology, 175, 35–48.
Rockenstein, E., Mallory, M., Hashimoto, M., Song, D., Shults, C. W., Lang, I., & Masliah, E. (2002). Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. Journal of Neuroscience Research, 68, 568–578.
Sanchez-Guajardo, V., Febbraro, F., Kirik, D., & Romero-Ramos, M. (2010). Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS One, 5, e8784.
Sato, H., Arawaka, S., Hara, S., Fukushima, S., Koga, K., Koyama, S., & Kato, T. (2011). Authentically phosphorylated alpha-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson’s disease. The Journal of Neuroscience, 31, 16884–16894.
Schell, H., Hasegawa, T., Neumann, M., & Kahle, P. J. (2009). Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience, 160, 796–804.
Sharon, R., Goldberg, M. S., Bar-Josef, I., Betensky, R. A., Shen, J., & Selkoe, D. J. (2001). Alpha-synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proceedings of the National Academy of Sciences of the United States of America, 98, 9110–9115.
Sharon, R., Bar-Joseph, I., Frosch, M. P., Walsh, D. M., Hamilton, J. A., & Selkoe, D. J. (2003). The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron, 37, 583–595.
Shi, M., Bradner, J., Hancock, A. M., Chung, K. A., Quinn, J. F., Peskind, E. R., Galasko, D., Jankovic, J., Zabetian, C. P., Kim, H. M., Leverenz, J. B., Montine, T. J., Ginghina, C., Kang, U. J., Cain, K. C., Wang, Y., Aasly, J., Goldstein, D., & Zhang, J. (2011). Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Annals of Neurology, 69, 570–580.
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., Paisan-Ruiz, C., Lichtner, P., Scholz, S. W., Hernandez, D. G., Kruger, R., Federoff, M., Klein, C., Goate, A., Perlmutter, J., Bonin, M., Nalls, M. A., Illig, T., Gieger, C., Houlden, H., Steffens, M., Okun, M. S., Racette, B. A., Cookson, M. R., Foote, K. D., Fernandez, H. H., Traynor, B. J., Schreiber, S., Arepalli, S., Zonozi, R., Gwinn, K., van der Brug, M., Lopez, G., Chanock, S. J., Schatzkin, A., Park, Y., Hollenbeck, A., Gao, J., Huang, X., Wood, N. W., Lorenz, D., Deuschl, G., Chen, H., Riess, O., Hardy, J. A., Singleton, A. B., & Gasser, T. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nature Genetics, 41, 1308–1312.
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R., Lincoln, S., Crawley, A., Hanson, M., Maraganore, D., Adler, C., Cookson, M. R., Muenter, M., Baptista, M., Miller, D., Blancato, J., Hardy, J., & Gwinn-Hardy, K. (2003). Alpha-synuclein locus triplication causes Parkinson’s disease. Science, 302, 841.
Smith, W. W., Jiang, H., Pei, Z., Tanaka, Y., Morita, H., Sawa, A., Dawson, V. L., Dawson, T. M., & Ross, C. A. (2005a). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Human Molecular Genetics, 14, 3801–3811.
Smith, W. W., Margolis, R. L., Li, X., Troncoso, J. C., Lee, M. K., Dawson, V. L., Dawson, T. M., Iwatsubo, T., & Ross, C. A. (2005b). Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 25, 5544–5552.
Snyder, H., Mensah, K., Theisler, C., Lee, J. M., Matouschek, A., & Wolozin, B. (2003). Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. The Journal of Biological Chemistry, 278(14), 11753–11759.
Song, Y. J., Halliday, G. M., Holton, J. L., Lashley, T., O’Sullivan, S. S., McCann, H., Lees, A. J., Ozawa, T., Williams, D. R., Lockhart, P. J., & Revesz, T. R. (2009). Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. Journal of Neuropathology and Experimental Neurology, 68, 1073–1083.
Spillantini, M. G., & Goedert, M. (2000). The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Annals of the New York Academy of Sciences, 920, 16–27.
Su, X., Federoff, H. J., & Maguire-Zeiss, K. A. (2009). Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotoxicity Research, 16(3), 238–254.
Su, X., Maguire-Zeiss, K. A., Giuliano, R., Prifti, L., Venkatesh, K., & Federoff, H. J. (2008). Synuclein activates microglia in a model of Parkinson’s disease. Neurobiology of Aging, 29, 1690–1701.
Su, L. J., Auluck, P. K., Outeiro, T. F., Yeger-Lotem, E., Kritzer, J. A., Tardiff, D. F., Strathearn, K. E., Liu, F., Cao, S., Hamamichi, S., Hill, K. J., Caldwell, K. A., Bell, G. W., Fraenkel, E., Cooper, A. A., Caldwell, G. A., McCaffery, J. M., Rochet, J. C., & Lindquist, S. (2010). Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Disease Models & Mechanisms, 3, 194–208.
Sugeno, N., Takeda, A., Hasegawa, T., Kobayashi, M., Kikuchi, A., Mori, F., Wakabayashi, K., & Itoyama, Y. (2008). Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. The Journal of Biological Chemistry, 283, 23179–23188.
Tanaka, Y., Engelender, S., Igarashi, S., Rao, R. K., Wanner, T., Tanzi, R., Sawa, A. L., Dawson, V., Dawson, T. M., & Ross, C. A. (2001). Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Human Molecular Genetics, 10, 919–992.
Tehranian, R., Montoya, S. E., Van Laar, A. D., Hastings, T. G., & Perez, R. G. (2006). Alpha-synuclein inhibits aromatic amino acid decarboxylase activity in dopaminergic cells. Journal of Neurochemistry, 99, 1188–1196.
Tetzlaff, J. E., Putcha, P., Outeiro, T. F., Ivanov, A., Berezovska, O., Hyman, B. T., & McLean, P. J. (2008). CHIP targets toxic alpha-synuclein oligomers for degradation. The Journal of Biological Chemistry, 283, 17962–17968.
Theodore, S., Cao, S., McLean, P. J., & Standaert, D. G. (2008). Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. Journal of Neuropathology and Experimental Neurology, 67, 1149–1158.
Tofaris, G. K., Garcia Reitbock, P., Humby, T., Lambourne, S. L., O’Connell, M., Ghetti, B., Gossage, H., Emson, P. C., Wilkinson, L. S., Goedert, M., & Spillantini, M. G. (2006). Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1–120): Implications for Lewy body disorders. The Journal of Neuroscience, 26, 3942–3950.
Tokuda, T., Qureshi, M. M., Ardah, M. T., Varghese, S., Shehab, S. A., Kasai, T., Ishigami, N., Tamaoka, A., Nakagawa, M., & El-Agnaf, O. M. (2010). Detection of elevated levels of alpha-synuclein oligomers in CSF from patients with Parkinson disease. Neurology, 75, 1766–1772.
Tomas-Zapico, C., Diez-Zaera, M., Ferrer, I., Gomez-Ramos, P., Moran, M. A., Miras-Portugal, M. T., Diaz-Hernandez, M., & Lucas, J. J. (2011). Alpha-synuclein accumulates in huntingtin inclusions but forms independent filaments and its deficiency attenuates early phenotype in a mouse model of Huntington’s disease. Human Molecular Genetics, 21(3), 495–510.
Tsika, E., Moysidou, M., Guo, J., Cushman, M., Gannon, P., Sandaltzopoulos, R., Giasson, B. I., Krainc, D., Ischiropoulos, H., & Mazzulli, J. R. (2010). Distinct region-specific alpha-synuclein oligomers in A53T transgenic mice: Implications for neurodegeneration. Journal of Neuroscience, 30, 3409–3418.
Ubhi, K., Low, P., & Masliah, E. (2011). Multiple system atrophy: A clinical and neuropathological perspective. Trends in Neurosciences, 34, 581–590.
Ueda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., Otero, D. A., Kondo, J., Ihara, Y., & Saitoh, T. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America, 90, 11282–11286.
Ulusoy, A., Febbraro, F., Jensen, P. H., Kirik, D., & Romero-Ramos, M. (2010). Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. The European Journal of Neuroscience, 32, 409–422.
van der Putten, H., Wiederhold, K. H., Probst, A., Barbieri, S., Mistl, C., Danner, S., Kauffmann, S., Hofele, K., Spooren, W. P., Ruegg, M. A., Lin, S., Caroni, P., Sommer, B., Tolnay, M., & Bilbe, G. (2000). Neuropathology in mice expressing human alpha-synuclein. The Journal of Neuroscience, 20, 6021–6029.
Ved, R., Saha, S., Westlund, B., Perier, C., Burnam, L., Sluder, A., Hoener, M., Rodrigues, C. M., Alfonso, A., Steer, C., Liu, L., Przedborski, S., & Wolozin, B. (2005). Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. The Journal of Biological Chemistry, 280, 42655–42668.
Vitner, E. B., Platt, F. M., & Futerman, A. H. (2010). Common and uncommon pathogenic cascades in lysosomal storage diseases. The Journal of Biological Chemistry, 285, 20423–20427.
Vogiatzi, T., Xilouri, M., Vekrellis, K., & Stefanis, L. (2008). Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. The Journal of Biological Chemistry, 283, 23542–23556.
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., Meaney, D. F., Trojanowski, J. Q., & Lee, V. M. (2011). Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron, 72, 57–71.
von Coelln, R., Thomas, B., Andrabi, S. A., Lim, K. L., Savitt, J. M., Saffary, R., Stirling, W., Bruno, K., Hess, E. J., Lee, M. K., Dawson, V. L., & Dawson, T. M. (2006). Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy. The Journal of Neuroscience, 26, 3685–3696.
Wakamatsu, M., Ishii, A., Ukai, Y., Sakagami, J., Iwata, S., Ono, M., Matsumoto, K., Nakamura, A., Tada, N., Kobayashi, K., Iwatsubo, T., & Yoshimoto, M. (2007). Accumulation of phosphorylated alpha-synuclein in dopaminergic neurons of transgenic mice that express human alpha-synuclein. Journal of Neuroscience Research, 85, 1819–1825.
Wakamatsu, M., Ishii, A., Iwata, S., Sakagami, J., Ukai, Y., Ono, M., Kanbe, D., Muramatsu, S., Kobayashi, K., Iwatsubo, T., & Yoshimoto, M. (2008). Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha-synuclein in mice. Neurobiology of Aging, 29, 574–585.
Wang, L., Fleming, S. M., Chesselet, M. F., & Tache, Y. (2008). Abnormal colonic motility in mice overexpressing human wild-type alpha-synuclein. NeuroReport, 19, 873–876.
Wang, W., Perovic, I., Chittuluru, J., Kaganovich, A., Nguyen, L. T., Liao, J., Auclair, J. R., Johnson, D., Landeru, A., Simorellis, A. K., Ju, S., Cookson, M. R., Asturias, F. J., Agar, J. N., Webb, B. N., Kang, C., Ringe, D., Petsko, G. A., Pochapsky, T. C., & Hoang, Q. Q. (2011). A soluble alpha-synuclein construct forms a dynamic tetramer. Proceedings of the National Academy of Sciences of the United States of America, 108, 17797–17802.
Weinreb, P., Zhen, W., Poon, A., Conway, K., & Lansbury, P. J. (1996). NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry, 35, 13709–13715.
Weintraub, D., Doshi, J., Koka, D., Davatzikos, C., Siderowf, A. D., Duda, J. E., Wolk, D. A., Moberg, P. J., Xie, S. X., & Clark, C. M. (2011). Neurodegeneration across stages of cognitive decline in Parkinson disease. Archives of Neurology, 68, 1562–1568.
Wersinger, C., & Sidhu, A. (2003). Attenuation of dopamine transporter activity by alpha-synuclein. Neuroscience Letters, 340, 189–192.
Wood, S. J., Wypych, J., Steavenson, S., Louis, J. C., Citron, M., & Biere, A. L. (1999). Alpha-synuclein fibrillogenesis is nucleation dependent. Implications for the pathogenesis of Parkinson’s disease. The Journal of Biological Chemistry, 274, 19509–19512.
Xilouri, M., & Stefanis, L. (2011). Autophagic pathways in Parkinson disease and related disorders. Expert Reviews in Molecular Medicine, 13, e8.
Xu, J., Kao, S. Y., Lee, F. J., Song, W., Jin, L. W., & Yankner, B. A. (2002). Dopamine-dependent neurotoxicity of alpha-synuclein: A mechanism for selective neurodegeneration in Parkinson disease. Nature Medicine, 8, 600–606.
Yamada, M., Iwatsubo, T., Mizuno, Y., & Mochizuki, H. (2004). Overexpression of alpha-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of alpha-synuclein and activation of caspase-9: Resemblance to pathogenetic changes in Parkinson’s disease. Journal of Neurochemistry, 91, 451–461.
Yasuda, T., Miyachi, S., Kitagawa, R., Wada, K., Nihira, T., Ren, Y. R., Hirai, Y., Ageyama, N., Terao, K., Shimada, T., Takada, M., Mizuno, Y., & Mochizuki, H. (2007). Neuronal specificity of alpha-synuclein toxicity and effect of Parkin co-expression in primates. Neuroscience, 144, 743–753.
Yavich, L., Jakala, P., & Tanila, H. (2006). Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in alpha-synuclein knockout and A30P transgenic mice. Journal of Neurochemistry, 99, 724–732.
Yavich, L., Oksman, M., Tanila, H., Kerokoski, P., Hiltunen, M., van Groen, T., Puolivali, J., Mannisto, P. T., Garcia-Horsman, A., MacDonald, E., Beyreuther, K., Hartmann, T., & Jakala, P. (2005). Locomotor activity and evoked dopamine release are reduced in mice overexpressing A30P-mutated human alpha-synuclein. Neurobiology of Disease, 20, 303–313.
Yavich, L., Tanila, H., Vepsalainen, S., & Jakala, P. (2004). Role of alpha-synuclein in presynaptic dopamine recruitment. Journal of Neuroscience, 24, 11165–11170.
Yu, W. H., Matsuoka, Y., Sziraki, I., Hashim, A., Lafrancois, J., Sershen, H., & Duff, K. E. (2008). Increased dopaminergic neuron sensitivity to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in transgenic mice expressing mutant A53T alpha-synuclein. Neurochemical Research, 33, 902–911.
Zaccai, J., Brayne, C., McKeith, I., Matthews, F., & Ince, P. G. (2008). Patterns and stages of alpha-synucleinopathy: Relevance in a population-based cohort. Neurology, 70, 1042–1048.
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., Vidal, L., Hoenicka, J., Rodriguez, O., Atares, B., Llorens, V., Gomez Tortosa, E., del Ser, T., Munoz, D. G., & de Yebenes, J. G. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Annals of Neurology, 55, 164–173.
Zhou, W., & Freed, C. R. (2005). DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. The Journal of Biological Chemistry, 280, 43150–43158.
Zhou, W., Milder, J. B., & Freed, C. R. (2008). Transgenic mice overexpressing tyrosine-to-cysteine mutant human alpha-synuclein: A progressive neurodegenerative model of diffuse Lewy body disease. The Journal of Biological Chemistry, 283, 9863–9870.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this entry
Cite this entry
Kragh, C.L., Romero-Ramos, M., Halliday, G., Jensen, P.H. (2014). Alpha Synuclein in Parkinson’s Disease. In: Kostrzewa, R. (eds) Handbook of Neurotoxicity. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5836-4_14
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5836-4_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5835-7
Online ISBN: 978-1-4614-5836-4
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences