
Abstract
Many cases of autism spectrum disorder (ASD) are caused by rare, highly penetrant mutations. The terms “secondary autism” and “syndromic autism” refer to those with an ASD diagnosis associated with a defined genetic defect. Well-known examples of syndromic ASD include neurofibromatosis type 1, fragile X syndrome, and tuberous sclerosis (TS). As with all forms of ASD, the neurological phenotype of patients with syndromic ASD is highly variable and is frequently accompanied by mental retardation, attention-deficit hyperactivity disorder, and/or seizures, conditions frequently found in individuals with idiopathic ASD. Despite the genetic heterogeneity underpinning ASD, this chapter explains how pathway- and network-based methods can (i) integrate the current ASD pathological hypotheses, such as abnormal synaptic, secretory pathway, calcium signalling, or mitochondrial function, and (ii) be used to explain the high incidence of ASD in individuals with defined genetic disorders, such as TS. Data from these analyses also suggest that a single therapeutic drug may be able to target ASD in a broad subset of patients.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Autism Spectrum Disorder
- Autism Spectrum Disorder
- Tuberous Sclerosis
- Tuberous Sclerosis Complex
- Autism Spectrum Disorder Patient
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Autism spectrum disorder (ASD) is considered a complex genetic disorder that affects around 1 % of the population. A diagnosis of ASD is dependent on qualitative impairments in communication and social skills and the presence of repetitive behaviors and/or restricted interests. Single-gene mutations and defined chromosomal defects account for over 25 % of ASD cases (Miles 2011), with the remainder often being referred to as idiopathic ASD. Twin and family studies indicate ASD is highly heritable, with heritability estimates of 80–90 %, while spontaneous (de novo) genetic mutations are implicated in ∼10 % of cases (Ronald and Hoekstra 2011). At the cellular level, many of the genes implicated in ASD have direct or indirect roles in synapse function, and ASD is sometimes referred to as a “synapsopathy” (Dölen and Bear 2009) or “synaptopathy” (Brose et al. 2010) due to the abnormal synaptic morphology and function detected. However, in addition to the current diagnostic criteria of ASD, a variety of additional neurological phenotypes are frequently detected such as attention-deficit hyperactivity disorder (ADHD), epilepsy, mental retardation (MR), and obsessive–compulsive disorder (see Bishop et al. 2014). Furthermore, biochemical, gastrointestinal, and immune system abnormalities frequently co-occur with ASD (McDougle et al. 2005; Castellani et al. 2009; Buie et al. 2010; Brown and Mehl-Madrona 2011; Suzuki et al. 2011; Benach et al. 2012). As with other common complex genetic disorders, the phenotype of each individual varies and is dependent on which gene or genes are mutated, the individual genetic background, as well the in utero environment before birth (Bishop et al. 2014).
Autism Genetics
To date, hundreds of genetic mutations have been linked to the development of ASD, and a variety of databases have been established to collate information on emerging and defined “autism genes” (Table 1). This number of genes is typical of complex genetic disorders, including mental retardation (MR), epilepsy, and ADHD. Despite the large number of genes implicated in complex genetic disorders, analysis of key subsets of ASD genes (see Table 2) suggests that mutations associated with ASD converge on key shared pathways which, when disrupted, affect synaptic function (Bourgeron 2009; Bill and Geschwind 2009). Interactomic analysis is discussed in more detail elsewhere (Barth and Bishop 2014), and an example incorporating the ASD genes from Table 2 is provided here (Fig. 1), illustrating the complex interactions between ASD genes.

Emerging functional links between autism gene products. Physical (blue), predicted (brown), co-expression (grey), and genetic (dark green) interaction network show complex interactions between the genes from Table 2. Red circle = TSC1; green circle = TSC2; blue circle = mammalian TOR. Syndromic ASD gene products, NF1, PTEN, and FMRP/FMR1 are indicated in pink
Of note, many of the genes implicated in ASD also increase the risk of a variety of neurological and medical conditions (Bolton 2009; Lichtenstein et al. 2010; Betancur 2011; Talkowski et al. 2012), and it is clear from syndromic ASD cases that a single-gene mutation frequently leads to a phenotype of which ASD is just one part (Benvenuto et al. 2009; Caglayan 2010; Betancur 2011; Barth and Bishop 2014). Evidence from research into a variety of single-gene disorders also indicates different mutations in the same gene can lead to different phenotypes (Antonarakis and Beckmann 2006; Bishop et al. 2014). This phenomenon is due to factors such as the multiple binding partners and pleiotropic roles of many gene products (see Fig. 1). Studying gene product interactions, and the effects of gene products in multiple cellular pathways, is a key tool for increasing understanding of these phenomena.
In the field of ASD research, some genetic mutations are known to directly affect synapse development, morphology, and function (Bourgeron 2009; Bill and Geschwind 2009; Peça and Feng 2012). By contrast, other autism genes appear to indirectly lead to synaptic anomalies and include genes affecting secretory pathway function, calcium signalling, or impacting on poorly understood mitochondrial activities (Krey and Dolmetsch 2007; Bourgeron 2009; Gargus 2009; Palmieri and Persico 2010; Aziz et al. 2011a, 2011b, 2012; Peça and Feng 2012). This chapter discusses how, using specific examples, pathway and network analyses can integrate the functional effects of single-gene mutations with the current pathogenic models of ASD etiology.
Syndromic Autism and the Target of Rapamycin Pathway
The most common single-gene forms of ASD are FXS and tuberous sclerosis (TS), which account for an estimated 7 % and 4 % of ASD cases, respectively. In addition, syndromes associated with PTEN (phosphatase and tensin homolog) gene mutations are found in 1–5 % of ASD patients (Zhou and Parada 2012). Along with neurofibromatosis type 1 (NF1), which accounts for around 0.6 % of ASDs, these three disorders represent around 15 % of ASD cases. These key syndromic forms of ASD are caused by mutations in the FMR1 gene (FXS), TSC1 or TSC2 genes (TS), NF1 (NF1), and the PTEN-hamartoma tumor syndromes (PHTS) and autism with macrocephaly (AM) by mutations in the PTEN gene (see Table 2). The genes each localize to different human chromosomes, as indicated in Fig. 2. The TSC1 gene encodes a protein known as TSC1 or hamartin, while TSC2 encodes TSC2, also known as tuberin (Fig. 3). The FMR1 gene encodes the fragile X mental retardation protein (FMRP) and PTEN encodes the phosphatase, PTEN (phosphatase and tensin homolog) (see Fig. 4). The NF1 gene encodes a large protein, known as neurofibromin-1 (NF1) (Fig. 5), and each protein has multiple known motifs and domains that contribute to their function. TS, PHTS, and NF1 are autosomal-dominant neurocutaneous disorders, presenting with neurologic and dermatologic abnormalities, and the protein products are known to act as tumor suppressors (Lodish and Stratakis 2010). By contrast FXS is an X-linked disorder (see Fig. 2) and the gene is not known to have a role in tumorigenesis. Readers are referred to the Online Mendelian Inheritance in Man (OMIM) database, GeneCards, and/or UniProt (see Table 1) for additional genetic and phenotypic information.

Chromosome localization of key syndromic autism genes. Human TSC1 and TSC2 localize to chromosome 9, at position 9q34.13, and chromosome 16, position 16p13.3 (red lines), respectively. Human PTEN is found on chromosome 10, position 10q23.3, NF1 on chromosome 17, position 17q11.2, while FMR1 is an X-linked gene at position Xq27.3 (Data derived from GeneCards, www.genecards.org)

Schematic representation of the TSC1 and TSC2 gene products. TSC1 and TSC2 encode the 1,164 amino acid (aa) gene product TSC1 (hamartin), and the 1,807 aa gene product, TSC2 (tuberin), respectively. Key domains and motifs are indicated, with inhibitory serine (S) or threonine (T) kinase sites on TSC2 for PKB (alias AKT) indicated (arrows). Further details are available from the GeneCards database (www.genecards.org)

Schematic representation of the FMR1 and PTEN gene products. FMR1 and PTEN encode the 632 amino acid (aa) gene product FMRP (fragile X mental retardation protein), and the 403 aa gene product, PTEN (phosphatase and tensin-homolog). Key domains and motifs are indicated. Further details are available from the GeneCards database (www.genecards.org)

Schematic representation of the NF1 gene product. NF1 encodes a 2,839 amino acid (aa) gene product NF1 (neurofibromin 1), which acts as a negative regulator of Ras, due to its GTPase-activating protein (GAP) activity. The Sec14 domain is also known as the CRAL-TRIO domain, and the functional role of nuclear-targeted NF1 is unknown. Further details are available from the GeneCards database (www.genecards.org)
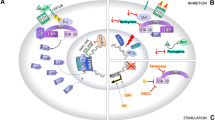
Strikingly, most of the genes causative of the syndromic forms of ASD discussed above (PTEN, NF1, TSC1, or TSC2) encode products functioning as upstream regulators of a cellular signalling pathway, known as the TOR (target of rapamycin) pathway (see Fig. 6). By contrast, the FMR1 gene product, FMRP, functions downstream in the TOR signalling pathway (Fig. 6). TS, or tuberous sclerosis complex disorder (TSCD), is of particular interest, as it is caused by the mutation of one of two genes. Mutations in either the TSC1 or TSC2 gene clearly give rise to a sufficiently similar phenotype that patients are diagnosed with the same disorder. This similarity in phenotype is due to the two gene products interacting with each other within cells to form a functional protein complex (Fig. 6). The resulting protein complex, known as the tuberous sclerosis complex (TSC), negatively regulates the kinase activity of the TOR complex, TORC1. The TSC regulates TORC1 activity by the activity of a mutual binding partner, Rheb (Ras homologue enriched in brain). Rheb can activate TORC1 when in the GTP-bound form (see Fig. 6), and this in turn is modulated by the GTPase-activating protein (GAP) domain of TSC2 (see Fig. 3), which is dependent on binding to TSC1.

Schematic of key components of the TSC–TOR pathway. Neuronal receptors and channels activate downstream signalling pathways leading to mammalian TOR complex 1 activation. TORC1 activation regulates several downstream effectors of translation: S6K, 4E-BP, and eEF2K. Presynaptic stimulation also leads to increased cytosolic levels of Ca2+ (from intracellular stores) and release of BDNF (brain-derived neurotrophic factor). PM plasma membrane; CaM Calmodulin (see Fig. 2)
As is well known for ASD, the neurodevelopmental phenotype of patients with TS, NF1, PTHS, and fragile X syndrome (FXS) is heterogeneous, and reasons underlying this heterogeneity are discussed elsewhere in this book (Bishop et al. 2014), and the mechanism underlying the association of ASD and these syndromes is also unclear. Current data on the etiology of TS, however, indicate that mutation of TSC1 or TSC2 directly influences the development of ASD, rather than it being a secondary effect of MR, epilepsy, or tumors in these patients (Smalley 1998; Bishop et al. 2014; Jülich and Sahin 2014). Therefore, ASD and TS are currently thought to share a pathobiological factor(s). Understanding the multiple downstream effects of the TOR pathway is essential, to explain how a single-gene mutation such as that of TSC2 or PTEN can lead to a complex phenotype of hamartomas, MR, epilepsy, and ASD. Based on current models of the pathobiology of ASD, it is expected that the downstream effects of TSC1, TSC2, NF1, PTEN, or FMR1 mutation must alter the function of the cell secretory pathway (including the ER and/or Golgi apparatus), calcium signalling, and/or mitochondrial function, ultimately leading to alterations in synapse function (Krey and Dolmetsch 2007; Bourgeron 2009; Gargus 2009; Palmieri and Persico 2010; Aziz et al. 2011a, b, 2014; Peça and Feng 2012). Evidence supporting these models of ASD pathogenesis is discussed next, with reference to TS and other syndromic forms of ASD.
Dendritic Spine Morphology and the TSC–TOR Pathway
Neurons are highly polarized cells, with a long axon for action potential propagation and release of neurotransmitters and several dendrites for responding to the signals from an adjoining axon. Dendrites and axons of mammalian neurons typically have a branching morphology of varying complexity, referred to as axonal or dendritic branching, or arborization, dependent on location. The exact morphology of neurons is highly regulated, and their structure, once thought static at maturation, is now known to be highly dynamic throughout life. Excitatory synapses in the brain are characterized by presynaptic axonal boutons (protrusions) apposing “spines” protruding from dendritic arbors. In particular, dendritic spines and excitatory synapses (such as glutamatergic brain synapses) are highly dynamic, and the term “synaptic plasticity” is used to describe the ability of synaptic connections between two neurons to change in “strength” in response to neuronal activity (Alvarez and Sabatini 2007). Dendritic spines change in number, size, and shape and therefore correlate with modifications in synaptic strength (Jan and Jan 2010).
Dendrite morphogenesis is dependent on a wide variety of cellular processes, including specific transcriptional regulators; factors affecting local translation in dendrites; cytoskeletal motors and regulators; secreted proteins, cell-surface receptors, and adhesins; regulators of the secretory and endocytic pathways; and signalling pathways that regulate these processes, including the TOR pathway (Kennedy and Ehlers 2006; Jan and Jan 2010; Troca-Marín et al. 2012). Dendritic spines are particularly dynamic structures. Although most dendritic spines are transient, long-term adjustments, such as the addition/loss of stable spines and synapses, are required for learning and memory. These long-term changes are referred to as long-term potentiation (LTP) and long-term depression (LTD) (Alvarez and Sabatini 2007). Of relevance to autism, abnormalities in dendritic spine size, shape, or number are found in many neurodevelopmental and neuropsychiatric disorders and have been documented in both idiopathic and syndromic cases of ASD (reviewed by Penzes et al. 2011).
While the TSC–TOR signalling pathway is well known as a regulator of cell growth and the formation of tumors, it has only recently been identified as an important pathway in nervous system development, by controlling factors such as cell migration, axon guidance, synaptic expansion, and dendritic arborization (Swiech et al. 2008). Nonetheless, differences in dendritic morphology and spine number have been reported in several syndromic forms of ASD, including those involved in TOR pathway processes. For example, in FXS an increase in dendritic spine density is detected, although these spines are long and immature. Likewise, in PHTS, increased dendritic spine density and hypertrophic dendritic arbors are detected, while in TS, aberrant dendrite and spine morphology is detected, specifically an increase in spine size, but a decrease in spine density (Peça and Feng 2012; Troca-Marín et al. 2012). The abnormal appearance of dendrites in patients is likewise found in animal models of syndromic forms of autism, and these morphological anomalies are associated with deficits in synaptic plasticity, including LTD and LTP, underpinning learning and memory (Penzes et al. 2011; Ehninger 2013; Gipson and Johnston 2012).
Glutamate Receptors and the TOR Pathway
As regulation of the number and density of neurotransmitter receptors is a key mechanism underlying various forms of synaptic plasticity, including LTP and LTD, a change in number of even a few receptors in dendritic spines can affect neurotransmission (Kennedy and Ehlers 2006). Efforts have been made to measure effects of mutations on neurotransmission and to quantify receptors within key dendritic regions, known as postsynaptic densities (due to their distinctive electron-dense appearance by microscopy). In FXS model mice, enhanced metabotropic glutamate receptor-dependent long-term depression (mGluR-LTD) has been detected, which has been attributed to effects of the Fmr1 mutation on cellular translation events essential for LTD, and the mutant phenotype in fragile X model organisms can be corrected by genetic or pharmacological inhibition of mGluR5 (Auerbach and Bear 2010). Surprisingly, the opposite has been detected in models of TS, as mGluR-LTD is impaired, not enhanced, indicating that TS and FXS have divergent synaptic plasticity phenotypes (Auerbach et al. 2011; Chévere-Torres et al. 2012).
Neuronal mGluR receptors are G-protein-coupled receptors (GPCRs) that are predominantly postsynaptic and, along with the ionotropic glutamate receptors, are activated by glutamate, the major excitatory amino acid neurotransmitter in the brain. The mGluRs, particularly mGluR5, have received considerable attention as key players in neuronal development, synaptic plasticity, LTP/LTD, and seizure activity. Stimulating cell-surface mGluRs leads to activation of phospholipase C (PLC) and the formation of the second messengers, InsP3 (inositol 1,4,5-trisphosphate) and DAG (diacyl glycerol). InsP3 production triggers Ca2+ release from the ER via InsP3 receptors (IP3Rs), while DAG activates protein kinase C (PKC), known to reduce oxidative stress and cell damage (Byrnes et al. 2009) (Fig. 6). Calcium released from the ER triggers further amplification of Ca2+ release, by activating ryanodine receptors (RyRs) on the ER, major cellular mediators of intracellular calcium-induced calcium release (CICR) (Mattson et al. 2008).
Interactomic and pathway analyses indicate that IP3R1 (ITPR1 gene product) interacts with Homer proteins and both are part of a multivalent complex which also includes mGluRs, Shank, and PSD-95 (postsynaptic density protein-95) (Tu et al. 1999; Xiao et al. 2000). These binding partners are key components of the postsynaptic density (see Fig. 6), the electron-dense protein network in dendritic spines. The interactions between mGluRs, ER, and PSD molecules are essential for the correct organization of Ca2+ sensors and effectors in the postsynapse and suggest that Ca2+ signalling in dendritic spines has a high degree of spatiotemporal organization (Tu et al. 1999). Shank and PSD-95 also provide links between mGluR and ionotropic glutamate receptor signalling.
Although coupled to the same InsP3 second-messenger pathway, mGluR1 and mGluR5 serve different physiological functions, which remain poorly understood. What is known, however, is that mGluR1 and mGluR5 traffic differently on interaction with Homer (Kuwajima et al. 2007) and a larger proportion of mGluR5 is found on intracellular membranes than at the cell surface (Kumar et al. 2012). Intracellular mGluR5 receptors remain targets for activation by glutamate, due to the presence of cell-surface glutamate transporters, known as EEATs (excitatory amino acid transporters, also known as glutamate transporters), part of the solute carrier family 1A (SLC1A) gene family, that facilitate glutamate uptake into the cytoplasm. Indeed, TORC1 signalling induces the upregulation of EAAT2, also known as glutamate transporter 1 (GLT1), while the TOR inhibition (by rapamycin) decreases EEAT2 protein and mRNA levels (Wu et al. 2010). Intracellular glutamate can then activate intracellular mGluR5, which induces a sustained calcium-response. By contrast, stimulation of cell-surface mGluR5 triggers a rapid, transient cytoplasmic calcium rise. These data indicate intracellular mGluR5 is critical for LTP and synaptic plasticity (Xiao et al. 2000; Kumar et al. 2012).
Therefore, in tuberous sclerosis, mGluR5 signalling via Ca2+ release from intracellular stores is impaired, leading to abnormal dendrite morphology and impairments in LTD. Mechanisms by which mGluR5 signalling may become impaired in patients with TSC1 or TSC2 mutations, which over-activates TORC1 signalling, will be discussed further below.
TSC Mutations: Effects on Calcium Homeostasis and Signalling
Intracellular Ca2+ storage and release has multiple cellular effects and affects several functions of the secretory pathway. It is important to note that IP3Rs are not only localized to the ER but are also found on the Golgi apparatus, and stimulation of cells with agonists, or IP3, results in significant Ca2+ release from Golgi stores (Lin et al. 1999). More recently, IP3R has also been localized to secretory vesicles, and IP3-mediated Ca2+ release from secretory granules has now been demonstrated in many cell types (Yoo 2010, 2011). The amount of IP3R localizing to each cellular compartment varies depending on the cell type, and in cells with a high secretory load, secretory granules contain the majority of intracellular calcium (Yoo 2010, 2011). For example, in the neuronal-like PC12 cells, secretory granules are responsible for the majority of IP3-mediated cellular Ca2+ release into the cytoplasm (Yoo 2010, 2011).
The unusually high calcium-storing capability of secretory granules (dense-core vesicles) is due to the presence of high concentrations of the Ca2+ storage proteins chromogranin A, chromogranin B, and chromogranin C. These proteins, abbreviated as CgA, CgB, and CgC, are also known as parathyroid secretory protein, secretogranin-I, and secretogranin-II and are encoded by the CHGA, CHGB, SCG2 genes, respectively. They are found in many secretory cell types, and it is well documented that neurons and astrocytes both have secretory granules containing chromogranins and IP3Rs (Yoo and Hur 2012). Most of the intra-vesicular calcium is bound to these proteins, which participate directly in secretory granule biogenesis, at least in part by facilitating condensation and packaging of proteins into secretory granules (Yoo 2010). Elevation of intracellular Ca2+ is known to stimulate the phosphorylation of CgA, which regulates protein packaging into secretory granules and subsequent secretion (Yanagihara et al. 1996). Of note, phosphorylation of CgA, CgB, and CgC is carried out by the Golgi casein kinase, an activity defective in a majority of ASD patients (Castagnola et al. 2008). Processing of proproteins, such as prohormones, in the secretory pathway is also regulated by luminal Ca2+ levels, as calcium is necessary for prohormone-processing enzyme activity (Austin and Shields 1996).
In addition to IP3R and SERCA, Calnuc, is another protein involved in regulating the secretory-pathway Ca2+ stores (Lin et al. 1999). Calnuc, also known as nucleobindin-1 and encoded by the NUCB1 gene, is a calcium-binding protein localizing to the lumen of the Golgi apparatus and secretory vesicles. Calnuc plays a key role in the Golgi lumen, controlling many aspects of the unfolded protein (UPR) response, as it regulates the activation of ATF6 (activating transcription factor) via proteolytic processing in the Golgi apparatus (Tsukumo et al. 2007). ATF6 is an ER membrane-anchored transcription factor, which is transported to the Golgi apparatus and cleaved, as part of the UPR. Calnuc also regulates proteolytic processing of other proproteins, occurring prior to secretion, such as that of APP (amyloid precursor protein) (Lin et al. 2007). Overexpression of Calnuc enhances regulated, but not constitutive or basal, secretion, e.g., secretion of ACTH (Lin et al. 2009). The related calcium-binding gene product, NUCB2, is similarly implicated in regulation of secretion by controlling the releasable Ca2+ store in the ER and Golgi apparatus, regulating signalling of GPCRs and mediating the exocytosis of secretory granules (Kalnina et al. 2009). The IP3-induced release of Ca2+ from secretory granules is sufficient to initiate exocytosis of secretory granules (Yoo 2010).
Given that Ca2+ and TOR both control many aspects of cell homeostasis, it not surprising that further links between the cellular processes regulated have recently been found. For example, TOR has been found not only to interact with, and phosphorylate, the IP3R but also to facilitate IP3R-mediated Ca2+ release (Frégeau et al. 2011). Indeed, cell treatment with rapamycin, or nutrient deprivation, which acts to inhibit TOR activity, leads to an increase in IP3-induced release of intracellular Ca2+ (Frégeau et al. 2011). These results clearly identify TOR as a modulator of intracellular Ca2+ signalling (see Fig. 6).
A central role for Ca2+ is also emerging at the earliest stages of the secretory pathway, where Ca2+ is emerging as a key regulator of ER function. It is well known that a Ca2+ concentration increase in the cytosol affects levels in the ER which, in turn, affects the function of a number chaperones within the ER lumen. ER chaperones, such as calreticulin, calnexin, BiP (alias Grp78), and endoplasmin (Grp94), are Ca2+-binding proteins, and changes in free Ca2+ concentration in the lumen of the ER profoundly affect their functional activity (Verkhratsky 2005). A decrease in luminal Ca2+ renders ER chaperones inactive and prevents correct protein folding and secretion. Under conditions of ER stress, chaperone expression is upregulated, while global translation is inhibited. Of these chaperones, calreticulin is the most abundant in neurons and contributes greatly to ER Ca2+-buffering capacity, as well as regulating SERCA pumps, in addition to its chaperone function. An important property of the SERCA2b calcium transporter (encoded by isoform 1 of the SERCA2 gene, which is expressed in brain), relevant for ER Ca2+ homeostasis, is that its activity is regulated by the free Ca2+ concentration in the ER lumen, which is largely mediated via calreticulin and ERp57 (the PDIA3 gene product, alias Grp58). Calreticulin “senses” luminal Ca2+ levels and binds SERCA2b, to activate Ca2+ uptake whenever luminal Ca2+ concentrations decrease. In turn, ERp57 regulates Ca2+ uptake by modulating the redox state of SERCA2b in a Ca2+-dependent manner and inhibits SERCA2b pump activity when luminal Ca2+ levels are high (Verkhratsky 2005). Of interest, both BiP and ERp57 are substrates of the Golgi casein kinase, implicated as defective in as many as 70 % of ASD patients (Castagnola et al. 2008), and phosphorylation of BiP is known to prevent substrate binding (Gaut 1997). Therefore, regulation of intracellular Ca2+ stores has multiple effects on cells, is modulated by TOR signalling, and also plays several roles in the regulation of exocytosis and protein secretion.
The discussions above indicate that Ca2+-regulated processes are expected to be disrupted in TS patients, similar to those reported in idiopathic ASD individuals (Krey and Dolmetsch 2007). While this is not a well-studied aspect in TS, very recently, CADPS2, an autism gene required for Ca2+-dependent secretion of neuropeptides, was found to be downregulated in cells from TS patients (Tyburczy and Kaminska 2012). These data support a role for dysregulated calcium signalling in TS.
TSC Mutations and Mitochondria
Neural stimulation induces the recruitment of organelles with important roles in secretion/exocytosis and/or calcium homeostasis: the ER, Golgi outposts, and mitochondria, to the relevant regions. These organelles help neurons respond and change in response to neural activity (Valenzuela et al. 2011). Mitochondrial fission and mitochondrial movement into dendritic spines are part of the intracellular phenomena required to regulate spine morphogenesis and regulate neuronal plasticity (Mattson 2007). Of note, these processes are also regulated by the TOR and AMPK signalling pathways, and inhibition of AMPK (AMP-activated protein kinase) leads to a TOR-dependent increase in mitochondrial biogenesis (D’Souza et al. 2007). AMPK is involved in signalling events that bridge calcium signalling pathways to the nutrient- and growth factor-sensing mTOR pathway (Shaw et al. 2004; Tamás et al. 2006). Recent research has identified novel TOR signalling mechanisms that modulate mitochondrial biogenesis, and TOR over-activation (which occurs in TS) leads to increased mitochondrial biogenesis, as well as cell growth (Dunlop and Tee 2009). Therefore, as detected in patients with idiopathic ASD, mitochondrial function may be indirectly affected in TS, and it is possible this may contribute to the neurological symptoms associated with tuberous sclerosis.
Mitochondrial biogenesis is controlled by the intimate relationship of mitochondria with the ER, which is mediated by regions known as MAM (mitochondria-associated membranes), and the interaction of the ER with mitochondria regulates mitochondrial metabolism and energy production. The GTPase, mitofusin-2 (MFN2), is important for the close apposition of mitochondria and the ER, and is enriched in the interacting membranous regions (de Brito and Scorrano 2009). Mfn2 ablation in mouse models causes alterations in mitochondrial morphology, mitochondrial dysfunction, as well as defective calcium homeostasis, leading to ER stress (Sebastián et al. 2012). The interactions between mitochondria and the ER are reversible and are regulated by physiological cytosolic Ca2+ levels (de Brito and Scorrano 2010). Indeed, the interaction between the ER and mitochondria is essential for both Ca2+ homeostasis and lipid biosynthesis. These findings have led to a growing understanding that a significant proportion of Ca2+ released from the ER is taken up by mitochondria via a “quasi-synaptic” mechanism between the two organelles (de Brito and Scorrano 2010). As mentioned above, when TSC is mutated, mGluR-LTP is impaired, less of the second-messenger IP3 is produced, and therefore less Ca2+ will be released from intracellular stores. Conversely, inhibition of TOR (by starvation) also causes morphological changes in mitochondria, specifically mitochondrial elongation (Gomes et al. 2011). Therefore the ER and mitochondria and Ca2+ signalling are intimately entwined and their relationship is essential for the correct cellular responses to neural activity. Therefore, secondary mitochondrial dysfunction would not be unexpected in TS patients.
Further confirming a role of TSC1 and TSC2 mutations in mitochondrial dysfunction, tissue from TS tumors contains cells with swollen mitochondria (Yamamoto et al. 2002), and increased numbers of mitochondria are found in postmortem brains of TS patients (Sarnat et al. 2011). Giant cells, the hallmark cells in the brain of tuberous sclerosis complex patients, have numerous lamellar mitochondria (Jozwiak et al. 2005). By contrast, TSC1 deficiency in T cells leads to decreased mitochondrial content and function, and this leads to a reduced numbers of both conventional and invariant natural killer T cells (O’Brien et al. 2011). These, albeit sparse, morphological data also indicate that disruption of the TSC–TOR pathway affects not only calcium signalling and ER function, but also mitochondrial function.
Cellular Exocytotic and Secretion Defects in Tuberous Sclerosis
The secretion of a number of peptide hormones is modulated by TSC–TOR signalling, and secretory defects are emerging as a key contributor to the multiple phenotypes characteristic of TS and in ASD pathogenesis. Effects on secretion/exocytosis are to be expected, in part due to effects of the TOR pathway on calcium homeostasis at multiple points, and also due to regulation of expression of key genes regulating exocytosis/secretion itself (Johnson et al. 2002). As discussed above, luminal ER, Golgi, and secretory vesicle Ca2+ levels regulate secretory pathway function and play an important role in the posttranslational processing, sorting, and packaging of secreted proteins. A rise in cytosolic Ca2+ concentration is also necessary to induce regulated secretion in most cell types, although in neurons, dense granule exocytosis requires a lower increase in cytosolic Ca2+ concentration for fusion than do synaptic vesicles (Scheenen et al. 1998). Hallmark giant TS cells are also characterized by a prominent Golgi apparatus, displaced rough ER, and large numbers of dense-core granules, suggestive of global changes in secretory traffic (Jozwiak et al. 2005). However, the effects of the TSC–TOR pathway on exocytosis/secretion remain the most poorly understood aspect of TSC–TOR signalling. Despite this scarcity of information, effects of the TSC–TOR pathway on five different secretory cargoes are known, as discussed next.
The first cargo for discussion is ghrelin, best known for effects on appetite and metabolism, but which also affects pituitary hormone secretion and sleep regulation (Portelli et al. 2012). Ghrelin is a peptide hormone, and emerging evidence suggests that decreased levels of ghrelin are associated with epilepsy and that ghrelin has anticonvulsant action (Portelli et al. 2012). It has also recently been suggested that ghrelin could be a therapeutic target for patients for both epilepsy and ASD, and ghrelin has been shown to promote LTP and memory formation (Portelli et al. 2012). Of relevance to TS, a reciprocal relationship has been found between TOR signalling and ghrelin. For example, in a mouse model, inhibition of TOR signalling by fasting leads to increased levels of ghrelin expression, increased gastric preproghrelin synthesis, and increased levels of secreted ghrelin. By contrast, activation of TOR signalling (which is the phenomenon occurring in TSC patients) decreases the expression of preproghrelin and the levels of secreted plasma ghrelin (Xu et al. 2009, 2010).
In the second example, the TSC–TOR pathway has been found to regulate secretion of neurotensin, another peptide hormone, which has been mostly intensively studied using cultured enteroendocrine cells, using nutrients to activate TOR. In this system, neurotensin secretion is negatively regulated in nutrient-rich medium in a TOR signalling-dependent manner (Li et al. 2011). By contrast, inhibition of TOR signalling, for example, using rapamycin, enhances both neurotensin expression and neurotensin secretion (Li et al. 2011). Of relevance, serum neurotensin levels in children with idiopathic ASD have recently been found to be higher than those of control children (Angelidou et al. 2010), and neurotensin has also been proposed as a potential target for novel ASD therapeutics (Ghanizadeh 2010).
Thirdly, in TSC2-deficient cells, intracellular trafficking of polycystin-1 (PC1), the product of the PKD1 gene, is disrupted, and PC1 is found sequestered within the Golgi apparatus, rather than being delivered to the cell surface. Re-expression of TSC2 restores the correct membrane localization of PC1 (Boletta 2009). Deletion of the PKD1 gene encoding PC1 frequently occurs in TS patients, due to its chromosomal location adjacent to TSC2. Deletion of PKD1 leads to a kidney disorder known as ADPCK (autosomal-dominant polycystic kidney disease) and 85 % of ADPCK patients also have tuberous sclerosis (Boletta 2009). However, ADPCK patients with deleted TSC2 have a more severe form of kidney disease, due to the additional TSC2 mutation modifying the ADPCK phenotype. This is because TSC2 loss affects the intracellular trafficking of PC1, leading to the retention of PC1 in the Golgi apparatus, rather than being delivered to the cell surface. Studies in animal models reveal that if Tsc2 function is restored, PC1 delivery to the cell surface is likewise restored (Boletta 2009). These data identify TSC2 as a determinant of PC1 function and, potentially, ADPKD severity.
Fourthly, caveolin-1, a membrane-associated scaffolding protein with multiple roles in signalling and traffic from the PM, is also mislocalized in cells lacking Tsc2 (Jones et al. 2004). In cells lacking TSC2, most caveolin-1 is displaced from the PM and is found on a Brefeldin-A-sensitive, post-Golgi compartment, distinct from the endosomes and lysosomes. Reintroduction of TSC2, but not a disease-causing mutant, reverses the caveolin-1 localization to the PM. Therefore, similar to the defect in PC1 delivery to the cell surface described above, when Tsc2 is deleted, caveolin-1 is retained in post-Golgi secretory vesicles and not delivered to the PM.
Finally, the vesicular stomatitis virus G protein (VSV-G), a viral glycoprotein usually targeted to the PM, is also retained in distinct post-Golgi vesicles and not transported to the PM in Tsc2-deficient cells (Jones et al. 2004). Together, these data suggest a role of TSC2 in regulating post-Golgi transport without affecting protein sorting, and the presence of mislocalized cell-surface proteins and secreted factors in TSC2-mutated cells is expected to contribute to the overall phenotype of TS. This has also been suggested as an etiological mechanism for idiopathic ASD (Aziz et al. 2014).
These findings are not unexpected, as a large body of evidence indicates that long-term synaptic plasticity, particularly of glutamatergic synapses, is critically dependent on cellular trafficking. For example, synaptic plasticity is crucially dependent on trafficking of metabotropic and ionotropic glutamate receptors, secretion of neuropeptides and hormones, while delivery of the postsynaptic density protein, PSD-95 (encoded by the DLG4 gene), to the synapse also requires vesicular transport (Yoshii et al. 2011). In addition, the well-characterized ASD genes encoding neurexins and neuroligins also highlight an expected role of secretory pathway deficits in other individuals with ASD. Originally, neurexins and neuroligins were considered to simply facilitate adhesion between the pre- and postsynapse; however, emerging evidence supports an essential role for neurexins in regulated secretion from both neurons and endocrine cells. Deletion of other “adhesion” genes, such as neural cell adhesion molecules (NCAMs) and cadherins (CDHs), also causes secretory deficits (Dudanova et al. 2006). These observations provide further support for the idea that cellular trafficking, including secretion and exocytosis, is of vital importance for long-term synaptic plasticity.
A comprehensive picture of how TSC1/TSC2 regulates the secretory pathway is still emerging, although regulation of calcium homeostasis via TOR signalling is likely to be a major contributor. In addition, some data indicates disruption of TSC1/TSC2 also affects the cytoskeletal network, which may inhibit secretory vesicle movement (Jones et al. 2004). More indirect effects of the TSC–mTOR pathway on the secretory pathway are also feasible. For example, TCS2 and Rheb (see Fig. 6) can be detected on ER and Golgi membranes (Wienecke et al.1996; Jones et al. 2004; Buerger et al. 2006), as can a large proportion of TOR (Drenan et al. 2004; Liu and Zheng 2007). Rheb also directly interacts with TOR (Fig. 6), and the Golgi localization of Rheb is essential for stimulation of TOR activity (Buerger et al. 2006). It appears that the ER and Golgi are crucial for signalling by TOR and central for TOR function and regulation of calcium homeostasis and cellular secretion. Therefore, TSC–TOR-dependent signalling pathways regulate protein synthesis, modulate metabolism, as well as control post-Golgi traffic of secretory cargo. This latter step will affect cellular secretion of neuropeptides, peptide hormones, and may affect delivery of transporters and receptors, to the cell surface.
Immune Deficits in TS and ASD
Recent evidence suggests the inflammatory immune response is significantly altered in TS patients, compared to controls (Haidinger et al. 2010). Ghrelin also plays an important role in cytokine secretion and immune function (Portelli et al. 2012). Cytokines, such as IL-1beta and/or IFN-gamma, in turn deplete the ER of Ca2+ and further activate the ER stress pathway (Cardozo et al. 2005; Matsuda et al. 2006) and can affect dendritic outgrowth and synapse formation (Kim et al. 2002; Ben Achour and Pascual 2010). Inhibition of TOR by rapamycin promotes production of proinflammatory cytokines, while deletion of TSC2 reverses this effect. In vivo, inhibition of TOR also regulates the inflammatory response and protects genetically susceptible mice against lethal infection (Weichhart et al. 2008). Therefore, the TSC–TOR pathway is a key regulator of innate immune homeostasis, and regulation of this pathway has clinical implications for infectious autoimmune diseases and cancer (Weichhart et al. 2008; Weichhart and Säemann 2008). Modification of cellular signalling via this pathway has broad implications for ASD, cancer etiology, infectious disease, and autoimmune disorders (Weichhart et al. 2008; Weichhart and Säemann 2008).
Of importance, these findings have parallels with patients with idiopathic ASD, where immune alterations are frequently detected, with multiple studies detecting increased levels of cytokines in serum and brains of individuals with ASD (Pardo et al. 2005; Vargas et al. 2005; Ashwood et al. 2011; Suzuki et al. 2011). These similarities further support the validity of studying syndromic forms of ASD to increase our understanding of idiopathic ASD. The abnormal levels of cytokine secretion detected in ASD patients may not only contribute to comorbid conditions in these individuals but may also contribute to the severity of ASD phenotype.
Gastrointestinal Dysfunction in TS and ASD
Gastrointestinal involvement has been reported in tuberous sclerosis (Moulis et al. 1992; Hizawa et al. 1994; Leung and Robson 2007) and in idiopathic ASD. While gastrointestinal dysfunction in TS may be due to cellular overgrowth, the dysregulated TOR signalling pathway also affects the secretory pathway (see above), nutrients are well known as activators of TORC1 activity and, overall, TORC1 signalling is known to have impact on digestion, gut motility, inflammation, appetite, and satiety signalling.
In the pancreas, TORC1 signalling regulates intestinal hormone secretion and the secretion of digestive enzymes from pancreatic acinar cells (Williams 2010; Xu et al. 2010; Li et al. 2011). For example, inhibition of TORC1 activity stimulates the expression of gastric ghrelin mRNA and protein and leads to an increase in concentration of plasma ghrelin, while gastrin synthesis and secretion are inhibited (Xu et al. 2010). In addition, hormones secreted from enteroendocrine cells, including ghrelin, leptin, and melatonin, also stimulate secretion of pancreatic digestive enzymes (Jaworek et al. 2010). The brain reciprocally regulates pancreatic exocrine function, and systemically circulating hormones have a complex interaction affecting the pancreas, gut, and the brain, with the latter often referred to as the “gut-brain axis” (Jaworek et al. 2010).
Another example of an enteroendocrine cell hormone whose secretion is regulated by TORC1 signalling is neurotensin (Li et al. 2011). Neurotensin regulates a number of digestive processes including gastrointestinal motility and pancreatic and biliary secretion. It exerts growth-promoting effects on normal gastrointestinal tissues and cancer cells and affects inflammatory mechanisms (Kalafatakis and Triantafyllou 2011). Neurotensin levels are elevated in patients with idiopathic ASD, and this has been proposed to stimulate immune cells, especially mast cells, and/or have direct effects on brain inflammation in ASD (Angelidou et al. 2010). Ghrelin likewise participates in crosstalk between the immune and neuroendocrine systems (Baatar et al. 2011). Therefore, the TOR pathway has multiple effects on gastrointestinal function.
From Disease Models to Therapeutic Agents: Implications for ASD
The new millennium has led to major breakthroughs in our understanding of the biology of ASDs and in progress towards effective therapeutics. To date, the most exciting findings have been those in animal models of ASD indicating that, even in adulthood, the phenotype is fully reversible (Ehninger et al. 2008; Silva and Ehninger 2009). Effective treatment of ASD in animal models has been achieved both with therapeutic drugs (see Barth and Bishop 2014) and by using “gene therapy” to change the expression of “modifier genes” that ameliorate the phenotype induced by the ASD mutation (see Bishop et al. 2014). Understanding the synaptic anomalies in TS, NF1, PTEN-associated ASD, FXS, and other syndromic forms of ASD has led to the development of animal models of disease, the testing of therapeutic drugs in these organisms, and has now proceeded to clinical trials. Many excellent review articles discuss the rationales behind these therapeutic approaches further (see Gipson and Johnston 2012; Hampson et al. 2012; Sahin 2012).
In the case of TS, the abnormal neuronal plasticity and memory defects in TSC2-deficient adult mice can be rescued by treating mice with rapamycin (known clinically as sirolimus) (Silva and Ehninger 2009). In humans, rapamycin was first used as an antifungal therapeutic but is now licenced for use as an immunosuppressant and antitumor agent (Ehninger and Silva 2011; Gipson and Johnston 2012). Rapamycin or analogs (“rapalogs”), such as everolimus, act on TOR signalling and partially compensate for the effect of TSC1/TSC2 mutation, which inappropriately activates TORC1 signalling (Fig. 6). Exciting recent research indicates the therapeutic benefits of rapamycin/rapalogs extend well beyond their roles as tumor suppressors, due to the pleiotropic effects induced by TORC1 hyperactivation. For example, in human cell-culture and mouse models, rapamycin therapy normalizes the innate immune-response deficits of TS (Weichhart et al. 2008), while in a mouse model of TS, a 3-day treatment of adult mice rescued synaptic plasticity and behavioral deficits (Ehninger et al. 2008). Several TS patients have been reported to show improvement in behavioral phenotypes on treatment with rapalogs (Chung et al. 2011), and a placebo-controlled, double-blind clinical trial is underway to assess the effects of everolimus on neurocognition, autistic features, epilepsy, and sleep habits (clinicaltrials.gov: NCT01289912). Therefore, therapeutics for TS and some other syndromic forms of ASD are looking promising. Of greater relevance, however, is the finding that many other ASD genes functionally interact with the TSC-TORC1 pathway (see Fig. 1), and indicates that therapies developed for syndromic ASD may be applicable to a subset(s) of other patients with ASD (discussed further in Barth and Bishop 2014).
Conclusion
In the past 20 years, impressive insights have been made into the genetic and cellular abnormalities of syndromic and nonsyndromic ASD. Understanding syndromic forms of ASD, such as TS, and the underlying dysregulation of the TOR pathway, dovetails with the many current models of ASD pathobiology, including dysregulated cellular calcium homeostasis, mitochondrial dysfunction, secretory pathway abnormalities, abnormal immunological findings, and gastrointestinal anomalies. Increased understanding of the TOR pathway and the contribution of the secretory pathway to neuronal function may lead to an improved understanding of the pathogenesis of ASD and illuminate novel targets for therapeutics. Furthermore, pathway analysis suggests a single therapeutic may be able to address both the core and noncore symptoms present in individuals with ASD. It is clear that significant progress has been made toward understanding the molecular pathways underlying TS and other syndromic forms of ASD; however many questions still need addressing if the links between genes, synapse function, and the ASD phenotype are to be fully understood.
Key Terms
-
Axons. Long protrusions from the main body of neuronal cells, which transmit signals to other cells.
-
Dendrites. Branched, treelike protrusions emanating from the main body of neuronal cells, which receive signals emitted from the axons of adjoining neurons.
-
Long-term potentiation. A long-lasting increase in synaptic signalling, thought to underpin learning and memory.
-
Long-term depression. A long-lasting decrease in synaptic signalling.
-
Synaptic plasticity. Changes in synaptic transmission leading to an increase or decrease in the efficacy of the synapse.
Key Facts of the TSC–TOR Pathway
-
The TSC1 and TSC2 gene products can interact to form the tuberous sclerosis complex (TSC).
-
Mutation in either the TSC1 or TSC2 gene causes the genetic disorder, tuberous sclerosis (TS), and is characterized by benign tissue growths, epilepsy, and autism.
-
The TSC regulates a lipid-anchored, membrane-associated GTPase, Rheb (Ras homolog enriched in brain).
-
TSC inactivation of Rheb leads to inactivation a serine/threonine protein kinase, referred to as TOR (target of rapamycin) or mTOR (mammalian TOR).
-
TOR is a key cellular nutrient, energy, and stress sensor and is part of two protein complexes: TORC1 or TORC2.
-
TORC1 is the only known target of activated Rheb.
-
TSC2 stimulates the conversion of Rheb–GTP (active) to Rheb–GDP (inactive), thereby inhibiting TORC1.
-
The best-characterized effect of TORC1 activation is an increase in protein translation due to phosphorylation of eIF4EBP1 (eukaryotic initiation factor 4E-binding protein 1) and p70S6K (p70 ribosomal S6 kinase).
-
TSC negatively regulates TORC1, and in TS patients TOR signalling via TORC1 is hyperactivated.
-
Of relevance to autism, TOR signalling pathways are not only linked to tumorigenesis but also affect synaptic plasticity and neuroendocrine function.
-
Mutations in genes affecting upstream and downstream TORC1 signalling molecules are also implicated in autism.
-
Rapamycin (sirolimus) inhibits TORC1 signalling and can reverse the effects of TSC1 or TSC2 mutations in model systems.
-
Rapamycin, widely used as an immunosuppressant, is now licenced for use as a cancer therapeutic and is in clinical trials to assess therapeutic benefits on epilepsy and autism.
Summary Points
-
Mutation of single genes causing syndromic ASD, such as those causing tuberous sclerosis (TS), affects synapse morphology and plasticity, a phenomenon common to other forms of ASD.
-
Understanding the cellular pathways and networks of interacting gene products can explain noncore phenotypes detected in individuals with syndromic forms of ASD.
-
The TSC-TORC1 pathway is dysregulated in TS and regulates many downstream cellular processes.
-
The TSC-TORC1 pathway regulates intracellular calcium signalling, and calcium-regulated processes are also reported to be affected in other types of ASD.
-
Mitochondrial morphology and function may also be aberrant in TS and may contribute to synaptic dysfunction in TS, as suggested for idiopathic ASD.
-
Secretion of several peptide hormones and targeting of cell-surface proteins is dysregulated in TS, and secretory pathway anomalies are also documented in idiopathic ASD.
-
The TSC-TORC1 pathway also regulates the innate immune response, another dysfunction reported in idiopathic ASD.
-
Gastrointestinal involvement has been reported in TS and idiopathic ASD and the TSC-TORC1 pathway is known to impact on digestion, gut motility, appetite, and satiety.
-
Therefore both ASD and a range of additional signs and symptoms can be caused by mutation in a single gene, because many genes act in complex cellular pathways.
-
Understanding the cellular pathways affected by ASD genes will help understand the noncore symptoms of individuals with ASD, the mechanisms causative of the core ASD phenotype, and in the design of effective therapeutics.
References
Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97.
Angelidou A, Francis K, Vasiadi M, et al. Neurotensin is increased in serum of children with autistic disorder. J Neuroinflammation. 2010;7:48.
Antonarakis SE, Beckmann JS. Mendelian disorders deserve more attention. Nat Rev Genet. 2006;7:277–82.
Ashwood P, Corbett BA, Kantor A, et al. In search of cellular immunophenotypes in the blood of children with autism. PLoS One. 2011;6:e19299.
Auerbach BD, Bear MF. Loss of FXMRP decouples metabotropic glutamate receptor dependent priming of long-term potentiation from protein synthesis. J Neurophysiol. 2010;104:1047–51.
Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–86.
Austin CD, Shields D. Prosomatostatin processing in permeabilized cells. Calcium is required for prohormone cleavage but not formation of nascent secretory vesicles. J Biol Chem. 1996;271:1194–9.
Aziz A, Harrop SP, Bishop N. DIA1R is an X-linked gene related to DIA1. PLoS One. 2011a;6:e14534.
Aziz A, Harrop SP, Bishop N. Characterization of the DIA1 protein family. PLoS One. 2011b;6:e14547.
Aziz A, Karmi T, Bishop N. Autism and the DIA1-family: role of the cellular secretory pathway. In: Patel VB, Preedy VR, Martin C, editors. Comprehensive guide to autism. New York: Springer; 2014.
Baatar D, Patel K, Taub DD. The effects of ghrelin on inflammation and the immune system. Mol Cell Endocrinol. 2011;340:44–58.
Barth C, Bishop N. Autism: comparative genomics and interactomics. In: Patel VB, Preedy VR, Martin C, editors. Comprehensive guide to autism. New York: Springer; 2014.
Ben Achour S, Pascual O. Glia: the many ways to modulate synaptic plasticity. Neurochem Int. 2010;57:440–5.
Benach JL, Li E, McGovern MM. A microbial association with autism. MBio. 2012;3:e00019–12.
Benvenuto A, Moavero R, Alessandrelli R, et al. Syndromic autism. World J Pediatr. 2009;5:169–76.
Betancur C. Etiological heterogeneity in autism spectrum disorders. Brain Res. 2011;1380:42–77.
Bill BR, Geschwind DH. Genetic advances in autism. Curr Opin Genet Dev. 2009;19:271–8.
Bishop N, Aziz A, Barth C. Understanding phenotypic variation in autism spectrum disorder: insights from syndromic forms of autism. In: Patel VB, Preedy VR, Martin C, editors. Comprehensive guide to autism. New York: Springer; 2014.
Boletta A. Emerging evidence of a link between the polycystins and the mTOR pathways. Pathogenetics. 2009;2:6.
Bolton PF. Medical conditions in autism spectrum disorders. J Neurodev Disord. 2009;1:102–13.
Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19:231–4.
Brose N, O’Connor V, Skehel P. Synaptopathy: dysfunction of synaptic function? Biochem Soc Trans. 2010;38:443–4.
Brown AC, Mehl-Madrona L. Autoimmune and gastrointestinal dysfunctions: does a subset of children with autism reveal a broader connection? Expert Rev Gastroenterol Hepatol. 2011;5:465–77.
Buerger C, DeVries B, Stambolic V. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem Biophys Res Commun. 2006;344:869–80.
Buie T, Fuchs GJ 3rd, Furata GT, et al. Recommendations for evaluation and treatment of common gastrointestinal problems in children with ASDS. Pediatrics. 2010;125(Suppl 1):S19–S29.
Byrnes KR, Loane DJ, Faden AI. Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics. 2009;6:94–107.
Caglayan AO. Genetic causes of syndromic and non-syndromic autism. Dev Med Child Neurol. 2010;52:130–8.
Cardozo AK, Ortis F, Storling J, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete ER Ca2+, leading to induction of ER stress. Diabetes. 2005;54:452–61.
Castagnola M, Messana I, Inzitari R, et al. Hypo-phosphorylation of salivary peptidome as a clue to the molecular pathogenesis of autism. J Proteome Res. 2008;7:5327–32.
Castellani ML, Conti CM, Kempuraj DJ, et al. Autism and immunity. Int J Immunopathol Pharmacol. 2009;22:15–9.
Chévere-Torres I, Kaphzan H, Bhattacharya A, et al. Metabotropic glutamate receptor-dependent long-term depression is impaired due to elevated ERK signaling in the ΔRG mouse model of tuberous sclerosis. Neurobiol Dis. 2012;45:1101–10.
Chung TK, Lynch ER, Fiser CJ, et al. Psychiatric comorbidity and treatment response in patients with tuberous sclerosis. Ann Clin Psychiatry. 2011;23:263–9.
D’Souza AD, Parikh N, Kaech SM, Shadel GS. Convergence of multiple signaling pathways is required to coordinately up-regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion. 2007;7:3743–85.
de Brito OM, Scorrano L. Mitofusin-2 regulates mitochondrial and ER morphology and tethering. Mitochondrion. 2009;9:222–6.
de Brito OM, Scorrano L. Spatial organization of the ER-mitochondria relationship. EMBO J. 2010;29:2715–23.
Dölen G, Bear MF. Fragile x syndrome and autism: from disease model to therapeutic targets. J Neurodev Disord. 2009;1:133–40.
Drenan RM, Liu X, Bertram PG, et al. FKBP12-rapamycin-associated protein or mTOR localization in the ER and Golgi apparatus. J Biol Chem. 2004;279:772–8.
Dudanova I, Sedej S, Ahmad M, et al. Important contribution of alpha-neurexins to Ca2+ triggered exocytosis of secretory granules. J Neurosci. 2006;26:10599–613.
Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1. Cell Signal. 2009;21:827–35.
Ehninger D. From genes to cognition in tuberous sclerosis. Neuropharmacology. 2013;68:97–105.
Ehninger D, Silva AJ. Rapamycin for treating tuberous sclerosis and ASDs. Trends Mol Med. 2011;17:78–87.
Ehninger D, Li W, Fox K, et al. Reversing neurodevelopmental disorders in adults. Neuron. 2008;60:950–60.
Frégeau MO, Régimbald-Dumas Y, Guillemette G. Positive regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by mTOR. J Cell Biochem. 2011;112:723–33.
Gargus JJ. Genetic calcium signaling abnormalities in the central nervous system: seizures, migraine, and autism. Ann NY Acad Sci. 2009;1151:133–56.
Gaut JR. Threonine phosphorylation of BiP maps to its protein binding domain. Cell Stress Chaperones. 1997;2:252–62.
Ghanizadeh A. Targeting neurotensin as a potential novel approach for the treatment of autism. J Neuroinflammation. 2010;7:58.
Gipson TT, Johnston MV. Plasticity and mTOR. Neural Plast. 2012;2012:486402.
Gomes LC, Di Benedetto G, Scorrano L. Essential amino acids and glutamine regulate induction of mitochondrial elongation during autophagy. Cell Cycle. 2011;10:2635–9.
Haidinger M, Hecking M, Weichhart T, et al. Sirolimus in renal transplant recipients with tuberous sclerosis complex. Transpl Int. 2010;23:777–85.
Hampson DR, Gholizadeh S, Pacey LK. Pathways to drug development for ASDs. Clin Pharmacol Ther. 2012;91:189–200.
Hizawa K, Iida M, Matsumoto T, et al. Gastrointestinal involvement in tuberous sclerosis. J Clin Gastroenterol. 1994;19:46–9.
Jan YN, Jan LY. Branching out: mechanisms of dendritic arborization. Nat Rev Neurosci. 2010;11:316–28.
Jaworek J, Nawrot-Porabka K, Leja-Szpak A, et al. Brain-gut axis in the modulation of pancreatic enzyme secretion. J Physiol Pharmacol. 2010;61:523–31.
Johnson JD, Klausen C, Habibi HR, Chang JP. Function-specific calcium stores selectively regulate growth hormone secretion, storage, and mRNA level. Am J Physiol Endocrinol Metab. 2002;282:E810–19.
Jones KA, Jiang X, Yamamoto Y, Yeung RS. Tuberin is a component of lipid rafts: role in post-Golgi transport. Exp Cell Res. 2004;295:512–24.
Jozwiak J, Jozwiak S, Skopinski P. Immunohistochemical and microscopic studies on giant cells in tuberous sclerosis. Histol Histopathol. 2005;20:1321–6.
Jülich K, Sahin M. Autism spectrum disorders in tuberous sclerosis. In: Patel VB, Preedy VR, Martin C, editors. Comprehensive guide to autism. New York: Springer; 2014.
Kalafatakis K, Triantafyllou K. Contribution of neurotensin in the immune and neuroendocrine modulation of enteric function. Regul Pept. 2011;170:7–17.
Kalnina Z, Silina K, Bruvere R, et al. Molecular characterisation and expression analysis of SEREX-defined antigen NUCB2 in gastric epithelium, gastritis and gastric cancer. Eur J Histochem. 2009;53:7–18.
Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu Rev Neurosci. 2006;29:325–62.
Kim IJ, Beck HN, Lein PJ, Higgins D. Interferon gamma induces retrograde dendritic retraction and inhibits synapse formation. J Neurosci. 2002;22:4530–9.
Krey JF, Dolmetsch RE. Molecular mechanisms of autism: a possible role for Ca2+ signaling. Curr Opin Neurobiol. 2007;17:112–19.
Kumar V, Fahey PG, Jong YJ, et al. Activation of intracellular metabotropic glutamate receptor 5 in striatal neurons leads to up-regulation of genes associated with sustained synaptic transmission. J Biol Chem. 2012;287:5412–25.
Kuwajima M, Dehoff MH, Furuichi T, et al. Localization and expression of group I metabotropic glutamate receptors in the mouse. J Neurosci. 2007;27:6249–62460.
Leung AK, Robson WL. Tuberous sclerosis complex. J Pediatr Health Care. 2007;21:108–14.
Li J, Liu J, Song J, et al. mTORC1 inhibition increases neurotensin secretion and expression. Am J Physiol Cell Physiol. 2011;301:C213–26.
Lichtenstein P, Carlström E, Råstam M, et al. The genetics of autism spectrum disorders and related disorders in childhood. Am J Psychiatry. 2010;167:1357–63.
Lin P, Yao Y, Hofmeister R, et al. Overexpression of Calnuc increases agonist and thapsigargin releasable Ca2+ storage in the Golgi. J Cell Biol. 1999;145:279–89.
Lin W, Bailey SL, Ho H, et al. The integrated stress response prevents demyelination. J Clin Invest. 2007;117:448–56.
Lin P, Fischer T, Lavoie C, et al. Calnuc plays a role in dynamic distribution of Galphai and modulates ACTH secretion. Mol Neurodegener. 2009;4:15.
Liu X, Zheng XF. Endoplasmic reticulum and Golgi localization sequences for mTOR. Mol Biol Cell. 2007;18:1073–82.
Lodish MB, Stratakis CA. Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes. Best Pract Res Clin Endocrinol Metab. 2010;24:439–49.
Matsuda T, Nagano T, Takemura M, Baba A. Topics on the Na+/Ca2+ exchanger. J Pharmacol Sci. 2006;102:22–6.
Mattson MP. Mitochondrial regulation of neuronal plasticity. Neurochem Res. 2007;32:707–15.
Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–66.
McDougle CJ, Erickson CA, Stigler KA, et al. Neurochemistry in the pathophysiology of autism. J Clin Psychiatry. 2005;66 Suppl 10:9–18.
Miles JH. Autism spectrum disorders – a genetics review. Genet Med. 2011;13:278–94.
Moulis H, Garsten JJ, Marano AR, Elser JM. Tuberous sclerosis complex: review of the gastrointestinal manifestations. Am J Gastroenterol. 1992;87:914–18.
O’Brien TF, Gorentla BK, Xie D, et al. Regulation of T-cell survival and mitochondrial homeostasis by TSC1. Eur J Immunol. 2011;41:3361–70.
Palmieri L, Persico AM. Mitochondrial dysfunction in autism: cause or effect? Biochim Biophys Acta. 2010;1797:1130–7.
Pardo CA, Vargas DL, Zimmerman AW. Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry. 2005;17:485–95.
Peça J, Feng G. Cellular and synaptic network defects in autism. Curr Opin Neurobiol. 2012;22:866–72.
Penzes P, Cahill ME, Jones KA, et al. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–93.
Portelli J, Michotte Y, Smolders I. Ghrelin: an emerging new anticonvulsant neuropeptide. Epilepsia. 2012;53:585–95.
Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:255–74.
Sahin M. Targeted treatment trials for tuberous sclerosis and autism: no longer a dream. Curr Opin Neurobiol. 2012;22:895–901.
Sarnat HB, Flores-Sarnat L, Hader W, et al. Mitochondrial ‘hypermetabolic’ neurons in paediatric epileptic foci. Can J Neurol Sci. 2011;38:909–17.
Scheenen WJ, Wollheim CB, Pozzan T, Fasolato C. Ca2+ depletion from granules inhibits exocytosis. J Biol Chem. 1998;273:19002–8.
Sebastián D, Hernández-Alvarez MI, Segalés J, et al. Mitofusin 2 links mitochondrial and ER function with insulin signaling. Proc Natl Acad Sci USA. 2012;109:5523–8.
Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–9.
Silva AJ, Ehninger D. Adult reversal of cognitive phenotypes in neurodevelopmental disorders. J Neurodev Disord. 2009;1:150–7.
Smalley SL. Autism and tuberous sclerosis. J Autism Dev Disord. 1998;28:407–14.
Suzuki K, Matsuzaki H, Iwata K, et al. Plasma cytokine profiles in subjects with high-functioning ASDs. PLoS One. 2011;6:e20470.
Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784:116–32.
Talkowski ME, Rosenfeld JA, Blumenthal I, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–37.
Tamás P, Hawley SA, Clarke RG, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med. 2006;203:1665–16670.
Troca-Marín JA, Alves-Sampaio A, Montesinos ML. Deregulated mTOR-mediated translation in intellectual disability. Prog Neurobiol. 2012;96(2):268–82.
Tsukumo Y, Tomida A, Kitahara O, et al. Nucleobindin 1 controls the unfolded protein response by inhibiting ATF6 activation. J Biol Chem. 2007;282:29264–72.
Tu JC, Xiao B, Naisbitt S, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–92.
Tyburczy ME, Kaminska B. Subependymal giant cell astrocytoma: gene expression profiling. In: Hayat MA, editor. Tumours of the central nervous system, vol. 5. Dordrecht: Springer; 2012.
Valenzuela JI, Jaureguiberry-Bravo M, Couve A. Neuronal protein trafficking: emerging consequences of ER dynamics. Mol Cell Neurosci. 2011;48:269–77.
Vargas DL, Nascimbene C, Krishnan C, et al. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81.
Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005;85:201–79.
Weichhart T, Säemann MD. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann Rheum Dis. 2008;67:iii70–4.
Weichhart T, Costantino G, Poglitsch M, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–77.
Wienecke R, Maize JC, Shoarinejad F, et al. Co-localization of the TSC2 product tuberin with its target Rap1 in the Golgi. Oncogene. 1996;13:913–23.
Williams JA. Regulation of acinar cell function in the pancreas. Curr Opin Gastroenterol. 2010;26:478–83.
Wu X, Kihara T, Akaike A, et al. PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochem Biophys Res Commun. 2010;393:514–851.
Xiao B, Tu JC, Worley PF. Homer: a link between neural activity and glutamate receptor function. Curr Opin Neurobiol. 2000;10:370–4.
Xu G, Li Y, An W, et al. Gastric mammalian target of rapamycin signaling regulates ghrelin production and food intake. Endocrinology. 2009;150:3637–44.
Xu G, Li Y, An W, et al. Regulation of gastric hormones by systemic rapamycin. Peptides. 2010;31:2185–92.
Yamamoto Y, Jones KA, Mak BC, et al. Multicompartmental distribution of the tuberous sclerosis gene products. Arch Biochem Biophys. 2002;404:210–17.
Yanagihara N, Oishi Y, Yamamoto H, et al. Phosphorylation of chromogranin A and catecholamine secretion stimulated by elevation of intracellular Ca2+. J Biol Chem. 1996;271:17463–8.
Yoo SH. Secretory granules in inositol 1,4,5-trisphosphate-dependent Ca2+ signaling in the cytoplasm of neuroendocrine cells. FASEB J. 2010;24:653–64.
Yoo SH. Role of secretory granules in inositol 1,4,5-trisphosphate-dependent Ca(2+) signaling. Cell. Cell Calcium. 2011;50:175–83.
Yoo SH, Hur YS. Enrichment of the inositol 1,4,5-trisphosphate receptor/Ca2+ channels in secretory granules and essential roles of chromogranins. Cell Calcium. 2012;51:342–50.
Yoshii A, Murata Y, Kim J, et al. TrkB and protein kinase Mζ regulate synaptic localization of PSD-95 in developing cortex. J Neurosci. 2011;31:11894–904.
Zhou J, Parada LF. PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol. 2012;22:873–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this entry
Cite this entry
Barth, C., Aziz, A., Bishop, N. (2014). Integrating Pathogenic Models of Autism: Pathway and Network Analysis. In: Patel, V., Preedy, V., Martin, C. (eds) Comprehensive Guide to Autism. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-4788-7_193
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4788-7_193
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-4787-0
Online ISBN: 978-1-4614-4788-7
eBook Packages: Behavioral ScienceReference Module Humanities and Social SciencesReference Module Business, Economics and Social Sciences