
Abstract
The past decade has witnessed a dramatic increase in the therapeutic options for the treatment of multiple myeloma (MM) with the introduction of novel biologically targeted agents which in turn have resulted in significantly improved outcomes. However, myeloma remains incurable, and ongoing efforts to identify novel therapeutic approaches remain an urgent priority. Newer agents that target tumour and stromal compartments can be categorized as those that target protein dynamics (such as ubiquitin–proteasome system and heat-shock protein 90), drugs modulating anti-MM immune response (IMiDs), antibodies targeting membrane-bound receptors (including CS1, CD38, CD138), epigenetic modulators (DNA methyltransferase and histone deacetylase inhibitors), intracellular-signalling kinase inhibitors (PI3k/Akt/mTOR, MAPK pathways) and compounds disrupting the cell-cycle molecular machinery (such as CDKIs and Aurora kinase inhibitors). This chapter focuses on new therapeutic targets which have shown promising preclinical results and early evidence of anti-MM activity in clinical studies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Multiple Myeloma
- Myeloma Cell
- Refractory Multiple Myeloma
- Suberoylanilide Hydroxamic Acid
- Farnesyltransferase Inhibitor
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Multiple myeloma (MM) represents a paradigm in drug development with an improved understanding of the biology and derived clinical trials translating into six new US Food and Drug Administration (FDA)-approved treatments over the past 10 years. The proteasome inhibitor, bortezomib, and the immunomodulatory drugs, thalidomide and lenalidomide, have been the cornerstone of the improvement in outcomes during the last decade [1, 2]. However, almost all patients with MM relapse and the outcome of patients who progress after therapy with the immunomodulatory drugs and bortezomib remain dismal [3]. Novel biologically based therapeutic approaches that target not only the MM cell but also the interaction with other cells and cytokines in the bone-marrow milieu have the potential to overcome resistance to conventional agents and improve patient outcomes in MM, with next generation targets now emerging [4, 5]. Here we will review novel targets in MM used either alone or in combination strategies.
2 Drug Combinations of Novel Agents in Myeloma
The introduction of thalidomide, lenalidomide and bortezomib has led to important changes in the management of patients with MM. Bortezomib received accelerated FDA approval for the treatment of patients with relapsed and refractory multiple myeloma in 2003 [6]. Subsequently, bortezomib also received full approval for the treatment of patients with relapsed multiple myeloma and as initial therapy on the basis of favorable results from phase III trials [7, 8]. The immunomodulatory drugs thalidomide, lenalidomide and pomalidomide target myeloma cells in the bone-marrow microenvironment. Specifically, these agents trigger caspase-8-mediated apoptosis, decrease binding of tumour cells to bone-marrow stromal cells, inhibit secretion of cytokines from the bone marrow (through both constitutive secretion as well as secretion induced by the binding of myeloma cells), inhibit angiogenesis and stimulate immunity against myeloma cells mediated by autologous natural killer cells, T cells or both [9, 10].
In the upfront setting, thalidomide with dexamethasone (thal/dex) and bortezomib (Velcade) in combination with melphalan and prednisone (MPV) increased the overall response rate (RR) and significantly prolonged time to progression (TTP) and are FDA-approved for this indication, [8, 11] with overall RRs for thal/dex of 64% and 71% with MPV. In the relapsed setting, bortezomib alone [6, 7] and the combinations of lenalidomide/dexamethasone (len/dex) [12, 13] and bortezomib and liposomal doxorubicin (Vel/Doxil) have all been approved [14]. Importantly, results of a phase III randomized trial suggest that lower doses of dex (40 mg weekly for 4 weeks) in combination with len provide a survival advantage mainly due to the decreased toxicity associated with lower doses of dex [15].
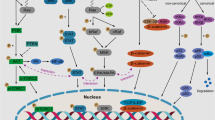
In order to improve upon current outcomes, optimal combinations of bortezomib, thal and len have been evaluated in phase II/III clinical trials, with the combination of lenalidomide–bortezomib–dexamethasone (RVD) showing particularly promising activity [16]. Preclinical data indicate synergistic cytotoxicity results from combining lenalidomide (which induces caspase-8-mediated apoptosis) with bortezomib (which induces predominantly caspase-9-mediated apoptosis) in in vitro models of myeloma (Fig. 9.1). Lenalidomide and bortezomib achieved 61% responses in patients with relapsed and refractory multiple myeloma and who were refractory to each agent alone [17]. In the setting of newly diagnosed disease, RVD produced an unprecedented overall RR of 100%, with 74% of patients achieving at least a very good partial response and 52% of patients showing complete or near-complete responses [16].

Rationale for combination therapies in multiple myeloma (adapted from Richardson et al. Br J Haematol 154(6):755–762)
3 Next Generation Novel Agents in Clinical Development
3.1 Monoclonal Antibodies
3.1.1 CS1-, CD38- and CD138-Targeting Antibodies
One of the major ongoing efforts is to identify MM cell-surface antigens and design-specific antibodies with cytotoxic properties. CS-1, CD38 and CD138 are multifunctional glycoproteins widely and highly expressed on MM cell surface. Elotuzumab (HuLuc63) is a CS1-targeting monoclonal antibody which triggers ADCC-mediated cell death in vitro and effectively reduces tumour growth in an in vivo MM model [18]. In relapsed and refractory MM patients, elotuzumab has a manageable toxicity profile, and stable disease was observed on a low-dose schedule with monotherapy [19]. Preliminary data indicate exciting results with the combination of elotuzumab with lenalidomide and dexamethasone [20], with efficacy evaluable patients, 22/26 (85%) achieving a confirmed or an unconfirmed response, including 31% VGPR/CR and the remaining 4/26 (15%) stable disease in one study.
In vitro, antibodies against CD38 induce antibody-dependent cell cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) against MM cells. There are ongoing clinical trials to further evaluate the CD38 antibodies with early results showing promise [21, 22].
Similarly, the maytansanoid toxin conjugated to an anti-CD138 monoclonal antibody has shown promising results in vitro, and xenograft models of human MM in mice have provided the framework for a clinical trial of this immunotoxin [23].
3.1.2 IL-6-Targeting Antibodies
Interleukin-6 (IL6) is an inflammatory cytokine that is both an autocrine and paracrine survival factor for malignant plasma cells. IL-6 is secreted by myeloma cells which also stimulate its production in the tumour niche by both bone-marrow stromal cells (BMSC) and osteoclasts (OC). In addition, IL-6 stimulates osteoclastogenesis [24]. CNTO328 is a novel human–mouse chimeric monoclonal antibody against IL6 currently undergoing clinical evaluation. CNTO328 enhances bortezomib-induced cytotoxicity on MM cells increasing the activation of the pro-apoptotic caspases 8, 9 and 3 [25], with stable disease and partial responses observed in MM patients treated with single-agent CNTO328, which in turn has led to combination studies of bortezomib-based therapy and CNTO328.
3.1.3 BAFF-Targeting Antibody
B-cell activating factor (BAFF) is a potent osteoclast (OC)-derived MM growth factor, and its inhibition reduces tumour burden as well as OCs and lytic lesions in in vivo models of myeloma bone disease [26]. Clinical trials of BAFF-neutralizing antibody in combination with bortezomib are currently ongoing to confirm the effects on bone lesions and tumour burden [NCT00689507].
3.1.4 Pomalidomide
CC-4047 (Pomalidomide) is a potent immunomodulatory analog (IMiDs), derived using the thalidomide backbone [27]. As mentioned above, IMiDs have multiple mechanisms of action beyond immunomodulation alone. Phase I clinical studies of pomalidomide in combination with low-dose dexamethasone showed activity in relapsed patients with MM who were resistant to other agents, including thalidomide, lenalidomide and bortezomib [28]. Pom/dex was found to be highly active and well tolerated including responses among patients who were lenalidomide and bortezomib refractory [29, 30]. No grade 3 neuropathy was seen, and thromboembolic events have been rare. Pom therefore appears to be a very promising agent in the therapy of MM and provides an alternative to patients who have received lenalidomide-, thalidomide- and bortezomib-based treatments (Table 9.1).
4 Modulators of Protein Homeostasis
Bortezomib, the first in class boronate peptide proteasome inhibitor, reversibly inhibits chymotrypsin-like activity of the 20S proteasome. Peripheral neuropathy, thrombocytopenia and gastrointestinal symptoms, although manageable, are important side effects. More potent inhibitors of chymotryptic activity, including carfilzomib and MLN 9708, have been noted to overcome bortezomib resistance in preclinical and early clinical trials. Carfilzomib, an irreversible proteasome inhibitor in the epoxyketone-category-induced partial response in approximately 23% of heavily pretreated relapsed and refractory MM patients, and importantly, the overall RR was noted to be 57% in a subset of bortezomib-naïve patients [31]. The toxicity profile was manageable, consisting mainly of myelosuppression and markedly reduced rates of neuropathy. Phase III clinical trials comparing carfilzomib–lenalidomide–dexamethasone with lenalidomide–dexamethasone in patients with relapsed multiple myeloma are now ongoing [32]. MLN 9708 is an oral proteasome inhibitor in the boronate peptide category [33] that has shown encouraging results in early phases I–II clinical trials both as a single agent and in combination with lenalidomide and dexamethasone [NCT00963820, NCT01383928]. ONX 0912, an oral epoxyketone proteasome inhibitor, is also now undergoing evaluation as a single agent in hematologic malignancies [NCT01416428] [34].
A broader and more potent proteasome inhibitor, NPI-0052 or marizomib, targets chymotryptic, tryptic and caspase-like activities and overcomes bortezomib resistance in preclinical studies and with early clinical trials confirming consistent activity in bortezomib-refractory patients [35–37]. Importantly, in preliminary results from a phase I study in patients with relapsed and refractory MM, NPI-0052 has not appeared to induce significant peripheral neuropathy or myelosuppression and was generally well tolerated and demonstrated unique safety profiles compared to bortezomib in spite of up to 100% proteasome inhibition [37].
Inhibitors of de-ubiquitinating enzymes located upstream of the proteasome, such as the USP-7 inhibitor P5091, have shown activity against multiple myeloma [38].
PR-924, an inhibitor of the LMP-7 immunoproteasome subunit, inhibits myeloma cells in vitro and in vivo. Owing to the selective expression of immunoproteasome subunits in malignant, but not in normal, haematological cells, inhibitors of the immunoproteasome should also have a favourable therapeutic index, and studies of these are awaited with interest [39].
In a similar context, NEDD8-activating-enzyme inhibitor MLN4924 targets the neddylation pathway upstream of the 20S proteasome, with downstream molecular sequelae which generates significant preclinical anti-myeloma activity that is distinct from that of established 20S proteasome inhibitors [40].
5 Histone Deacetylase Inhibitors
Histone deacetylase (HDAC) inhibitors are novel antineoplastic agents that correct the transcriptional deregulation of genes involved in the induction of apoptosis and cell-cycle arrest. They have multiple mechanisms of action, including mediating tumour cell death via caspase-dependent and non-caspase-dependent apoptosis as well as autophagy. They also block the aggresome complex which represents a protein–scavenger system that mediates protein degradation in the event of either proteasome overload or inhibition. Intriguingly, the high protein turnover characteristic of plasma cells and MM cells requires aggresome formation, and so the synergistic activity seen in combination with the proteasome inhibitor, bortezomib, is particularly promising. Specifically, HDAC inhibitors suppress proteasome activity, decrease expression of proteasome subunits and critically inhibit the aggresome. For example, inhibition of this pathway via tubacin, a specific HDAC6 inhibitor, synergizes with proteasome inhibition achieved with bortezomib. The HDAC6 inhibitors also have the potential of reduced toxicity, and the HDAC6-specific inhibitor, ACY 1215, is currently being studied in a phase I clinical trial. HDAC inhibitors have been shown to be effective anticancer agents in both in vitro and in vivo studies [41, 42].
Other HDACi which have been developed in the clinical setting include SAHA (suberoylanilide hydroxamic acid, vorinostat), LBH589 (panobinostat) and romidepsin with both vorinostat and romidepsin FDA-approved in cutaneous lymphomas. The multitude of effects of these compounds are complex, with the transcriptional signature of SAHA, for example, revealing downregulation of IGF-1R/AKT and IL6R/STAT3-signalling pathways, as well as DNA synthesis and repair enzymes [43].
The effects and toxicities of HDACi differ according to the specific compound, the formulation and schedule of administration. Intravenous doses of SAHA cause myelosuppression and thrombocytopenia, while with the oral formulation, fatigue, diarrhoea and dehydration are more common. Adverse effects of oral LBH589 consist of thrombocytopenia and neutropenia. HDACi have now been assessed also in combination strategies with novel anti-MM agents, including bortezomib and len with considerable promise shown with both panobinostat and bortezomib, vorinostat and lenalidomide and romidepsin and bortezomib [44–48].
The combination of HDACi and bortezomib in vivo not only effectively reduced tumour burden but also improved osteolytic lesions in a mouse model of bone disease [49].
6 HSP90 Inhibitors
Heat-shock protein 90 (HSP90) is a chaperone protein that regulates protein folding and translocation into the different cellular compartments. Studies demonstrate that bortezomib treatment of MM cells in vitro induces death signalling, downregulates survival signalling and upregulates both ubiquitin/proteasome and stress response gene transcripts. In vitro studies show that Hsp90 inhibitor 17AAG can block the Hsp90 stress response induced by bortezomib and thereby increase MM cell apoptosis. These studies therefore provided the framework for a clinical trial coupling of these agents in MM with favourable tolerability and encouraging responses seen in relapsed and refractory patients [50, 51]. As a result of production difficulties with 17 AAG, studies of this compound are no longer going forward. However, other HSP 90s are now under study.
7 PI3K/Akt Inhibitors
Cytokine-induced activation of Akt has been reported to induce growth and survival advantage to MM cells and mediate dex-resistance in MM cells in the context of the BM microenvironment [52]. Agents targeting PI3K/Akt network directly, in particular the pleiotropic Akt inhibitor perifosine, the PKC inhibitor enzastaurin and the mTOR inhibitors RAD001 and CCI-779, have been examined in MM preclinical models.
The novel oral Akt inhibitor perifosine (Keryx Biopharmaceuticals) triggers cytotoxicity against MM cells, both in vitro and in vivo [53, 54]. Molecular studies revealed that perifosine-induced inhibition of Akt phosphorylation and its downstream molecules (GSK)-3β and FKHRL1 was associated with c-jun NH2-terminal kinase activation. Perifosine treatment also triggered the formation of the death-inducing signalling complex as well as the recruitment of TRAIL-R1/DR4 and TRAIL-R2/DR5, resulting in potent apoptosis [55].
Preclinical data has also been reported on bortezomib-induced activation of Akt as a putative mechanism of resistance, which in turn has been completely blocked by perifosine, while bortezomib successfully abrogated perifosine-induced ERK phosphorylation [54]. This blockade of both Akt and ERK signalling cascades by perifosine and bortezomib enhances JNK phosphorylation, caspase/PARP cleavage and apoptosis. Results of a phases I–II trial with the combination of perifosine and bortezomib showed durable responses, even in the setting of bortezomib refractoriness. In 73 evaluable patients, an overall response rate (ORR; defined as minimal response or better) of 41% was demonstrated with this combination, including an ORR of 65% in patients who relapsed following bortezomib treatment and 32% in bortezomib-refractory patients. Median PFS was 6.4 months, with an encouraging median overall survival of 25 months (and 22.5 months in bortezomib-refractory patients) [56]. A phase III clinical trial of bortezomib versus bortezomib with perifosine in patients with relapsed multiple myeloma is ongoing [NCT01002248].
8 mTOR Inhibitors
PI3K/Akt/mTOR kinase cascade plays a critical role in cell proliferation, survival and development of drug resistance in MM [57]. Rapamycin is a universal inhibitor of mTORC1-dependent S6K1 phosphorylation [58, 59]. Rapamycin-induced cytotoxicity is predominantly triggered as a consequence of autophagy (programmed cell death type II) via excessive cell digestion. Therefore, activated Akt can be a key upstream inhibitor of two cell death-inducing events: autophagy via mTOR activation and apoptosis via phosphorylation of BAD and inhibition of the catalytic subunit of caspase-9. In vitro and in vivo preclinical studies have demonstrated anti-MM activity of rapamycin and its analogs (CCI-779 and RAD001) [59, 60].
However, resistance to rapamycin results from a strong positive feedback loop from mTOR/S6K1 to Akt with consequent Akt activation [61, 62]. This effect in some cancer types is due to rapamycin activity only on mTORC1 complex, whereas mTORC2, the one responsible for Akt activation, remains unaffected. Promising data reported on combined targeting of mitogen-activated protein kinase (MAPK) and PI3K/mTOR pathways by rapamycin with len [59] have been translated to clinical trials. In the phase I study of RAD001 with lenalidomide, stable disease or better was observed in 68% of patients (13/19–90%, CI: 30–76%) with grade 3/4 adverse events (5%) included thrombocytopenia (11%) and neutropenia (22%) [63]. In the phase 2 study of the combination of temsirolimus with bortezomib in heavily pretreated, advanced MM patients, the proportion of patients with a partial response or better was robust at 33% (14 of 43; 90% CI 21–47) [64].
There are ongoing and planned trials with dual inhibitors of mTORC1/2-INK 128 and AZD 8055 [65, 66] and the composite mTORC1/2 and PI3-kinase inhibitors NVP-BEZ235 [67].
9 Cyclin-Dependent Kinase Inhibitors
Dysregulated and/or increased expression of cyclin D1, D2 or D3 occurs as an early, unifying event in MM pathogenesis, predisposing MM cells to proliferative stimuli, and is frequently seen in relapsed patients with poor prognosis [68]. Specific inhibition of Cdk4/6 by PD 0332991, an orally bioavailable small-molecule Cdk inhibitor, has demonstrated only growth arrest in MM cells [69], suggesting that selective cyclin-dependent kinase (CDK) inhibition may not be sufficient in inducing MM cell death. Rather, effective MM cytotoxicity may be best achieved when multiple CDKs are inhibited concurrently, as demonstrated in preclinical studies with multi-targeted CDK inhibitors such as AT7519 [70]. Additionally, they target CDK complexes that phosphorylate RNA pol II resulting in inhibition of RNA pol II phosphorylation and transcriptional inhibition and also modulate expression/activity of multiple signalling pathways critical for MM cell proliferation and survival in the context of the bone-marrow microenvironment. AT7519, independent of its potent inhibitory effects on CDKs, effectively induces the dephosphorylation of glycogen synthase kinase (GSK)-3β [71], another important target in MM therapy. AT7519 is being evaluated in combination with bortezomib and dexamethasone in patients with relapsed and/or refractory MM [NCT01183949].
10 Aurora Kinase Inhibitors
The aurora kinases regulate cell-cycle transit from G2 through to cytokinesis. Myeloma is characterized by genetic instability and disruption of cell-cycle checkpoints which renders myeloma cells suspectible to induction of apoptotic death in mitosis. Aurora kinase inhibitors have been shown to inhibit the growth of MM cell lines and primary myeloma samples at nanomolar concentrations with minimal effect on proliferating lympocytes and hematopoietic cells [72–75]. Phase I/II studies of MLN8237, an aurora kinase inhibitor, are now ongoing in multiple myeloma [NCT01034553].
11 Telomerase Inhibitors (GRN163L)
Telomerase is a reverse transcriptase that protects chromosome endings and therefore expands cell lifespan. It is expressed at high levels in cancer cells, including MM, while almost no expression detected in normal somatic cells. Targeting telomerase via a novel inhibitor, GRN163L, results in MM cell death in vitro. In vivo studies demonstrated that GRN163L impaired tumour growth and enhanced animal survival [76]. There is a completed phase I study of the telomerase inhibitor GRN163L alone and in combination activity with bortezomib and dexamethasone in patients with relapsed or refractory MM and an ongoing phase II study of GRN 163 L (Imetelstat) currently under way [NCT00594126, NCT00718601].
12 Farnesyltransferase Inhibitors
Mutations of Ras are commonly encountered and are associated with disease progression and decreased survival [77]. Because Ras and other proteins require farnesylation, a lipid posttranslational modification, for malignant transformation activity, farnesyltransferase inhibitors (FTIs) were studied as potential anticancer drugs. In a phase II trial of patients with advanced MM, disease stabilization was achieved in 64% of patients treated with FTI5777 (Zarnestra) [78]. Preclinical evaluation of the combination of the specific FTI, tipifarnib, and bortezomib revealed synergistic anti-MM activity. This combination has been shown to enhance the ER-stress-induced apoptosis and overcome the CAM-DR phenotype, therefore delineating a treatment strategy that specifically targets microenvironment-mediated drug resistance [79]. Based upon these observations, a phase I trial combining escalating doses of tipifarnib (100–400 mg/BID) with bortezomib (1.0 mg/m2) in patients with relapsed MM was initiated, and encouraging preliminary data reported stabilization of disease or better seen among 7/16 patients with 2 of the 7 achieving an MR; no serious drug-related toxicities were noted, including the absence of cardiac events or DVT [80]. Future studies are anticipated with interest.
13 Conclusions and Future Directions
The availability of several classes of agents targeting biologically relevant pathways and proteins in MM remains remarkably exciting and productive. Patients with MM now have increasing therapeutic options with agents active alone and in combination. Future studies will focus on biologic risk stratification and optimizing drug combinations relevant to specific patient profiles, including adverse cytogenetics and extramedullary disease. Given that several of these agents have different toxicity profiles, the future holds promise in terms of novel drug combinations with improved efficacy and tolerability with rational combination strategies derived from both preclinical models and clinical experience, providing real hope for further improving patient outcome [81, 82].
References
Brenner H, Gondos A, Pulte D (2008) Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood 111:2521–2526
Kumar SK, Rajkumar SV, Dispenzieri A et al (2008) Improved survival in multiple myeloma and the impact of novel therapies. Blood 111:2516–2520
Kumar SK, Lee JH, Lahuerta JJ et al (2011) Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia 26(1):149–157
Anderson KC (2011) Oncogenomics to target myeloma in the bone marrow microenvironment. Clin Cancer Res 17:1225–1233
Palumbo A, Anderson K (2011) Multiple myeloma. N Engl J Med 364:1046–1060
Richardson PG, Barlogie B, Berenson J et al (2003) A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 348:2609–2617
Richardson PG, Sonneveld P, Schuster MW et al (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352:2487–2498
San Miguel JF, Schlag R, Khuageva NK et al (2008) Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 359:906–917
Hideshima T, Chauhan D, Shima Y et al (2000) Thalidomide and its analogs overcome drug resistance of human multiple myeloma cells to conventional therapy. Blood 96:2943–2950
Hideshima T, Richardson PG, Anderson KC (2006) Current therapeutic uses of lenalidomide in multiple myeloma. Expert Opin Investig Drugs 15:171–179
Rajkumar SV, Rosinol L, Hussein M et al (2008) Multicenter, randomized, double-blind, placebo-controlled study of thalidomide plus dexamethasone compared with dexamethasone as initial therapy for newly diagnosed multiple myeloma. J Clin Oncol 26:2171–2177
Dimopoulos M, Spencer A, Attal M et al (2007) Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med 357:2123–2132
Weber DM, Chen C, Niesvizky R et al (2007) Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med 357:2133–2142
Orlowski RZ, Nagler A, Sonneveld P et al (2007) Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol 25:3892–3901
Rajkumar SV, Jacobus S, Callander N et al (2007) A randomized trial of lenalidomide plus high-dose dexamethasone (RD) versus lenalidomide plus low-dose dexamethasone (Rd) in newly diagnosed multiple myeloma (E4A03): a trial coordinated by the Eastern Cooperative Oncology Group. ASH Annual Meeting Abstracts 110:74
Richardson PG, Weller E, Lonial S et al (2010) Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood 116:679–686
Richardson PG, Weller E, Jagannath S et al (2009) Multicenter, phase I, dose-escalation trial of lenalidomide plus bortezomib for relapsed and relapsed/refractory multiple myeloma. J Clin Oncol 27:5713–5719
Tai YT, Dillon M, Song W et al (2008) Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 112:1329–1337
Zonder JA, Mohrbacher AF, Singhal S et al (2011) A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 120(3):552–559
Richardson PG, Moreau P, Jakubowiak AJ et al (2010) Elotuzumab In combination with lenalidomide and dexamethasone in patients with relapsed multiple myeloma: interim results of a phase 2 study. ASH Annual Meeting Abstracts 116:986
de Weers M, Tai YT, van der Veer MS et al (2011) Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol 186:1840–1848
Gimsing P, Plesner T, Nahi H et al (2011) A phase I/II, dose-escalation study of daratumumab, A CD38 Mab in patients with multiple myeloma – preliminary safety data. ASH Annual Meeting Abstracts 118:1873
Ikeda H, Hideshima T, Fulciniti M et al (2009) The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin Cancer Res 15:4028–4037
Kurihara N, Bertolini D, Suda T, Akiyama Y, Roodman GD (1990) IL-6 stimulates osteoclast-like multinucleated cell formation in long term human marrow cultures by inducing IL-1 release. J Immunol 144:4226–4230
Voorhees PM, Chen Q, Kuhn DJ et al (2007) Inhibition of interleukin-6 signaling with CNTO 328 enhances the activity of bortezomib in preclinical models of multiple myeloma. Clin Cancer Res 13:6469–6478
Neri P, Kumar S, Fulciniti MT et al (2007) Neutralizing B-cell activating factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin Cancer Res 13:5903–5909
Bartlett JB, Dredge K, Dalgleish AG (2004) The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 4:314–322
Lacy MQ, Hayman SR, Gertz MA et al (2009) Pomalidomide (CC4047) plus low-dose dexamethasone as therapy for relapsed multiple myeloma. J Clin Oncol 27:5008–5014
Lacy MQ, Allred JB, Gertz MA et al (2011) Pomalidomide plus low-dose dexamethasone in myeloma refractory to both bortezomib and lenalidomide: comparison of 2 dosing strategies in dual-refractory disease. Blood 118:2970–2975
Richardson PG, Siegel DS, Vij R et al (2011) Randomized, open label phase 1/2 study of pomalidomide (POM) alone or in combination with low-dose dexamethasone (LoDex) in patients (Pts) with relapsed and refractory multiple myeloma who have received prior treatment that includes lenalidomide (LEN) and bortezomib (BORT): phase 2 results. ASH Annual Meeting Abstracts 118:634
Vij R, Wang M, Orlowski R et al (2008) Initial results of PX-171-004, an open-label, single-arm, phase II study of carfilzomib (CFZ) in patients with relapsed myeloma (MM). ASH Annual Meeting Abstracts 112:865
US National Library of Medicine (2011) ClinicalTrials.gov. [online], http://clinicaltrials.gov/ct2/show/NCT01080391
Chauhan D, Tian Z, Zhou B et al (2011) In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res 17:5311–5321
Chauhan D, Singh AV, Aujay M et al (2010) A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 116:4906–4915
Singh AV, Palladino MA, Lloyd GK, Potts BC, Chauhan D, Anderson KC (2010) Pharmacodynamic and efficacy studies of the novel proteasome inhibitor NPI-0052 (marizomib) in a human plasmacytoma xenograft murine model. Br J Haematol 149:550–559
Chauhan D, Singh AV, Ciccarelli B, Richardson PG, Palladino MA, Anderson KC (2010) Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood 115:834–845
Richardson PG, Spencer A, Cannell P et al (2011) Phase 1 clinical evaluation of twice-weekly marizomib (NPI-0052), a novel proteasome inhibitor, in patients with relapsed/refractory multiple myeloma (MM). ASH Annual Meeting Abstracts 118:302
Chauhan D, Tian Z, Nicholson B et al (2009) Deubiquitylating enzyme USP-7, a novel therapeutic target in multiple myeloma. ASH Annual Meeting Abstracts 114:610
Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ (2009) Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 113:4667–4676
McMillin DW, Hunter Z, Delmore J et al (2010) MLN4924, a novel investigational NEDD8 activating enzyme inhibitor, exhibits preclinical activity in multiple myeloma and Waldenstrom’s macroglobulinemia through mechanism distinct from existing proteasome inhibitors. ASH Annual Meeting Abstracts 116:2988
Hideshima T, Bradner JE, Wong J et al (2005) Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci USA 102:8567–8572
Santo L, Hideshima T, Kung AL et al (2010) Selective inhibition of HDAC6 with a new prototype inhibitor (ACY-1215) overcomes bortezomib resistance in multiple myeloma (MM). ASH Annual Meeting Abstracts 116:2997
Mitsiades CS, Mitsiades NS, McMullan CJ et al (2004) Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci USA 101:540–545
Wolf JL, Siegel D, Matous J et al (2008) A phase II study of oral panobinostat (LBH589) in adult patients with advanced refractory multiple myeloma. ASH Annual Meeting Abstracts 112:2774
Weber DM, Jagannath S, Sobecks R et al (2008) Combination of vorinostat plus bortezomib for the treatment of patients with multiple myeloma who have previously received bortezomib. ASH Annual Meeting Abstracts 112:3711
Richardson P, Weber D, Mitsiades CS et al (2010) A phase I study of vorinostat, lenalidomide, and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: excellent tolerability and promising activity in a heavily pretreated population. ASH Annual Meeting Abstracts 116:1951
Siegel DS, Jagannath S, Hajek R et al (2010) Vorinostat combined with bortezomib in patients with relapsed or relapsed and refractory multiple myeloma: update on the Vantage Study Program. ASH Annual Meeting Abstracts 116:1952
Harrison SJ, Quach H, Link E et al (2011) A high rate of durable responses with romidepsin, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma. Blood 118:6274–6283
Deleu S, Lemaire M, Arts J et al (2009) Bortezomib alone or in combination with the histone deacetylase inhibitor JNJ-26481585: effect on myeloma bone disease in the 5T2MM murine model of myeloma. Cancer Res 69:5307–5311
Richardson P, Chanan-Khan AA, Lonial S et al (2006) A multicenter phase 1 clinical trial of tanespimycin (KOS-953) + bortezomib (BZ): encouraging activity and manageable toxicity in heavily pre-treated patients with relapsed refractory multiple myeloma (MM). ASH Annual Meeting Abstracts 108:406
Richardson PG, Chanan-Khan AA, Lonial S et al (2011) Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: results of a phase 1/2 study. Br J Haematol 153:729–740
Tai YT, Podar K, Catley L et al (2003) Insulin-like growth factor-1 induces adhesion and migration in human multiple myeloma cells via activation of beta1-integrin and phosphatidylinositol 3’-kinase/AKT signaling. Cancer Res 63:5850–5858
Hideshima T, Catley L, Raje N et al (2007) Inhibition of Akt induces significant downregulation of survivin and cytotoxicity in human multiple myeloma cells. Br J Haematol 138:783–791
Hideshima T, Catley L, Yasui H et al (2006) Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood 107:4053–4062
Gajate C, Mollinedo F (2007) Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 109:711–719
Richardson PG, Wolf J, Jakubowiak A et al (2011) Perifosine plus bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma previously treated with bortezomib: results of a multicenter phase I/II trial. J Clin Oncol 29:4243–4249
Hideshima T, Chauhan D, Richardson P, Anderson KC (2005) Identification and validation of novel therapeutic targets for multiple myeloma. J Clin Oncol 23:6345–6350
Shi Y, Hsu JH, Hu L, Gera J, Lichtenstein A (2002) Signal pathways involved in activation of p70S6K and phosphorylation of 4E-BP1 following exposure of multiple myeloma tumor cells to interleukin-6. J Biol Chem 277:15712–15720
Raje N, Kumar S, Hideshima T et al (2004) Combination of the mTOR inhibitor rapamycin and CC-5013 has synergistic activity in multiple myeloma. Blood 104:4188–4193
Frost P, Moatamed F, Hoang B et al (2004) In vivo antitumor effects of the mTOR inhibitor CCI-779 against human multiple myeloma cells in a xenograft model. Blood 104:4181–4187
Wan X, Harkavy B, Shen N, Grohar P, Helman LJ (2007) Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 26:1932–1940
Sun SY, Rosenberg LM, Wang X et al (2005) Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 65: 7052–7058
Mahindra A, Richardson PG, Hari P et al (2010) Updated results of a phase I study of RAD001 in combination with lenalidomide in patients with relapsed or refractory multiple myeloma with pharmacodynamic and pharmacokinetic analysis. ASH Annual Meeting Abstracts 116:3051
Ghobrial IM, Weller E, Vij R et al (2011) Weekly bortezomib in combination with temsirolimus in relapsed or relapsed and refractory multiple myeloma: a multicentre, phase 1/2, open-label, dose-escalation study. Lancet Oncol 12:263–272
US National Library of Medicine. http://clinicaltrials.gov/ct2/show/NCT01118689 [Dose Escalation Study of INK128 in Relapsed or Refractory Multiple Myeloma or Waldenstrom Macroglobulinemia].
Cirstea D, Hideshima T, Santo L et al (2010) Disruption of DEPTOR/mTORC1/mTORC2 signaling cascade using a novel selective mtor kinase inhibitor azd8055 results in growth arrest and apoptosis in multiple myeloma cells. ASH Annual Meeting Abstracts 116:791
McMillin DW, Ooi M, Delmore J et al (2009) Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer Res 69:5835–5842
Bergsagel PL, Kuehl WM (2005) Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol 23:6333–6338
Baughn LB, Di Liberto M, Wu K et al (2006) A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res 66:7661–7667
Santo L, Vallet S, Hideshima T et al (2010) AT7519, A novel small molecule multi-cyclin-dependent kinase inhibitor, induces apoptosis in multiple myeloma via GSK-3beta activation and RNA polymerase II inhibition. Oncogene 29:2325–2336
Santo L, Vallet S, Hideshima T et al (2008) AT7519, a novel small molecule multi-cyclin dependent kinase inhibitor, induces apoptosis in multiple myeloma VIA GSK3{beta}. ASH Annual Meeting Abstracts 112:251
Dutta-Simmons J, Zhang Y, Gorgun G et al (2009) Aurora kinase A is a target of Wnt/beta-catenin involved in multiple myeloma disease progression. Blood 114:2699–2708
Evans RP, Naber C, Steffler T et al (2008) The selective Aurora B kinase inhibitor AZD1152 is a potential new treatment for multiple myeloma. Br J Haematol 140:295–302
Hose D, Reme T, Meissner T et al (2009) Inhibition of aurora kinases for tailored risk-adapted treatment of multiple myeloma. Blood 113:4331–4340
Negri JM, McMillin DW, Delmore J et al (2009) In vitro anti-myeloma activity of the Aurora kinase inhibitor VE-465. Br J Haematol 147(5):672–676
Shammas MA, Koley H, Bertheau RC et al (2008) Telomerase inhibitor GRN163L inhibits myeloma cell growth in vitro and in vivo. Leukemia 22:1410–1418
Bezieau S, Devilder MC, Avet-Loiseau H et al (2001) High incidence of N and K-Ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis. Hum Mutat 18:212–224
Alsina M, Fonseca R, Wilson EF et al (2004) Farnesyltransferase inhibitor tipifarnib is well tolerated, induces stabilization of disease, and inhibits farnesylation and oncogenic/tumor survival pathways in patients with advanced multiple myeloma. Blood 103:3271–3277
Yanamandra N, Colaco NM, Parquet NA et al (2006) Tipifarnib and bortezomib are synergistic and overcome cell adhesion-mediated drug resistance in multiple myeloma and acute myeloid leukemia. Clin Cancer Res 12:591–599
Lonial S, Francis D, Karanes C et al (2008) A phase I MMRC clinical trial testing the combination of bortezomib and tipifarnib in relapsed/refractory multiple myeloma. ASH Annual Meeting Abstracts 112:3706
Richardson PG, Mitsiades C, Schlossman R, Munshi N, Anderson K (2007) New drugs for myeloma. Oncologist 12:664–689
Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K (2012) Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol 9:135–143
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Mahindra, A., Laubach, J., Mitsiades, C., Richardson, P. (2013). Novel Agents in Multiple Myeloma. In: Munshi, N., Anderson, K. (eds) Advances in Biology and Therapy of Multiple Myeloma. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5260-7_9
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5260-7_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5259-1
Online ISBN: 978-1-4614-5260-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)