
Abstract
An important challenge for TB investigators in the postgenomic era is to integrate distinct functional strategies to study the molecular mechanism of Mycobacterium tuberculosis (Mtb) virulence. However, the biological function of the majority of Mtb genes is unknown. This has revealed the need for an approach to convert raw genome sequence data into functional information. In the past decade, the yeast two-hybrid system (Y2H) has contributed significantly towards studying TB virulence and persistence, but has several drawbacks. Recently, several mycobacterial protein–protein interaction (PPI) technologies have been reported that helped propose functions for unknown proteins through “guilt by association” and will be discussed in this chapter. We will examine the advantages, disadvantages and limitations of these systems and how these technologies can be used to dissect signaling, drug resistance, and virulence pathways. We will also discuss how mycobacterial PPI technologies can be exploited to force proteins to interact and for the discovery of small-molecule inhibitors against protein complexes. In sum, by characterizing Mtb PPIs on a genomic scale, it will be possible to assemble physiologically relevant protein pathways in mycobacteria, the outcome of which will be invaluable for determining virulence mechanisms and the function of previously uncharacterized proteins.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Mycobacterium tuberculosis is an extremely successfully pathogen due to its ability to persist, and to latently infect more than one-third of the world’s population [1, 2]. Annually, there are approximately eight million new cases of TB and two million deaths worldwide. The increase in multidrug-resistant (MDR), extensively drug-resistant (XDR) and super XDR Mycobacterium tuberculosis (Mtb) strains, together with the synergy with HIV infection is a frightening development [2, 3] and poses significant problems in the treatment and control of TB.
Genome-scale molecular networks such as protein interaction and gene regulatory pathways are taking a center stage in the emerging disciplines of systems biology and biocomplexity. As a result, an important challenge for TB investigators in the postgenomic era is to integrate functional strategies such as allelic replacement techniques [3–5], signature tagging mutagenesis [6, 7], in vivo expression technology [8, 9], proteomics, [10, 11], DNA microarrays [12–16], deep-genome sequence strategies [17, 18] and protein–protein interaction (PPI) approaches [19, 20], to study the molecular mechanism of Mtb virulence.
To fulfill their biological function in cells, most proteins function in association with protein partners or as large molecular assemblies. Not surprisingly, virulence pathways are also mediated by molecular connections that require PPIs. The rationale for studying PPI in bacterial pathogens such as Mtb is several fold. First, in dissecting these pathways, it has been established that physical association between a protein of unknown function and a known protein suggests that the former often has a function related to that of the latter. This “guilty by association” principle has led to the functional annotations of numerous proteins of unknown function. Since over the past decade more than 1,000 microbial genomes have been sequenced it is anticipated that the focus on genes of unknown function will continue to increase, as it is these genes (of unknown function), which make the particular microbe unique. Second, an important feature of PPI networks is that most proteins associate with multiple interacting partners, suggesting that they fulfill multiple functions. Third, elucidation of PPI can rapidly provide detailed mechanistic information about a specific biological question. The above-mentioned approaches usually attempt to identify new drug targets, or to achieve a better understanding of the mechanistic basis of Mtb virulence.
While substantial efforts focused on prediction of protein–protein association by in silico analysis using phylogenetic profiles [21], domain fusion [22], and gene clustering methods [23, 24], these types of analyses must be supported by biological experimentation. Not surprisingly, due to the large number of PPIs studies over the past 20 years, a large number of protein interaction databases such as HPRD, DRP, MIPS, STRING, BIND [25] have been generated.
Mtb is a genetically intractable microbe and there is an urgent need to develop effective genome-wide tools to study protein–protein association in mycobacterial cells. By characterizing PPIs on a genomic scale it will be possible to assemble physiologically relevant protein pathways in mycobacteria, the outcome of which will be invaluable for determining the function of previously uncharacterized proteins and virulence mechanisms.
Thus far, despite the development of bacterial systems (BacterioMatch [BM] and bacterial adenylate cyclase two-hybrid [BACTH] and the mammalian two-hybrid system [M2H]), Saccharomyces cerevisiae is the most exploited surrogate host and represents the current standard. The first large-scale yeast two-hybrid (Y2H) interaction network was performed with the Escherichia coli bacteriophage T7 [26] and was rapidly followed by a whole-genome analysis of S. cerevisiae [27–30], Drosophila [31, 32], Arabidopsis [33], and C. elegans [34]. These studies predicted the function of a multitude of proteins and revealed numerous novel interactions, thereby allowing investigators to link biological functions together into larger cellular processes.
In this review, we will provide an overview about the different PPI techniques that have successfully been exploited to study Mtb. We will discuss different mycobacterial PPI technologies, how it could be exploited for the discovery of new antimycobacterial drugs, potential pitfalls of PPI technologies, and in silico methods for predicting PPI.
2 Microbial PPI Systems
2.1 The Y2H System
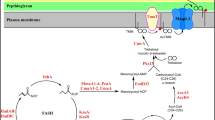
In the original Y2H assay, a bait protein is fused to the GAL4 DNA-binding domain (DNA-BD), and a library of prey proteins are expressed as fusions to the GAL4 activation domain (AD) [35] (Fig. 5.1a). When the “bait” protein interacts with a “prey” protein from the library, the DNA-BD and AD are brought into proximity to activate transcription of several reporter genes (e.g., ADE2, HIS3, MEL1, and AUR1). The Y2H system is an effective tool use to identify novel protein interactions, confirm putative interactions, and define interacting domains and residues. Subsequent to the development of the original Y2H system, the reverse Y2H [36] and yeast three-hybrid (Y3H) [37] systems were developed.

Conceptual basis of PPI methods used to study protein function in mycobacteria and other pathogens. In the (a) Y2H, (b) BM, (c) BACTH, and (d) M-PFC systems the two interacting proteins (bait [B] and prey [P]) are independently fused to either a DNA-AD (e.g., Gal4-AD, α-subunit of RNAP) and DNA-BD (e.g., Gal4-BD, λcI) (a, b) or to two enzymatic subunits (c, d) that reconstitute enzymatic activity (e.g., AC or hDHFR). However, in the case of RAP-inducible M-PFC (e), rapamycin functions as a bridge that forces FKBP12 and FRB to “interact,” thereby functionally reconstituting the reporter system consisting of F-[1,2] and F-[3] to generate TrimR mycobacterial clones. Note that in case of e, F-[1,2] and F-[3] can be replaced by any two proteins that the investigator wishes to force to interact. Although the above systems examine bimolecular protein interactions, (a), (b), and (d) have been modified to examine tri-molecular protein interactions (see text for detail). UAS upstream activating sequence, cAMP-CAP prom cAMP-CAP promoter, λcI oper λcI operator, T18 and T25 adenylate cyclase enzymatic domains
2.2 The E. coli BacterioMatch System
Similar to the Y2H system, the BM two-hybrid system is designed to examine PPIs between a pair of proteins cloned into separate “bait” and “prey” vectors. The bait protein is fused to the full-length bacteriophage λ repressor protein (λcI) containing the amino terminal DNA binding domain and the carboxyl terminal dimerization domain (Fig. 5.1b). When produced inside cell, the bait fusion is tethered to the operator sequence upstream of the reporter promoter through the DNA-BD of λcI [38]. The target or prey protein is fused to the N-terminal domain of the α-subunit of RNA polymerase (RNAP). When the bait and prey proteins associate, they recruit and stabilize binding of RNAP at the promoter and activate transcription of the HIS3 and aadA (confers streptomycin resistance) reporter genes [39].
Very recently, the BM system was modified to study ternary mycobacterial protein complexes in E. coli [40]. Using this three-hybrid system, it was demonstrated that the interaction between CFP-10 and Rv3871 was strengthened in the presence of Esat-6. Lastly, the BM system was also used to examine PPIs between Mtb proteins and approximately 8,000 novel interactions were discovered [41]. Notably, validation of PPI using overexpression and surface plasmon resonance analyses demonstrated a success rate of approximately 60 %. Important findings include demonstrating a link between the Mtb ESX1 and ESX5 protein secretion systems, and that the Fe–S cluster proteins WhiB3 and WhiB7 are highly connected [41].
2.3 The Bacterial Adenylate Cyclase Two-Hybrid System
In the BACTH system, proteins of interest are fused with two fragments of the catalytic domain of the Bordetella pertussis adenylate cyclase (AC) and co-expressed in an E. coli ∆cya strain (Fig. 5.1c). Interaction of the two proteins results in the functional complementation between the two AC subunits, leading to cAMP synthesis and subsequent activation of catabolic operons [42] or the expression of the lacZ gene. Using BACTH, it was demonstrated that Msm PsPpm2 interacts with MsPpm1 to stabilize the synthase MsPpm1 in the bacterial membrane [43]. The BACTH system was also successfully used to examine the interactions between Mtb ClpX and FtsZ [44].
2.4 Protein Fragment Complementation
Recently, a different experimental system, coined protein fragment complementation (PFC), was shown to be highly effective in studying PPIs in a variety of organisms [45–48]. In PCF, a particular reporter enzyme is rationally dissected into two fragments and fused with two interacting proteins. Interaction among the two proteins results in active refolding and reconstitution of the enzyme activity of the two fragments. Since nuclear translocation of interacting proteins is not required, membrane proteins can also be analyzed.
For example, using human dihydrofolate reductase (hDHFR), any two proteins (X and Y) thought to interact are fused to two rationally dissected DHFR fragments called F-[1,2] and F-[3]. In vivo reassembly due to the interacting proteins X and Y, and subsequent reconstitution of hDHFR domains X-F-[1,2] and Y-F-[3] into active hDHFR can be monitored in vivo by cell survival under methotrexate selection, by fluorescence detection of fluorescein-conjugated methotrexate binding to reconstituted hDHFR, or by trimethoprim (Trim) resistance (Fig. 5.1d). hDHFR is a small 21-kDa monomeric protein that contains three structural fragments (F-1, F-2, and F-3) containing two domains; an adenine-binding domain (F-2) and a discontinuous domain (F-[1] and F-[3]). Previously, it has been shown that disruption of the disordered loop at the junction between F-[2] and F-[3] has no significant effect on activity [49]. This property was exploited to develop a eukaryotic DHFR PFC system to analyze reassembly of murine dihydrofolate reductase (hDHFR) fragments [47, 50, 51]. Using eukaryotic DHFR PFC, 148 combinations of 35 different PPIs in the RTK/FRAP signal transduction pathway were studied with no false-positive interactions observed among the pairs tested [47]. Importantly, the DHFR PFC system (albeit eukaryotic) is the only system that could validate the interactions through pharmacological perturbation of the interactions—even if the site of action of the perturbant is distant from the interaction studied [47].
The concept of PFC using hDHFR fragments was successfully exploited to develop a mycobacterial PFC system termed mycobacterial PFC (M-PFC) [52] (Fig. 5.1d). Using this system, the interactions between the two-component proteins DevS and DevR, and KdpD and KdpE were demonstrated. In addition, several previously undiscovered proteins were shown to interact with Mtb Cfp-10. Notably, proteins complexes were identified that form only in mycobacteria and not in the Y2H system [52]. It is likely that many interacting Mtb proteins will require the mycobacterial cytoplasmic environment to associate and is an important consideration in a PPI screen. In an independent study, M-PFC identified a strong interaction between Pup and the proteasome substrate FabD (malonyl coenzyme A acyl carrier protein), whereas this interaction was not detected using E. coli as surrogate host [53]. M-PFC was successful in demonstrating interaction between an essential DNA-binding protein (IdeR) and the enzymatic complexes (LeuC/LeuD) [54]. Lastly, M-PFC was also used to demonstrate interaction between Mtb ClpX and FtsZ [44].
The split-Trp system is another PFC assay that monitors the enzymatic reconstitution of tryptophan biosynthesis in a tryptophan autotrophic microbe. This system was originally developed in S. cerevisiae [55] and shown to be effective for examining the Mtb protein complexes Esat-6/CFP-10, RegX3 homodimerization, self-association of Rv3782 (galactosyl transferase), and the coiled-coil peptides C1 and C2 [56].
3 Shared Properties of Microbial Interaction Systems
The bacterial and Y2H systems (or variations thereof) have several properties in common that can profoundly affect postscreen analyses. For example, all systems have relatively strong promoters (Y2H: ADH1 p or GAL10; M2H: CMV; BM: lac-UV5; BACT: lac-UV5 and M-PFC: hsp60), and all PPI systems are based upon fusion technologies (Y2H; GAL4 AD and BD or LexA; M2H; JAK and GP130; BM; α-subunit of RNAP and λcI; BACT; AC; and M-PFC; DHFR). Lastly, all systems rely on unique peptide detection tags (HA, cMyC, FLAG, His, GP120, etc., or the reporter domains itself) to enable specific detection.
The two most widely used protein interaction validation approaches are to fuse a detection tag (e.g., GST) to the “bait” protein, in vitro transcribe/translate the “prey” protein followed by incubation of the mixtures and assessment of binding/elution of the labeled prey protein. A second widely-used validation experiment includes in vivo co-affinity purification in which one protein is tagged, overexpressed in E. coli (or native host) followed by a pull-down of the prey from the extract.
Important differences between the BM and PFC systems are that: (1) in PFC, protein interaction does not need to take place near the transcription machinery, (2) PFC is better suited for studying interactions among membrane proteins, (3) PFC requires no other host-specific processes or enzymes, (4) the structure of DHFR is known thereby allowing control over the way interactions can occur, and (5) it is advantageous to employ PFC in the native host rather than surrogate hosts such as yeast or E. coli wherein protein interactions are determined in the native host where they function in the context of other native proteins.
4 Is Yeast the Optimal Host for Studying Mycobacterial PPIs?
As is described in the section below, the Y2H system has been used successfully to study Mtb biology and pathogenesis. While in some cases it might be beneficial to use yeast as surrogate host, the Y2H system does have certain limitations. For example, (1) protein interactions occur in the nucleus, (2) membrane proteins are not fully compatible with the conventional Y2H system, (3) bacterial proteins do not undergo appropriate post-translational modification, (4) self-activation of bait proteins can occur, and finally, (5) high G+C DNA is sometimes not well tolerated in the Y2H system. A well-known class of Y2H false positives is “anti-sense” clones that contain anti-sense DNA fragments cloned in the library vector that when translated produce a nonphysiological peptide that associates with the bait protein. False positives are inherently present in all large-scale Y2H screens and are extensively documented in the literature.
5 Specificity and False Positives of PPI Technologies
In large-scale PPI studies, technical and biological false positives are typically being considered. Technical false positives resulting in experimental errors can be avoided. However, in order to eliminate false positives (e.g., those interacting clones that are genuinely observed in more than one assay, but do not occur in vivo) and to increase the verification rate, the following factors are taken in consideration during large-scale protein interaction screens: (1) overlapping (interacting) clones increases the confidence score [28, 31, 57], (2) literature curated interactions increases the confidence score [31, 57], (3) membrane proteins are underrepresented and negatively affect the confidence score [31], (4) post-translational modifications (e.g., phosphorylation) may be required for many interactions, (5) verification with other independent techniques increase the confidence score [27], (6) “masking” of bait or prey proteins and “self-activation” affect the screens [28, 31], (7) logistic regression models increase the probability of interactions [31], and (8) most studies validate interactions using detection tags or reporter fusions for “pull-down assays” [57].
6 How Can PPI Technologies Help Us Understand Mtb Virulence?
PPI technologies are flexible approaches that typically allow investigators to address previously unanswered questions. This is important to the mycobacteriology field as Mtb is a genetically intractable microbe for which few novel tools to determine virulence mechanisms are available. A widely cited rationale for exploiting PPI technologies in microbes is to ascribe function to genes of unknown function (e.g., those genes that are unique to the organism). It can be speculated that these genes distinguish the particular species from all other species and play a unique role in the biology of the microbe. Other areas in which PPI technologies can play an timportant role include the identification and dissection of virulence pathways, linking virulence pathways with each other, and examining the components of signaling cascades and drug resistance pathways. Particularly relevant to the study of Mtb is the effect of in vivo environmental conditions implicated in Mtb persistence (e.g., temperature, pH, NO, superoxide, etc.) on protein–protein association. Other important areas include the effect of post-translational modifications on Mtb PPI, screening for drugs that disrupt PPI, and construction of a complete protein linkage map of the Mtb proteome.
7 Impact of the Y2H System on Mtb Research
Over the past decade, PPI technologies have filled an important void in the mycobacterial field and opened up new avenues of TB research. The original discovery of Mtb WhiB3 in 2002 using the Y2H system [19] is a particularly good example for how PPI technologies can advance a particular research area.
7.1 Mtb WhiB3
It was previously established that a single-point mutation in the 4.2 region of the principal σ-factor rpoV causes loss of virulence in Mycobacterium bovis (Mbov), a member of the Mtb complex [58]. This mutation, known to result in an Arg515-His change, was originally suggested to influence recognition of the –35 promoter region that abolished or altered expression of a gene or subsets of genes essential for virulence. However, it was hypothesized that this mutation might alter the interaction of RpoV with a transcription factor responsible for regulating the expression of one or more genes involved in virulence. An abundance of data have shown that mutations in, or close to the helix-turn-helix motif in region 4.2 of bacterial δ70-type sigma factors results in either positive or negative effects on activation by transcription factors. Subsequently, it was hypothesized that the 4.2 domain of Mtb SigA interacts with a regulatory protein that controls a subset of genes involved in virulence. To screen for proteins that interact exclusively with the 4.2 region of SigA in which the Arg515-His mutation is localized, a sigA DNA fragment (spanning region 4.2) was screened against a Mtb library using the Y2H system. Several clones contained in-frame fusions with the full-length open reading frame of Mtb whiB3 (Rv3416) [19]. Since it was initially hypothesized that the Arg515-His mutation abolished or reduced interaction of an unknown transcription factor with the 4.2 region of SigA, it was shown that SigAR515-H does not interact with WhiB3, suggesting that the single Arg515-His mutation abolishes the interaction of WhiB3 with SigA. Knock-out studies have shown that the Mtb whiB3 mutant behaved identically to the wild-type strain with respect to its ability to replicate in mice, but was attenuated in terms of host survival. In addition, the whiB3 mutant strain showed much reduced lung pathology, compared to wild type infected mice [19]. Intriguingly, a whiB3 mutant of virulent Mbov was completely impaired for growth in guinea pigs. These mutants define a new class (“path”; pathology) of virulence genes in Mtb and Mbov. It is notable that this virulence gene would not have been detected using conventional screens such as signature-tagged mutagenesis, which screen primarily for mutants defective in growth and not virulence. Notably, these findings led to the identification of WhiB3 as a 4Fe–4S cluster protein that reacts with NO and O2 [59], and is implicated in the metabolic switchover from using glucose as carbon source to fatty acids. Mtb WhiB3 was also shown to regulate virulence lipid production, function as an intracellular redox sensor [60] and prevent the bacillus from experiencing reductive stress during infection of macrophages [61] (for a recent review on Mtb WhiB3, see [62]). The above findings illustrate the power of PPI technologies to study virulence mechanisms in a genetically intractable pathogen.
7.2 Secretion
Mtb Esat-6 and Cfp-10 are important secreted antigens that are part of the ESX-1 secretion system, which delivers virulence proteins during infection of host cells [63]. These small proteins interact strongly with each other as well as several other Mtb proteins. In recent years, the Y2H system has been particularly effective in mapping Mtb ESX PPIs [64], and identifying and characterizing the individual components of the ESX-1 secretion system [64–68], which has led to new testable hypotheses. A substantial advance in our understanding of Mtb protein secretion was the discovery of a C-terminal signal sequence in Cfp-10 using the Y2H system. This C-terminal signal sequence was shown to be necessary for targeting Cfp-10 and Esat-6 for secretion. Besides, the C-terminal seven amino acid signal sequence was sufficient for targeting unrelated proteins such as ubiquitin for secretion [65].
7.3 Mtb Two-Component Signaling Proteins and Sigma Factors
Two-component signal transduction pathways are typically comprised of a membrane bound histidine kinase and its cognate cytoplasmic response regulator. In response to a signal, auto-phosphorylation occurs at a conserved residue of the histidine kinase and subsequently the phosphate group is transferred to the conserved aspartate residue of the response regulator. Even though these interactions are likely transient the Y2H system was effective in examining these interactions. Interactions among different domains of Mtb HK1 (Rv0600c), HK2 (Rv0601c), and TcrA (Rv0602c) were examined using the Y2H system [69]. It was found that HK2, but not HK1 or TcrA self-interacted, and that HK2 interacted with HK1 and TcrA. Lastly, the conserved receiver domain of TcrA was shown to interact with HK2, but not HK1 [69]. In another study the Y2H system was used to identify proteins that interact with the sensing domain of the Mtb histidine kinase, KdpD. Two membrane lipoproteins, LprJ and LprF, were identified that specifically associated with KdpD [20].
Mtb contains 13 sigma factors that can associate with one or more components of RNAP (RpoB, RpoB′, α-subunit) under distinct environmental conditions. In addition, anti-anti-sigma factors can interact with anti-sigma factors (e.g., RsbW) or sigma factors. In extensive Y2H studies, it was shown that most anti-sigma factor antagonists interact with either RsbW or SigF or both [70]. In a separate study, it was shown that SigK positively regulates expression of the antigenic proteins MBP70 and MBP83 [71]. High-level expression of sigK was associated with a mutated Rv0444c, and Y2H analysis demonstrated that the N-terminal region of Rv0444c interacted with SigK. The authors concluded that Rv0444c functions as a regulator of SigK (RskA) that modulates MPT70/MPT83 expression [71]. As described earlier the principle sigma factor, SigA was used in Y2H screen to identify the virulence factor WhiB3 [19].
7.4 DNA Repair
The Y2H system has been effectively exploited to study DNA damage and repair in Mtb [72–75]. For example, in a genome wide screen UvrD1 was identified as a novel interacting partner of Ku, suggesting potential cross-talk between components of nonhomologous end-joining and nucleotide excision repair pathways [74]. In another study that examined the role of Mtb DinB homologs in DNA damage, Y2H analyses showed that DinB1, but not DinB2 interacts with the mycobacterial β clamp, which is consistent with its C-terminal DNA-binding motif [73]. In a related study Y2H analysis showed that ImuB interact with ImuA′ and DnaE2 as well as with the β clamp [75].
7.5 Other
The Y2H system has also been used to identify and characterize interacting partners of Mtb WhiB1, an iron–sulfur cluster protein [76], the Mtb SUF machinery [77, 78], components of FASII (KasA, KasB, mtFabH, InhA, and MabA) [79, 80], the ABC transporter Rv1747 [81], resuscitation promoting factors (Rpfs) [82], a GTP binding protein (Obg) [83], and VapBC toxin-antitoxin modules [84].
8 Protein–Protein Interaction in Other Pathogens
PPI networks of bacteria have not yet reached the same comprehensive level as their yeast counterpart. An exception is the protein network of the human gastric pathogen Helicobacter pylori [85]. A high-throughput Y2H systems was used to screen 261 H. pylori proteins against a highly complex library of genome-encoded polypeptides and yielded over 1,200 interactions; connecting 46.6 % of the proteome [85]. The success of this approach in detecting new protein interactions and assignment of previously un-annotated proteins to new pathways lead to many such studies using the Y2H system to develop PPI maps for Plasmodium falciparum [86], Rickettsia sibirica [87], Bacillus anthracis, Francisella tularensis, Yersinia pestis [88], Campylobacter jejuni [89], Treponema pallidum [90] and viruses including HIV and HCV [91]. Unfortunately, these high-throughput screens are plagued by many drawbacks including false positives and negatives, and the temporal or spatial requirement of expression and post-translational modifications. Consequently, high-throughput PPI approaches have been augmented by the addition of techniques such as protein arrays and mass spectrometry [92–94].
While evaluating intra-bacterial PPIs provides a unique resource to identify essential cell processes and protein targets for drug screens against pathogenic bacteria, assessing interactions between host and bacterial proteins are imperative for understanding the mechanism of disease pathogenesis. Using a high-throughput Y2H screen, extensive host–pathogen PPIs have been identified for the pathogens B. anthracis, F. tularensis, and Y. pestis. Though the three pathogens cause different diseases (anthrax, lethal acute pneumonic disease and bubonic plague, respectively), PPIs pointed to similar mechanisms of immune modulation. For example, both B. anthracis and Y. pestis proteins interact with host major histocompatibility complex proteins, whereas TGF-β1 was shown to interact with Y. pestis and F. tularensis proteins. In sum, a network of 3,073 human-B. anthracis, 1,383 human-F. tularensis, and 4,059 human-Y. pestis PPIs were identified. The networks included 304 uncharacterized proteins from B. anthracis, 52 from F. tularensis, and 330 from Y. pestis [88].
Using three datasets that include physical interaction assays, genome-wide RNA interference (RNAi) screens, and microarray assays, the first draft of the mosquito PPI network was developed for the Dengue virus (DENv) carrier. This PPI network included 4,214 Aedes aegypti proteins with 10,209 interactions [95]. The study identified 714 putative DENv-associated mosquito proteins, and RNAi-mediated gene silencing of some of the highly interconnected proteins reduced the dengue viral titer in mosquito midgets. This observation further underscores the importance of identifying critical host–pathogen PPIs, which can provide an immense resource for identifying prospective antimicrobial drug targets.
In an attempt to characterize essential cellular process in Bacillus subtilis, a PPI network was generated that comprised 793 interactions that connected 287 proteins. Further evaluation of these hubs provided insights into distinct subgroups of PPI corresponding to protein networks or regulatory pathways differentially expressed under diverse conditions [96]. These PPI network data are a valuable resource for the functional annotation of genes of unknown function and integration of cellular pathways.
In addition to the Y2H system, high-throughput pull-down strategies combined with quantitative proteomics have also been used to decipher interacting circuits in methicillin-resistant Staphylococcus aureus [97]. Several highly connected hub proteins were identified. Notably, examination of the PPI network of S. aureus drug targets indicated that most of the clinical or experimental drugs targets lie at the periphery of the interacting circuit with few interacting partners. In contrast, the proteins that lie at the network hub, which could logically serve as a better target, were overlooked as drug targets [97].
9 Considerations for Mycobacterial PPIs
9.1 Some Mycobacterial Proteins Interact Exclusively in Their Native Environment
Although many bacterial PPIs have been identified in the Y2H system, it is logical to expect that some bacterial protein interactions may require the native cytoplasmic or membrane environment. For example, using M-PFC some Mtb proteins were shown to only interact in mycobacterial cytoplasmic environment, but not in yeast [52]. In an independent study, detection of interactions between Pup and other Mtb proteasome components in E. coli was unsuccessful. However, using M-PFC and therefore Msm as host, a strong interaction was observed between Pup and the proteasome substrate FabD [53].
9.2 Some Mycobacterial Proteins Require More Than One Protein for Interaction
Since all in vivo PPI methods (with the exception of the Y3H system) are binary systems that can detect interaction between only two proteins, interacting partners that require the presence of two or more proteins might be missed. In a recent study that made use of a modified M-PFC system, the Mtb ESX secretory system was examined by using a single fusion protein comprised of EsxB and EsxA as bait [98]. Three novel prey proteins, Rv3869, Rv3884 and Rv3885 were identified, whereas the single bait protein EsxB was unable to interact with any of these three proteins [98]. Exploiting fusion proteins that naturally associated in mycobacteria as bait has broad implications for the characterization of Mtb protein complexes, and may open new avenues of research.
The Y3H system was also exploited to delineate the molecular interactions between two membrane proteins and the Mtb two-component sensor kinases KdpD [20]. In this system, a third protein acts as a bridge between two proteins and can stabilize, enhance, or prevent interaction between proteins. The third protein is under the control of the inducible methionine promoter that is positively regulated in media lacking methionine. In this study LprJ and LprF were shown to modulate the interaction between N-KdpD and C-KdpD, and it was speculated that it is this ternary protein complex that modulates the KdpE-specific phosphatase activity of KdpD to regulate the expression of the KdpFABC system [20].
9.3 Post-translational Modification Can Affect PPI in the Y2H System
In 2003, a Y2H assay was developed to examine nitric oxide (NO)-dependent PPI [99]. Deleting yeast hemoglobin, which consumes NO very efficiently, was essential to the success of this approach. In this study, the authors screened a library of proteins that interact with procaspase-3 only in the presence of NO and identified four clones, iNOS, ASM, IRG and PGM [99]. These findings suggest that S-nitrosylation regulates PPI and may profoundly influence cellular signaling.
In another study, in vitro proteomic analysis identified numerous thioredoxin (TRX) targets. However, in vivo approaches failed to identify the expected number of TRX targets [100]. This problem was solved by constructing a specific yeast strain that contains deletions of genes encoding cytosolic TRX1 and TRX2. Subsequently, numerous TRX interacting partners were identified, whereas the same interactions could not be detected in the classic Y2H strain [100]. The above findings are highly relevant for studying mycobacterial PPIs, and illustrate the fundamental concepts that (1) proteins only interact when functionally required, (2) essential genes can be studied since genetic knockouts are not required, and (3) genes that are transcriptionally switched off can be studied since constitutive promoters are being used in the PPI systems.
10 Molecules That Dissociate or Force Protein–Protein Interaction
Despite recent successes [101–103], no new effective anti-tuberculosis drugs have been developed in the past 40 years. As a result, a high priority of the Global Alliance for TB Drug Development is the generation of new drugs with activity against dormant bacilli as well as the discovery of agents which could shorten or simplify the treatment of active TB. TB can be cured with existing drugs; however, the 6–9 months of treatment lead to patient noncompliance, which enhances drug resistance. Approximately 50 million people are already infected with MDR-TB [2]. While drug-sensitive TB can be cured with isoniazid, rifampin, ethambutol and pyrazinamide following a 6-month regimen, treatment of MDR-TB can exceed 2 years, thus dramatically increasing costs.
How can PPI in pathogenic microbes contribute to the discovery of new drugs? Tightly regulated PPIs are required for cellular functions in all living systems. The necessity for proper protein placement within enzymatic and receptor–ligand complexes, cell signaling pathways, and PPIs lend to the appeal of disruption of critical PPIs as therapeutic intervention. However, it should be noted that PPIs that participate in virulence or persistence pathways may only be induced in vivo and therefore, not be susceptible to drugs in in vitro screens and must be identified through other means. PPIs in particular share complimentary interfaces and “hot spots” with one another [104, 105], in which the primary forces that drive two proteins to interact are: van der Waal’s forces, electrostatic interactions, hydrogen bonds, and hydrophobic interactions [106–110]. Successful “disruption” of these interactions by an inhibitor, while not necessarily always in the context of protein–protein separation would be considered as any compound which modulates a protein interacting complex to achieve a desired therapeutic outcome and/or downstream effect.
Among several well-known inhibitors that modulate PPI, common mechanisms of action have emerged: prevention of PPI via protein binding, allosteric inhibition and, forced dissociation and association. More importantly, their method of action differs from drugs that prevent substrates from binding to active sites on enzyme complexes, as these sites are often marked with clear, defined pockets [111]. PPI inhibitors can include peptides, drugs, and small molecule compounds. These PPI inhibitors exert their functions over a range of target protein complexes in different cell types and have been reviewed over recent years [112–115].
Many inhibitors that prevent protein interaction have been shown to bind with amino acid residues that comprise “hot spots” at the protein–protein interface. Inhibitors form a complex with a protein at the binding site to structurally alter or prevent natural association of the cognate partner protein. For example, structural biology studies revealed that nutlins, a series of cis-imidazoline analogs identified via high-throughput screening, act by binding to three dominant residues of the p53 binding site on MDM2 and display in vitro and in vivo antitumor activity [116, 117]. Virstatin is an example of a compound that targets the dimerization domain of ToxT, a homodimer that regulates the production of cholera toxin and toxin co-regulated pilus in Vibrio cholera. Bacterial two-hybrid assays with ToxT truncation mutants demonstrated that virstatin specifically targets the N-terminal dimerization domain of ToxT [118, 119].
PPI can also be modulated when compounds bind distally to the protein interaction interface, that cause structural changes that prevent PPI without competition for protein binding sites. Such allosteric modifications have been documented for compounds that inhibit iNOS dimerization [120, 121]. PPA250, BBS-2, and clotrimazole are compounds that bind to the heme cofactor in the protein active site, which subsequently distort the α-helices [120] or the 8b and 9b β-strands [121] to prevent iNOS dimerization. In addition, other examples of allosteric inhibitors have been demonstrated for CBFβ-RUNX1, LFA-1-ICAM-1, and β-lactamase [122–124].
Inhibitors that dissociate preexisting protein complexes are functionally different from those that prevent protein dimerization. The most notable example of this method is TNF-α, whose active complex is maintained as a homotrimer when bound to its receptor. He et al. [125] demonstrated that at low TNF-α concentrations, the compound SPD00000034 bound to the pre-associated TNF-α trimer and promoted the dissociation of the active complex into dimer and monomer subunits. Similarly, previous studies have shown that the GroEL multimeric complex can exist in an “open” state, which allows 4,4′-dithiodipyridene to bind to an otherwise inaccessible Cys458, leading to GroEL subunit disassembly [126]. More recently, a proof-of-concept quantitative HTS screen was developed to screen for small-molecule inhibitors of Mtb PPI [54], which demonstrated the versatility of M-PFC.
Finally, several compounds modulate PPI by inducing the formation of previously unassociated complexes or by stabilizing protein complexes. Chemical inducers exist for the p66 and p51 subunits of HIV-1 reverse transcriptase [127]. However, the most well-known example of forced protein association comes from studies involving a physical relationship between immunophilins, their ligands, and their target [128]. FK506, rapamycin, and cyclosporine, are examples of hydrophobic, immunosuppressive ligands that contain two protein binding surfaces which mediate interactions between FKBP12 or cyclophilin [128, 129] and their corresponding target protein. In mammalian cells, the FKBP12–FK506 complex binds to and inhibits calcineurin phosphatase activity [130]. The FKBP12–rapamycin complex binds to the rapamycin binding domain (FRB) of FRAP [131]. The resulting complexes affect different immune responses and can lead to programmable physiological responses. Furthermore, these binding partners have led many researchers to exploit forcible ligand binding of effector molecules for the development of inducible PFC assays (PCA) [132–135]. In mycobacteria, a rapamycin inducible mycobacterial-PFC (RAP-inducible M-PFC) assay was developed as proof-of-concept to show forced interaction in bacterial cells, where FKBP12 and FRB were independently fused to the DHFR reporter fragments F-[1,2] and F-[3], respectively. Association of FKBP12 and FRB could only be detected in the presence of the selective drug trimethoprim and nanomolar concentrations of the rapamycin ligand [54]. Taken together, the M-PFC and the RAP-inducible M-PFC systems are powerful methods used to identify interacting proteins in protein networks, where in future studies, vehicles like FKBP12-ligand binding can be designed and utilized to manipulate PPIs in mycobacteria (Fig. 5.1e). In short, the ability of these effector molecules to bridge or induce dimeric and multimeric complexes paves the way for potential applications in controlling protein pathways for therapeutic and experimental studies [129, 136, 137].
11 In Silico Methods for Predicting PPI
Over the past few decades, knowledge of PPI has been generated primarily from biochemical and genetic experimentation approaches such as Y2H systems, pull-down assays, mass spectrometry, co-related mRNA expression, and protein arrays. However, despite the best attempts to collect experimental data on different organisms, the rate of discovery remains slow (e.g., approximately <10 % of interactions in humans have been experimentally characterized). With the advent of the genomic era, several computational and bioinformatics-based approaches have been developed to infer PPI. These in silico approaches exploit annotated information from established observations and use the structural, genomic, and biological context of proteins and genes from completely sequenced genomes to predict protein interaction networks [138, 139]. These in silico methods may rely on information gleaned from protein structure, gene sequence and the presence or absence of genes across numerous genomes, conserved gene neighborhoods across different species, co-expression of genes in transcriptome studies, involvement of proteins in a common metabolic pathway, curation of published literature or a combination of these datasets [140–145]. High-resolution three-dimensional (3D) structures of interacting proteins provide the best source of information with atomic description of the binding interfaces based on hydrophobicity, charge, and thermodynamic constraints of the interaction [140, 146]. Several approaches have been developed that include computational modeling of homologous proteins based on previously known structures, or domain or sequence signature analysis if the complete structure of a homologous protein is not available [147, 148]. For example, Inter PreTS (EMBL Heidelberg) is a popular resource, which for any pair of query sequences first searches for homologues in a database of interacting domains of known 3D complex structures [149]. Pairs of sequences homologous to a known interacting pair are scored for how well they preserve the atomic contacts at the interaction interface and a priority ranking is used to score for possible interacting partners.
A number of structure-based computational methods have been developed for the prediction of PPIs, which utilize advances in the field of genomics. One such popular approach, known as phylogenetic profiling [150], is based on the pattern of the presence or absence of a given gene in a given set of genomes. This method could, for example, ascertain the distribution of a specific gene in different species [151]. Any similarity of phylogenetic profiles might then be interpreted as being indicative of the functional need for corresponding proteins to be present simultaneously to perform a given function together. This approach stems from the idea that functionally linked proteins would co-occur in genomes and that the phylogenetic trees for known interacting protein families tend to show a higher degree of similarity than trees for noninteracting proteins. In several cases, the similarity in topology of phylogenetic trees has been considered as a positive indication towards establishing the likelihood of interacting proteins pairs, especially in the case of protein partners that may have co-evolved (mirror tree approach) [150]. Likewise, co-localization-based approaches are based on the notion that physically interacting (or functionally associated) proteins must co-evolve to preserve their ability to interact with one another [152]. This is especially relevant in the case of prokaryotes, which have operonic transcription units.
Genomic context-based approaches also exploit gene fusion events, which can be considered as the ultimate form of co-localization as the fusion of two independent genes to encode a single unrelated polypeptide (called a Rosetta stone protein) retains the physical proximity of the two peptides, but also makes them a single entity [142]. Publically available databases that provide support for gene context and co-localization analyses include FUSION DB, STRING and PHYDBAC.
Another robust tool implementing genome context-based analysis is based on the Integrated Microbial Genomes (IMG) database. The IMG provides one of the largest genome integrations, containing ∼7,000 complete and draft genomes across all three domains of life [153]. Similarly, an in silico two-hybrid method has been proposed, based on the study of correlated mutations in multiple sequence alignments. In this method, pairs of multiple sequence alignments with a distinctive co-variation signal are analyzed based on the hypothesis that co-adaptation of interacting proteins can be detected by the presence of a distinctive number of compensatory mutations in the corresponding proteins of different species [154].
Similar to wet lab-based approaches, most computational approaches have intrinsic limitations [155]. For example, the success of most of sequence and genomic context-based approaches requires extensive analysis of completely sequenced genomes, whereas the success of phylogenetic tree-based methods depends on the number and distribution of genomes used to build the tree [156]. Similarly, gene fusion-based methods may be confounded by errors caused by the occurrence of lateral gene transfer events in prokaryotes and the longer multi-gene architecture of eukaryotes [157]. Likewise, despite providing the highest quality information on PPI, protein structure-based approaches are restricted in their scope because of the limited availability of high quality protein structures in the databases and the high cost associated with determination of protein structures.
There is a clear need to unify genome sequencing and functional genomics data using computational tools to minimize the discrepancies associated with the use of a single approach. Several worthy attempts have been made in this direction. In addition, there is an encouraging community-driven initiative in the form of guidelines such as “MIMIx” and “MIAPE, which are the minimum information required for reporting a molecular interaction experiment” or a proteomics experiment, respectively [158, 159]. Under this initiative, a checklist of information has been provided, which every scientist must furnish when describing experimental molecular interaction data in an article, displaying data on a website or depositing data directly into a public database.
12 Integrative Physiology: The Emergence of Systems Biology?
Proteins are the catalytic effectors that carry out the intent of the microbial cell, but protein levels do not necessarily correlate with gene expression. For example, a lack of correlation was found between mRNA level and the corresponding protein level in Haemophilus influenza exposed to antibiotics [160], increased cell density in E. coli cultures [161], Bacillus subtilis exposed to peroxide stress [162], exponentially growing S. cerevisiae cells [163], and S. cerevisiae exposure to lithium [164]. This demonstrates the challenges of correlating mRNA expression levels with protein levels, and highlights the role of post-transcriptional regulatory control. Furthermore, some studies have observed a disparity between gene expression profiles and metabolic flux. This was elegantly demonstrated by analysis of the transcriptome, metabolome, and fluxome of Corynebacterium glutamicum [165]. Integrating PPI data with complementary high-throughput techniques such as transcriptomes, proteomics, metabolomics, and fluxomics represent unique opportunities to study and predict Mtb protein function through systems biology (Fig. 5.2).

Integrated analyses methodology depicting the role of PPI in TB systems biology. Towards this end, regulatory networks (gene expression arrays), proteomics, fluxomics and PPI networks have already begun to be established, but are commonly represented as static set of nodes to represent the components of the network (mRNA, proteins, metabolites, etc.). The ultimate goal will be to develop, test, and validate mathematical models that represent cellular components and their interactions to eventually predict cellular function. TAP tandem affinity purification, IP immunoprecipitation, SPR surface plasmon resonance
13 Conclusions
Tuberculosis research is primarily driven by the quest for a better understanding of how Mtb causes disease. The past decade, the Y2H system and mycobacterial PPI technologies gave mechanistic insight into distinct aspects of Mtb virulence, pathogenesis and have stimulated antimycobacterial drug discovery efforts. PPI studies are particularly powerful to provide information about the function of genes with unknown function through “guilt by association.” Not surprisingly, it is anticipated that the integration of functional data from PPI networks with the emerging discipline of systems biology could prove particularly useful to provide a better understanding of Mtb persistence. Although mycobacterial PPI networks have already begun to be established, the current focus is still on high-throughput PPI tool development, which is still lacking for mycobacteria. In addition, despite the generation of a single Mtb PPI map using E. coli as surrogate host, the more important stage of data interpretation, validation and integration with mycobacterial physiology is lacking. A future challenge would be to interconnect increasing amounts of mycobacterial PPI data with the PPI networks of other bacterial pathogens and its integration with other genome-wide databases, which should lead to new testable hypotheses. The generation of high-throughput global datasets will be an expensive venture that requires detailed knowledge about mycobacterial physiology, metabolism, pathogenesis, and computer modeling, which will contribute to a goal understanding of Mtb pathogenesis.
Abbreviations
- BACTH:
-
Bacterial adenylate cyclase two-hybrid
- BM:
-
BacterioMatch
- hDHFR:
-
Human dihydrofolate reductase
- Mbov :
-
Mycobacterium bovis
- M-PFC:
-
Mycobacterial protein fragment complementation
- Msm :
-
Mycobacterium smegmatis
- Mtb :
-
Mycobacterium tuberculosis
- NO:
-
Nitric oxide
- PFC:
-
Protein fragment complementation
- PPI:
-
Protein–protein interaction
- RNAi:
-
RNA interference
- RNAP:
-
RNA polymerase
- TRX:
-
Thioredoxin
- Y2H:
-
Yeast two-hybrid system
- Y3H:
-
Yeast three-hybrid system
References
Arno PS, Murray CJ, Bonuck KA, Alcabes P (1993) The economic impact of tuberculosis in hospitals in New York City: a preliminary analysis. J Law Med Ethics 21(3–4):317–323
Murray CJ, Salomon JA (1998) Modeling the impact of global tuberculosis control strategies. Proc Natl Acad Sci USA 95(23):13881–13886
Pelicic V, Jackson M, Reyrat JM, Jacobs WR Jr, Gicquel B, Guilhot C (1997) Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 94(20):10955–10960
Bardarov S, Kriakov J, Carriere C, Yu S, Vaamonde C, McAdam RA, Bloom BR, Hatfull GF, Jacobs WR Jr (1997) Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc Natl Acad Sci USA 94(20):10961–10966
Bardarov S, Bardarov S Jr, Pavelka MS Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR Jr (2002) Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148(Pt 10):3007–3017
Cox JS, Chen B, McNeil M, Jacobs WR Jr (1999) Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402(6757):79–83
Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C (1999) Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol 34(2):257–267
Dubnau E, Fontan P, Manganelli R, Soares-Appel S, Smith I (2002) Mycobacterium tuberculosis genes induced during infection of human macrophages. Infect Immun 70(6):2787–2795
Dubnau E, Chan J, Mohan VP, Smith I (2005) Responses of Mycobacterium tuberculosis to growth in the mouse lung. Infect Immun 73(6):3754–3757
Mollenkopf HJ, Grode L, Mattow J, Stein M, Mann P, Knapp B, Ulmer J, Kaufmann SH (2004) Application of mycobacterial proteomics to vaccine design: improved protection by Mycobacterium bovis BCG prime-Rv3407 DNA boost vaccination against tuberculosis. Infect Immun 72(11):6471–6479
Mattow J, Schaible UE, Schmidt F, Hagens K, Siejak F, Brestrich G, Haeselbarth G, Muller EC, Jungblut PR, Kaufmann SH (2003) Comparative proteome analysis of culture supernatant proteins from virulent Mycobacterium tuberculosis H37Rv and attenuated M. bovis BCG Copenhagen. Electrophoresis 24(19–20):3405–3420
Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK (2003) Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med 198(5):693–704
Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK (2001) Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha-crystallin. Proc Natl Acad Sci USA 98(13):7534–7539
Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik GK (2003) Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med 198(5):705–713
Voskuil MI, Visconti KC, Schoolnik GK (2004) Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis (Edinb) 84(3–4):218–227
Sassetti CM, Boyd DH, Rubin EJ (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48(1):77–84
Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, Mohaideen N, Ioerger TR, Sacchettini JC, Lipsitch M (2011) Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet 43(5):482–486
Ioerger TR, Koo S, No EG, Chen X, Larsen MH, Jacobs WR Jr, Pillay M, Sturm AW, Sacchettini JC (2009) Genome analysis of multi-and extensively-drug-resistant tuberculosis from KwaZulu-Natal, South Africa. PLoS One 4(11):e7778
Steyn AJ, Collins DM, Hondalus MK, Jacobs WR Jr, Kawakami RP, Bloom BR (2002) Mycobacterium tuberculosis WhiB3 interacts with RpoV to affect host survival but is dispensable for in vivo growth. Proc Natl Acad Sci USA 99(5):3147–3152
Steyn AJ, Joseph J, Bloom BR (2003) Interaction of the sensor module of Mycobacterium tuberculosis H37Rv KdpD with members of the Lpr family. Mol Microbiol 47(4):1075–1089
Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO (1999) Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA 96(8):4285–4288
Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D (1999) A combined algorithm for genome-wide prediction of protein function. Nature 402(6757):83–86
Overbeek R, Fonstein M, D’Souza M, Pusch GD, Maltsev N (1999) The use of gene clusters to infer functional coupling. Proc Natl Acad Sci USA 96(6):2896–2901
Dandekar T, Snel B, Huynen M, Bork P (1998) Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci 23(9):324–328
De Las RJ, De Luis A (2004) Interactome data and databases: different types of protein interaction. Comp Funct Genomics 5(2):173–178
Bartel PL, Roecklein JA, SenGupta D, Fields S (1996) A protein linkage map of Escherichia coli bacteriophage T7. Nat Genet 12(1):72–77
Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y (2001) A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci USA 98(8):4569–4574
Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwin B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403(6770):623–627
Schwikowski B, Uetz P, Fields S (2000) A network of protein-protein interactions in yeast. Nat Biotechnol 18(12):1257–1261
Ito T, Tashiro K, Muta S, Ozawa R, Chiba T, Nishizawa M, Yamamoto K, Kuhara S, Sakaki Y (2000) Toward a protein-protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc Natl Acad Sci USA 97(3):1143–1147
Formstecher E, Aresta S, Collura V, Hamburger A, Meil A, Trehin A, Reverdy C, Betin V, Maire S, Brun C, Jacq B, Arpin M, Bellaiche Y, Bellusci S, Benaroch P, Bornens M, Chanet R, Chavrier P, Delattre O, Doye V, Fehon R, Faye G, Galli T, Girault JA, Goud B, de Gunzburg J, Johannes L, Junier MP, Mirouse V, Mukherjee A, Papadopoulo D, Perez F, Plessis A, Rosse C, Saule S, Stoppa-Lyonnet D, Vincent A, White M, Legrain P, Wojcik J, Camonis J, Daviet L (2005) Protein interaction mapping: a Drosophila case study. Genome Res 15(3):376–384
Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, Vijayadamodar G, Pochart P, Machineni H, Welsh M, Kong Y, Zerhusen B, Malcolm R, Varrone Z, Collis A, Minto M, Burgess S, McDaniel L, Stimpson E, Spriggs F, Williams J, Neurath K, Ioime N, Agee M, Voss E, Furtak K, Renzulli R, Aanensen N, Carrolla S, Bickelhaupt E, Lazovatsky Y, DaSilva A, Zhong J, Stanyon CA, Finley RL Jr, White KP, Braverman M, Jarvie T, Gold S, Leach M, Knight J, Shimkets RA, McKenna MP, Chant J, Rothberg JM (2003) A protein interaction map of Drosophila melanogaster. Science 302(5651):1727–1736
de Folter S, Immink RG, Kieffer M, Parenicova L, Henz SR, Weigel D, Busscher M, Kooiker M, Colombo L, Kater MM, Davies B, Angenent GC (2005) Comprehensive interaction map of the Arabidopsis MADS box transcription factors. Plant Cell 17(5):1424–1433
Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, Goldberg DS, Li N, Martinez M, Rual JF, Lamesch P, Xu L, Tewari M, Wong SL, Zhang LV, Berriz GF, Jacotot L, Vaglio P, Reboul J, Hirozane-Kishikawa T, Li Q, Gabel HW, Elewa A, Baumgartner B, Rose DJ, Yu H, Bosak S, Sequerra R, Fraser A, Mango SE, Saxton WM, Strome S, Van Den Heuvel S, Piano F, Vandenhaute J, Sardet C, Gerstein M, Doucette-Stamm L, Gunsalus KC, Harper JW, Cusick ME, Roth FP, Hill DE, Vidal M (2004) A map of the interactome network of the metazoan C. elegans. Science 303(5657):540–543
Fields S, Song O (1989) A novel genetic system to detect protein protein interactions. Nature 340(6230):245–246
Vidal M, Brachmann RK, Fattaey A, Harlow E, Boeke JD (1996) Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc Natl Acad Sci 93(19):10315
SenGupta DJ, Zhang B, Kraemer B, Pochart P, Fields S, Wickens M (1996) A three-hybrid system to detect RNA-protein interactions in vivo. Proc Natl Acad Sci 93(16):8496
Dove SL, Joung JK, Hochschild A (1997) Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature 386(6625):627–630
Dove SL, Hochschild A (2004) A bacterial two-hybrid system based on transcription activation. Methods Mol Biol 261:231–246
Tharad M, Samuchiwal SK, Bhalla K, Ghosh A, Kumar K, Kumar S, Ranganathan A (2011) A three-hybrid system to probe in vivo protein-protein interactions: application to the essential proteins of the RD1 complex of M. tuberculosis. PLoS One 6(11):e27503
Wang Y, Cui T, Zhang C, Yang M, Huang Y, Li W, Zhang L, Gao C, He Y, Li Y (2010) A global protein-protein interaction network in the human pathogen Mycobacterium tuberculosis H37Rv. J Proteome Res 9(12):6665–6677
Karimova G, Dautin N, Ladant D (2005) Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol 187(7):2233–2243
Baulard AR, Gurcha SS, Engohang-Ndong J, Gouffi K, Locht C, Besra GS (2003) In vivo interaction between the polyprenol phosphate mannose synthase Ppm1 and the integral membrane protein Ppm2 from Mycobacterium smegmatis revealed by a bacterial two-hybrid system. J Biol Chem 278(4):2242
Dziedzic R, Kiran M, Plocinski P, Ziolkiewicz M, Brzostek A, Moomey M, Vadrevu IS, Dziadek J, Madiraju M, Rajagopalan M (2010) Mycobacterium tuberculosis ClpX interacts with FtsZ and interferes with FtsZ assembly. PLoS One 5(7):e11058
Galarneau A, Primeau M, Trudeau LE, Michnick SW (2002) Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat Biotechnol 20(6):619–622
Nyfeler B, Michnick SW, Hauri HP (2005) Capturing protein interactions in the secretory pathway of living cells. Proc Natl Acad Sci USA 102(18):6350–6355
Remy I, Michnick SW (2001) Visualization of biochemical networks in living cells. Proc Natl Acad Sci USA 98(14):7678–7683
Subramaniam R, Desveaux D, Spickler C, Michnick SW, Brisson N (2001) Direct visualization of protein interactions in plant cells. Nat Biotechnol 19(8):769–772
Gegg CV, Bowers KE, Matthews CR (1997) Probing minimal independent folding units in dihydrofolate reductase by molecular dissection. Protein Sci 6(9):1885–1892
Remy I, Michnick SW (1999) Clonal selection and in vivo quantitation of protein interactions with protein-fragment complementation assays. Proc Natl Acad Sci USA 96(10):5394–5399
Remy I, Galarneau A, Michnick SW (2002) Detection and visualization of protein interactions with protein fragment complementation assays. Methods Mol Biol 185:447–459
Singh A, Mai D, Kumar A, Steyn AJ (2006) Dissecting virulence pathways of Mycobacterium tuberculosis through protein-protein association. Proc Natl Acad Sci USA 103(30):11346–11351
Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH (2008) Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322(5904):1104
Mai D, Jones J, Rodgers JW, Hartman JL, Kutsch O, Steyn AJC (2011) A screen to identify small molecule inhibitors of protein-protein interactions in Mycobacteria. Assay Drug Dev Technol 9(3):299–310
Tafelmeyer P, Johnsson N, Johnsson K (2004) Transforming a ([beta]/[alpha]) 8-barrel enzyme into a split-protein sensor through directed evolution. Chem Biol 11(5):681–689
O’Hare H, Juillerat A, Dianiskova P, Johnsson K (2008) A split-protein sensor for studying protein-protein interaction in mycobacteria. J Microbiol Methods 73(2):79–84
Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437(7062):1173–1178
Collins DM, Kawakami RP, de Lisle GW, Pascopella L, Bloom BR, Jacobs WR Jr (1995) Mutation of the principal sigma factor causes loss of virulence in a strain of the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA 92(17):8036–8040
Singh A, Guidry L, Narasimhulu KV, Mai D, Trombley J, Redding KE, Giles GI, Lancaster JR, Steyn AJC (2007) Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc Natl Acad Sci 104(28):11562
Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, Steyn AJC (2009) Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog 5(8):e1000545
Farhana A, Guidry L, Srivastava A, Singh A, Hondalus MK, Steyn AJC (2010) Reductive stress in microbes: implications for understanding Mycobacterium tuberculosis disease and persistence. Adv Microb Physiol 57:43–117
Saini V, Farhana A, Steyn AJ (2012) Mycobacterium tuberculosis WhiB3: a novel iron-sulfur cluster protein that regulates redox homeostasis and virulence. Antioxid Redox Signal 16(7):687–697
Abdallah AM, van Pittius NCG, Champion PADG, Cox J, Luirink J, Vandenbroucke-Grauls CMJE, Appelmelk BJ, Bitter W (2007) Type VII secretion: mycobacteria show the way. Nat Rev Microbiol 5(11):883–891
Teutschbein J, Schumann G, Möllmann U, Grabley S, Cole ST, Munder T (2009) A protein linkage map of the ESAT-6 secretion system 1 (ESX-1) of Mycobacterium tuberculosis. Microbiol Res 164(3):253–259
Champion PADG, Stanley SA, Champion MM, Brown EJ, Cox JS (2006) C-terminal signal sequence promotes virulence factor secretion in Mycobacterium tuberculosis. Science 313(5793):1632
DiGiuseppe Champion PA, Champion MM, Manzanillo P, Cox JS (2009) ESX-1 secreted virulence factors are recognized by multiple cytosolic AAA ATPases in pathogenic mycobacteria. Mol Microbiol 73(5):950–962
Lightbody KL, Renshaw PS, Collins ML, Wright RL, Hunt DM, Gordon SV, Hewinson RG, Buxton RS, Williamson RA, Carr MD (2004) Characterisation of complex formation between members of the Mycobacterium tuberculosis complex CFP-10/ESAT-6 protein family: towards an understanding of the rules governing complex formation and thereby functional flexibility. FEMS Microbiol Lett 238(1):255–262
MacGurn JA, Raghavan S, Stanley SA, Cox JS (2005) A non-RD1 gene cluster is required for Snm secretion in Mycobacterium tuberculosis. Mol Microbiol 57(6):1653–1663
Shrivastava R, Ghosh AK, Das AK (2009) Intra-and intermolecular domain interactions among novel two-component system proteins coded by Rv0600c, Rv0601c and Rv0602c of Mycobacterium tuberculosis. Microbiology 155(3):772
Parida BK, Douglas T, Nino C, Dhandayuthapani S (2005) Interactions of anti-sigma factor antagonists of Mycobacterium tuberculosis in the yeast two-hybrid system. Tuberculosis 85(5):347–355
Saïd-Salim B, Mostowy S, Kristof AS, Behr MA (2006) Mutations in Mycobacterium tuberculosis Rv0444c, the gene encoding anti-SigK, explain high level expression of MPB70 and MPB83 in Mycobacterium bovis. Mol Microbiol 62(5):1251–1263
Handa P, Acharya N, Thanedar S, Purnapatre K, Varshney U (2000) Distinct properties of Mycobacterium tuberculosis single-stranded DNA binding protein and its functional characterization in Escherichia coli. Nucleic Acids Res 28(19):3823
Kana BD, Abrahams GL, Sung N, Warner DF, Gordhan BG, Machowski EE, Tsenova L, Sacchettini JC, Stoker NG, Kaplan G (2010) Role of the DinB homologs Rv1537 and Rv3056 in Mycobacterium tuberculosis. J Bacteriol 192(8):2220
Sinha KM, Stephanou NC, Gao F, Glickman MS, Shuman S (2007) Mycobacterial UvrD1 is a Ku-dependent DNA helicase that plays a role in multiple DNA repair events, including double-strand break repair. J Biol Chem 282(20):15114
Warner DF, Ndwandwe DE, Abrahams GL, Kana BD, Machowski EE, Venclovas Ä, Mizrahi V (2010) Essential roles for imuA′- and imuB-encoded accessory factors in DnaE2-dependent mutagenesis in Mycobacterium tuberculosis. Proc Natl Acad Sci 107(29):13093–13098
Garg S, Alam MS, Bajpai R, Kishan KVR, Agrawal P (2009) Redox biology of Mycobacterium tuberculosis H37Rv: protein-protein interaction between GlgB and WhiB1 involves exchange of thiol-disulfide. BMC Biochem 10(1):1
Huet G, Castaing JP, Fournier D, Daffé M, Saves I (2006) Protein splicing of SufB is crucial for the functionality of the Mycobacterium tuberculosis SUF machinery. J Bacteriol 188(9):3412–3414
Huet G, Daffa M, Saves I (2005) Identification of the Mycobacterium tuberculosis SUF machinery as the exclusive mycobacterial system of [Fe-S] cluster assembly: evidence for its implication in the pathogen’s survival. J Bacteriol 187(17):6137
Veyron-Churlet R, Guerrini O, Mourey L, Daffa M, Zerbib D (2004) Protein-protein interactions within the Fatty Acid Synthase-II system of Mycobacterium tuberculosis are essential for mycobacterial viability. Mol Microbiol 54(5):1161–1172
Veyron-Churlet R, Bigot S, Guerrini O, Verdoux S, Malaga W, Daffe M, Zerbib D (2005) The biosynthesis of mycolic acids in Mycobacterium tuberculosis relies on multiple specialized elongation complexes interconnected by specific protein-protein interactions. J Mol Biol 353(4):847–858
Curry JM, Whalan R, Hunt DM, Gohil K, Strom M, Rickman L, Colston MJ, Smerdon SJ, Buxton RS (2005) An ABC transporter containing a forkhead-associated domain interacts with a serine-threonine protein kinase and is required for growth of Mycobacterium tuberculosis in mice. Infect Immun 73(8):4471–4477
Hett EC, Chao MC, Steyn AJ, Fortune SM, Deng LL, Rubin EJ (2007) A partner for the resuscitation-promoting factors of Mycobacterium tuberculosis. Mol Microbiol 66(3):658–668
Sasindran S, Saikolappan S, Scofield V, Dhandayuthapani S (2011) Biochemical and physiological characterization of the GTP-binding protein Obg of Mycobacterium tuberculosis. BMC Microbiol 11(1):43
Ahidjo BA, Kuhnert D, McKenzie JL, Machowski EE, Gordhan BG, Arcus V, Abrahams GL, Mizrahi V (2011) VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS One 6(6):e21738
Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schachter V, Chemama Y, Labigne A, Legrain P (2001) The protein-protein interaction map of Helicobacter pylori. Nature 409(6817):211–215
LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C (2005) A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 438(7064):103–107
Malek JA, Wierzbowski JM, Tao W, Bosak SA, Saranga DJ, Doucette-Stamm L, Smith DR, McEwan PJ, McKernan KJ (2004) Protein interaction mapping on a functional shotgun sequence of Rickettsia sibirica. Nucleic Acids Res 32(3):1059–1064
Dyer MD, Neff C, Dufford M, Rivera CG, Shattuck D, Bassaganya-Riera J, Murali TM, Sobral BW (2010) The human-bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis. PLoS One 5(8):e12089
Parrish JR, Yu J, Liu G, Hines JA, Chan JE, Mangiola BA, Zhang H, Pacifico S, Fotouhi F, DiRita VJ (2007) A proteome-wide protein interaction map for Campylobacter jejuni. Genome Biol 8(7):R130
Titz B, Rajagopala SV, Goll J, Häuser R, McKevitt MT, Palzkill T, Uetz P (2008) The binary protein interactome of Treponema pallidum: the syphilis spirochete. PLoS One 3(5):e2292
Zhang L, Villa NY, Rahman MM, Smallwood S, Shattuck D, Neff C, Dufford M, Lanchbury JS, LaBaer J, McFadden G (2009) Analysis of vaccinia virus-host protein-protein interactions: validations of yeast two-hybrid screenings. J Proteome Res 8(9):4311–4318
Gavin AC, Basche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415(6868):141–147
Gerber D, Maerkl SJ, Quake SR (2008) An in vitro microfluidic approach to generating protein-interaction networks. Nat Methods 6(1):71–74
Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415(6868):180–183
Xiang G, Yao X, Guowu B, Yan X, Zhiyong X (2010) Response of the mosquito protein interaction network to dengue infection. BMC Genomics 11:380
Marchadier E, Carballidoa-Lapez R, Brinster S, Fabret C, Mervelet P, Bessiares P, Noirota-Gros MF, Fromion V, Noirot P (2011) An expanded protein-protein interaction network in Bacillus subtilis reveals a group of hubs: exploration by an integrative approach. Proteomics 11(15):2981–2991
Cherkasov A, Hsing M, Zoraghi R, Foster LJ, See RH, Stoynov N, Jiang J, Kaur S, Lian T, Jackson L (2011) Mapping the protein interaction network in methicillin-resistant Staphylococcus aureus. J Proteome Res 10(3):1139–1150
Callahan B, Nguyen K, Collins A, Valdes K, Caplow M, Crossman DK, Steyn AJC, Eisele L, Derbyshire KM (2010) Conservation of structure and protein-protein interactions mediated by the secreted mycobacterial proteins EsxA, EsxB, and EspA. J Bacteriol 192(1):326
Matsumoto A, Comatas KE, Liu L, Stamler JS (2003) Screening for nitric oxide-dependent protein-protein interactions. Science 301(5633):657
Vignols F, Brahalin C, Surdin-Kerjan Y, Thomas D, Meyer Y (2005) A yeast two-hybrid knockout strain to explore thioredoxin-interacting proteins in vivo. Proc Natl Acad Sci USA 102(46):16729
Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307(5707):223–227
Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE 3rd (2006) Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 103(2):431–436
Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405(6789):962–966
Bogan AA, Thorn KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280(1):1–9
Clackson T, Wells JA (1995) A hot spot of binding energy in a hormone-receptor interface. Science 267(5196):383
Tsai CJ, Lin SL, Wolfson HJ, Nussinov R (1997) Studies of protein-protein interfaces: a statistical analysis of the hydrophobic effect. Protein Sci 6(1):53–64
Xu D, Tsai CJ, Nussinov R (1997) Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng 10(9):999–1012
McCoy AJ, Chandana Epa V, Colman PM (1997) Electrostatic complementarity at protein/protein interfaces. J Mol Biol 268(2):570–584
Jones S, Thornton JM (1996) Principles of protein-protein interactions. Proc Natl Acad Sci USA 93(1):13–20
Gadek TR, Nicholas JB (2003) Small molecule antagonists of proteins. Biochem Pharmacol 65(1):1–8
Laskowski RA, Luscombe NM, Swindells MB, Thornton JM (1996) Protein clefts in molecular recognition and function. Protein Sci 5(12):2438–2452
Loregian A, Palu G (2005) Disruption of protein-protein interactions: towards new targets for chemotherapy. J Cell Physiol 204(3):750–762
Wells JA, McClendon CL (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450(7172):1001–1009
Arkin MR, Wells JA (2004) Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov 3(4):301–317
Strosberg AD (2007) Protein-protein interactions as targets for novel therapeutics. Drug Discov
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303(5659):844–848
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274(5289):948–953
Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ (2005) Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310(5748):670–674
Shakhnovich EA, Hung DT, Pierson E, Lee K, Mekalanos JJ (2007) Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc Natl Acad Sci USA 104(7):2372–2377
McMillan K, Adler M, Auld DS, Baldwin JJ, Blasko E, Browne LJ, Chelsky D, Davey D, Dolle RE, Eagen KA, Erickson S, Feldman RI, Glaser CB, Mallari C, Morrissey MM, Ohlmeyer MH, Pan G, Parkinson JF, Phillips GB, Polokoff MA, Sigal NH, Vergona R, Whitlow M, Young TA, Devlin JJ (2000) Allosteric inhibitors of inducible nitric oxide synthase dimerization discovered via combinatorial chemistry. Proc Natl Acad Sci USA 97(4):1506–1511
Sennequier N, Wolan D, Stuehr DJ (1999) Antifungal imidazoles block assembly of inducible NO synthase into an active dimer. J Biol Chem 274(2):930–938
Gorczynski MJ, Grembecka J, Zhou Y, Kong Y, Roudaia L, Douvas MG, Newman M, Bielnicka I, Baber G, Corpora T, Shi J, Sridharan M, Lilien R, Donald BR, Speck NA, Brown ML, Bushweller JH (2007) Allosteric inhibition of the protein-protein interaction between the leukemia-associated proteins Runx1 and CBFbeta. Chem Biol 14(10):1186–1197
Last-Barney K, Davidson W, Cardozo M, Frye LL, Grygon CA, Hopkins JL, Jeanfavre DD, Pav S, Qian C, Stevenson JM, Tong L, Zindell R, Kelly TA (2001) Binding site elucidation of hydantoin-based antagonists of LFA-1 using multidisciplinary technologies: evidence for the allosteric inhibition of a protein-protein interaction. J Am Chem Soc 123(24):5643–5650
Horn JR, Shoichet BK (2004) Allosteric inhibition through core disruption. J Mol Biol 336(5):1283–1291
He MM, Smith AS, Oslob JD, Flanagan WM, Braisted AC, Whitty A, Cancilla MT, Wang J, Lugovskoy AA, Yoburn JC, Fung AD, Farrington G, Eldredge JK, Day ES, Cruz LA, Cachero TG, Miller SK, Friedman JE, Choong IC, Cunningham BC (2005) Small-molecule inhibition of TNF-alpha. Science 310(5750):1022–1025. doi:10.1126/science.1116304
Bochkareva E, Safro M, Girshovich A (1999) Interaction of 4,4′-dithiodipyridine with Cys(458) triggers disassembly of GroEL. J Biol Chem 274(30):20756–20758
Tachedjian G, Orlova M, Sarafianos SG, Arnold E, Goff SP (2001) Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc Natl Acad Sci USA 98(13):7188–7193
Crabtree GR, Schreiber SL (1996) Three-part inventions: intracellular signaling and induced proximity. Trends Biochem Sci 21(11):418–422
Michnick SW (2000) Chemical biology beyond binary codes. Chem Biol 7(12):R217–221, pii: S1074-5521(00)00040-5
Liu J, Farmer JD Jr, Lane WS, Friedman J, Weissman I, Schreiber SL (1991) Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66(4):807–815, pii: 0092-8674(91)90124-H
Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369(6483):756–758. doi:10.1038/369756a0
Williams DJ, Puhl HL, Ikeda SR, Brezina V (2009) Rapid modification of proteins using a rapamycin-inducible tobacco etch virus protease system. PLoS One 4(10):e7474
Remy I, Michnick SW (2006) A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods 3(12):977–979
Janse DM, Crosas B, Finley D, Church GM (2004) Localization to the proteasome is sufficient for degradation. J Biol Chem 279(20):21415–21420
Joshi PB, Hirst M, Malcolm T, Parent J, Mitchell D, Lund K, Sadowski I (2007) Identification of protein interaction antagonists using the repressed transactivator two-hybrid system. Biotechniques 42(5):635–644
Kley N (2004) Chemical dimerizers and three-hybrid systems: scanning the proteome for targets of organic small molecules. Chem Biol 11(5):599–608
Veselovsky AV, Ivanov YD, Ivanov AS, Archakov AI, Lewi P, Janssen P (2002) Protein-protein interactions: mechanisms and modification by drugs. J Mol Recognit 15(6):405–422
Valencia A, Pazos F (2002) Computational methods for the prediction of protein interactions. Curr Opin Struct Biol 12(3):368–373
Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D (1999) Detecting protein function and protein-protein interactions from genome sequences. Science 285(5428):751
Bock JR, Gough DA (2001) Predicting protein-protein interactions from primary structure. Bioinformatics 17(5):455–460
Eisenberg D, Marcotte EM, Xenarios I, Yeates TO (2000) Protein function in the post-genomic era. Nature 405(6788):823–826
Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA (1999) Protein interaction maps for complete genomes based on gene fusion events. Nature 402(6757):86–90
Fraser HB, Hirsh AE, Wall DP, Eisen MB (2004) Coevolution of gene expression among interacting proteins. Proc Natl Acad Sci USA 101(24):9033
Blaschke C, Andrade MA, Ouzounis C, Valencia A (1999) Automatic extraction of biological information from scientific text: protein-protein interactions. Proc Int Conf Intell Syst Mol Biol 1999:60–67
Cusick ME, Yu H, Smolyar A, Venkatesan K, Carvunis AR, Simonis N, Rual JF, Borick H, Braun P, Dreze M (2008) Literature-curated protein interaction datasets. Nat Methods 6(1):39–46
Russell RB, Alber F, Aloy P, Davis FP, Korkin D, Pichaud M, Topf M, Sali A (2004) A structural perspective on protein-protein interactions. Curr Opin Struct Biol 14(3):313–324
Neuvirth H, Raz R, Schreiber G (2004) ProMate: a structure based prediction program to identify the location of protein-protein binding sites. J Mol Biol 338(1):181–199
Stein A, Caol A, Aloy P (2011) 3did: identification and classification of domain-based interactions of known three-dimensional structure. Nucleic Acids Res 39(suppl 1):D718
Aloy P, Russell RB (2003) InterPreTS: protein interaction prediction through tertiary structure. Bioinformatics 19(1):161
Pazos F, Valencia A (2001) Similarity of phylogenetic trees as indicator of protein-protein interaction. Protein Eng 14(9):609
Pagel P, Wong P, Frishman D (2004) A domain interaction map based on phylogenetic profiling. J Mol Biol 344(5):1331–1346
Shoemaker BA, Panchenko AR (2007) Deciphering protein-protein interactions. Part II. Computational methods to predict protein and domain interaction partners. PLoS Comput Biol 3(4):e43
Mavromatis K, Chu K, Ivanova N, Hooper SD, Markowitz VM, Kyrpides NC (2009) Gene context analysis in the Integrated Microbial Genomes (IMG) data management system. PLoS One 4(11):e7979
Pazos F, Valencia A (2002) In silico two-hybrid system for the selection of physically interacting protein pairs. Proteins 47(2):219–227
Pazos F, Valencia A (2008) Protein co-evolution, co-adaptation and interactions. EMBO J 27(20):2648–2655
Friedberg I (2006) Automated protein function prediction: the genomic challenge. Brief Bioinform 7(3):225–242
Snitkin E, Gustafson A, Mellor J, Wu J, DeLisi C (2006) Comparative assessment ofperformance and genome dependence among phylogenetic profiling methods. BMC Bioinformatics 7(1):420
Orchard S, Salwinski L, Kerrien S, Montecchi-Palazzi L, Oesterheld M, Stümpflen V, Ceol A, Chatr-aryamontri A, Armstrong J, Woollard P (2007) The minimum information required for reporting a molecular interaction experiment (MIMIx). Nat Biotechnol 25(8):894–898
Taylor CF, Paton NW, Lilley KS, Binz PA, Julian RK, Jones AR, Zhu W, Apweiler R, Aebersold R, Deutsch EW (2007) The minimum information about a proteomics experiment (MIAPE). Nat Biotechnol 25(8):887–893
Gmuender H, Kuratli K, Di Padova K, Gray CP, Keck W, Evers S (2001) Gene expression changes triggered by exposure of Haemophilus influenzae to novobiocin or ciprofloxacin: combined transcription and translation analysis. Genome Res 11(1):28–42
Yoon SH, Han MJ, Lee SY, Jeong KJ, Yoo JS (2003) Combined transcriptome and proteome analysis of Escherichia coli during high cell density culture. Biotechnol Bioeng 81(7):753–767
Mostertz J, Scharf C, Hecker M, Homuth G (2004) Transcriptome and proteome analysis of Bacillus subtilis gene expression in response to superoxide and peroxide stress. Microbiology 150(2):497
Gygi SP, Rochon Y, Franza BR, Aebersold R (1999) Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19(3):1720–1730
Bro C, Regenberg B, Lagniel G, Labarre J, Montero-Lomelí M, Nielsen J (2003) Transcriptional, proteomic, and metabolic responses to lithium in galactose-grown yeast cells. J Biol Chem 278(34):32141–32149
Kramer JO, Sorgenfrei O, Klopprogge K, Heinzle E, Wittmann C (2004) In-depth profiling of lysine-producing Corynebacterium glutamicum by combined analysis of the transcriptome, metabolome, and fluxome. J Bacteriol 186(6):1769–1784
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Steyn, A.J.C., Mai, D., Saini, V., Farhana, A. (2013). Protein–Protein Interaction in the -Omics Era: Understanding Mycobacterium tuberculosis Function. In: McFadden, J., Beste, D., Kierzek, A. (eds) Systems Biology of Tuberculosis. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-4966-9_5
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4966-9_5
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-4965-2
Online ISBN: 978-1-4614-4966-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)