
Abstract
Tuberous sclerosis complex (TSC) is a genetic, multisystem disorder that affects the skin, kidneys, heart, and lungs, and, in the vast majority of cases, the brain. It is caused by mutations of either the TSC1 or TSC2 gene. These genes function as inhibitors of the mTOR Complex 1 (mTORC1) pathway, a master regulator in the cell that integrates growth factor, energy, and nutrient signals to control cellular growth and protein translation. The TSC/mTORC1 pathway is also involved in various steps of neuronal development and maturation, such as neuronal polarization, axon guidance, synaptic plasticity, and myelination. A loss-of-function mutation in either of the TSC genes results in constitutive overactivation of mTORC1 and a loss of control over downstream events.
Neurological symptoms of TSC can include epilepsy, cognitive disabilities, and behavioral abnormalities such as attention deficit hyperactivity disorder (ADHD), autism, and autism spectrum disorders (ASD). About 50 % of TSC patients are diagnosed with ASD, with a male-to-female ratio of 1:1. How TSC causes this phenomenon is not yet clear. It was originally thought that the benign hamartomas called cortical tubers were responsible for autism and other neurological symptoms of this disorder. However, research from the past decade, including rodent models with learning deficits and abnormal social approach behavior in the absence of a tuber-like brain pathology, indicates that microstructural abnormalities such as axon connectivity and myelination, as well as altered glutamate and GABAergic neurotransmission, render individuals with TSC highly susceptible to autism.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

Keywords
- Autism Spectrum Disorder
- Autism Spectrum Disorder
- Attention Deficit Hyperactivity Disorder
- Diffusion Tensor Imaging
- Tuberous Sclerosis Complex
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Tuberous sclerosis complex (TSC) is a genetic disorder caused by heterozygous mutations of the TSC1 or TSC2 genes and the loss of function of their respective gene products, TSC1 (tuberin) and TSC2 (hamartin). It is characterized by the growth of benign tumors (hamartomas) in multiple organ systems. In addition to manifestations of tumors in the kidneys, skin, heart, lungs, liver, and brain, more than 90 % of patients develop neurological symptoms such as epilepsy, cognitive disabilities, sleep disorders, autism, autism spectrum disorders (ASD), and other behavioral problems such as attention deficit hyperactivity disorder (ADHD).
Historical Background
The first descriptions of patients with symptoms resembling those of TSC date back to 1835, when the French physician Pierre Rayer reported patients with skin abnormalities resembling facial angiofibromas seen in TSC. However, it was not until 1880 that the French neurologist Désiré-Magloire Bourneville recognized as a pathological entity the dense structures he discovered in brains of patients suffering from epilepsy and intellectual disability. He referred to this new entity as “tuberous sclerosis” (“sclérose tubéreuse des convolutions cérébrales”) due to the tuberous appearance of the structures on macroscopic examination (Fig. 1). He and his colleague, Édouard Brissaud, later described a patient with brain tubers, kidney tumors, and skin abnormalities, prompting a surge of articles describing new symptoms and findings over the following decades.

Brain tubers by Dres. Bourneville and Brissaud. “Encéphalite ou sclérose tubéreuse des circonvolutions cérébrales” (Illustration from Bourneville and Brissaud (1881), Archives de Neurologie, not in copyright)
ASD-like features in patients with TSC were first recognized in 1932 by MacDonald Critchley and Charles J. C. Earl, long before the term “autism” was used by Leo Kanner to describe patients with similar symptoms. A cohort of 29 TSC patients whom they tended were described as having ASD-like features such as social withdrawal, impaired social skills and communication, and stereotypical behaviors (For a more extensive view on the history of TSC, please see Whittemore 2010).
Diagnostic Criteria
TSC is a multisystem disorder with wide phenotypic variability. No single sign or symptom is specific to TSC, making diagnosis a matter of careful clinical evaluation. In 1998 the Tuberous Sclerosis Consensus Conference published revised diagnostic criteria based on the presence of major and/or minor features. A clinical diagnosis of TSC requires TSC-associated features in two or more organ systems or two or more different features in the same organ (Table 1) (Roach et al. 1998). Revised diagnostic criteria are being decided upon and will be announced shortly.
Genetics
Tuberous sclerosis complex is caused by heterozygous mutations in either the TSC1 or the TSC2 gene. TSC2 is located on chromosome 16 (16p13.3) and was discovered in 1993. TSC1, located on chromosome 9 (9q34), was described several years later (European Chromosome 16 Tuberous Sclerosis Consortium 1993; van Slegtenhorst et al. 1997). More than 1,500 mutations have been reported (please refer to the TSC mutation database, http://chromium.liacs.nl/LOVD2/TSC/home.php).
TSC occurs with a frequency of 1:6,000 (Osborne et al. 1991), and mutations can be detected in about 85 % of patients with a definite clinical diagnosis of TSC. Seventy percent of patients carry de novo germline mutations, while 30 % inherit the mutation in an autosomal dominant fashion. Penetrance is thought to be complete, but phenotypic variety is wide. Mutations in the TSC2 gene occur more frequently de novo and result in a more severe phenotype compared to mutations in TSC1, with a higher likelihood of seizures and moderate to severe mental disability as well as a higher tuber count. Genotype-phenotype analyses have shown a larger percentage – about 90 % – of frameshift or nonsense mutations (resulting in a truncated Tsc protein or no protein at all) in patients with TSC1 mutations compared to patients with TSC2 mutations, who often carry missense mutations. This finding is in accordance with clinical data showing that patients carrying TSC1 mutations have a milder phenotype – TSC1 missense mutations presumably cause a subtle phenotype and hence might often be missed. Apart from this, genotype-phenotype analyses have failed to show further correlations, possibly due to the broad phenotype and genotype variability (reviewed in Au and Northrup 2010; Kwiatkowski 2010).
Tsc1/Tsc2 Signaling
The mTOR Pathway
mTOR (mammalian target of rapamycin) is a ubiquitously expressed kinase that functions in two distinct complexes, mTORC1 (mTOR Complex 1) and mTORC2 (mTOR Complex 2). mTORC2 mainly regulates the actin cytoskeleton, while mTORC1 acts as a central signaling pathway that integrates growth factor, energy, and nutrient signals to control cellular growth and protein translation (for a review of the mTOR pathway, see Laplante and Sabatini 2012). In addition to mTOR and other proteins, mTORC1 consists of the protein raptor (regulatory associated protein of mTOR), which acts as a scaffold to recruit substrates to the catalytic domain of mTOR.
The gene products TSC1 (tuberin) and TSC2 (hamartin) exert an inhibitory control on the mTORC1 pathway by forming a heterodimeric complex in which TSC1 prevents the degradation of TSC2, while TSC2 acts as a GTPase-activating protein on the small G-protein RHEB (Ras homologue enriched in brain), resulting in its inactivation and subsequent mTORC1 inhibition. Mutation of either the TSC1 or TSC2 gene results in tuberous sclerosis complex disease, so it is clear that both are required for proper complex function. Inactivating mutations in TSC1 or TSC2 results in overactivation of mTORC1 and uncontrolled signaling to its downstream targets.
While mTORC2 is largely insensitive to rapamycin treatment, mTORC1 kinase activity is sensitive to the compound, which was originally used as an immunosuppressant after kidney transplantations. When bound to FKBP12, rapamycin competes with raptor for binding to mTOR. Binding of a growth factor to its cell surface receptor activates PI3 kinase and, in turn, the kinase AKT, which then phosphorylates TSC2, leading to the subsequent dissociation of the TSC1: TSC2 complex. Growth factor-activated phosphorylation of TSC2 also occurs through the RAS/MAP kinase pathway and through extracellular signal-regulated kinase (ERK). Complex dissociation releases the inhibitory effect of Tsc2 on RHEB, which allows mTORC1 to phosphorylate its substrates, including the best-studied S6 kinase 1 (S6K1) and eIF-4E binding proteins (4E-BPs) that mediate the control of cell size and protein synthesis through regulation of translation initiation.
Another upstream signaling pathway regulating TSC1/2 activity is the AMPK pathway, which activates TSC2 in response to a low cellular energy status and thereby halts mTORC1-mediated protein translation and cellular growth.
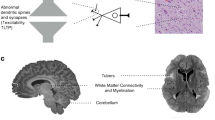
In addition to regulating cell growth and protein translation, the mTORC1 pathway has also been shown to be important for many steps in neuronal development and maturation, such as neuronal polarity, axon guidance, dendritic spine density, synaptogenesis, and myelination. TSC1 and TSC2 expression levels remain upregulated after embryogenesis in the brain in contrast to most other tissues, which suggests that TSC-mediated regulation of the mTOR pathway plays an important role not only in developing but also in mature neurons. Indeed, mTOR has also been shown to be important for functions in the mature brain, such as synaptic plasticity (Fig. 2) (reviewed by Hoeffer and Klann 2010).

The TSC/mTOR pathway and ASD. Schematic description of the mTOR pathway. Growth factors bind to receptors and lead to sequential activation of PI3 kinase and AKT, which inhibits TSC-mediated negative control on mTORC1. Mutation of TSC1 or TSC2 results in hyperactivation of mTORC1 and uncontrolled downstream signaling that can be blocked by the drug rapamycin. Stars mark the targets in the mTORC1 pathway that have been associated with autism or autism spectrum disorders
Autism and the mTOR Pathway
Dysregulation of the mTOR pathway is seen in several syndromes commonly associated with ASD (Fig. 2). Upstream regulators of mTOR activity that have been linked to ASD include PTEN (phosphatase and tensin homolog), which inhibits PI3 kinase activity; NF1 (neurofibromin 1), a regulator of RAS/MAP kinase signaling involved in neurofibromatosis type I; and common variants of the receptor tyrosine kinase MET, which signal through PI3 kinase. One downstream effector thought to be associated with ASD is eIF4E, a eukaryotic translation initiation factor involved in directing ribosomes to the cap structure of mRNAs. Its activity is indirectly regulated by mTOR through the 4E-BPs, which upon phosphorylation release eIF4E and enable protein translation initiation (reviewed by Ehninger and Silva 2011).
Tuberous Sclerosis Complex and Autism Spectrum Disorders
Even though neurological and behavioral abnormalities are not part of the criteria required for TSC diagnosis, these symptoms are very common in TSC and often have a major effect on the daily lives of patients. Table 2 gives an overview of common cognitive and behavioral problems associated with TSC. A consensus guideline published in 2005 recommends that cognitive and behavioral profiles of individuals with TSC should be assessed at regular intervals (de Vries et al. 2005).
The prevalence of autism in TSC patients is dramatically higher than in the general population. Up to 50 % of TSC patients are diagnosed with autism spectrum disorder (ASD), and TSC accounts for 1–4 % of overall autism cases (Bolton et al. 2002; Smalley 1998). Autistic features such as impaired communication and social interaction or poor eye contact are often evident early, even at 18 months of age (Jeste et al. 2008). While the general population has a male-to-female ratio of 4:1, the sex ratio of autism in TSC is 1:1, indicating that a TSC1/2 mutation is a high and gender-independent risk factor for developing autism.
Association of ASD with Other Neurological Symptoms
The majority of TSC patients with autism or ASD have seizures as well, and the onset of seizures usually precedes the emergence of autistic features. Epilepsy is the most common neurological symptom of TSC, affecting up to 90 % of patients. Complex partial and primary generalized seizures are the most common seizure types, and 50 % of patients also suffer from infantile spasms.
Thirty percent of children with ASD who do not have TSC have epilepsy, raising the question of whether early seizure activity might promote the development of autism later in life. However, many children with infantile spasms never develop autism, and more importantly, some TSC patients with autism never have seizures, which makes a clear-cut cause-effect relationship of seizures in infancy leading to autism unlikely.
Infantile spasms in TSC respond to treatment with vigabatrin, a drug that raises the levels of the inhibitory neurotransmitter GABA in the brain. Abnormalities in GABAergic interneurons have also been proposed to be involved in the pathogenesis of autism in TSC, suggesting that infantile spasms and autism may be due to a similar pathological mechanism rather than a result of each other (reviewed in Curatolo et al. 2010; Ess 2009; Paciorkowski et al. 2011).
Despite the fact that approximately half of TSC patients have an IQ in the normal range, the disease shows a bimodal IQ distribution. Thirty percent are severely affected with IQs in the very low range, while the remaining 70 % show a normal yet slightly left-shifted IQ distribution (de Vries and Howe 2007; Joinson et al. 2003). There is a significant correlation between autism and cognitive impairment in TSC patients, as children with TSC and autism have on average higher cognitive impairment than those without autism. However, 20 % of TSC patients with autism have an IQ in the normal range (de Vries and Howe 2007; Jeste et al. 2008).
Association of ASD with Neuropathological Abnormalities
Tubers
Structural abnormalities of the central nervous system (CNS) are very common in TSC. Hallmark features include the so-called tubers (the correlate of hamartomas in the brain) that form during embryonal development and are typically localized at the junction of gray and white matter. Many patients also have subependymal nodules (SENs) in the lateral ventricles, which may transform into subependymal giant cell astrocytomas (SEGAs) that can cause obstruction at the foramen of Monro.
The mechanism behind the formation of tubers remains unclear. Some TSC manifestations require loss of both alleles of TSC1 or TSC2 (loss of heterozygosity or LOH) as a “second hit” in order to develop. This has been observed in angiomyolipomas, rhabdomyomas, lymphangioleiomyomatosis (LAM), and SEGAs but not consistently in tubers, which arise as cortical or subcortical malformations during early brain development and contain large cells expressing markers of neuronal and glia cell differentiation (Crino 2004).
Previously, a high tuber count and/or frontal and occipital tuber location was thought to be associated with poor intellectual outcome (Goodman et al. 1997; Jambaque et al. 1991). More recent studies have shown that tuber size and overall tuber volume rather than tuber number correlate with cognitive disabilities (Jansen et al. 2008). However, even though tubers often are associated with neurological symptoms, TSC patients can have symptoms in the absence of tubers and vice versa, which suggests that abnormalities other than tubers may account for the neurological phenotypes.
In contrast, ASDs in TSC patients have been associated with temporal lobe abnormalities such as tubers or epileptiform activity (Bolton et al. 2002) as well as cerebellar lesions (Bolton et al. 2002; Eluvathingal et al. 2006; Weber et al. 2000). Cerebellar abnormalities have also been found in many patients with ASD without TSC, including reduced number of Purkinje cells and abnormalities of the deep cerebellar nuclei (reviewed by Fatemi et al. 2012). Because ASD patients can develop cerebellar abnormalities with or without TSC, it seems likely that there is a connection between the mechanisms leading to ASDs and cerebellar abnormalities. A recently generated mouse model demonstrates that loss of Tsc1 gene function in cerebellar Purkinje cells is sufficient to induce autistic-like behaviors in mice, providing further support for a role of the cerebellum in the neuronal circuitry underlying autism (Tsai et al. 2012).
White Matter Abnormalities and Abnormal Neuronal Connectivity
White matter abnormalities, particularly abnormalities of the corpus callosum, which represents the major interhemispheric connection in the brain, have been reported in autism patients in large studies using different imaging techniques and pathological studies (reviewed by Fatemi et al. 2012).
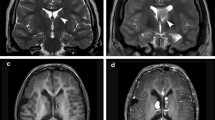
In contrast, white matter appears largely normal in the brains of TSC patients on conventional MRIs. Recent advances using diffusion tensor imaging (DTI), a technique based on the ability to measure water diffusion in tissues, revealed aberrant connectivity and other microstructural abnormalities likely due to abnormal myelination and/or astrogliosis in brains of TSC patients (reviewed in Tsai and Sahin 2011). One study reported microstructural abnormalities of corpus callosum tracts that were present only in TSC patients with ASD, not in those without ASD (Fig. 3, Peters et al. 2012), supporting a model of white matter disconnectivity in TSC patients with autism.

Aberrant white matter connectivity in TSC patients with autism. Illustration of projections of the corpus callosum and average fractional anisotropy (AFA) values show aberrant white matter connectivity in TSC patients with autism compared to healthy controls or TSC patients without autism: (a) a 5.3-year-old healthy control, AFA = 0.53; (b) a 4.7-year-old patient with tuberous sclerosis complex (TSC) but no autism spectrum disorder (ASD), AFA = 0.54; and (c) a 4.9-year-old patient with TSC and ASD, AFA = 0.34. Average fractional anisotropy (AFA) is used as a measure of directionality of diffusion (Illustration from Peters et al. (2012), with reprint permission from Elsevier)
Neurologic Phenotype in Animal Models
So far, no animal model exists that fully replicates the human pathology of TSC. However, several animal models exhibit cognitive and behavioral abnormalities similar to these aspects of TSC.
Mice with homozygous inactivation of Tsc1 or Tsc2 have a more severe phenotype than heterozygous mice. Germline Tsc1 and Tsc2 deletions are lethal in early embryonic stages (Kobayashi et al. 1999, 2001), while conditional neuronal Tsc1 inactivation results in mice that are viable perinatally but have severe epilepsy and die at 4–8 weeks of age (Meikle et al. 2007). Tsc2 deletion in radial glia cells and Tsc1 deletion in astrocytes also result in seizures, failure to thrive, and death at an early age (Uhlmann et al. 2002; Way et al. 2009).
Animals with heterozygous inactivating Tsc1 or Tsc2 mutations seem to be a more promising model for understanding the mechanisms that lead to cognitive disabilities and social impairment in TSC. These animals have memory and behavioral deficits in the absence of seizures or tubers, suggesting that neither seizures nor tubers are required for inducing this phenotype.
Germline Tsc1 +/− and Tsc2 +/− heterozygous mice both display deficits in hippocampal-dependent learning (Goorden et al. 2007; Ehninger et al. 2008). In Tsc2 +/− mice, abnormal hippocampal long-term potentiation (LTP) leads to deficits in spatial learning and contextual discrimination.
Tsc1 +/− mice show abnormal social approach behavior, and alterations in mother-pup interaction as measured by ultrasonic vocalization have been observed in the Tsc2 +/− line (Goorden et al. 2008; Young et al. 2010). Similarly, a naturally occurring Tsc2 +/− model, the Eker rat, displays decreased novel object and social exploration as well as reduced open field rearing (Waltereit et al. 2010), which are consistent with autistic features in TSC patients. Artificial induction of seizures in the Eker rat results in deterioration of these features, suggesting that seizures might contribute to a worse outcome (all animal models are reviewed in Tsai and Sahin 2011).
Neurobiological Aspects
CNS neurons develop into a polarized structure consisting of a single axon and multiple dendrites. This highly organized structure is required for proper information transmission and processing. Preclinical evidence from tissue culture and animal models has shown that disinhibited mTORC1 signaling likely contributes to neurological phenotypes, because mTORC1 regulates not only cell growth and protein translation but also axon guidance, neuronal polarity and migration, dendritic spine morphology, myelination, and, in mature neurons, synaptic plasticity. Evidence that aberrant Tsc/mTORC1 signaling causes neuronal dysfunction comes from the following studies:
-
During early development, components of the Tsc/mTORC1 pathway are expressed predominantly in nascent axons. Overexpression of Tsc1 or Tsc2 suppresses axon formation, and loss of Tsc1 or Tsc2 results in abnormal neuronal polarity. Haploinsufficiency of Tsc2 leads to aberrant axon guidance resulting in aberrant neuronal projections (Choi et al. 2008; Nie et al. 2010).
-
Evidence exists for neuronal migration deficits such as cortical cytoarchitectural disorganization, cortical heterotopias, and lamination defects in TSC patients. Astrocyte-specific Tsc1 knockout in mice has demonstrated cortical and hippocampal lamination defects as well as hippocampal heterotopias (Way et al. 2009).
-
The postsynaptic density of dendritic spines is important for proper synaptic communication. Postmitotic loss of Tsc1 in neurons results in increased spine length and reduced spine density. Shank proteins function to stabilize postsynaptic density, and mutations of Shank genes have been found in ASD patients. There is evidence for shared binding partners of Shank proteins and Tsc1/2 (reviewed in Peça and Feng 2012).
-
Neuronal Tsc1 as well as glial Tsc2 inactivation in mice results in myelination defects in the CNS, which is consistent with microstructural white matter abnormalities seen in TSC patients (Meikle et al. 2007; Way et al. 2009).
-
Evidence for postmitotic functions of mTORC1 signaling in neurons results from impaired long-term potentiation (LTP) in Tsc1-deficient cells and abnormally low thresholds for stabilization of synaptic plasticity in Tsc2 +/− (Ehninger et al. 2008).
-
Many studies suggest abnormal neurotransmitter signaling in autism disorders. GABA and glutamate neurotransmission is not only crucial for signal transmission between synapses but also important for neuronal migration and cortical development. There is also evidence for dysregulated GABA and glutamate signals in TSC. In TSC patients, increased GABA levels as well as altered glutamate-receptor patterns in cortical tubers have been reported in several studies (Taki et al. 2009; Talos et al. 2008). Astrocyte-specific Tsc1 knockout mice show reduced expression levels of the glutamate transporter-1 (Glt-1) and, as a consequence, high extracellular glutamate levels. This likely contributes to their seizures as treatment with ceftriaxone, a drug that enhances Glt-1 expression, reduces glutamate levels and seizure incidence (Zeng et al. 2010).
Taken together, these findings in TSC patients as well as in animal models and tissue culture support the model that TSC is a disorder of aberrant neuronal connectivity and signal transmission in the CNS and suggest that its neurocognitive and behavioral symptoms result from disturbed mTORC1 signaling leading to erroneous neuronal development and network functions (Fig. 4).

Model for TSC/mTORC1 contribution to autism in TSC. Mutation of the TSC1 or TSC2 gene triggers a cascade of events altering the developmental program and abnormal development of the neural circuitry. Hyperactivation of mTORC1 signaling may be directly responsible for the spectrum of central nervous system abnormalities and susceptibility to autism (Modified from Curatolo et al. (2010), with reprint permission from Sage)
Therapeutic Options and Outlook
In the past, treatment options for TSC were based on symptomatic therapies, often with limited success. Recent studies have helped to uncover neurobiological mechanisms in TSC, and inhibition of the Tsc/mTORC1 pathway in tissue culture and mouse models has proven promising for future clinical applications.
The drug rapamycin, an allosteric mTORC1 inhibitor, and its derivatives have already been used in many trials to treat TSC manifestations such as SEGAs and renal angiomyolipomas. Additionally, the rapamycin analogue everolimus became the first mTORC1 inhibitor to receive approval from the FDA for the treatment of inoperable SEGAs associated with TSC. Everolimus also reduced seizure frequency in these patients and improved white matter microarchitecture (as measured by DTI) (reviewed by Kohrman 2012).
While clinical studies examining the effect of mTOR inhibitors on cognitive and behavioral symptoms are currently under way, preclinical evidence of research conducted in animal models has yielded promising results. Postnatal treatment of neuronal or glial Tsc1 knockout mice with the mTORC1 inhibitor rapamycin substantially improved survival. Rapamycin prevented the onset of epilepsy when given before seizures developed and reduced seizure frequency when given after the onset of symptoms. However, some neuropathological abnormalities remained, such as abnormal neuronal polarity, while others improved, such as myelination, indicating that myelination deficits largely account for neurological symptoms in these mice. In the astrocyte-specific Tsc1 knockout mouse model, rapamycin increased Glt-1 expression and reduced glutamate levels, which possibly contributed to the reduction of seizures. In Tsc2 +/− mice that lack seizures but display learning and memory deficits, rapamycin normalized synaptic plasticity by restoring long-term potentiation to normal levels (reviewed by Ehninger and Silva 2011). These results implicate that mTOR inhibition might not only halt tumor/hamartoma growth, but also might rescue features such as disturbed neural circuit functions.
In summary, tuberous sclerosis complex is a multisystem disorder that frequently presents with autism and other behavioral problems. Growing evidence highlighting the pathobiology of TSC suggests that aberrant neuronal connectivity and network functions, likely caused by uninhibited mTOR signaling, contribute to the neurological phenotype in these patients. Studies with mTOR inhibitors such as rapamycin and its analogues have shown promising results in animal models and are currently under way in patients. However, important issues still require resolution, such as the variability of neurologic phenotypes in TSC. Why do some TSC patients develop autism while others do not? What other factors might contribute to the development of autism in TSC in addition to a Tsc mutation? Are symptoms preventable or reversible in humans through treatment with mTOR inhibitors, and would chronic therapy with these drugs be necessary or feasible? Translational research has provided valuable insights into the neurobiology of autism and TSC and will continue to address these questions in the future.
Key Terms
-
TSC. Tuberous sclerosis complex, a genetic multisystem disorder with a high susceptibility of autism or autism spectrum disorders and other cognitive or behavioral problems. Hallmark features are benign tumors, the so-called hamartomas.
-
TSC1/TSC2. The genes and their respective gene products. TSC1/TSC2 forms a heterodimeric complex to inhibit mTORC1 signaling. Mutation in either one results in the dissociation of the complex, an uninhibited mTORC1 cascade, and tuberous sclerosis complex disease.
-
mTOR. A central signal integration point in cells. mTOR (mammalian target of rapamycin) is part of two complexes with distinct functions, mTORC1 and mTORC2. mTORC1 is negatively regulated by the TSC1/TSC2 complex. It has been shown to be involved in many steps of neuronal development and maturation.
-
Rapamycin. A macrolide drug that was originally isolated from a strain of S. hygroscopicus on Easter Island (Rapa Nui). Rapamycin is an allosteric mTOR inhibitor that has been widely used as an immunosuppressant after organ transplantations and as a chemotherapeutic drug for malignancies.
-
Knockout Animals. Knockout animals are genetically engineered animals in which the gene of interest has been “knocked out,” i.e., disrupted or replaced. A conditional knockout uses a cell type-/differentiation-specific promoter, which will only inactivate the gene of interest in a specific subset of cells.
-
Synaptic Plasticity. Synaptic plasticity describes the ability of a synapse to adapt the strength of a signal in response to use/nonuse. Long-term potentiation (LTP) describes long-lasting enhancement in signal transmission between two neurons that results from synchronous stimulation and is considered a model for learning and memory.
-
Neuronal Polarity. During development CNS neurons form a highly polarized structure consisting (for most neurons) of one axon and multiple dendrites. Signal transduction occurs via synapses, which are connections between axons (presynaptic) and dendrites (postsynaptic).
-
Axon Guidance. Growing axons receive signals (guidance cues) through receptors on their growth cones, located at the tip of the axon, that guide their growth.
-
Neuronal Migration. Cortical neurons migrate from their site of origin, the periventricular germinal matrix, to their final destination in the cortex, where they form the gray matter consisting of six layers. Failure to properly do so results in heterotopia (ectopic gray matter) or cortical disorganization (aberrant distribution within the cortex).
-
Myelination. Oligodendrocytes in the CNS (Schwann cells in the PNS) produce the myelin sheath, which “insulates” the axon and ensures fast impulse transmission.
-
DTI. Diffusion tensor imaging is an MRI technique that measures the diffusion of water molecules in tissues. It is particularly useful for evaluation of white matter abnormalities.
-
GABA. GABA (g-aminobutyric acid) acts as a neurotransmitter at the synapse and has mostly inhibitory functions.
-
Glutamate. Glutamate acts as a neurotransmitter at the synapse and has mostly excitatory functions.
Key Facts
-
Tuberous sclerosis complex (TSC) is a neurocutaneous disorder characterized by lesions of the skin and the central nervous system.
-
It is caused by a heterozygous mutation of either TSC1 or TSC2 and has a prevalence of 1:6,000.
-
In addition to the hallmark skin and nervous system abnormalities, many other organs are commonly involved. Skin and brain are affected in 90–95 % of patients, followed by kidney (80–85 %), heart (50 %), eye (50 %), lung (30 % of female patients), and liver and gastrointestinal tract involvement (25 %).
-
Other examples of neurocutaneous disorders include neurofibromatosis I and II, Sturge-Weber syndrome, von Hippel-Lindau disease, and ataxia telangiectasia.
Summary Points
-
Tuberous sclerosis complex (TSC) is a genetic disorder that is associated with autism and autism spectrum disorders (ASD) in 50 % of cases with a male-to-female ratio of 1:1.
-
Autistic features in TSC are evident at an early age and have a major impact on the lives of patients and their families.
-
Mutation of the TSC1 or TSC2 gene results in hyperactivation of the mTORC1 pathway, a central signal integration point in cells that has been shown to be involved in neuronal development and maturation.
-
Recent evidence suggests that autism in TSC is caused by interplay of aberrant white matter connectivity, synapse anomalies, myelination defects, and altered neurotransmitter signaling.
-
Animal models exist that replicate features of autism such as abnormal social approach, repetitive behaviors, and alterations in mother-pup interaction associated ultrasonic vocalizations.
-
Clinical trials based upon inhibition of the mTORC1 pathway in TSC-associated tumors have shown promising results, and trials addressing cognitive and behavioral symptoms in TSC are under way.
References
Au KS, Northrup H. Genotype-phenotype studies in TSC and molecular diagnostics. In: Kwiatkowski DJ, Whittemore VH, Thiele EA, editors. Tuberous sclerosis complex. 1st ed. Weinheim: Wiley-VCH; 2010. p. 61–84.
Bolton PF, Park RJ, Higgins JN, et al. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain. 2002;125:1247–55.
Bourneville DM, Brissaud E. Encéphalite ou sclérose tubéreuse des circonvolutions cérébrales. Arch Neurol. 1881;1:390–412.
Choi YJ, Di Nardo A, Kramvis I, et al. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–95.
Crino PB. Molecular pathogenesis of tuber formation in tuberous sclerosis complex. J Child Neurol. 2004;19:716–25.
Curatolo P, Napolioni V, Moavero R. Autism spectrum disorders in tuberous sclerosis: pathogenetic pathways and implications for treatment. J Child Neurol. 2010;25:873–80.
de Vries PJ, Howe CJ. The tuberous sclerosis complex proteins – a GRIPP on cognition and neurodevelopment. Trends Mol Med. 2007;13:319–26.
de Vries P, Humphrey A, McCartney D, et al. Consensus clinical guidelines for the assessment of cognitive and behavioural problems in tuberous sclerosis. Eur Child Adolesc Psychiatry. 2005;14:183–90.
Ehninger D, Silva AJ. Rapamycin for treating tuberous sclerosis and autism spectrum disorders. Trends Mol Med. 2011;17:78–87.
Ehninger D, Han S, Shilyansky C, et al. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–8.
Eluvathingal TJ, Behen ME, Chugani HT, et al. Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J Child Neurol. 2006;21:846–51.
Ess KC. Tuberous sclerosis complex: everything old is new again. J Neurodev Disord. 2009;1:141–9.
European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–15.
Fatemi SH, Aldinger KA, Ashwood P, et al. Consensus paper: pathological role of the cerebellum in autism. Cerebellum. 2012;11(3):777–807.
Goodman M, Lamm SH, Engel A, et al. Cortical tuber count: a biomarker indicating neurologic severity of tuberous sclerosis complex. J Child Neurol. 1997;12:85–90.
Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/- mice in the absence of cerebral lesions and seizures. Ann Neurol. 2007;62:648–655.
Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75.
Jambaque I, Cusmai R, Curatolo P, et al. Neuropsychological aspects of tuberous sclerosis in relation to epilepsy and MRI findings. Dev Med Child Neurol. 1991;33:698–705.
Jansen FE, Vincken KL, Algra A, et al. Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology. 2008;70:916–23.
Jeste SS, Sahin M, Bolton P, et al. Characterization of autism in young children with tuberous sclerosis complex. J Child Neurol. 2008;23:520–5.
Joinson C, O’Callaghan FJ, Osborne JP, et al. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med. 2003;33:335–44.
Kobayashi T, Minowa O, Kuno J, Mitani H, Hino O, Noda, T. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res. 1999;59:206–1211.
Kobayashi T, Minowa O, Sugitani Y, Takai S, Mitani H, Kobayashi E, Noda T, Hino O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc Natl Acad Sci USA. 2001;98:8762–8767.
Kohrman MH. Emerging treatments in the management of tuberous sclerosis complex. Pediatr Neurol. 2012;46:267–75.
Kwiatkowski DJ. Genetics of tuberous sclerosis complex. In: Kwiatkowski DJ, Whittemore VH, Thiele EA, editors. Tuberous sclerosis complex. 1st ed. Weinheim: Wiley-VCH; 2010. p. 29–60.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93.
Meikle L, Talos DM, Onda H, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–58.
Nie D, Di Nardo A, Han JM, et al. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci. 2010;13:163–72.
Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:125–7.
Paciorkowski AR, Thio LL, Dobyns WB. Genetic and biologic classification of infantile spasms. Pediatr Neurol. 2011;45:355–67.
Peça J, Feng G. Cellular and synaptic network defects in autism. Curr Opin Neurobiol. 2012;22:1–7.
Peters JM, Sahin M, Vogel-Farley VK, et al. Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Acad Radiol. 2012;19:17–25.
Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19:643–9.
Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13:624–8.
Smalley SL. Autism and tuberous sclerosis. J Autism Dev Disord. 1998;28:407–14.
Taki MM, Harada M, Mori K, et al. High gamma-aminobutyric acid level in cortical tubers in epileptic infants with tuberous sclerosis complex measured with the MEGA-editing J-difference method and a three-Tesla clinical MRI instrument. Neuroimage. 2009;47:1207–14.
Talos DM, Kwiatkowski DJ, Cordero K, et al. Cell-specific alterations of glutamate receptor expression in tuberous sclerosis complex cortical tubers. Ann Neurol. 2008;63:454–65.
Tsai P, Sahin M. Mechanisms of neurocognitive dysfunction and therapeutic considerations in tuberous sclerosis complex. Curr Opin Neurol. 2011;24:106–13.
Tsai PT, Hull C, Greene-Colozzi E, Sadowski A, Leech J, Steinberg J, Crawley JN, Regehr WG, Sahin M. Autistic behavior and cerebellar dysfunction in Purkinje cell Tsc1 mutants. Nature. 2012;488(7413)647–51.
Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada K, Gutmann DH. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296.
van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805–8.
Waltereit R, Japs B, Schneider M, de Vries PJ, Bartsch D. Epilepsy and Tsc2 haploinsufficiency lead to autistic-like social deficit behaviors in rats. Behav Genet. 2010.
Way SW, McKenna 3rd J, Mietzsch U, et al. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Hum Mol Genet. 2009;18:1252–65.
Weber AM, Egelhoff JC, McKellop JM, et al. Autism and the cerebellum: evidence from tuberous sclerosis. J Autism Dev Disord. 2000;30:511–17.
Whittemore VH. The history of tuberous sclerosis complex. In: Kwiatkowski DJ, Whittemore VH, Thiele EA, editors. Tuberous sclerosis complex. Weinheim: Wiley-VCH; 2010. p. 3–8.
Young DM, Schenk AK, Yang SB, Jan YN, Jan LY. Altered ultrasonic vocalizations in a tuberous sclerosis mouse model of autism. Proc Natl Acad Sci USA. 2010;107:11074–11079.
Zeng LH, Bero AW, Zhang B, et al. Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2010;37:764–71.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this entry
Cite this entry
Julich, K., Sahin, M. (2014). Autism Spectrum Disorders in Tuberous Sclerosis. In: Patel, V., Preedy, V., Martin, C. (eds) Comprehensive Guide to Autism. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-4788-7_184
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4788-7_184
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-4787-0
Online ISBN: 978-1-4614-4788-7
eBook Packages: Behavioral ScienceReference Module Humanities and Social SciencesReference Module Business, Economics and Social Sciences