
Abstract
During bacterial pneumonias, neutrophils are usually the first leukocytes recruited from the circulation to the lung, where they protect the host by killing microbial pathogens through phagocytosis and the release of antimicrobial products. In addition to their role in eliminating pathogens, the role of neutrophils in shaping the immune response and resolution of inflammation is now increasingly appreciated. Patients with defects in neutrophil production or function suffer from recurrent microbial infections, thus illustrating the critical role of neutrophils in host defense. However, the very characteristics and functions that make neutrophils useful to the host can also injure host tissues, and neutrophil-mediated tissue damage has been implicated in the pathogenesis of a number of serious disorders, including ALI and ARDS, in which both these beneficial and harmful effects are integrated.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Introduction
During bacterial pneumonias, neutrophils are usually the first leukocytes recruited from the circulation to the lung, where they protect the host by killing microbial pathogens through phagocytosis and release of antimicrobial products. In addition to their role in eliminating pathogens, the role of neutrophils in shaping the immune response and resolution of inflammation is now increasingly appreciated. Patients with defects in neutrophil production or function suffer from recurrent microbial infections, thus illustrating the critical role of neutrophils in host defense. However, the very characteristics and functions that make neutrophils useful to the host can also injure host tissues, and neutrophil mediated tissue damage has been implicated in the pathogenesis of a number of serious disorders, including ALI and ARDS, in which both these beneficial and harmful effects are integrated. This article focuses on the role of neutrophils during bacterial pneumonias (Fig. 4.1). The first sections focus on recruitment of neutrophils from the bone marrow and from the blood. The later sections focus on neutrophil functions in the lung. There is clearly much work remaining to understand these processes, in order to find and develop ways to treat disease and modulate the inflammatory and immune response.

Neutrophil kinetics at steady state and during inflammation. In the bone marrow, neutrophils are produced from precursors, mature and are stored until release. G-CSF is a major cytokine that regulates both neutrophil production and release from the bone marrow. CXCR4/CXCL12 (SDF-1) signaling retains cells within the bone marrow. Upon release into the circulation, intravascular neutrophils may circulate or enter the marginating pool in the liver, lungs, and other organs. In the absence of inflammation, intravascular neutrophils senesce and become apoptotic and are cleared by the reticuloendothelial system in the liver, spleen, bone marrow, and possibly the lung. During inflammation, chemokines and other mediators induce neutrophil migration into tissue as well as enhancing release from the bone marrow. Emigrated neutrophils can die through one of the several death pathways. Apoptotic neutrophils are taken up by tissue macrophages, which then release IL-23, inducing IL-17 which in turn induces G-CSF
4.2 Neutrophil Production, Maturation, and Release from the Bone Marrow and Trafficking
Interactions between the lungs and the bone marrow are important in neutrophil homeostasis in healthy individuals and during many lung diseases. During homeostasis, communication between the lungs and the bone marrow contributes to regulating the circulating neutrophil count, as illustrated by the changes in circulating neutrophil numbers in response to chronic smoking and the inhalation of air pollutants (van Eeden and Hogg 2000, 2002; Corre et al. 1971). During the inflammatory response, mediators produced in the lungs play a critical role in controlling neutrophil production and release from the bone marrow. For example, the time required for maturation of neutrophils, as measured by their transit time in the bone marrow, is shortened during pneumonia induced by Streptococcus pneumoniae (Terashima et al. 1996). GM-CSF, granulocyte-colony stimulating factor (G-CSF), chemokines, and cytokines are produced in the lungs during inflammation and have effects on neutrophil production and release from the bone marrow, as will be described in subsequent sections. Thus, the bone marrow has an important impact on the inflammatory response, and the interaction between the lungs and the bone marrow is critical in the number and maturity of neutrophils that reach the lungs, both of which influence the effects of neutrophils in pulmonary inflammation.
Neutrophil release from the bone marrow is particularly significant for the lungs because the pulmonary microvasculature is the first capillary bed that the newly released neutrophils will traverse. Newly released neutrophils were preferentially retained in the lung capillaries during endotoxemia and pneumococcal infections, and migrated more slowly into the lung parenchyma (van Eeden et al. 1997; Sato et al. 1998a, b; Lawrence et al. 1996). Newly released circulating neutrophils had decreased deformability as measured by filter assays in vitro, which may contribute to their preferential retention in the lung for reasons discussed in subsequent sections (van Eeden et al. 1997, 2000). The accumulation of immature neutrophils in the lungs is exacerbated by shortened transit time of neutrophils in the bone marrow, for example, in S. pneumoniae pneumonia (Terashima et al. 1996). Morphologically mature neutrophils and band cells in human bone marrow exhibit functional deficiencies in degranulation, respiratory burst, and phagocytosis compared with blood neutrophils (Berkow and Dodson 1986), and may thus be less capable of defending the host against infection. Furthermore, prolonged sequestration and activation of these immature neutrophils within the vasculature may result in damage to the endothelium.
4.2.1 Neutrophil Production and Circulation in Health
The bone marrow is the site of granulopoiesis in mature animals. Neutrophils are ultimately derived from a multipotent hematopoietic stem cell (HSC) that gives rise to all lineages of hematopoietic cells. Neutrophil progenitors are able to divide up to the myelocyte stage; post-mitotic neutrophil precursors undergo a process of maturation that lasts several days and involves changes in cell surface molecule expression, biomechanical and structural properties, and granule content (Bainton 1999). A large number of mature neutrophils reside in the bone marrow stroma presumably ready for release into the circulation, and there is a marginating pool of neutrophils within the vasculature of the bone marrow (Fig. 4.2).

Complex interplay of signals mediated by G protein-coupled receptors and their ligands, and G-CSF regulate neutrophil retention in the bone marrow and release into the circulation. Binding of SDF-1 (CXCL12) to its receptor CXCR4 mediates neutrophil retention in the bone marrow. Binding of the chemokines MIP-2 or KC to CXCR2 during inflammation induce neutrophil shape changes and release into the circulation. Binding of other inflammatory mediators (such as complement fragments or the bacterial peptide fMLP) to G protein-coupled receptors also induce shape changes and rapid release of neutrophils into the circulation. G-CSF disrupts the CXCR4/SDF-1 signaling axis and enhances release induced by MIP-2 or KC. Crosstalk between CXCR4 and CXCR2 regulates neutrophil release during inflammation through mechanisms that are not yet completely clear
The cytokine granulocyte-colony stimulating factor (G-CSF) is a major regulator of neutrophil production and release from the bone marrow. G-CSF acts by binding to its receptor G-CSFR, which induces Jak/STAT signaling through sequential phosphorylation of the Janus family of tyrosine kinases (Jaks), phosphorylation of the G-CSFR by Jaks, and recruitment of STATs to the phosphorylated G-CSFR and phosphorylation of STATs by Jaks. Jak/STAT signaling, as well as other signaling cascades including Ras/MAP kinase and PI3K/Akt, promote proliferation and differentiation toward mature neutrophils and regulate neutrophil release and function, both at steady state and in conditions of stress (Panopoulos and Watowich 2008; Nicholson et al. 1994, 1995; Shimoda et al. 1994). Mice that are deficient in G-CSF or G-CSFR have low circulating counts and decreased numbers of neutrophils and their precursors in the bone marrow (Liu et al. 1996; Lieschke et al. 1994; Basu et al. 2002; Richards et al. 2003). However, mice deficient in G-CSF or G-CSFR do have neutrophils, albeit in reduced numbers, suggesting that G-CSF-independent pathways for neutrophil production partially compensate for a lack of G-CSF signaling. G-CSF is also important for neutrophil trafficking into the circulation both at baseline and during pulmonary bacterial infection (Semerad et al. 2002; Gregory et al. 2007).
After release from the bone marrow, neutrophils circulate in the blood. The concentration of neutrophils in the blood varies depending on the site within the vasculature. For example, differences in the neutrophil concentrations in tail, eye, and heart have been identified (Nemzek et al. 2001). In general, concentrations increase in vascular beds where blood flow and pressure are less and the diameters and branching geometry of the bed favors increased concentration. This increased concentration is often described as a marginated pool, but these pools are unlikely to be stagnant and in disequilibrium with the circulating pool. The mechanisms for the increased concentration in the pulmonary capillaries are discussed in a subsequent section.
In animals without an inflammatory focus, the circulating half-life of neutrophils is 4–14 h (Basu et al. 2002; Dancey et al. 1976; Deubelbeiss et al. 1975; Eash et al. 2009; Gomez et al. 2008; Lord et al. 1991; Price et al. 1996). Neutrophils that do not migrate into inflammatory sites become senescent, in which they are unresponsive to stimuli that would otherwise induce degranulation or respiratory burst, and die by apoptosis. The mechanisms that regulate senescence are not well understood, and may be regulated by microRNAs (Ward et al. 2011). At steady state, apoptotic neutrophils accumulate in the liver, bone marrow, and spleen, suggesting these are the sites of clearance for effete neutrophils (Furze and Rankin 2008a, b; Suratt et al. 2001). Constitutive removal of apoptotic neutrophils requires macrophages in the bone marrow stroma and marginal zone of the spleen (Gordy et al. 2011). The capillaries of the lungs may also be a site of clearance, since intravascular neutrophils that get trapped in narrow capillary segments may undergo apoptosis or other forms of cell death (Bicknell et al. 1994). The recognition systems whereby the reticuloendothelial system recognizes effete neutrophils are becoming clearer, although numerous mechanisms are likely to contribute, including increased expression of CXCR4 (Eash et al. 2009, 2010; Martin et al. 2003; Suratt et al. 2004), an important molecule in the retention of neutrophils within the bone marrow that is discussed below. A recent study reported that expression of the anti-inflammatory phospholipid-binding protein Annexin A1 by mouse bone marrow macrophages is critical for the uptake of apoptotic neutrophils (Dalli et al. 2012).
Constant exposure to the outside world, either through inhalation, ingestion, cutaneous routes, or other means, cannot always be managed by first-line organ-specific host defense mechanisms. An exposure can result in mild inflammatory responses that are unlikely to be experienced by the host as the five cardinal signs of inflammation, rubor (redness), calor (increased heat), tumor (swelling), dolor (pain), and function laesa (loss of function), and thus is not detected clinically. However, these common responses may influence circulating neutrophils by inducing efflux of neutrophils from the blood stream or even in altering production by the bone marrow, as may occur during more chronic noxic exposures. The lungs are an important route of exposure, even though airflow patterns through the nasopharynx, the nasopharyngeal surfaces themselves, and the mucociliary clearance mechanisms in the bronchi and trachea are highly functional first-line defense mechanisms. For example, a reduction in inhaled atmospheric particulates, as occurs upon relocation from an urban level of particular matter to that found in Antarctica, is associated with a reduction in the numbers of mature and immature neutrophils circulating in the blood, and these numbers increase upon return to the urban environment (Sakai et al. 2004). Similarly, circulating neutrophil counts in cigarette smokers are higher than in age-matched nonsmokers in the same environment (van Eeden and Hogg 2000; Corre et al. 1971).
Thus, in healthy subjects, the number of circulating neutrophils reflects a dynamic balance between production and continual release from the bone marrow, the marginating pools, the clearance of neutrophils from the circulation, and neutrophil migration from the circulation into the tissues in response to a constant barrage of stimuli, where they usually perform any needed function and are cleared by resident macrophages. A clinically relevant injury or infection will alter this equilibrium.
4.2.2 Mechanisms of Neutrophil Release from the Bone Marrow During Inflammation
In response to injury or infection, the number of neutrophils in the circulation can undergo a rapid increase that largely reflects the release of neutrophils from the bone marrow. Marginating pools of mature or nearly mature neutrophils are present both within the stroma of the bone marrow and within the venous sinusoids. Many inflammatory mediators that are chemotactic agents or activators of neutrophils can cause this acute release. For example, infusion of the chemotactic bacterial peptide fMLP (N-formyl-l-methionyl-l-leucyl-l-phenylalanine, also abbreviated fMLF), the chemokine interleukin-8 (IL-8), or its murine homolog MIP-2, or complement protein fragments (particularly C5a) induce neutrophil release from the venous sinusoids and/or from the marginating pool of mature neutrophils in the bone marrow stroma into the sinusoids and then the circulation (Jagels et al. 1995; Jagels and Hugli 1992, 1994; Burdon et al. 2008). Neutrophil egress from the stroma into the venous sinusoids usually occurs through the endothelial cells, rather than between them. For example, neutrophils were observed to squeeze through pores in the bone marrow endothelium in response to MIP-2. In perfused rat femurs, the endothelial cells presented a barrier, and neutrophil mobilization by MIP-2 required p38 MAPK activity but was not affected by the presence of a nonspecific matrix metalloprotease inhibitor (Burdon et al. 2008). Thus, neutrophil mobilization is an active process that requires the integration of many signals and pathways involving neutrophils as well as other cells in the bone marrow.
4.2.2.1 The Importance of Biomechanical Properties of Neutrophils in Their Production and Release
Neutrophils appear to undergo changes in their biomechanical properties as they mature (Fig. 4.2). Early studies showed that myelocytes were stiffer than mature neutrophils (Lichtman 1970), and that mature granulocytes were better able to transit through smaller pore diameters and respond to a chemoattractant than less mature cells (Giordano and Lichtman 1973). Studies using magnetic twisting cytometry showed that mature neutrophils isolated from the bone marrow are stiffer than those isolated from circulating blood (Saito et al. 2002). This increased stiffness was associated with the presence of an f-actin rim beneath the cell membrane, and disruption of the actin cytoskeleton by treatment with cytochalasin D reduced the stiffness of bone marrow neutrophils to the level seen in circulating neutrophils (Saito et al. 2002). The greater stiffness of immature neutrophils may facilitate their retention within the bone marrow, by preventing or slowing the passage of these cells through the stroma or pores in the sinusoidal wall, along with other factors such as CXCR4/SDF-1 signaling and adhesivity (discussed below). Furthermore, it is also possible that the process of neutrophil migration through sinusoidal endothelial cells alters their biomechanical properties to decrease their stiffness.
During an inflammatory stimulus, bone marrow neutrophils deform in order to transit toward and enter into the venous sinusoids. Their biomechanical properties may be an important factor in their retention or release. Chemokines and other mediators that act through serpentine receptors (also called heptahelical receptors or G protein-coupled receptors) share the ability to alter the cytoskeleton of neutrophils, which is often evaluated as changes in f-actin in neutrophils. This rearrangement of actin appears initially to result in f-actin beneath the cell membrane, inducing an increase in stiffness, which further remodels after 1–2 min to a flattening out and cytoskeletal changes that vary within regions of the neutrophils. In fact, the initial stiffening may cause neutrophils to round up and come off the stroma or sinusoidal endothelium (Luscinskas et al. 1992; Hechtman et al. 1991), which could also facilitate release. Studies show that stimulation by fMLP or complement fragments in vitro caused an increase in stiffness in both circulating and bone marrow neutrophils and an increase in f-actin beneath the cell membrane (Saito et al. 2002). Pretreatment with cytochalasin D prevented the stiffening induced by inflammatory mediators, suggesting that f-actin rearrangement was responsible for the increased stiffness. Infusion of intravascular fMLP and complement fragments induced an extremely rapid release (within seven minutes) of mature neutrophils from the bone marrow into the circulation (Saito et al. 2002; Kubo et al. 1998). The fMLP-induced increase in circulating neutrophil counts was not prevented by pretreatment with colchicine, indicating that microtubule rearrangements were not required for this process (Saito et al. 2002). These observations suggest that structural and mechanical changes induced by circulating mediators may facilitate the release of bone marrow neutrophils into the circulation.
4.2.2.2 The Role of CXCR4 and SDF-1 in Bone Marrow Release of Neutrophils
Disruption of G protein signaling by treatment with pertussis toxin led to leukocytosis and mobilization of hematopoietic stem/progenitor cells from the bone marrow (Papayannopoulou et al. 2003), indicating that signaling through pertussis toxin-sensitive G protein-coupled receptors is important in retaining primitive as well as more mature hematopoietic cells within the bone marrow. One of these G protein-coupled receptors, CXCR4, has been studied extensively and is expressed on a wide variety of hematopoietic cells, including neutrophils (Broxmeyer 2008). Its ligand is SDF-1 (CXCL12), a CXC chemokine produced by stromal cells in the bone marrow and a key signal for retaining and maintaining cells in the bone marrow through its binding to CXCR4 (Broxmeyer 2008). Ma and colleagues showed that mice whose bone marrow was reconstituted with CXCR4-deficient fetal liver cells had reduced levels of granulocytic cells in the bone marrow and elevated numbers of circulating mature and immature granulocytes (Ma et al. 1999). The critical role of CXCR4/SDF-1 signaling in retaining neutrophils is also supported by studies of WHIM syndrome, in which mutations that lead to prolonged signaling or hyperactivation of CXCR4 result in immune abnormalities and failure of bone marrow release of mature neutrophils, resulting in severe neutropenia, myeloid hyperplasia, and apoptosis of mature neutrophils within the bone marrow (myelokathexis) (Kawai and Malech 2009). Other studies interfering with CXCR4 signaling have shown that treatment of mice with a CXCR4 antagonist (Martin et al. 2003) or with CXCR4 blocking antibody (Suratt et al. 2004) mobilized neutrophils from the bone marrow. Selective deletion of CXCR4 in myeloid cells caused increased numbers of circulating neutrophils with no increase in immature forms and elevation in the ratio of circulating to bone marrow neutrophils, indicating a cell-autonomous requirement for CXCR4 in neutrophil retention in the bone marrow (Eash et al. 2009). Thus, many approaches have supported the concept that SDF-1/CXCR4 signaling serves as a retention signal for neutrophils in the bone marrow. However, the molecular mechanisms initiated by SDF-1 binding to CXCR4 that are responsible for this retention are not yet clear.
Disruption of CXCR4/SDF-1 signaling in the bone marrow may be a common feature of neutrophil release induced by some chemokines or G-CSF. Mobilization of bone marrow neutrophils induced by the CXC chemokine KC (CXCL1) was enhanced by treatment with a CXCR4 antagonist (Martin et al. 2003) or blocking antibody (Suratt et al. 2004), whereas Cxcr2−/− neutrophils were not mobilized by transiently inhibiting CXCR4 (Eash et al. 2010). Pretreatment of murine neutrophils with KC led to a decrease in the calcium flux in response to SDF-1, suggesting that KC and other chemokines may mobilize neutrophils in the bone marrow by disrupting SDF-1 signaling in the bone marrow (Suratt et al. 2004). Conversely, SDF-1 attenuated neutrophil responses to KC in vitro (Martin et al. 2003). These studies suggest that signaling through CXCR4 and CXCR2 act in opposite ways to regulate neutrophil release.
The interplay between CXCR4/SDF-1 signaling, CXCR chemokines and G-CSF is complex. Neutrophil mobilization in response to pulmonary infection with P. aeruginosa was reduced in mice lacking G-CSF receptor, indicating that G-CSF signaling may be critical for neutrophil recruitment in this infection (Gregory et al. 2007). Antibody neutralization of G-CSF resulted in fewer neutrophils within the lungs at 48 h of S. pneumoniae pneumonia by less than but had no effect at 24 h (Knapp et al. 2004). G-CSF may induce neutrophil release from the bone marrow either directly by reducing expression of CXCR4 on myeloid cells (Kim et al. 2006) or by disrupting CXCR4 signaling through reducing levels of SDF-1 in the bone marrow (Semerad et al. 2005; Christopher et al. 2009). Giving KC, MIP-2 or G-CSF intravenously resulted in increased neutrophil numbers in the blood and decreased numbers in the bone marrow of mice, and infusion of G-CSF and KC together mobilized a significantly greater number of neutrophils compared with either mediator alone in a perfused femoral bone marrow system (Wengner et al. 2008). Antibody neutralization of G-CSF, MIP-2, KC, or MIP-2 and KC together reduced neutrophil mobilization from the bone marrow and recruitment to the peritoneum of mice in thioglycollate-induced acute peritonitis, but inhibition of G-CSF did not alter the response to a selective CXCR4 antagonist (Wengner et al. 2008). G-CSF-induced neutrophil mobilization is attenuated in Cxcr2−/− mice or in wild type (WT) mice given blocking antibody to CXCR2, and G-CSF treatment induces CXCR2 ligands in the bone marrow (Eash et al. 2010; Kohler et al. 2011). Notably, Kohler and colleagues found that G-CSF did not directly induce CXCR2 ligands, but rather induced thrombopoietin, which induced CXCL1 production from megakaryocytes (Kohler et al. 2011). Taken together, these studies suggest that neutrophil release is regulated by a complex interplay between CXCR4/SDF-1 signaling in the bone marrow and mobilization signals induced by G-CSF and inflammatory mediators.
CXCR4/SDF-1 signaling in the periphery may play a role in neutrophil mobilization and recruitment to tissue during inflammation. In LPS-induced pneumonitis, SDF-1 expression was upregulated in lung epithelium and SDF-1 blockade prevented neutrophil recruitment to the airspace at 24 h after injury (Petty et al. 2007). A recent study reported that SDF-1 blockade prevented the increase in circulating neutrophils and decrease in BM neutrophils induced by sepsis at 12 h (Delano et al. 2011). Taken together these studies suggest that generation of an SDF-1 gradient in the periphery is required for mobilization. Notably, neutrophil mobilization in the murine model of polymicrobial sepsis did not require MyD88, IFNα/βR, TRIF, or TLR4, and was not inhibited by CXCR2 blockade, suggesting that other pathways can modulate CXCR4/SDF-1 signaling to induce mobilization (Delano et al. 2011).
4.2.2.3 The Functions of Rac2
The small GTPase Rac2 is expressed in leukocytes and activated by signaling downstream of many receptors, including G protein-coupled receptors of chemokines/chemoattractants and the β2 integrin, CD11/CD18 expressed on leukocytes. It regulates a wide variety of functions in neutrophils, including cytoskeletal organization and rearrangements, superoxide production, chemotaxis, phagocytosis, transcription, and cell growth and proliferation (Bokoch 2005). Rac2−/− mice have circulating neutrophil counts that are several times those seen in wild type animals and a slight increase in marrow granulopoiesis, which persist in otherwise asymptomatic mutant mice (Roberts et al. 1999). Rac2−/− mice have higher numbers of circulating HSC/Ps at baseline and after G-CSF treatment compared to WT littermates despite having similar numbers of HSC/Ps in the bone marrow (Yang et al. 2001), suggesting that Rac2 may be important in the retention and mobilization of these cells from the bone marrow. Rac2−/− HSC/P adhered less to bone marrow stromal cells in vitro and exhibited growth defects in stroma-dependent cultures than wild type cells, indicating that Rac2 in hematopoietic cells is required for optimal growth and development (Jansen et al. 2005). Neutrophil kinetic studies suggested that Rac2 modulates the time from the last mitosis to release of neutrophils into the circulation and does not prolong their circulating half-life (Gomez et al. 2008). Lethally irradiated wild type mice reconstituted with a mixture of wild type and Rac2−/− stem cells were protected from neutrophilia, and neutrophils constituted a greater percentage of the Rac2−/− leukocytes than wild type leukocytes in these mice (Gomez et al. 2008). These findings are consistent with the role of Rac2 in transducing signals downstream of integrin activation (please see next section). However, whereas Rac2 deficiency in hematopoietic cells alone resulted in increased neutrophil production in the bone marrow, Rac2 deficiency in both hematopoietic and nonhematopoietic cells was required for the increase in circulating neutrophil counts, suggesting a role for Rac2 in nonhematopoietic cells in regulating bone marrow release of neutrophils (Gomez et al. 2008).
4.2.2.4 Adhesion Molecules and the Regulation of Neutrophil Production and Release
Adhesion of hematopoietic cells to stromal cells or to the extracellular matrix within the bone marrow plays an important role in retaining hematopoietic cells within the bone marrow and in mediating the return of some hematopoietic cells to the bone marrow from the circulation (“homing”). Conversely, adhesive interactions may be important in mobilizing hematopoietic cells to enter the circulation, by allowing hematopoietic cells to crawl through the bone marrow stroma toward the sinusoids and cross the sinusoidal endothelium. Bone marrow neutrophils express the integrin VLA-4 (α4β1 or CD49d/CD29) which binds to VCAM-1 expressed by bone marrow stromal cells and endothelium (Petty et al. 2009). Blocking antibodies against VLA-4 or VCAM-1 given intravascularly caused neutrophilia at 4 h, in the absence of any inflammatory stimulus (Petty et al. 2009). Evidence of interaction between CXCR4 and VCAM-1 in neutrophil mobilization was observed (Petty et al. 2009). In contrast, baseline neutrophil release in a model of rat perfused femoral bone marrow was not inhibited by blocking CD49d (the alpha subunit of VLA-4), and an anti-CD49d antibody partially prevented neutrophil mobilization induced by MIP-2 (Burdon et al. 2005). The basis for this discrepancy is not yet clear, but these data underline the complexities of VLA-4/VCAM-1, CXCR4/SDF-1, and CD11/CD18/ICAM-1 axes and signaling. Light may be shed on these complexities by observations made in immature bone marrow hematopoietic cells, where VLA-4 plays an important role in mediating homing to and mobilization from the bone marrow by binding to VCAM-1 (Bonig et al. 2006; Priestley et al. 2006; Scott et al. 2003). Interestingly, lacking CXCR4 expression in hematopoietic cells abolished G-CSF-induced HSC/P mobilization in mice, but had no effect on mobilization induced by blocking VLA-4 (Christopher et al. 2009), suggesting multiple independent or redundant mechanisms of HSC/P release that may also operate in neutrophils.
Integrins of the β2 integrin subfamily (CD11/CD18 complex) are the main integrins expressed on the surface of leukocytes. Patients with heterogeneous mutations in the gene that encodes CD18 suffer from the clinical syndrome Leukocyte Adhesion Deficiency type I (LAD I). LAD I is characterized by extremely high levels of circulating neutrophils, recurrent bacterial infections, impaired wound healing, and functional defects in neutrophils. Mice that are completely deficient in CD18 (Itgb2-/-) express no functional leukocyte β2 integrins, including LFA-1 (CD11a/CD18 or αLβ2) and Mac-1 (CD11b/CD18 or αMβ2) and have a phenotype similar to human LAD I patients, including granulocytosis, spontaneous infections, and myeloid hyperplasia (Scharffetter-Kochanek et al. 1998). Mice deficient in CD11a/CD18 (LFA-1) (Ding et al. 1999) or the CD11/CD18 receptor ICAM-1 (Sligh et al. 1993) have modestly elevated circulating neutrophil counts.
The neutrophilia in CD18 deficiency is present within the first few days of life, and occurs even when mice were housed in clean, specific pathogen-free facilities and in the absence of discernible infections. When lethally irradiated wild type (WT) mice are given a 1:1 mixture of WT and CD18-deficient fetal liver cells, neutrophilia is inhibited by greater than 95 % compared with WT mice given CD18-deficient stem cells alone (Horwitz et al. 2001), indicating that the neutrophilia seen in CD18 deficiency can be largely corrected by the presence of WT hematopoietic cells. Weinmann and colleagues demonstrated that circulating neutrophils in CD18-deficient mice show decreased apoptosis, and the delay in apoptosis of CD18-deficient neutrophils is abolished in the presence of WT leukocytes (Weinmann et al. 2003). These studies led to the hypothesis that neutrophilia in CD18 deficiency is mainly due to the inability of mutant leukocytes to defend the host from microbial pathogens, resulting in chronic infection and subsequent chronic stimulation of the bone marrow. The presence of WT neutrophils is thus postulated to remove the stimuli that increase neutrophil production in the bone marrow by conferring protection to the host.
Cell-intrinsic mechanisms that were not corrected by the presence of wild type cells contributed to the granulocytosis observed in CD18 deficiency, because a mild granulocytosis developed in mice that received a mixture of wild type and CD18-deficient stem cells (Horwitz et al. 2001). In chimeric mice with both wild type and CD18-deficient bone marrow, a larger than expected fraction of the circulating neutrophils are CD18-deficient and a much larger proportion of the CD18-deficient circulating leukocytes are neutrophils, even in the presence of normal numbers of wild type neutrophils in the blood and bone marrow (Horwitz et al. 2001). The proportion of apoptotic Gr-1+ cells in both the bone marrow of chimeric animals and in vitro cultures of wild type and CD18-deficient HSCs was lower in CD18-deficient than in wild type cells (Gomez and Doerschuk 2010). These data suggest that CD18 can directly regulate neutrophil production, in part by limiting the survival of neutrophils and their precursors.
A novel mechanism through which the defects of CD18-deficient neutrophils in host defense result in neutrophilia was suggested by Forlow and colleagues, who showed that CD18-deficient mice have increased levels of circulating IL-17 and G-CSF and that blocking IL-17 or G-CSF suppressed the neutrophilia in these mice (Forlow et al. 2001). The cytokine IL-17 is produced by T cells and induces G-CSF production, and is itself induced by IL-23 produced by macrophages and dendritic cells (von Vietinghoff and Ley 2008). Stark and colleagues demonstrated a novel feedback loop in which phagocytosis of apoptotic neutrophils by tissue-resident macrophages and dendritic cells inhibits the production of IL-23, thus shutting down the IL-17A/G-CSF signaling axis and preventing increased granulopoiesis in the bone marrow (Stark et al. 2005).
Similar concepts may underlie neutrophilia found in mice deficient in signaling through CXCR2. Cxcr2−/− mice have increased numbers of neutrophils in the bone marrow and circulation, and Cxcl5−/− (LIX-deficient) mice have a similar but milder phenotype (Mei et al. 2012). The authors showed that the phenotype was due to increased IL-17A in the ileum, and the treatment with anti-IL17A or antibiotics resulted marked reduction in neutrophils in the blood and bone marrow (Mei 2012). The authors propose that CXCL5 (LIX) produced by enterocytes attract circulating neutrophils into the gut where they feedback into the IL-17A/G-CSF signaling axis. Thus, the failure of neutrophils to migrate into the tissues may underlie the reactive neutrophilia observed in mice deficient in adhesion molecules or CXCR2 signaling. Interestingly, uptake of apoptotic neutrophils by bone marrow-derived macrophages induces G-CSF production, whereas LPS-induced G-CSF production by peritoneal macrophages is inhibited (Furze and Rankin 2008a), suggesting that neutrophil uptake may induce distinct signaling pathways depending on the macrophage population and the microenvironment, with IL-17 signaling playing a role in neutrophil kinetics during inflammation by inducing G-CSF.
4.3 Neutrophil Margination and Sequestration in Pulmonary Microvasculature and Recruitment into Lung Tissue
4.3.1 Neutrophil Trafficking and Margination in the Normal Pulmonary Microvasculature
Within the capillaries of the pulmonary circulation in healthy lungs, there is an increased concentration of neutrophils relative to other vessels. This increased concentration of neutrophils has been termed the marginated pool, although the implication that this pool is stagnant is unlikely to be true (Hogg 1987). Many studies have shown that this increased concentration is explained by increased transit time of neutrophils through the pulmonary capillary bed, compared to the rapid transit times of erythrocytes. This longer transit time is likely due to constraints set by the diameters of the capillaries and the neutrophils, and the biomechanical properties of the neutrophils (Brumwell et al. 1991; Doerschuk et al. 1993; Hogg et al. 1994; Lien et al. 1987, 1990; Wiggs et al. 1994; Yoder et al. 1990; MacNee et al. 1989; Selby et al. 1991). Capillary segments are often narrower than the diameter of spherical neutrophils, indicating that neutrophils must stop and deform in order to traverse these narrow segments. Video microscopy demonstrated that in contrast to the continuous movement of the discoid erythrocytes that can fold, leukocytes move in hops, stopping once or more in transiting through the capillary bed from a pulmonary arteriole to a venule (Lien et al. 1990). The prolonged transit time of neutrophils and other leukocytes through the pulmonary capillaries may be important for host defense, by allowing neutrophils to sample their microenvironment for the presence of inflammatory stimuli and to respond appropriately. The observation that transit times are longer has become controversial of late (Summers et al. 2010), but the numerous ways in which neutrophil transit times have been studied (imaging, morphometry, computational modeling), the many species (human, dog, rabbit, mice), the many ways in which neutrophils have been isolated, and the numerous studies showing the effect of epinephrine and respiratory maneuvers on the release of this pool suggest that neutrophil transit times are longer than erythrocyte transits, resulting in an increased concentration of neutrophils in the pulmonary capillary blood that is likely important for host defense in the lungs.
4.3.2 Neutrophil Sequestration in the Lungs During Inflammation
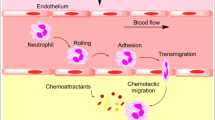
Neutrophils migrate from the circulation to the tissues in response to inflammatory stimuli. The recruitment of neutrophils and other leukocytes to the tissues during inflammation occurs in a sequence of events that includes leukocyte recognition of the inflammatory site, sequestration within the microvasculature, firm adhesion, transmigration through the endothelium, and migration in the tissues (Fig. 4.3).

Postulated pathway of neutrophil recruitment during inflammation in the lungs. Complex signaling processes occurring in and between neutrophils and endothelial cells facilitate the early stages of neutrophil recruitment from the circulation. Asterisk depending on the stimulus, neutrophils migrate into the airspaces of the lungs using CD11/CD18-dependent or -independent mechanisms. Double asterisks most transendothelial migration occurs paracellularly as depicted; a minority of cells migrate through endothelial cells (transcellularly). Please see text for details
The mechanisms through which neutrophils are sequestered at a site of inflammation within the alveolar spaces appear quite different in the pulmonary microcirculation compared to the systemic microcirculation. In the systemic microcirculation, the initial steps of leukocyte capture and rolling occur within the post-capillary venules and are mediated by selectins, a family of calcium-dependent lectins that includes L-selectin expressed on leukocytes, P-selectin expressed on platelets and endothelial cells, and E-selectin expressed on endothelial cells. Neutrophils express L-selectin and several ligands that can bind to selectins expressed on leukocytes, platelets, and endothelial cells. Among the best characterized selectin ligands on neutrophils are PSGL-1, ESL-1, and CD44. Neutrophil PSGL-1 can bind to all three selectins, whereas ESL-1 and CD44 can bind to E-selectin expressed on activated endothelial cells. Binding of E-selectin to PSGL-1, ESL-1, or CD44 on neutrophils elicits distinct signals that correlate with their distribution on the cell surface (Hidalgo et al. 2007). The biology of selectin adhesion has been extensively studied in vitro and within the systemic post-capillary venules.
In contrast, the major site of neutrophil sequestration within the pulmonary microvasculature in response to an inflammatory stimulus is the pulmonary capillaries, and this sequestration does not require rolling. Rolling does not occur in the pulmonary capillaries, because of the spatial restraints arising from the diameter of many pulmonary capillaries being narrower than those of round neutrophils (Doerschuk et al. 1993; Gebb et al. 1995; Lien et al. 1991). Rather, mediator-induced changes in the biomechanical properties of neutrophils appear to underlie their sequestration in the pulmonary capillaries (Doerschuk 2001). The binding of inflammatory mediators present at the inflammatory site to G protein-coupled receptors on leukocytes leads to changes in the biomechanical properties of neutrophils that decrease their ability to deform and change shape. In normal lungs as mentioned above, the transit times of neutrophils are longer than for erythrocytes, allowing them 2–20 s or more to assess their environment. The binding of neutrophil receptors by chemokines or bacterial products induces an f-actin rim to form beneath the cell membrane within seconds, which causes neutrophils to become stiffer and less deformable (Downey et al. 1991; Inano et al. 1992). This increased stiffness appears to prevent them from deforming and passing into the pulmonary venules, resulting in their sequestration at inflammatory sites. During bacterial pneumonias in rats, neutrophils with f-actin rims were preferentially retained in the lungs over those without f-actin rims at a time when sequestration was actively ongoing (Yoshida et al. 2006). CD18 blockade had no effect on this preferential sequestration of f-actin rimmed neutrophils, and L-selectin expression or platelet binding made no difference in which neutrophils were preferentially retained. Furthermore, L-selectin-deficient mice had no defect in neutrophil sequestration within lung capillaries induced by intravascular activated complement or intra-alveolar S. pneumoniae (Doyle et al. 1997).
Although selectins appear to play no role in the immediate recruitment of neutrophils in the pulmonary capillaries, this family of adhesion molecules was required for prolonged retention of neutrophils in the lungs in response to intravascular complement fragments (Kubo et al. 1999). During bacterial pneumonia, the requirement for selectins may depend upon the bacterial species. For example, neutrophil sequestration and migration in response to S. pneumoniae does not appear to require selectins, whereas L-selectin does contribute to emigration induced by Escherichia coli (Doyle et al. 1997; Mizgerd et al. 1996). This requirement may reflect a role of selectins in integrin activation (see below) and other signaling processes in neutrophils and other cells. For example, platelet-derived P-selectin-mediated platelet-neutrophil interactions, and P-selectin blockade abrogated neutrophil recruitment and lung injury in a model of acid-respiration induced lung injury (Zarbock et al. 2006). Thromboxane A2 released by platelet-neutrophil aggregates increased the expression of ICAM-1, the receptor for the major leukocyte integrin, on endothelial cells and adhesion of neutrophils to endothelial cells (Zarbock et al. 2006).
4.3.3 Neutrophil Adhesion to Endothelial Cells and Migration into the Lungs
4.3.3.1 The Biology of Integrin Activation
Firm adhesion of leukocytes is mediated by activated integrins present on the cell surface. Integrins expressed on resting leukocytes generally have low affinity for their ligands. Integrin activation caused by binding of various agonists to receptors on the cell surface induces structural changes in integrins that result in increased affinity for ligands (inside-out signaling) and clustering of integrins on the cell surface. Integrins on leukocytes can be rapidly activated by the binding of chemokines and other inflammatory mediators to cognate G protein-coupled receptors on the leukocyte surface. Binding of agonists to the N terminal extracellular portion of their cognate G protein-coupled receptors results in activation of the associated cytoplasmic G protein, which dissociates into the GTP-bound Gα subunit and Gβγ dimers. The G protein subunits activate downstream effectors, including phospholipase C, the small GTPase Rap, GEFs including CALDAG-GEF1, and the cytoskeletal protein talin, that mediate integrin activation to allow high affinity binding to ligands. Upon binding ligand, integrins transmit signals that control a wide variety of cellular processes, including structural and mechanical changes, adhesion, and migration (outside-in signaling). Integrin structure and signaling are discussed in detail in recent reviews (Abram and Lowell 2009; Luo et al. 2007).
The functional importance of integrin activation is apparent in patients with LAD type 3/type 1 variant (LAD III/1V), who manifest defects in integrin activation in leukocytes and platelets that lead to immune deficiencies and bleeding disorders. The disease is due to mutations in the gene FERMT3 that encodes Kindlin-3 (Malinin et al. 2009; Moser et al. 2009; Svensson et al. 2009; Kuijpers et al. 2009). Kindlin-3 binds to the distal portion of the cytoplasmic tail of β1 and β3 integrins on platelets and β2 integrins on leukocytes and mediates inside-out activation (Moser et al. 2008, 2009). Kindlin-3-deficient neutrophils do not firmly adhere to and spread on β2 ligands ICAM-1 and iC3b after stimulation, and show defects in adhesion to endothelial cells and extravasation in response to inflammatory mediators (Moser et al. 2009).
In addition to rapid activation by GPCRs, integrins on neutrophils can also be activated by engagement of L-selectin by glycoproteins, or by binding of selectin ligands on neutrophils by E-selectin (expressed by stimulated endothelial cells) or P-selectin (expressed on platelets and endothelial cells). The transition of integrins from low to high affinity states can be bridged by an intermediate affinity state triggered by rolling on selectins, which can be modulated by hemodynamic shear forces (Alon and Ley 2008). Engagement of PSGL-1 on neutrophils by E-selectin on endothelial cells can activate rolling on ICAM-1 by LFA-1, via Syk, the Src family kinase Fgr, and the immunotyrosine activation motifs (ITAM)-containing adaptors FcRγ and DAP12 (Zarbock et al. 2007a, 2008). PSGL-1 binding to P-selectin expressed by platelets and endothelial cells activated LFA-1 and Mac-1 through a pathway that involved phosphorylation of Naf1 by Src family kinases and subsequent activation of PI(3)K (Wang et al. 2007). Yago showed that E-selectin engages CD44 or PSGL-1 to activate slow rolling on LFA-1 through the Src kinases Hck, Fgr, and Lyn, the adaptors FcRγ and DAP12, Syk, Btk, and p38 MAPK (Yago et al. 2010). Thus binding of endothelial selectins to neutrophil ligands induces signaling pathways that result in activation of β2 integrins.
In vivo, GPCR and selectin-dependent pathways likely cooperate to induce integrin activation. Very little information is available about the integration of these signaling pathways to regulate adhesion and migration in the lungs, although there are hints that this is occurring. In venules of cremaster muscles inflamed by TNF-α, both pertussis-toxin inhibitable G protein-coupled receptors including CXCR2 and E-selectin contributed to neutrophil adhesion (Smith et al. 2004). Similarly, during thioglycollate-induced peritonitis, defects in neutrophil recruitment were observed only when E-selectin-deficient mice were treated with pertussis toxin or when CXCR2-deficient mice were given a blocking anti-E-selectin antibody (Smith et al. 2004). Other studies are pursuing an understanding of the complexities of integrin activation during neutrophil recruitment, including roles for Syk, Galphai2, the Src kinase Fgr, and the adaptors FcR gamma and DAP12 (Zarbock et al. 2007b, 2008; Van Ziffle and Lowell 2009). The contributions of G protein-coupled receptors and selectin ligands to integrin activation are very likely to depend on the stimulus and the recruitment site.
4.3.3.2 Integrin Activation in the Lungs and Integrin-Independent Adhesion and Migration
As discussed in detail above, much of the recruitment of neutrophils to the distal airways and the alveoli occurs through the pulmonary capillaries, which are too narrow to allow rolling (Fig. 4.3). Thus, the selectin- and integrin-mediated processes that mediate neutrophil rolling and tethering in the systemic circulation may not be required for the initial processes of neutrophil sequestration. However, integrin activation by GPCR and/or selectin signaling may be required for neutrophil adhesion and migration along the endothelium, transendothelial cell migration and travel into the tissues, as well as carrying out their effector function. GPCR signaling is required for neutrophil recruitment and lung injury in a number of models of acute lung injury. Deficiency of the GPCR CXCR2 or blockade of CXCR2 ligand interactions resulted in reduction in neutrophil recruitment and lung injury in a mouse model of ventilation-induced lung injury (Belperio et al. 2002). Signaling through CXCR2 expressed on neutrophils and nonhematopoietic cells mediated neutrophil recruitment and lung injury induced by inhaled LPS (Reutershan et al. 2006). Gαi2 in neutrophils was required for KC-induced neutrophil arrest and for neutrophil recruitment into the lung induced by LPS inhalation (Zarbock et al. 2007b). Engagement of ESL-1 on neutrophils by E-selectin led to activation of the integrin CD11b/CD18 (αMβ2) on microdomains in neutrophils, capture of platelets and release of reactive oxygen species (ROS) in cremasteric venules in a model of transfusion-related acute lung injury (Hidalgo et al. 2009). Blocking E-selectin or CD11b/CD18 but not P-selectin prevented lung injury in this model, suggesting that heterotypic interactions may be occurring in the lung as well.
In the post-capillary venules of the systemic circulation, neutrophil adhesion usually requires the CD11/CD18 adhesion complex. However, neutrophil migration from the pulmonary capillaries to inflammatory sites in the airspaces of the lung utilizes either CD11/CD18-dependent or CD11/CD18-independent mechanisms. Even in CD11/CD18 dependent emigration, inhibition of this adhesion complex blocks only 70–80 % of neutrophils from migrating, and 20–30 % of neutrophils still migrate. The adhesion pathway depends on the stimulus. For example, in the lungs, stimuli that induce primarily CD18-dependent emigration include E. coli, E. coli lipopolysaccharide, P. aeruginosa, IgG immune complexes, IL-1, and phorbol myristate acetate (PMA). Those inducing CD11/CD18-independent neutrophil emigration include S. pneumoniae, group B Streptococcus, Staphylococcus aureus, hydrochloric acid, hyperoxia, pulmonary PMN sequestration early in the course of ventilator-induced lung injury, C5a, and the chemokine KC (Doerschuk 2000; Mackarel et al. 2000; Ridger et al. 2001; Doerschuk et al. 1990; Choudhury et al. 2004). In vitro, fMLP-induced CD18-dependent migration of neutrophils across either pulmonary arterial endothelial cells or HUVECs, whereas IL-8, LTB4, and sputum from patients with purulent bronchiectasis induced CD18-independent neutrophil transendothelial migration (Doerschuk 2000; Mackarel et al. 2000; Ridger et al. 2001; Doerschuk et al. 1990; Morland et al. 2000).
The mechanisms underlying CD18-independent neutrophil recruitment into the lung are poorly understood. CD11/CD18-independent leukocyte migration has also been reported in liver sinusoids, a site where selectin-dependent rolling and tethering also do not occur (reviewed in (Lee and Kubes 2008)) and where binding of CD44 to hyaluronan contributes to neutrophil sequestration in inflamed liver sinusoids (McDonald et al. 2008). However, in the lungs, CD44 had no role in neutrophil migration into the airspaces of the lungs when induced by S. pneumoniae, a stimulus that induces neutrophil recruitment through predominantly CD18-independent mechanisms (Wang et al. 2002; van der Windt et al. 2011). However, curiously, in pneumonias induced by E. coli and Klebsiella pneumoniae, stimuli which induce CD18-dependent neutrophil emigration, CD44 deficiency enhanced neutrophil recruitment (Wang et al. 2002; van der Windt et al. 2010). Studies addressing the adhesion molecules and signaling pathways mediating CD18-independent migration of neutrophils in the lungs have suggested a partial and sometimes small contribution of VLA-4 (CD29d) in mediating neutrophil recruitment during S. pneumoniae pneumonia (Tasaka et al. 2002) and for both CD29b and CD29d during KC-induced pulmonary inflammation (Ridger et al. 2001). The mechanisms mediating CD18-independent adhesion in the lungs remain unclear, despite their obvious importance. The remainder of this section will focus on CD18-dependent neutrophil recruitment and signaling into the endothelium.
4.3.3.3 Neutrophil Trafficking from the Capillaries to the Alveoli
Thus, during an acute inflammatory response in the lung, neutrophils rapidly sequester in the pulmonary capillary bed (Fig. 4.3). Endogenous or exogenous chemoattractants alter the biomechanical properties of circulating neutrophils, resulting in a prolonged transit time for a neutrophil to cross the pulmonary capillary bed and neutrophil sequestration. As an inflammatory response progresses, cytokines are produced to activate lung parenchymal cells including the endothelium. Chemokines are induced, which are necessary to activate the integrins on neutrophils and to further signal the neutrophils to stop in the lung. In addition, adhesion molecules such as ICAM-1 are expressed and activated on endothelial cells, and these molecules mediate firm adhesion of neutrophils to the endothelium. Neutrophils then spread and crawl on the endothelial surface until they reach the site for transmigration. Intravascular crawling is mediated by CD11b/CD18 on the neutrophils and ICAM-1 on endothelial cells. This spreading and crawling implies that the adhesive bonds between integrins and their ligands must be displaced while new bonds are formed. This requires coordinated signaling from chemokines, integrins, and ICAM-1. Besides regulating neutrophil adhesion and locomotion on endothelial cells, signaling during adhesion also modulates endothelial cell junctions, and induces expression of inflammatory genes. These responses likely play important roles in the progression of an inflammatory response by modulating endothelial cell permeability, neutrophil transmigration and production of inflammatory mediators. Neutrophil transmigration through the endothelium can occur at endothelial cell borders (paracellular transmigration) or through endothelial cells (transcellular migration) in vitro and in vivo. During paracellular migration another set of adhesion molecules are engaged that include platelet endothelial cell adhesion molecule (PECAM)-1, VE-cadherin, junctional adhesion molecules and CD99 (Muller 2009).
In vivo studies suggest that most neutrophils sequestered within the capillaries of inflamed lungs reenter the circulation, while only a small fraction transmigrate to reach the extravascular space (Doerschuk et al. 1994). Whether this is a random process or whether there are localized responses that guide neutrophils to reach the site of transmigration and how these responses occur remain important questions.
The path neutrophils take from the capillary to the alveoli has been described elsewhere (Burns et al. 2003). In other tissues and often in vitro, whether a sequestered neutrophil will take a paracellular or transcellular route to migrate through the endothelium appears to depend on the nature of the inflammatory stimulus, the tissue microenvironment, the level of ICAM-1 expression, and the localization of ICAM-1 (Yang et al. 2005; Carman and Springer 2004; Feng et al. 1998; Cinamon et al. 2004; Millan et al. 2006; Nieminen et al. 2006; Woodfin et al. 2010). However, in the pulmonary microvasculature, the majority of the neutrophils (and perhaps nearly all) take the paracellular route between endothelial cells (Burns et al. 2003; Walker et al. 1995; Behzad et al. 1996). Fibroblasts in the interstitium may guide the migrating neutrophils from the endothelial cell junctions to discontinuities at the basal aspect of the epithelial borders. Neutrophils then migrate into the alveolar space through tricellular corners at the junctions of type I and type II epithelial cells (Burns et al. 2003; Walker et al. 1995; Behzad et al. 1996). These sites may be preferred due to the discontinuous nature of the tight junctions. A role for both the leukocyte adhesion complex CD11/CD18 and the β1 integrin CD29 are important in neutrophil migration through the pulmonary interstitium toward the alveolar space (Ridger et al. 2001; Ong et al. 2003).
Once in the alveolar space, neutrophils adhere to the apical surface of epithelial cells, possibly through ICAM-1 interactions. Interestingly, a soluble form of ICAM-1 (sICAM-1) is present in the plasma and in the lung lining fluid. sICAM may be generated through alternative splicing or by proteolytic cleavage of the membrane-bound form (mICAM-1). The mechanisms of sICAM-1 production are different in type I alveolar epithelial cells (AEC) and pulmonary microvascular endothelial cells (PVEC). High baseline release of sICAM-1 in AEC and presence in the alveolar epithelial lining fluid were observed. TNFα or LPS had little effect on sICAM-1 expression in AEC, but greatly increased sICAM-1 from PVEC (Mendez et al. 2008). AEC sICAM-1 shedding was inhibited by a serine protease inhibitor; whereas protease inhibitors had no effect on PVEC sICAM-1 expression. These differences may reflect the roles of sICAM-1 in the vasculature versus the air spaces. Overexpression of sICAM-1 in the alveolar space distal lung resulted in decreased survival after intranasal infection with K. pneumoniae (Mendez et al. 2011). At 24 h, a greater percentage of the transgenic mice (SPC-sICAM-1) had bacteria in the spleen compared with WT mice, while bacterial burden in the lungs was similar, and the number of neutrophils in the BALF was threefold greater in the SPC-sICAM-1 mice. These data suggest that sICAM-1 modulates host defense toward pathogens in the lung.
4.4 Neutrophil Functions in the Lung
4.4.1 Bactericidal Functions
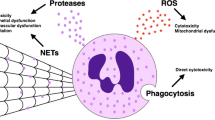
Upon arrival at the site of infection, neutrophils contribute to microbial killing by binding and phagocytosing pathogens, and subsequently releasing highly toxic granule contents and radicals into the phagosome (Fig. 4.4). Oxidant production and other effector functions is enhanced by priming neutrophils with inflammatory mediators or chemokines and cytokines, including TNF-α, IL-8, or IFN-γ, or by the processes of adhesion and migration. The recognition of microbes or microbial products may be mediated by pattern recognition receptors, including toll-like receptors, C-type lectin receptors, NLRs, and RIG-I helicase receptors. Human neutrophils have been demonstrated to express all the TLRs except TLR3, and also express CLEC7A (dectin 1), CLEC2 (CLEC1B), RIG-I, MDA5, NOD1, and FPR1 (reviewed in (Mantovani et al. 2011)).

Neutrophil effector functions during inflammation. Release of granule contents and reactive oxygen species into phagosomes or into the surrounding environment occurs upon stimulation. Granule contents and ROS have direct antimicrobial effects, as well as modulating the inflammatory response. Other neutrophils functions include secretion of soluble signaling molecules, extracellular pathogen killing through NETs, and regulating host defense through direct interactions with cells present in the inflammatory milieu, including other leukocytes
Uptake of bacteria and other particles by phagocytes (neutrophils, macrophages, and to a lesser extent dendritic cells) is markedly enhanced by coating the particle surface with opsonins, which include antibodies, complement fragments, pentraxins, and collectins such as mannan- binding lectin and in the lung, SP-A and SP-D (Greenberg and Grinstein 2002). Targets coated with antibodies or complement fragments bind phagocytic receptors expressed on the surface of phagocytes (Lee et al. 2003; Flannagan et al. 2009). The main phagocytic receptors in neutrophils are Fcγ receptors that bind particles coated with IgG, and complement receptors that recognize particles coated with complement fragments (Greenberg and Grinstein 2002; Lee et al. 2003). The low affinity Fc receptors FcγRIIIB and FcγRIIA are constitutively expressed in human neutrophils, whereas the high affinity receptor FcγRI is induced by the treatment with G-CSF or IFN-γ (Repp et al. 1991; Cassatella et al. 1990; Nimmerjahn and Ravetch 2006). FcγRIIA has ITAM in its cytoplasmic domain which become phosphorylated upon ligand binding. FcγRIIIB is GPI-anchored to the cell membrane and may signal intracellularly by acting in concert with FcγRIIa in lipid microdomains (Chuang et al. 2000; Marois et al. 2011). FcγRI has a short cytoplasmic tail and requires an associated gamma-chain to induce signaling. Binding of ligands to Fc receptors leads to receptor clustering and recruitment of Src family kinases, which phosphorylate tyrosine residues in the ITAM domains, leading to recruitment of the tyrosine kinase Syk and activation of small GTPases, and triggering a signaling cascade that mediates target engulfment (Lee et al. 2003; Flannagan et al. 2009).
Complement receptor 3 (CR3) is the leukocyte integrin CD11b/CD18 (Mac-1), and it binds particles coated with C3bi, a cleavage product of C3b which is in turn produced by proteolysis of C3 during complement activation. Whereas phagocytosis of IgG-coated particles is characterized by the extension of pseudopods that surround and engulf the target, complement-coated targets are observed to sink into the cell, indicating distinct mechanisms of engulfment (Lee et al. 2003). Crosstalk between the Fc receptors and complement receptors modulate their respective activities. For example, in human monocytes, CR3-mediated phagocytosis is inhibited by binding FcγRI and enhanced by binding FcγRII (Huang et al. 2011).
The nascent phagosome undergoes a complex process of maturation, whereby it acquires the machinery and materials for microbial killing and degradation (reviewed in (Lee et al. 2003; Flannagan et al. 2009; Nordenfelt and Tapper 2011)). The process culminates in the release of granule contents into the phagosome and the assembly and activation of NADPH oxidase on the membrane. In contrast to macrophages, phagocytosis in neutrophils occurs in seconds rather than minutes, maturation involves the fusion of preformed granules with the phagosome rather than the endocytic maturation pathway in macrophages, large amounts of oxidants are produced through NADPH oxidase activity, and the phagosomal pH is neutral rather than acidic (Nordenfelt and Tapper 2011; Bianchi et al. 2009).
Neutrophil granule contents are synthesized and packaged during neutrophil development in the bone marrow (Borregaard and Cowland 1997; Borregaard et al. 2007; Faurschou and Borregaard 2003). Neutrophil granules are generally classified based on the timing of their synthesis and their major contents. Primary (azurophil) granules are made earliest and contain myeloperoxidase, as well as serine proteases, defensins, and bactericidal permeability-increasing protein. Specific or secondary granules are peroxidase-negative and contain lactoferrin, as well as the cathelicidin hCAP-18 (precursor for the antimicrobial peptide LL-37), whereas tertiary granules contain gelatinase. The membrane of specific granules contains the cytochrome b558 moiety of NADPH oxidase which is incorporated into the phagosome and cell membrane upon neutrophil activation. Secretory vesicles are made during the terminal stages of maturation and enriched in receptors including the complement receptors CR1 (CD35) and CR3 (CD11b/CD18), and albumin. The membranes of secretory vesicles are incorporated into the plasma membrane in response to chemotactic factors and early during leukocyte recruitment, thus supplying membrane proteins including adhesion receptors that are critical for neutrophil function. Using a calcium ionophore or the chemotactic peptide fMLF to induce degranulation, the contents of granules are released in the opposite order to their synthesis: secretory vesicles are mobilized most readily, the threshold for exocytosis of gelatinase granules, and specific granules are progressively higher (Sengelov et al. 1993).
Non-oxidative killing of microbes by neutrophils is effected by antimicrobial peptides and proteases stored in granules and released into the phagosome, including serine proteases, matrix metalloproteinases, and various antimicrobial peptides. In addition to their role in killing pathogens in the phagosome, neutrophil granule contents may also kill pathogens extracellularly in neutrophil extracellular traps (NETs, described in the following section).
The neutrophil serine proteases neutrophil elastase (NE), cathepsin G (CG), and proteinase-3 are structurally related, abundantly expressed enzymes stored in the azurophilic granules (Pham 2006, 2008). Before being packaged into primary granules, the proenzyme form of these serine proteases are processed by dipeptidyl peptidase (DPPI, or cathepsin C) to yield the active forms. Serine proteases can cleave a large variety of substrates, including bacterial constituents, components of the extracellular matrix, plasma proteins, cytokines, and growth factors (Pham 2006, 2008). The diversity of substrates indicates that neutrophil serine proteases may play many roles in the inflammatory response.
Neutrophil serine proteases have been shown to kill a variety of microbial pathogens in vitro. For example, purified NE or CG kill S. pneumoniae in vitro, and inhibitors of serine proteases abrogate this microbicidal activity (Standish and Weiser 2009). NE degrades virulence factors of Gram-negative enterobacteria Shigella, Salmonella, and Yersinia (Weinrauch et al. 2002). Serine proteases can regulate host defense by proteolytic modification of cytokines and chemokines which results in either enhanced activity or inactivation of their targets, and by activation of specific cellular receptors (Pham 2006, 2008). For example, serine proteases process IL-8 to more active truncated forms (Padrines et al. 1994), NE induces apoptosis of lung epithelial cells through PAR-1 activation (Suzuki et al. 2005, 2009), and CG and NE cleave IL-33 into active mature forms (Lefrancais et al. 2012).
The role of serine proteases in infection models have been tested in vivo using mice that are deficient in one or a combination of these enzymes, or mice deficient in DPPI. DPPI-deficient mice have no defect in clearance of the fungus Aspergillus fumigatus from the lung, and studies of bacterial clearance remain to be pursued (Vethanayagam et al. 2011). Surprisingly, DPPI deficiency was protective in a murine model of sepsis due to increased levels of IL-6 (Mallen-St Clair et al. 2004). Humans with Papillon–Lefevre syndrome due to DPPI deficiency have pyogenic liver abscesses, but no clear defect in neutrophil killing of S. aureus or E. coli (Pham et al. 2004; Almuneef et al. 2003). NE-deficient mice exhibit impaired host defense following intraperitoneal infection with K. pneumoniae and E. coli but not S. aureus (Belaaouaj et al. 1998). NE null mice have increased susceptibility and impaired bacterial killing in P. aeruginosa pneumonia (Hirche et al. 2008). However, NE is not required for neutrophil recruitment into the lungs or peritoneum in response to P. aeruginosa or LPS (Hirche et al. 2004). Purified CG does not inhibit the growth of S. aureus, K. pneumoniae, or E. coli, and no defect in clearance or survival upon challenge in vivo with any of these organisms (MacIvor et al. 1999). In a model of pneumonia induced by S. pneumoniae, mice deficient in CG or in both CG and NE had reduced survival and increased bacterial load (Hahn et al. 2011). These studies indicate that the serine proteases have site- and organism-specific and often nonredundant roles in host defense against microbes.
The matrix metalloproteinases are a family of zinc-dependent endopeptidases expressed in many cell types, including neutrophils, that degrade extracellular matrix components and are thus implicated in tissue remodeling, but also regulate host defense by targeting cell adhesion molecules, cytokines, and growth factors (Greenlee et al. 2007). MMP-8 (neutrophil collagenase) and MMP9 (gelatinase B) are stored in secondary granules and secreted or expressed on the surface upon activation. The levels of MMP-8 and MMP-9 were elevated in lung lavage fluid and plasma from patients with hospital-acquired pneumonia (Hartog et al. 2003), suggesting that these MMPs may play a role during pulmonary infections. Membrane-expressed MMP8 has recently been shown to cleave MIP-1α and attenuate injury in LPS-induced ALI (Quintero et al. 2010). IL-8 binding to CXCR2 stimulates the release of MMP9 (gelatinase B) which processes IL-8 to increase its activity (Van den Steen et al. 2000; Chakrabarti and Patel 2005). A recent study demonstrated that MMP9 (gelatinase B) was critical for efficient phagocytosis and superoxide production by neutrophils, and cleaved IL-17A in vitro (Hong et al. 2011). MMP2/9 double-deficient mice have more neutrophils in the lungs, greater bacterial load, and are more susceptible to S. pneumoniae-induced acute pneumonia (Hong et al. 2011). Clearly MMP-8 and MMP-9 can modulate the host response by processing some cytokines directly, but their effect can also be indirect, for example collagen digestion by MMP-8, MMP-9, and prolyl endopeptidase generates proline-glycine-proline fragments which are potent neutrophil chemoattractants (Gaggar et al. 2008; Weathington et al. 2006).
The antimicrobial peptides defensins and cathelicidins are small cationic peptides with antimicrobial and immune regulatory properties (Yang et al. 2004). There are four human neutrophil α-defensins, human neutrophil peptides 1–4, which are stored in primary granules and released upon neutrophil activation (Lehrer and Lu 2012). The human cathelicidin hCAP18 is cleaved by serine proteases to yield the antimicrobial fragment LL-37. Defensins and cathelicidins can kill a broad range of pathogens through permeabilization of target membranes. In addition to their antimicrobial effects, these antimicrobial peptides can modulate immune responses by serving as alarmins to recruit and activate immune cells (Chertov et al. 1996; Grigat et al. 2007).
In resting neutrophils, the NADPH oxidase complex is separated into cytoplasmic and membrane-bound components. When neutrophils are activated by a variety of stimuli, including inflammatory mediators, adhesion via integrins or binding of opsonized particles, the cytoplasmic components of the phagocyte NADPH oxidase (p40, p47, p67, and the small GTPase Rac) associate with the membrane bound cytochrome b558 heterodimer of gp91 and p22 to assemble the functional enzyme that catalyzes the production of superoxide from molecular oxygen (Quinn and Gauss 2004; Nauseef 2004; Babior 2004). Superoxide anion is converted spontaneously or enzymatically into hydrogen peroxide, which is converted by myeloperoxidase into the potent microbicide hypochlorous acid. Superoxide and other ROS formed downstream can interact with a large variety of molecules and alter their target structure and function. Fusion and release of granule contents into the phagosome is coincident with NADPH oxidase activity, so that the engulfed bacterium is exposed to high ROS levels, proteases, and a plethora of antimicrobial proteins. NADPH oxidase-generated oxidants are generally thought to be major effectors of bacterial killing. An alternative hypothesis has implicated protease activation in the phagosome by ion fluxes generated during NADPH oxidase activation, but this remains controversial (Segal 2005; Nauseef 2007). It seems clear, however, that oxidase activity is required for host defense caused by some organisms. For example, in mice with pulmonary A. fumigatus or systemic Burkholderia cepacia infection, NADPH oxidase activity rather than serine proteases was required for protection (Vethanayagam et al. 2011). Interestingly, NADPH oxidase appears to downmodulate inflammation in certain conditions (Marriott et al. 2008; Morgenstern et al. 1997; Segal et al. 2010), most clearly through ROS-mediated inactivation of chemokines, cytokines, and other inflammatory mediators. Consistent with both the microbicidal functions and the inflammatory downmodulatory effects, patients with chronic granulomatous disease characterized by having defects in NADPH oxidase function often have B. cepacia and A. fumigatus infections and over-exuberant sterile inflammation.
ROS can modify the function of many signaling molecules by targeting thiols on cysteine residues. Signaling molecules that are targeted by ROS leading to changes in their signaling pathways include the protein tyrosine phosphatases, the small GTPase Rho, and Src kinases. Notably, a recent paper showed that NADPH oxidase deficiency or inhibiting NADPH oxidase led to defects in chemotaxis in human neutrophils, implicating NADPH oxidase-generated oxidants as regulators of neutrophil chemotaxis (Hattori et al. 2010). NADPH oxidase is also required for the production of IFN-γ by neutrophils during S. pneumoniae pneumonia (Yamada et al. 2011). NADPH oxidase is required for elaboration of NETs, as discussed in the following section.
4.4.2 NET Formation
NETs were first described as extracellular structures composed of decondensed chromatin with histones and granular contents that are able to bind and kill bacteria (Brinkmann et al. 2004). Inducers of NET formation include bacteria and their components, fungi, PMA, cytokines, and chemokines including IL-8 and IFN-γ (von Kockritz-Blickwede and Nizet 2009). The formation of NETs is initially marked by chromatin decondensation, perhaps through posttranslational histone modification (Neeli et al. 2008, 2009; Li et al. 2010) or through cleavage by neutrophil elastase translocated to the nucleus (Papayannopoulos et al. 2010). This is followed by breakdown of the nuclear and granular membranes and contact between granular contents and nuclear material, and subsequently the cell ruptures and NETs are released extracellularly (Fuchs et al. 2007). NET formation requires NADPH oxidase (Bianchi et al. 2009; Fuchs et al. 2007) and is regulated by neutrophil elastase and myeloperoxidase (Papayannopoulos et al. 2010; Metzler et al. 2011). This pathway of NET formation usually occurs over a span of several hours and is likely a form of cell death (Fuchs et al. 2007; Remijsen et al. 2011a). Rapid NET formation has also been induced by platelet binding to neutrophils after TLR4 stimulation (Clark et al. 2007). Other pathways of NET formation occur more rapidly and may not require lytic cell death. Live GM-CSF-primed neutrophils form extracellular nets in response to TLR4 or C5a signaling by extruding their mitochondrial DNA (Yousefi et al. 2009). Release of chromatin in vesicles from intact neutrophils induced by S. aureus, distinct from later lytic release of DNA, has been reported as an oxidant-independent pathway of NET production (Pilsczek et al. 2010). The Raf-MEK-ERK pathway was identified by a chemical inhibitor screen as a critical step for NET formation, likely upstream of NADPH oxidase (Hakkim et al. 2011). Notably, PKC inhibition by staurosporine led to a marked decrease in NET induction by PMA and PAF but not by Helicobacter pylori, indicating some redundancy in NET signaling pathways depending on the stimulus.
NETs have been reported to kill bacteria, fungi, and protozoa in vitro (Brinkmann et al. 2004; Fuchs et al. 2007; Guimaraes-Costa et al. 2009; Parker et al. 2012; Urban et al. 2006, 2009; Young et al. 2011). The antimicrobial functions of NETs are due to killing by histones, oxygen radicals, and microbicidal granular contents and to trapping of pathogens in the chromatin mesh (Brinkmann et al. 2004; Clark et al. 2007; Parker et al. 2012; Urban et al. 2006, 2009; Papayannopoulos and Zychlinsky 2009). NETs from human neutrophils stimulated with PMA contained molecules with known antimicrobial properties: granular proteins (leukocyte elastase, lactotransferrin, azurocidin, cathepsin G, myeloperoxidase, leukocyte proteinase-3, lysozyme C, and neutrophil defensins 1 and 3), nuclear components (histones), and the cytoplasmic calprotectin complex (Urban et al. 2009).
Determining the specific contribution of NETs in host defense in vivo is complicated because many of the neutrophil constituents present in NETs and the processes required in NET formation are involved in other antimicrobial processes (Fig. 4.4). Supporting an antimicrobial role for NETs in vivo is the observation that bacterial pathogens appear to have evolved mechanisms to counter them. Pathogenic group A streptococci possess endonucleases that allow them to escape from NETs (Buchanan et al. 2006; Walker et al. 2007). Pneumococcal strains express an endonuclease that allows them to escape from NETs, and mutant pneumococci lacking endonuclease have impaired ability to spread to the lung and blood in mice infected intranasally (Beiter et al. 2006). Pneumococci also modify their surface charge to repulse antimicrobial peptides present in NETs and synthesize a capsule that enables them to evade entrapment (Wartha et al. 2007).
NETs may have deleterious effects by entangling or activating host immune cells, or by directly damaging tissues through contact with NET contents. NETs have been implicated in the pathogenesis of autoimmune diseases (Hakkim et al. 2010; Kessenbrock et al. 2009; Leffler et al. 2012; Garcia-Romo et al. 2011; Lande et al. 2011), gout (Mitroulis et al. 2011), sepsis (Clark et al. 2007), and venous thrombosis (von Bruhl et al. 2012; Brill et al. 2012; Fuchs et al. 2010). In vitro, NETs damage endothelial cells and the lung epithelial cancer cell line (A549) likely through the activities of proteases and histones (Clark et al. 2007; Saffarzadeh et al. 2012; Gupta et al. 2010).
NETs may play a role in host defense in the lung during bacterial pneumonias. NETs were induced in a murine model of acute pneumonia induced by K. pneumoniae (Papayannopoulos et al. 2010) and S. pneumoniae (Yamada et al. 2011; Beiter et al. 2006; Wartha et al. 2007). Notably fewer NETs were observed when pneumonia was induced by E. coli (Yamada et al. 2011). IFN-γ production by neutrophils was required for NET formation and decreased bacterial load (Yamada et al. 2011).
4.4.3 Recruitment of More Neutrophils from the Bone Marrow and Blood
Inflammatory mediators emanating from the inflammatory stimulus itself (bacterial constituents such as fMLP, for example), or produced by resident cells and cells recruited to the lung during the infection can recruit neutrophils into the lungs from the blood and bone marrow. Alveolar macrophages, epithelial cells, and endothelial cells produce mediators that recruit neutrophils. Among these mediators are complement fragments, lipid mediators such as LTB4, cytokines such as IL-1β and TNF-α, and chemokines such as IL-8. Neutrophils contribute to this process in several ways, primarily by producing mediators themselves. For example, human neutrophils make lipid mediators and IL-8, and are thus able to attract more neutrophils into the site of inflammation. LL-37, a cathelicidin released upon degranulation, is chemotactic for neutrophils, monocytes, and T lymphocytes (De et al. 2000). Neutrophil proteases may also contribute to this process by modifying chemokines and their receptors, or cleaving collagen to generate the chemoattractant PGP (discussed previously).
4.4.4 Immunoregulatory Functions
The complex and critical role of neutrophils in regulating innate and adaptive immunity is increasingly being appreciated (Mantovani et al. 2011). In addition to their well-described roles in pathogen killing and clearance, neutrophils regulate the immune response by producing and releasing molecules with immunoregulatory activities (Fig. 4.4). Proteases, antimicrobial peptides, and radicals that are released upon neutrophil activation can regulate the functions of surrounding cells, recruit immune cells, or act on signaling molecules in the surrounding milieu, as described in the previous sections. Neutrophils can also produce soluble cell signaling molecules, including potent cytokines such as IFN-γ and TNF-α, chemokines such as IL-8, growth factors such as G-CSF, pattern recognition molecules that enhance pathogen recognition by neutrophils and other cells, and lipid-derived mediators (Mantovani et al. 2011). For example, neutrophils make IFN-γ in pneumonias induced by S. pneumoniae and S. aureus, but not by E. coli or P. aeruginosa (Yamada et al. 2011). Production of IFN-γ by neutrophils requires NADPH oxidase. IFN-γ modulates NET production and improves bacterial clearance (Yamada et al. 2011).
Direct contact between neutrophils and other cells can modulate their respective functions. For example, engulfment of apoptotic neutrophils modifies macrophage function, as described below. Crosstalk through direct interactions between neutrophils and monocytes, dendritic cells, or T cells, leading to modulation of the subsequent immune response, has also been demonstrated. For example, CD49d-expressing neutrophils induce FcεRI expression on lung dendritic cells, facilitating the recruitment of Th2 cells in a murine model of post-viral asthma (Cheung et al. 2010). Finally, neutrophils are critical for resolving inflammation or setting the stage for the immune response, as described in the next section.
4.4.5 Resolution of Inflammatory Response or Transition from Inflammatory to Innate Immune Response
Resolution is not merely the passive cessation of pro-inflammatory responses, but rather requires an active response from the host, resulting in tissue repair and return to homeostasis. Ideally, neutrophilic inflammation is self-limiting and resolves with the proper removal of apoptotic neutrophils by scavenging macrophages and tissue repair. Several classes of lipid-derived mediators of resolution have been identified including lipoxins, D- and E-series resolvins, protectins, and maresins. Resolvins bind G protein-coupled receptors, decreasing neutrophil recruitment and downregulating production of inflammatory mediators and ROS, while promoting apoptotic neutrophil uptake by macrophages (Uddin and Levy 2011). For example, in a model of acid aspiration-induced lung injury and bacterial challenge with E. coli, prophylactic treatment with RvE1 decreased neutrophil accumulation in the lungs and bacterial load, and improved survival (Seki et al. 2010). These were associated with decreased levels of pro-inflammatory mediators IL-1beta, IL-6, HMGB-1, MIP-1alpha, MIP-1beta, keratinocyte-derived chemokine, and MCP-1 (Seki et al. 2010). RvD1 and RvD2 target leukocytes and reduce neutrophil recruitment (Spite et al. 2009).
A role for CD44 has also recently been described in the process of resolution. Interestingly, deficiency of CD44 results in improved clearance of either S. pneumoniae or K. pneumoniae (van der Windt et al. 2010, 2011), suggesting that CD44 facilitates bacterial growth and dissemination. However, CD44-deficient mice also show delayed resolution of the inflammatory process (van der Windt et al. 2010, 2011) suggesting that CD44 contributes to resolution through its interactions with hyaluronan or other ligands. Studies investigating bleomycin-induced lung inflammation and fibrosis also show that CD44 is important in resolving the inflammatory process (Teder et al. 2002).
In addition to their immunoregulatory and pro-resolution functions described previously, neutrophils can modulate the transition from inflammation to innate immune response by activating antigen presenting cells through alarmins. Lactoferrin, α-defensins, cathelicidin, and HMGB-1 are considered alarmins for their ability to serve as endogenous danger signals that alert the immune system (Yang and Oppenheim 2009). Neutrophils can also modulate monocyte recruitment by releasing granule proteins that activate endothelial cells or modify chemokines by proteolysis, and neutrophils and monocytes/macrophages interact and regulate each other’s functions (Soehnlein and Lindbom 2010; Soehnlein et al. 2009). For example, uptake of apoptotic neutrophils modulates macrophage function, as described below.
4.5 Clearance of Neutrophils
4.5.1 Apoptosis
The fate of emigrated neutrophils can be one of the several death pathways with varying inflammatory and immunogenic consequences for the host; apoptosis, NETosis and necrosis (Fig. 4.1) (Bratton and Henson 2011). Neutrophil apoptosis occurs via the intrinsic and extrinsic pathways resulting in death without release of cellular contents that may otherwise be released into the surrounding tissue and amplify inflammation (Bratton and Henson 2011; Witko-Sarsat et al. 2011; Fox et al. 2010). As described in several recent reviews (Witko-Sarsat et al. 2011; Fox et al. 2010), the distinct features of neutrophil apoptosis include the key role of the prosurvival factor Mcl-1, the dual role of TNFα which can induce apoptosis or prolong the lifespan of neutrophils, the proapoptotic role of ROS and NADPH oxidase activation, and the role of the granule protein cathepsin D which can activate caspase-8 (Conus et al. 2008, 2012) or process the pro-apoptotic Bcl-2 protein Bid to promote apoptosis (Blomgran et al. 2007). These and other pathways may be modified during the inflammatory response to prolong neutrophil lifespan. For example, cathepsin D release in the cytosol is blocked during inflammation, inhibiting its activation of caspase 8 (Conus et al. 2008). These reviews and the studies described therein highlight the complex nature of apoptosis programs in neutrophils, and the differences between spontaneous versus inflammation-induced apoptosis, indicating that regulation of neutrophil life span may be a therapeutic target.
Apoptotic neutrophils are removed by scavenging macrophages (efferocytosis) that are attracted to the site by “find me” signals and recognize the so-called “eat me” signals on the surface of apoptotic neutrophils (Bratton and Henson 2011). Diffusible find me signals include extracellular nucleotides (ATP and UTP), while the most studied “eat me” signal is phosphatidylserine, which can directly engage receptors on macrophages or bind soluble pattern recognition molecules that are recognized by macrophage receptors (Bratton and Henson 2011). In the inflammatory microenvironment, pattern recognition molecules that coat apoptotic target cells facilitate efferocytosis (Litvack and Palaniyar 2010). For example, the lung collectins SP-A and SP-D regulate inflammatory mediator production and efferocytosis in contrasting ways, depending on the signaling complexes bound and the phagocyte subset (Janssen et al. 2008; Gardai et al. 2003).
During inflammation, the induction of neutrophil apoptosis and clearance of apoptotic neutrophils limit tissue injury and promote resolution (Fox et al. 2010; El Kebir and Filep 2010; Elliott and Ravichandran 2010). Apoptotic neutrophils are generally functionally spent, and uptake of apoptotic neutrophils by macrophages prevents the release of toxic neutrophil contents into the tissues. Phagocytosis of apoptotic cells by macrophages suppresses macrophage release of pro-inflammatory mediators such as TNFα, and increases macrophage release of anti-inflammatory mediators such as IL-10 and TGFβ (Fox et al. 2010; El Kebir and Filep 2010; Elliott and Ravichandran 2010). Neutrophil apoptosis as well as clearance of apoptotic neutrophils is regulated by pro-inflammatory cytokines and toll-like receptor agonists. Pro-inflammatory mediators such as TNF-α, high mobility group protein-1 and toll-like receptor agonists such as LPS, peptidoglycans, and CpG DNA can rescue neutrophils from apoptosis and inhibit apoptotic cell clearance in the lung (Borges et al. 2009; Liu et al. 2008; Michlewska et al. 2009; Banerjee et al. 2011).
4.5.2 NETosis
NETosis was coined to describe the death of neutrophils associated with the production of NETs, described above (Steinberg and Grinstein 2007). Morphologically, NETosis proceeds in the following stages: initially, the nuclei lose their lobules and chromatin begins to decondense; then, the nuclear envelope vesiculates and chromatin is decondensed, subsequently the granules disappear, and the chromatin mixes with cytoplasmic and granular contents, as evidenced by colocalization of elastase with chromatin (Fuchs et al. 2007). Compared with apoptosis and necrosis, the distinguishing morphological features of NETosis are the mixing of nuclear and cytoplasmic constituents after the nuclear membrane disintegrates and the disappearance of internal membranes and cytoplasmic organelles (Fuchs et al. 2007). In contrast to apoptosis, neutrophils undergoing NETosis do not exhibit chromatin condensation or DNA fragmentation, do not express PS on the outer surface of the cell membrane prior to cell rupture, and are resistant to pan-caspase inhibitor (Fuchs et al. 2007; Remijsen et al. 2011a, b). Notably, NETs may be produced without undergoing NETosis, as in the case of neutrophils that extrude mitochondrial DNA (Yousefi et al. 2009) or neutrophils that release chromatin in vesicles (Pilsczek et al. 2010).
4.5.3 Necrosis
Primary necrosis occurs when emigrated neutrophils simply disintegrate at the site of inflammation, releasing their contents into the surrounding environment, with potentially deleterious effects due to the cytotoxic properties of their contents and to increased inflammation. Secondary necrosis occurs when apoptotic neutrophils are not taken up by scavenging phagocytes because the scavenging capacity is exceeded by a large number of apoptotic cells or the macrophages themselves are damaged or targeted by pathogens (Silva 2010a, b). Secondary necrosis may be particularly relevant in the pneumonia, where accumulation of apoptotic neutrophils leads to increased inflammation without increased bacterial load (Silva 2010b; Haslett 1999). A pathway of programmed necrosis or necroptosis occurring via death receptor signaling and requiring the kinases RIP1 or RIP3 has also been described (Vandenabeele et al. 2010; Han et al. 2011). Neutrophils exposed to Shigella flexneri underwent death by this pathway rather than apoptosis (Francois et al. 2000).
References
Abram CL, Lowell CA (2009) The ins and outs of leukocyte integrin signaling. Annu Rev Immunol 27:339–362
Almuneef M, Al Khenaizan S, Al Ajaji S, Al-Anazi A (2003) Pyogenic liver abscess and papillon-lefevre syndrome: not a rare association. Pediatrics 111:e85–88
Alon R, Ley K (2008) Cells on the run: shear-regulated integrin activation in leukocyte rolling and arrest on endothelial cells. Curr Opin Cell Biol 20:525–532
Babior BM (2004) NADPH oxidase. Curr Opin Immunol 16:42–47
Bainton DF (1999) Developmental biology of neutrophils and eosinophils. In: Gallin JI, Snyderman R (eds) Inflammation: Basic principles and clinical correlates. Lippincott Williams & Wilkins, Philadelphia, PA, pp 13–34
Banerjee S, de Freitas A, Friggeri A, Zmijewski JW, Liu G, Abraham E (2011) Intracellular hmgb1 negatively regulates efferocytosis. J Immunol 187:4686–4694
Basu S, Hodgson G, Katz M, Dunn AR (2002) Evaluation of role of g-csf in the production, survival, and release of neutrophils from bone marrow into circulation. Blood 100:854–861
Behzad AR, Chu F, Walker DC (1996) Fibroblasts are in a position to provide directional information to migrating neutrophils during pneumonia in rabbit lungs. Microvasc Res 51:303–316
Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B (2006) An endonuclease allows streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol 16:401–407
Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD (1998) Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med 4:615–618
Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, Phillips RJ, Strieter RM (2002) Critical role for cxcr2 and cxcr2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest 110:1703–1716
Berkow RL, Dodson RW (1986) Purification and functional evaluation of mature neutrophils from human bone marrow. Blood 68:853–860
Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, Reichenbach J (2009) Restoration of net formation by gene therapy in cgd controls aspergillosis. Blood 114:2619–2622
Bicknell S, van Eeden S, Hayashi S, Hards J, English D, Hogg JC (1994) A non-radioisotopic method for tracing neutrophils in vivo using 5’-bromo-2’-deoxyuridine. Am J Respir Cell Mol Biol 10:16–23
Blomgran R, Zheng L, Stendahl O (2007) Cathepsin-cleaved bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J Leukoc Biol 81:1213–1223
Bokoch GM (2005) Regulation of innate immunity by rho gtpases. Trends Cell Biol 15:163–171
Bonig H, Priestley GV, Papayannopoulou T (2006) Hierarchy of molecular-pathway usage in bone marrow homing and its shift by cytokines. Blood 107:79–86
Borges VM, Vandivier RW, McPhillips KA, Kench JA, Morimoto K, Groshong SD, Richens TR, Graham BB, Muldrow AM, Heule LV, Henson PM, Janssen WJ (2009) Tnf{alpha} inhibits apoptotic cell clearance in the lung, exacerbating acute inflammation. Am J Physiol Lung Cell Mol Physiol 297:L586–595
Borregaard N, Cowland JB (1997) Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89:3503–3521
Borregaard N, Sorensen OE, Theilgaard-Monch K (2007) Neutrophil granules: a library of innate immunity proteins. Trends Immunol 28:340–345
Bratton DL, Henson PM (2011) Neutrophil clearance: when the party is over, clean-up begins. Trends Immunol 32:350–357
Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD (2012) Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost 10:136–144
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Broxmeyer HE (2008) Chemokines in hematopoiesis. Curr Opin Hematol 15:49–58
Brumwell ML, MacNee W, Doerschuk CM, Wiggs B, Hogg JC (1991) Neutrophil kinetics in normal and emphysematous regions of human lungs. Ann N Y Acad Sci 624:30–39
Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, Feramisco J, Nizet V (2006) Dnase expression allows the pathogen group a streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 16:396–400
Burdon PC, Martin C, Rankin SM (2005) The cxc chemokine mip-2 stimulates neutrophil mobilization from the rat bone marrow in a cd49d-dependent manner. Blood 105:2543–2548
Burdon PC, Martin C, Rankin SM (2008) Migration across the sinusoidal endothelium regulates neutrophil mobilization in response to elr+cxc chemokines. Br J Haematol 142:100–108
Burns AR, Smith CW, Walker DC (2003) Unique structural features that influence neutrophil emigration into the lung. Physiol Rev 83:309–336
Carman CV, Springer TA (2004) A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol 167:377–388
Cassatella MA, Flynn RM, Amezaga MA, Bazzoni F, Vicentini F, Trinchieri G (1990) Interferon gamma induces in human neutrophils and macrophages expression of the mrna for the high affinity receptor for monomeric igg (fc gamma r-i or cd64). Biochem Biophys Res Commun 170:582–588
Chakrabarti S, Patel KD (2005) Regulation of matrix metalloproteinase-9 release from il-8-stimulated human neutrophils. J Leukoc Biol 78:279–288
Chertov O, Michiel DF, Xu L, Wang JM, Tani K, Murphy WJ, Longo DL, Taub DD, Oppenheim JJ (1996) Identification of defensin-1, defensin-2, and cap37/azurocidin as t-cell chemoattractant proteins released from interleukin-8-stimulated neutrophils. J Biol Chem 271:2935–2940
Cheung DS, Ehlenbach SJ, Kitchens RT, Riley DA, Thomas LL, Holtzman MJ, Grayson MH (2010) Cutting edge: Cd49d+neutrophils induce fcepsilonri expression on lung dendritic cells in a mouse model of postviral asthma. J Immunol 185:4983–4987
Choudhury S, Wilson MR, Goddard ME, O’Dea KP, Takata M (2004) Mechanisms of early pulmonary neutrophil sequestration in ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 287:L902–910
Christopher MJ, Liu F, Hilton MJ, Long F, Link DC (2009) Suppression of cxcl12 production by bone marrow osteoblasts is a common and critical pathway for cytokine-induced mobilization. Blood 114:1331–9
Chuang FY, Sassaroli M, Unkeless JC (2000) Convergence of fc gamma receptor iia and fc gamma receptor iiib signaling pathways in human neutrophils. J Immunol 164:350–360
Cinamon G, Shinder V, Shamri R, Alon R (2004) Chemoattractant signals and {beta}2 integrin occupancy at apical endothelial contacts combine with shear stress signals to promote transendothelial neutrophil migration. J Immunol 173:7282–7291
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P (2007) Platelet tlr4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13:463–469
Conus S, Perozzo R, Reinheckel T, Peters C, Scapozza L, Yousefi S, Simon HU (2008) Caspase-8 is activated by cathepsin d initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med 205:685–698
Conus S, Pop C, Snipas SJ, Salvesen GS, Simon HU (2012) Cathepsin d primes caspase-8 activation by multiple intra-chain proteolysis. J Biol Chem 287:21142–51
Corre F, Lellouch J, Schwartz D (1971) Smoking and leucocyte-counts. Results of an epidemiological survey. Lancet 2:632–634
Dalli J, Jones CP, Cavalcanti DM, Farsky SH, Perretti M, Rankin SM (2012) Annexin a1 regulates neutrophil clearance by macrophages in the mouse bone marrow. FASEB J 26:387–396
Dancey JT, Deubelbeiss KA, Harker LA, Finch CA (1976) Neutrophil kinetics in man. J Clin Invest 58:705–715
De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O (2000) Ll-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (fprl1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and t cells. J Exp Med 192:1069–1074
Delano MJ, Kelly-Scumpia KM, Thayer TC, Winfield RD, Scumpia PO, Cuenca AG, Harrington PB, O’Malley KA, Warner E, Gabrilovich S, Mathews CE, Laface D, Heyworth PG, Ramphal R, Strieter RM, Moldawer LL, Efron PA (2011) Neutrophil mobilization from the bone marrow during polymicrobial sepsis is dependent on cxcl12 signaling. J Immunol 187:911–918
Deubelbeiss KA, Dancey JT, Harker LA, Finch CA (1975) Neutrophil kinetics in the dog. J Clin Invest 55:833–839
Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, Smith CW, Ballantyne CM (1999) Relative contribution of lfa-1 and mac-1 to neutrophil adhesion and migration. J Immunol 163:5029–5038
Doerschuk CM (2000) Leukocyte trafficking in alveoli and airway passages. Respir Res 1:136–140
Doerschuk CM (2001) Mechanisms of leukocyte sequestration in inflamed lungs. Microcirculation 8:71–88
Doerschuk C, Winn R, Coxson H, Harlan J (1990) Cd18-dependent and -independent mechanisms of neutrophil emigration in the pulmonary and systemic microcirculation of rabbits. J Immunol 144:2327–2333
Doerschuk CM, Beyers N, Coxson HO, Wiggs B, Hogg JC (1993) Comparison of neutrophil and capillary diameters and their relation to neutrophil sequestration in the lung. J Appl Physiol 74:3040–3045
Doerschuk CM, Markos J, Coxson HO, English D, Hogg JC (1994) Quantitation of neutrophil migration in acute bacterial pneumonia in rabbits. J Appl Physiol 77:2593–2599
Downey GP, Elson EL, Schwab B 3rd, Erzurum SC, Young SK, Worthen GS (1991) Biophysical properties and microfilament assembly in neutrophils: modulation by cyclic amp. J Cell Biol 114:1179–1190
Doyle NA, Bhagwan SD, Meek BB, Kutkoski GJ, Steeber DA, Tedder TF, Doerschuk CM (1997) Neutrophil margination, sequestration, and emigration in the lungs of l-selectin-deficient mice.J Clin Invest 99:526–533
Eash KJ, Means JM, White DW, Link DC (2009) Cxcr4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood 113:4711–4719
Eash KJ, Greenbaum AM, Gopalan PK, Link DC (2010) Cxcr2 and cxcr4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest 120:2423–2431
El Kebir D, Filep JG (2010) Role of neutrophil apoptosis in the resolution of inflammation. TheScientificWorldJournal 10:1731–1748
Elliott MR, Ravichandran KS (2010) Clearance of apoptotic cells: implications in health and disease. J Cell Biol 189:1059–1070
Faurschou M, Borregaard N (2003) Neutrophil granules and secretory vesicles in inflammation. Microbes Infect 5:1317–1327
Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM (1998) Neutrophils emigrate from venules by a transendothelial cell pathway in response to fmlp. J Exp Med 187:903–915
Flannagan RS, Cosio G, Grinstein S (2009) Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7:355–366
Forlow SB, Schurr JR, Kolls JK, Bagby GJ, Schwarzenberger PO, Ley K (2001) Increased granulopoiesis through interleukin-17 and granulocyte colony-stimulating factor in leukocyte adhesion molecule-deficient mice. Blood 98:3309–3314
Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG (2010) Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun 2:216–227
Francois M, Le Cabec V, Dupont MA, Sansonetti PJ, Maridonneau-Parini I (2000) Induction of necrosis in human neutrophils by shigella flexneri requires type iii secretion, ipab and ipac invasins, and actin polymerization. Infect Immun 68:1289–1296
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–241
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA 107:15880–15885
Furze RC, Rankin SM (2008a) The role of the bone marrow in neutrophil clearance under homeostatic conditions in the mouse. FASEB J 22:3111–3119
Furze RC, Rankin SM (2008b) Neutrophil mobilization and clearance in the bone marrow. Immunology 125:281–288
Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, Rowe SM, Clancy JP, Blalock JE (2008) A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol 180:5662–5669
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V (2011) Netting neutrophils are major inducers of type i ifn production in pediatric systemic lupus erythematosus. Sci Transl Med 3:73ra20
Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, Henson PM (2003) By binding sirpalpha or calreticulin/cd91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 115:13–23
Gebb SA, Graham JA, Hanger CC, Godbey PS, Capen RL, Doerschuk CM, Wagner WW Jr (1995) Sites of leukocyte sequestration in the pulmonary microcirculation. J Appl Physiol 79:493–497
Giordano GF, Lichtman MA (1973) Marrow cell egress. The central interaction of barrier pore size and cell maturation. J Clin Invest 52:1154–1164
Gomez JC, Doerschuk CM (2010) The role of cd18 in the production and release of neutrophils from the bone marrow. Lab Invest 90:599–610
Gomez JC, Soltys J, Okano K, Dinauer MC, Doerschuk CM (2008) The role of rac2 in regulating neutrophil production in the bone marrow and circulating neutrophil counts. Am J Pathol 173:507–517
Gordy C, Pua H, Sempowski GD, He YW (2011) Regulation of steady-state neutrophil homeostasis by macrophages. Blood 117:618–629
Greenberg S, Grinstein S (2002) Phagocytosis and innate immunity. Curr Opin Immunol 14:136–145
Greenlee KJ, Werb Z, Kheradmand F (2007) Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev 87:69–98
Gregory AD, Hogue LA, Ferkol TW, Link DC (2007) Regulation of systemic and local neutrophil responses by g-csf during pulmonary pseudomonas aeruginosa infection. Blood 109:3235–3243
Grigat J, Soruri A, Forssmann U, Riggert J, Zwirner J (2007) Chemoattraction of macrophages, T lymphocytes, and mast cells is evolutionarily conserved within the human alpha-defensin family. J Immunol 179:3958–3965
Guimaraes-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao-Silva F, Saraiva EM (2009) Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci USA 106:6748–6753
Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, Resink TJ (2010) Activated endothelial cells induce neutrophil extracellular traps and are susceptible to netosis-mediated cell death. FEBS Lett 584:3193–3197
Hahn I, Klaus A, Janze AK, Steinwede K, Ding N, Bohling J, Brumshagen C, Serrano H, Gauthier F, Paton JC, Welte T, Maus UA (2011) Cathepsin g and neutrophil elastase play critical and nonredundant roles in lung-protective immunity against streptococcus pneumoniae in mice. Infect Immun 79:4893–4901
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A (2010) Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA 107:9813–9818
Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H (2011) Activation of the raf-mek-erk pathway is required for neutrophil extracellular trap formation. Nat Chem Biol 7:75–77
Han J, Zhong CQ, Zhang DW (2011) Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol 12:1143–1149
Hartog CM, Wermelt JA, Sommerfeld CO, Eichler W, Dalhoff K, Braun J (2003) Pulmonary matrix metalloproteinase excess in hospital-acquired pneumonia. Am J Respir Crit Care Med 167:593–598
Haslett C (1999) Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med 160:S5–11
Hattori H, Subramanian KK, Sakai J, Jia Y, Li Y, Porter TF, Loison F, Sarraj B, Kasorn A, Jo H, Blanchard C, Zirkle D, McDonald D, Pai SY, Serhan CN, Luo HR (2010) Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc Natl Acad Sci USA 107:3546–3551
Hechtman DH, Cybulsky MI, Fuchs HJ, Baker JB, Gimbrone MA Jr (1991) Intravascular il-8. Inhibitor of polymorphonuclear leukocyte accumulation at sites of acute inflammation. J Immunol 147:883–892
Hidalgo A, Peired AJ, Wild MK, Vestweber D, Frenette PS (2007) Complete identification of e-selectin ligands on neutrophils reveals distinct functions of psgl-1, esl-1, and cd44. Immunity 26:477–489
Hidalgo A, Chang J, Jang J-E, Peired AJ, Chiang EY, Frenette PS (2009) Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med 15:384–391
Hirche TO, Atkinson JJ, Bahr S, Belaaouaj A (2004) Deficiency in neutrophil elastase does not impair neutrophil recruitment to inflamed sites. Am J Respir Cell Mol Biol 30:576–584
Hirche TO, Benabid R, Deslee G, Gangloff S, Achilefu S, Guenounou M, Lebargy F, Hancock RE, Belaaouaj A (2008) Neutrophil elastase mediates innate host protection against pseudomonas aeruginosa. J Immunol 181:4945–4954
Hogg JC (1987) Neutrophil kinetics and lung injury. Physiol Rev 67:1249–1295
Hogg JC, Coxson HO, Brumwell ML, Beyers N, Doerschuk CM, MacNee W, Wiggs BR (1994) Erythrocyte and polymorphonuclear cell transit time and concentration in human pulmonary capillaries. J Appl Physiol 77:1795–1800
Hong JS, Greenlee KJ, Pitchumani R, Lee SH, Song LZ, Shan M, Chang SH, Park PW, Dong C, Werb Z, Bidani A, Corry DB, Kheradmand F (2011) Dual protective mechanisms of matrix metalloproteinases 2 and 9 in immune defense against streptococcus pneumoniae. J Immunol 186:6427–6436
Horwitz BH, Mizgerd JP, Scott ML, Doerschuk CM (2001) Mechanisms of granulocytosis in the absence of cd18. Blood 97:1578–1583
Huang ZY, Hunter S, Chien P, Kim MK, Han-Kim TH, Indik ZK, Schreiber AD (2011) Interaction of two phagocytic host defense systems: Fcgamma receptors and complement receptor 3. J Biol Chem 286:160–168
Inano H, English D, Doerschuk CM (1992) Effect of zymosan-activated plasma on the deformability of rabbit polymorphonuclear leukocytes. J Appl Physiol 73:1370–1376
Jagels MA, Hugli TE (1992) Neutrophil chemotactic factors promote leukocytosis. A common mechanism for cellular recruitment from bone marrow. J Immunol 148:1119–1128
Jagels MA, Hugli TE (1994) Mechanisms and mediators of neutrophilic leukocytosis. Immunopharmacology 28:1–18
Jagels MA, Chambers JD, Arfors KE, Hugli TE (1995) C5a- and tumor necrosis factor-alpha-induced leukocytosis occurs independently of beta 2 integrins and l-selectin: differential effects on neutrophil adhesion molecule expression in vivo. Blood 85:2900–2909
Jansen M, Yang FC, Cancelas JA, Bailey JR, Williams DA (2005) Rac2-deficient hematopoietic stem cells show defective interaction with the hematopoietic microenvironment and long-term engraftment failure. Stem Cells 23:335–346
Janssen WJ, McPhillips KA, Dickinson MG, Linderman DJ, Morimoto K, Xiao YQ, Oldham KM, Vandivier RW, Henson PM, Gardai SJ (2008) Surfactant proteins a and d suppress alveolar macrophage phagocytosis via interaction with sirp alpha. Am J Respir Crit Care Med 178:158–167
Kawai T, Malech HL (2009) Whim syndrome: congenital immune deficiency disease. Curr Opin Hematol 16:20–26
Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, Grone HJ, Brinkmann V, Jenne DE (2009) Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 15:623–625
Kim HK, De La uz Sierra M, Williams CK, Gulino AV, Tosato G (2006) G-csf down-regulation of cxcr4 expression identified as a mechanism for mobilization of myeloid cells. Blood 108:812–820
Knapp S, Hareng L, Rijneveld AW, Bresser P, van der Zee JS, Florquin S, Hartung T, van der Poll T (2004) Activation of neutrophils and inhibition of the proinflammatory cytokine response by endogenous granulocyte colony-stimulating factor in murine pneumococcal pneumonia. J Infect Dis 189:1506–1515
Kohler A, De Filippo K, Hasenberg M, van den Brandt C, Nye E, Hosking MP, Lane TE, Mann L, Ransohoff RM, Hauser AE, Winter O, Schraven B, Geiger H, Hogg N, Gunzer M (2011) G-csf-mediated thrombopoietin release triggers neutrophil motility and mobilization from bone marrow via induction of cxcr2 ligands. Blood 117:4349–4357
Kubo H, Graham L, Doyle NA, Quinlan WM, Hogg JC, Doerschuk CM (1998) Complement fragment-induced release of neutrophils from bone marrow and sequestration within pulmonary capillaries in rabbits. Blood 92:283–290
Kubo H, Doyle NA, Graham L, Bhagwan SD, Quinlan WM, Doerschuk CM (1999) L- and p-selectin and cd11/cd18 in intracapillary neutrophil sequestration in rabbit lungs. Am J Respir Crit Care Med 159:267–274
Kuijpers TW, van de Vijver E, Weterman MA, de Boer M, Tool AT, van den Berg TK, Moser M, Jakobs ME, Seeger K, Sanal O, Unal S, Cetin M, Roos D, Verhoeven AJ, Baas F (2009) Lad-1/variant syndrome is caused by mutations in FERMT3. Blood 113:4740–4746
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M (2011) Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 3:7319
Lawrence E, Van Eeden S, English D, Hogg JC (1996) Polymorphonuclear leukocyte (pmn) migration in streptococcal pneumonia: comparison of older pmn with those recently released from the marrow. Am J Respir Cell Mol Biol 14:217–224
Lee WY, Kubes P (2008) Leukocyte adhesion in the liver: distinct adhesion paradigm from other organs. J Hepatol 48:504–512
Lee WL, Harrison RE, Grinstein S (2003) Phagocytosis by neutrophils. Microbes Infect 5:1299–1306
Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, Bengtsson AA, Blom AM (2012) Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol 188:3522–3531
Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, Cayrol C (2012) Il-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin g. Proc Natl Acad Sci USA 109:1673–1678
Lehrer RI, Lu W (2012) Alpha-defensins in human innate immunity. Immunol Rev 245:84–112
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y (2010) Pad4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207:1853–1862
Lichtman MA (1970) Cellular deformability during maturation of the myeloblast. Possible role in marrow egress. N Engl J Med 283:943–948
Lien DC, Wagner WW Jr, Capen RL, Haslett C, Hanson WL, Hofmeister SE, Henson PM, Worthen GS (1987) Physiological neutrophil sequestration in the lung: visual evidence for localization in capillaries. J Appl Physiol 62:1236–1243
Lien DC, Worthen GS, Capen RL, Hanson WL, Checkley LL, Janke SJ, Henson PM, Wagner WW Jr (1990) Neutrophil kinetics in the pulmonary microcirculation. Effects of pressure and flow in the dependent lung. Am Rev Respir Dis 141:953–959
Lien DC, Henson PM, Capen RL, Henson JE, Hanson WL, Wagner WW Jr, Worthen GS (1991) Neutrophil kinetics in the pulmonary microcirculation during acute inflammation. Lab Invest 65:145–159
Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR (1994) Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 84:1737–1746
Litvack ML, Palaniyar N (2010) Review: soluble innate immune pattern-recognition proteins for clearing dying cells and cellular components: implications on exacerbating or resolving inflammation. Innate Immun 16:191–200
Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC (1996) Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity 5:491–501
Liu G, Wang J, Park YJ, Tsuruta Y, Lorne EF, Zhao X, Abraham E (2008) High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol 181:4240–4246
Lord BI, Molineux G, Pojda Z, Souza LM, Mermod JJ, Dexter TM (1991) Myeloid cell kinetics in mice treated with recombinant interleukin-3, granulocyte colony-stimulating factor (csf), or granulocyte-macrophage csf in vivo. Blood 77:2154–2159
Luo BH, Carman CV, Springer TA (2007) Structural basis of integrin regulation and signaling. Annu Rev Immunol 25:619–647
Luscinskas FW, Kiely JM, Ding H, Obin MS, Hebert CA, Baker JB, Gimbrone MA Jr (1992) In vitro inhibitory effect of il-8 and other chemoattractants on neutrophil-endothelial adhesive interactions. J Immunol 149:2163–2171
Ma Q, Jones D, Springer TA (1999) The chemokine receptor cxcr4 is required for the retention of b lineage and granulocytic precursors within the bone marrow microenvironment. Immunity 10:463–471
MacIvor DM, Shapiro SD, Pham CT, Belaaouaj A, Abraham SN, Ley TJ (1999) Normal neutrophil function in cathepsin g-deficient mice. Blood 94:4282–4293
Mackarel AJ, Russell KJ, Brady CS, FitzGerald MX, O’Connor CM (2000) Interleukin-8 and leukotriene-b4, but not formylmethionyl leucylphenylalanine, stimulate cd18-independent migration of neutrophils across human pulmonary endothelial cells in vitro. Am J Respir Cell Mol Biol 23:154–161
MacNee W, Wiggs B, Belzberg AS, Hogg JC (1989) The effect of cigarette smoking on neutrophil kinetics in human lungs. N Engl J Med 321:924–928
Malinin NL, Zhang L, Choi J, Ciocea A, Razorenova O, Ma YQ, Podrez EA, Tosi M, Lennon DP, Caplan AI, Shurin SB, Plow EF, Byzova TV (2009) A point mutation in kindlin3 ablates activation of three integrin subfamilies in humans. Nat Med 15:313–318
Mallen-St Clair J, Pham CT, Villalta SA, Caughey GH, Wolters PJ (2004) Mast cell dipeptidyl peptidase i mediates survival from sepsis. J Clin Invest 113:628–634
Mantovani A, Cassatella MA, Costantini C, Jaillon S (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11:519–531
Marois L, Pare G, Vaillancourt M, Rollet-Labelle E, Naccache PH (2011) Fc gammariiib triggers raft-dependent calcium influx in igg-mediated responses in human neutrophils. J Biol Chem 286:3509–3519
Marriott HM, Jackson LE, Wilkinson TS, Simpson AJ, Mitchell TJ, Buttle DJ, Cross SS, Ince PG, Hellewell PG, Whyte MK, Dockrell DH (2008) Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am J Respir Crit Care Med 177:887–895
Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM (2003) Chemokines acting via cxcr2 and cxcr4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 19:583–593
McDonald B, McAvoy EF, Lam F, Gill V, de la Motte C, Savani RC, Kubes P (2008) Interaction of cd44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J Exp Med 205:915–927
Mei J, Liu Y, Dai N, Hoffmann C, Hudock KM, Zhang P, Guttentag SH, Kolls JK, Oliver PM, Bushman FD, Worthen GS (2012) Cxcr2 and cxcl5 regulate the il-17/g-csf axis and neutrophil homeostasis in mice. J Clin Invest 122:974–986
Mendez MP, Morris SB, Wilcoxen S, Du M, Monroy YK, Remmer H, Murphy H, Christensen PJ, Paine R 3rd (2008) Disparate mechanisms of sicam-1 production in the peripheral lung: contrast between alveolar epithelial cells and pulmonary microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol 294:L807–814
Mendez MP, Monroy YK, Du M, Preston AM, Tolle L, Lin Y, VanDussen KL, Samuelson LC, Standiford TJ, Curtis JL, Beck JM, Christensen PJ, Paine R 3rd (2011) Overexpression of sicam-1 in the alveolar epithelial space results in an exaggerated inflammatory response and early death in gram negative pneumonia. Respir Res 12:12
Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, Wahn V, Papayannopoulos V, Zychlinsky A (2011) Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 117:953–959
Michlewska S, Dransfield I, Megson IL, Rossi AG (2009) Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for tnf-{alpha}. FASEB J 23:844–854
Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ (2006) Lymphocyte transcellular migration occurs through recruitment of endothelial icam-1 to caveola- and f-actin-rich domains. Nat Cell Biol 8:113–123
Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, Drosos GI, Boumpas DT, Ritis K (2011) Neutrophil extracellular trap formation is associated with il-1beta and autophagy-related signaling in gout. PLoS One 6:e29318
Mizgerd JP, Meek BB, Kutkoski GJ, Bullard DC, Beaudet AL, Doerschuk CM (1996) Selectins and neutrophil traffic: margination and streptococcus pneumoniae-induced emigration in murine lungs. J Exp Med 184:639–645
Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC (1997) Absence of respiratory burst in x-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med 185:207–218
Morland CM, Morland BJ, Darbyshire PJ, Stockley RA (2000) Migration of cd18-deficient neutrophils in vitro: evidence for a cd18-independent pathway induced by il-8. Biochim Biophys Acta 1500:70–76
Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R (2008) Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med 14:325–330
Moser M, Bauer M, Schmid S, Ruppert R, Schmidt S, Sixt M, Wang HV, Sperandio M, Fassler R (2009) Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat Med 15:300–305
Muller WA (2009) Mechanisms of transendothelial migration of leukocytes. Circ Res 105:223–230
Nauseef WM (2004) Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol 122:277–291
Nauseef WM (2007) How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev 219:88–102
Neeli I, Khan SN, Radic M (2008) Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol 180:1895–1902
Neeli I, Dwivedi N, Khan S, Radic M (2009) Regulation of extracellular chromatin release from neutrophils. J Innate Immun 1:194–201
Nemzek JA, Bolgos GL, Williams BA, Remick DG (2001) Differences in normal values for murine white blood cell counts and other hematological parameters based on sampling site. Inflamm Res 50:523–527
Nicholson SE, Oates AC, Harpur AG, Ziemiecki A, Wilks AF, Layton JE (1994) Tyrosine kinase jak1 is associated with the granulocyte-colony-stimulating factor receptor and both become tyrosine-phosphorylated after receptor activation. Proc Natl Acad Sci USA 91:2985–2988
Nicholson SE, Novak U, Ziegler SF, Layton JE (1995) Distinct regions of the granulocyte colony-stimulating factor receptor are required for tyrosine phosphorylation of the signaling molecules jak2, stat3, and p42, p44mapk. Blood 86:3698–3704
Nieminen M, Henttinen T, Merinen M, Marttila-Ichihara F, Eriksson JE, Jalkanen S (2006) Vimentin function in lymphocyte adhesion and transcellular migration. Nat Cell Biol 8:156–162
Nimmerjahn F, Ravetch JV (2006) Fcgamma receptors: old friends and new family members. Immunity 24:19–28
Nordenfelt P, Tapper H (2011) Phagosome dynamics during phagocytosis by neutrophils. J Leukoc Biol 90:271–284
Ong ES, Gao XP, Xu N, Predescu D, Rahman A, Broman MT, Jho DH, Malik AB (2003) E. coli pneumonia induces cd18-independent airway neutrophil migration in the absence of increased lung vascular permeability. Am J Physiol Lung Cell Mol Physiol 285:L879–888
Padrines M, Wolf M, Walz A, Baggiolini M (1994) Interleukin-8 processing by neutrophil elastase, cathepsin g and proteinase-3. FEBS Lett 352:231–235
Panopoulos AD, Watowich SS (2008) Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine 42:277–288
Papayannopoulos V, Zychlinsky A (2009) Nets: a new strategy for using old weapons. Trends Immunol 30:513–521
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191:677–691
Papayannopoulou T, Priestley GV, Bonig H, Nakamoto B (2003) The role of g-protein signaling in hematopoietic stem/progenitor cell mobilization. Blood 101:4739–4747
Parker H, Albrett AM, Kettle AJ, Winterbourn CC (2012) Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol 91:369–376
Petty JM, Sueblinvong V, Lenox CC, Jones CC, Cosgrove GP, Cool CD, Rai PR, Brown KK, Weiss DJ, Poynter ME, Suratt BT (2007) Pulmonary stromal-derived factor-1 expression and effect on neutrophil recruitment during acute lung injury. J Immunol 178:8148–8157
Petty JM, Lenox CC, Weiss DJ, Poynter ME, Suratt BT (2009) Crosstalk between cxcr4/stromal derived factor-1 and vla-4/vcam-1 pathways regulates neutrophil retention in the bone marrow. J Immunol 182:604–612
Pham CT (2006) Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 6:541–550
Pham CT (2008) Neutrophil serine proteases fine-tune the inflammatory response. Int J Biochem Cell Biol 40:1317–1333
Pham CT, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ (2004) Papillon-lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin c/dipeptidyl peptidase i deficiency in humans. J Immunol 173:7277–7281
Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, Kubes P (2010) A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 185:7413–7425
Price TH, Chatta GS, Dale DC (1996) Effect of recombinant granulocyte colony-stimulating factor on neutrophil kinetics in normal young and elderly humans. Blood 88:335–340
Priestley GV, Scott LM, Ulyanova T, Papayannopoulou T (2006) Lack of alpha4 integrin expression in stem cells restricts competitive function and self-renewal activity. Blood 107:2959–2967
Quinn MT, Gauss KA (2004) Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol 76:760–781
Quintero PA, Knolle MD, Cala LF, Zhuang Y, Owen CA (2010) Matrix metalloproteinase-8 inactivates macrophage inflammatory protein-1 alpha to reduce acute lung inflammation and injury in mice. J Immunol 184:1575–1588
Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P (2011a) Vanden Berghe T. Dying for a cause: Netosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ 18:581–588
Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P (2011b) Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 21:290–304
Repp R, Valerius T, Sendler A, Gramatzki M, Iro H, Kalden JR, Platzer E (1991) Neutrophils express the high affinity receptor for igg (fc gamma ri, cd64) after in vivo application of recombinant human granulocyte colony-stimulating factor. Blood 78:885–889
Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, Ley K (2006) Critical role of endothelial cxcr2 in LPS-induced neutrophil migration into the lung. J Clin Invest 116:695–702
Richards MK, Liu F, Iwasaki H, Akashi K, Link DC (2003) Pivotal role of granulocyte colony-stimulating factor in the development of progenitors in the common myeloid pathway. Blood 102:3562–3568
Ridger VC, Wagner BE, Wallace WAH, Hellewell PG (2001) Differential effects of cd18, cd29, and cd49 integrin subunit inhibition on neutrophil migration in pulmonary inflammation. J Immunol 166:3484–3490
Roberts AW, Kim C, Zhen L, Lowe JB, Kapur R, Petryniak B, Spaetti A, Pollock JD, Borneo JB, Bradford GB, Atkinson SJ, Dinauer MC, Williams DA (1999) Deficiency of the hematopoietic cell-specific rho family gtpase rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity 10:183–196
Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT (2012) Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 7:e32366
Saito H, Lai J, Rogers R, Doerschuk CM (2002) Mechanical properties of rat bone marrow and circulating neutrophils and their responses to inflammatory mediators. Blood 99:2207–2213
Sakai M, Sato Y, Sato S, Ihara S, Onizuka M, Sakakibara Y, Takahashi H (2004) Effect of relocating to areas of reduced atmospheric particulate matter levels on the human circulating leukocyte count. J Appl Physiol 97:1774–1780
Sato Y, van Eeden SF, English D, Hogg JC (1998a) Bacteremic pneumococcal pneumonia: bone marrow release and pulmonary sequestration of neutrophils. Crit Care Med 26:501–509
Sato Y, Van Eeden SF, English D, Hogg JC (1998b) Pulmonary sequestration of polymorphonuclear leukocytes released from bone marrow in bacteremic infection. Am J Physiol 275:L255–261
Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, Smith CW, Montgomery CA, Rich S, Beaudet AL (1998) Spontaneous skin ulceration and defective t cell function in cd18 null mice. J Exp Med 188:119–131
Scott LM, Priestley GV, Papayannopoulou T (2003) Deletion of alpha4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol Cell Biol 23:9349–9360
Segal AW (2005) How neutrophils kill microbes. Annu Rev Immunol 23:197–223
Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML, Blackwell TS (2010) NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One 5:e9631
Seki H, Fukunaga K, Arita M, Arai H, Nakanishi H, Taguchi R, Miyasho T, Takamiya R, Asano K, Ishizaka A, Takeda J, Levy BD (2010) The anti-inflammatory and proresolving mediator resolvin e1 protects mice from bacterial pneumonia and acute lung injury. J Immunol 184:836–843
Selby C, Drost E, Lannan S, Wraith PK, MacNee W (1991) Neutrophil retention in the lungs of patients with chronic obstructive pulmonary disease. Am Rev Respir Dis 143:1359–1364
Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC (2002) G-csf is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity 17:413–423
Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, Levesque JP, Chappel J, Ross FP, Link DC (2005) G-csf potently inhibits osteoblast activity and cxcl12 mrna expression in the bone marrow. Blood 106:3020–3027
Sengelov H, Kjeldsen L, Borregaard N (1993) Control of exocytosis in early neutrophil activation. J Immunol 150:1535–1543
Shimoda K, Iwasaki H, Okamura S, Ohno Y, Kubota A, Arima F, Otsuka T, Niho Y (1994) G-csf induces tyrosine phosphorylation of the jak2 protein in the human myeloid g-csf responsive and proliferative cells, but not in mature neutrophils. Biochem Biophys Res Commun 203:922–928
Silva MT (2010a) Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett 584:4491–4499
Silva MT (2010b) Bacteria-induced phagocyte secondary necrosis as a pathogenicity mechanism. J Leukoc Biol 88:885–896
Sligh JE Jr, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL (1993) Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc Natl Acad Sci USA 90:8529–8533
Smith ML, Olson TS, Ley K (2004) Cxcr2- and e-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med 200:935–939
Soehnlein O, Lindbom L (2010) Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol 10:427–439
Soehnlein O, Lindbom L, Weber C (2009) Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 114:4613–4623
Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN (2009) Resolvin d2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461:1287–1291
Standish AJ, Weiser JN (2009) Human neutrophils kill streptococcus pneumoniae via serine proteases. J Immunol 183:2602–2609
Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K (2005) Phagocytosis of apoptotic neutrophils regulates granulopoiesis via il-23 and il-17. Immunity 22:285–294
Steinberg BE, Grinstein S (2007) Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE 2007:11
Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER (2010) Neutrophil kinetics in health and disease. Trends Immunol 31:318–324
Suratt BT, Young SK, Lieber J, Nick JA, Henson PM, Worthen GS (2001) Neutrophil maturation and activation determine anatomic site of clearance from circulation. Am J Physiol Lung Cell Mol Physiol 281:L913–921
Suratt BT, Petty JM, Young SK, Malcolm KC, Lieber JG, Nick JA, Gonzalo JA, Henson PM, Worthen GS (2004) Role of the cxcr4/sdf-1 chemokine axis in circulating neutrophil homeostasis. Blood 104:565–571
Suzuki T, Moraes TJ, Vachon E, Ginzberg HH, Huang TT, Matthay MA, Hollenberg MD, Marshall J, McCulloch CA, Abreu MT, Chow CW, Downey GP (2005) Proteinase-activated receptor-1 mediates elastase-induced apoptosis of human lung epithelial cells. Am J Respir Cell Mol Biol 33:231–247
Suzuki T, Yamashita C, Zemans RL, Briones N, Van Linden A, Downey GP (2009) Leukocyte elastase induces lung epithelial apoptosis via a par-1-, nf-kappab-, and p53-dependent pathway. Am J Respir Cell Mol Biol 41:742–755
Svensson L, Howarth K, McDowall A, Patzak I, Evans R, Ussar S, Moser M, Metin A, Fried M, Tomlinson I, Hogg N (2009) Leukocyte adhesion deficiency-iii is caused by mutations in kindlin3 affecting integrin activation. Nat Med 15:306–312
Tasaka S, Richer SE, Mizgerd JP, Doerschuk CM (2002) Very late antigen-4 in cd18-independent neutrophil emigration during acute bacterial pneumonia in mice. Am J Respir Crit Care Med 166:53–60
Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW (2002) Resolution of lung inflammation by cd44. Science 296:155–158
Terashima T, Wiggs B, English D, Hogg JC, van Eeden SF (1996) Polymorphonuclear leukocyte transit times in bone marrow during streptococcal pneumonia. Am J Physiol 271:L587–592
Uddin M, Levy BD (2011) Resolvins: natural agonists for resolution of pulmonary inflammation. Prog Lipid Res 50:75–88
Urban CF, Reichard U, Brinkmann V, Zychlinsky A (2006) Neutrophil extracellular traps capture and kill candida albicans yeast and hyphal forms. Cell Microbiol 8:668–676
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against candida albicans. PLoS Pathog 5:e1000639
Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G (2000) Neutrophil gelatinase b potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades ctap-iii, pf-4, and gro-alpha and leaves rantes and mcp-2 intact. Blood 96:2673–2681
van der Windt GJ, Florquin S, de Vos AF, Van’t Veer C, Queiroz KC, Liang J, Jiang D, Noble PW, van der Poll T (2010) Cd44 deficiency is associated with increased bacterial clearance but enhanced lung inflammation during gram-negative pneumonia. Am J Pathol 177:2483–2494
van der Windt GJ, Hoogendijk AJ, de Vos AF, Kerver ME, Florquin S, van der Poll T (2011) The role of cd44 in the acute and resolution phase of the host response during pneumococcal pneumonia. Lab Invest 91:588–597
van Eeden SF, Hogg JC (2000) The response of human bone marrow to chronic cigarette smoking. Eur Respir J 15:915–921
van Eeden SF, Hogg JC (2002) Systemic inflammatory response induced by particulate matter air pollution: the importance of bone-marrow stimulation. J Toxicol Environ Health A 65:1597–1613
van Eeden SF, Kitagawa Y, Klut ME, Lawrence E, Hogg JC (1997) Polymorphonuclear leukocytes released from the bone marrow preferentially sequester in lung microvessels. Microcirculation 4:369–380
van Eeden SF, Lawrence E, Sato Y, Kitagawa Y, Hogg JC (2000) Neutrophils released from the bone marrow by granulocyte colony-stimulating factor sequester in lung microvessels but are slow to migrate. Eur Respir J 15:1079–1086
Van Ziffle JA, Lowell CA (2009) Neutrophil-specific deletion of syk kinase results in reduced host defense to bacterial infection. Blood 114:4871–4882
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714
Vethanayagam RR, Almyroudis NG, Grimm MJ, Lewandowski DC, Pham CT, Blackwell TS, Petraitiene R, Petraitis V, Walsh TJ, Urban CF, Segal BH (2011) Role of NADPH oxidase versus neutrophil proteases in antimicrobial host defense. PLoS One 6:e28149
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D,Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S (2012) Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 209:819–835
von Kockritz-Blickwede M, Nizet V (2009) Innate immunity turned inside-out: antimicrobial defense by phagocyte extracellular traps. J Mol Med (Berl) 87:775–783
von Vietinghoff S, Ley K (2008) Homeostatic regulation of blood neutrophil counts. J Immunol 181:5183–5188
Walker DC, Behzad AR, Chu F (1995) Neutrophil migration through preexisting holes in the basal laminae of alveolar capillaries and epithelium during streptococcal pneumonia. Microvasc Res 50:397–416
Walker MJ, Hollands A, Sanderson-Smith ML, Cole JN, Kirk JK, Henningham A, McArthur JD, Dinkla K, Aziz RK, Kansal RG, Simpson AJ, Buchanan JT, Chhatwal GS, Kotb M, Nizet V (2007) Dnase sda1 provides selection pressure for a switch to invasive group a streptococcal infection. Nat Med 13:981–985
Wang Q, Teder P, Judd NP, Noble PW, Doerschuk CM (2002) Cd44 deficiency leads to enhanced neutrophil migration and lung injury in escherichia coli pneumonia in mice. Am J Pathol 161:2219–2228
Wang HB, Wang JT, Zhang L, Geng ZH, Xu WL, Xu T, Huo Y, Zhu X, Plow EF, Chen M, Geng JG (2007) P-selectin primes leukocyte integrin activation during inflammation. Nat Immunol 8:882–892
Ward JR, Heath PR, Catto JW, Whyte MK, Milo M, Renshaw SA (2011) Regulation of neutrophil senescence by micrornas. PLoS One 6:e15810
Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, Henriques-Normark B (2007) Capsule and d-alanylated lipoteichoic acids protect streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol 9:1162–1171
Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE (2006) A novel peptide cxcr ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 12:317–323
Weinmann P, Scharffetter-Kochanek K, Forlow SB, Peters T, Walzog B (2003) A role for apoptosis in the control of neutrophil homeostasis in the circulation: insights from cd18-deficient mice. Blood 101:739–746
Weinrauch Y, Drujan D, Shapiro SD, Weiss J, Zychlinsky A (2002) Neutrophil elastase targets virulence factors of enterobacteria. Nature 417:91–94
Wengner AM, Pitchford SC, Furze RC, Rankin SM (2008) The coordinated action of g-csf and elr+cxc chemokines in neutrophil mobilization during acute inflammation. Blood 111:42–49
Wiggs BR, English D, Quinlan WM, Doyle NA, Hogg JC, Doerschuk CM (1994) Contributions of capillary pathway size and neutrophil deformability to neutrophil transit through rabbit lungs. J Appl Physiol 77:463–470
Witko-Sarsat V, Pederzoli-Ribeil M, Hirsch E, Sozzani S, Cassatella MA (2011) Regulating neutrophil apoptosis: new players enter the game. Trends Immunol 32:117–124
Woodfin A, Voisin MB, Nourshargh S (2010) Recent developments and complexities in neutrophil transmigration. Curr Opin Hematol 17:9–17
Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, Coggeshall KM, McEver RP (2010) E-selectin engages psgl-1 and cd44 through a common signaling pathway to induce integrin alphalbeta2-mediated slow leukocyte rolling. Blood 116:485–494
Yamada M, Gomez JC, Chugh PE, Lowell CA, Dinauer MC, Dittmer DP, Doerschuk CM (2011) Interferon-gamma production by neutrophils during bacterial pneumonia in mice. Am J Respir Crit Care Med 183:1391–1401
Yang D, Oppenheim JJ (2009) Alarmins and antimicrobial immunity. Med Mycol 47(suppl 1):S146–153
Yang FC, Atkinson SJ, Gu Y, Borneo JB, Roberts AW, Zheng Y, Pennington J, Williams DA (2001) Rac and cdc42 gtpases control hematopoietic stem cell shape, adhesion, migration, and mobilization. Proc Natl Acad Sci USA 98:5614–5618
Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ (2004) Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu Rev Immunol 22:181–215
Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW (2005) Icam-1 regulates neutrophil adhesion and transcellular migration of tnf-{alpha}-activated vascular endothelium under flow. Blood 106:584–592
Yoder MC, Checkley LL, Giger U, Hanson WL, Kirk KR, Capen RL, Wagner WW Jr (1990) Pulmonary microcirculatory kinetics of neutrophils deficient in leukocyte adhesion-promoting glycoproteins. J Appl Physiol 69:207–213
Yoshida K, Kondo R, Wang Q, Doerschuk CM (2006) Neutrophil cytoskeletal rearrangements during capillary sequestration in bacterial pneumonia in rats. Am J Respir Crit Care Med 174:689–698
Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR, Nichols DP, Taylor-Cousar JL, Saavedra MT, Randell SH, Vasil ML, Burns JL, Moskowitz SM, Nick JA (2011) Neutrophil extracellular trap (net)-mediated killing of pseudomonas aeruginosa: evidence of acquired resistance within the cf airway, independent of cftr. PLoS One 6:e23637
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU (2009) Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 16:1438–1444
Zarbock A, Singbartl K, Ley K (2006) Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest 116:3211–3219
Zarbock A, Lowell CA, Ley K (2007a) Spleen tyrosine kinase syk is necessary for e-selectin-induced alpha(l)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity 26:773–783
Zarbock A, Deem TL, Burcin TL, Ley K (2007b) Galphai2 is required for chemokine-induced neutrophil arrest. Blood 110:3773–3779
Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K (2008) Psgl-1 engagement by e-selectin signals through src kinase fgr and itam adapters dap12 and fcr gamma to induce slow leukocyte rolling. J Exp Med 205:2339–2347
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Gomez, J.C., Wang, Q., Doerschuk, C.M. (2013). Neutrophils in Acute Bacterial Pneumonia. In: Prince, A. (eds) Mucosal Immunology of Acute Bacterial Pneumonia. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5326-0_4
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5326-0_4
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5325-3
Online ISBN: 978-1-4614-5326-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)