
Abstract
Dendritic cells (DC) are antigen presenting cells that play a crucial role in initiating anti-tumor immunity. DC capture antigens, process them, and migrate to the draining lymph nodes where they can induce an antigen-specific T cell response. A promising strategy to induce a potent, specific, and lasting anti-tumor response is to target tumor antigens to DC in vivo. This represents a clinically generally applicable and cost-effective approach to DC-based vaccination against cancer. Here, an overview is provided of the different delivery vehicles (e.g., viruses, proteins, liposomes, and nanobodies) that are currently being explored for the development of therapeutic cancer vaccines and considerations for their successful application as well as future developments.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Dendritic Cell
- Herpes Simplex Virus
- Prostate Specific Antigen
- Dendritic Cell Vaccine
- Dendritic Cell Subset
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction: The Case for DC-Targeted Vaccines
Dendritic cells (DC) are generally regarded as the most powerful antigen-presenting cells (APC) with a singular ability to prime naive T cells and thus initiate adaptive immunity. They form a crucial link between the innate and the adaptive arms of the immune system and are central regulators in numerous immune processes. They stem from a common CD34+ bone marrow (BM)-derived precursor and can differentiate into various subsets, which can be myeloid (conventional DC) or more lymphoid in nature (plasmacytoid DC). From the blood, DC precursors home to peripheral tissues where they develop into immature DC. Immature DC patrol all tissues of the body and carry specialized receptors to bind and detect pathogen- or danger-associated molecular patterns (PAMP and DAMP, respectively). Upon their activation through these infectious and/or pro-inflammatory stimuli, they reach end-stage maturation, at which point they acquire the ability to migrate to secondary lymphoid organs and activate (naïve) T cells in an antigen-specific manner, thus starting an adaptive immune response1 , 2.
Numerous clinical trials have been carried out and are currently underway to study the efficacy of DC vaccines. A common strategy in the DC-based immunotherapy of cancer is the ex vivo generation of autologous DC from blood-derived DC precursors, which are then loaded with proteins or peptides that carry known T cell epitopes from tumor-associated antigens (TAA). Such DC vaccines are subsequently readministered to the patient. Alternatively, TAA-encoding genes can be transferred to DC. Genetic modification of DC for immunotherapy has distinct advantages. In contrast to the use of proteins, a genetic TAA vaccine provides a long-lived and continuous source of antigen, facilitating durable presentation of TAA-derived epitopes to both cytotoxic T lymphocytes (CTL) and helper T cells (Th). Endogenous TAA expression resulting from gene transfer ensures access to the MHC class I processing pathway, which is essential for subsequent activation of specific CTL, the proposed main effector cells of anti-tumor immunity. Although vaccination with ex vivo generated autologous DC has led to some clinical successes, its wide-scale implementation is hindered by limitations with respect to logistics, costs, and standardization. Indeed, there is a general consensus that this methodology requires optimization to improve therapeutic efficacy and alternative tumor vaccination approaches are actively being pursued3.
An ever expanding knowledge of DC biology has led to a new generation of genetically modified vaccines that can specifically target DC in vivo. By simultaneously ensuring proper DC activation these generally applicable DC-targeted vaccines may ultimately render the ex vivo generation and loading of DC redundant. Indeed, vaccines based on the targeting and triggering of tissue-resident DC can be designed to exploit the physiological processes already in place to facilitate DC activation, migration, lymph node homing, and subsequent T cell activation. Additionally, the presence of different DC subsets in peripheral tissues allows the targeting of specific DC subsets that have been demonstrated to hold potent immunostimulatory capacities. For example, for anti-tumor immunization it may prove beneficial to target Langerhans cells specifically, i.e., the DC subset residing in the epidermis and other epithelial surfaces, as these have been implicated in the selective generation of cell-mediated immunity1 , 3.
2 DC Targeting Motifs
In mouse studies, in vivo immunotargeting of protein antigens to DC-restricted markers, such as DEC-205, was shown to induce strong immune responses4 , 5. However, for effective CTL-mediated anti-tumor immunity, DC targeting alone may not be enough; additional activation is required. For instance, it has been shown by Steinman and colleagues that targeting of model tumor-, or HIV-derived antigens to the DEC-205 receptor (or other DC-associated C-type lectin receptors [CLR], like Langerin and Clec9A) on DC led to specific T cell unresponsiveness within 7 days after immunization6 , 7. This unresponsiveness was only overcome after the co-injection of a CD40 agonistic antibody or activating ligands of toll-like receptors (TLR), like CpG oligodeoxynucleotides. Therefore, in selecting DC targeting motifs two important factors should be considered: selectivity and activation.
The choice of molecules to target for DC-specific gene transfer is closely related to the subset, the maturation state, and the anatomical location of the DC in question. The most attractive targets should (a) be only expressed on DC, (b) be rapidly internalized upon binding, (c) route internalized antigens into MHC class I and II processing pathways, and (d) induce DC maturation and migration upon binding, to allow for optimal CTL and other immune cell activation. DC express many different antigen-capture and PAMP or DAMP-binding molecules at their surface, collectively referred to as pattern recognition receptors (PRR). PRR are by definition attractive targets, because it is their natural function to internalize antigens and mediate their routing to antigen processing pathways in order to facilitate generation of a T cell response (reviewed in reference 8). PRR include CLR, TLR, scavenger receptors (SR) and NOD-like receptors. Upon infection and/or tissue damage, DC bind PAMPs and/or DAMPs, leading to endocytosis, processing, and presentation of associated antigens. Their ability to capture and process antigens for subsequent T cell activation, coupled to the capacity of some to also induce DC maturation, make PRR attractive candidate targeting motifs for in vivo DC vaccination. In addition, their differential expression on DC subsets may allow for targeting of specific subsets with specialized functions: for example, Langerin for Langerhans cells (CTL activation); MR, TLR2, and DC-SIGN for dermal or interstitial DC (B- and T cell activation); or CD141 and Clec9A for enhanced cross-priming in specific DC subsets3. Upon binding of their ligand(s), some PRR (e.g., TLR) can activate DC, whereas others do not (e.g., most CLR), necessitating the incorporation of DC-activating signals in the vaccine formulation. Alternatively, DC-activating receptors that are (relatively) over-expressed on DC might be directly targeted to achieve simultaneous DC targeting and activation. Members of the family of TNF receptors are attractive candidates in this respect. For instance, we found that CD40-targeted adenovirus-mediated TAA gene transfer resulted in selective DC transduction in human skin explants and skin-draining lymph nodes and simultaneously induced their activation, leading to the high-efficiency priming and activation of tumor-reactive CTL9 , 10.
3 Targeting DC In Vivo: Delivery Vehicles
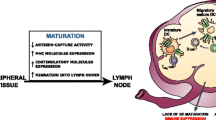
DC-targeted vaccines generally consist of antigenic proteins or genetic material encoding antigenic sequences (Fig. 6.1). Immunogenic DC targeting of either modality has to adhere to a separate and specific set of requirements. For protein targeting it is essential that upon binding to the DC surface motif, the protein is efficiently endocytosed and routed to MHC processing pathways for subsequent presentation to T cells. In contrast, for viral transduction, binding to a DC-specific docking molecule per se might be sufficient, as most viruses have co-receptors (e.g., integrins) and other mechanisms in place for the induction of subsequent uptake and release from endosomes into the cytoplasm. Various vehicles are now available for targeted in vivo gene and/or protein transfer to DC, both viral and nonviral (see Fig. 6.1). An overview is given below.

Representation of the different mechanisms to target dendritic cells (DC). Tumor associated antigens can be targeted to DC residing in the skin via several mechanisms. The antigen itself can be coupled to an antibody specific for DC. Another option is to inject DNA or RNA encoding the antigen of choice directly into the skin or incorporate the DNA/RNA in a viral vector, liposome, or a nanobody manipulated in such a way that it will only bind to DC (such as through specific glycosylation motifs or antibodies). A last option is to use synthetic long peptides that will be taken up and processed by the DC. Upon capture, the DC will start to mature and migrate towards the draining lymph nodes where the mature DC can induce an antigen specific T cell response against antigen-derived epitopes.
4 Viruses
There are some major advantages to the use of viral vectors for gene delivery to DC:
-
(1)
Many viruses exhibit a natural tropism for DC (e.g., lentiviruses) that might be utilized for DC-targeted vaccination.
-
(2)
Viruses have a natural ability to infect target cells, e.g., to be efficiently endocytosed by DC.
-
(3)
Viruses have developed mechanisms to transfer their genetic cargo efficiently to the host cytoplasm and/or nucleus to take over the host replication and/or transcription machinery and thus ensure high-level expression of the transgenes they carry.
These characteristics make viruses extremely attractive vaccine vehicles, despite regulatory restrictions that complicate their clinical implementation and concerns about preexisting or induced neutralizing antibody responses. Nonviral vehicles often need to be chemically altered to achieve the above listed advantageous traits for DC-targeted vaccination that viruses often possess naturally.
5 DNA Viruses
5.1 Adenovirus
One of the most commonly used gene transfer vectors for DC is the adenovirus serotype 5 (Ad5). Advantages of adenoviruses over other delivery vehicles, such as retroviral vectors, are that Ad vectors can efficiently infect both dividing and nondividing cells, that they can be produced at high titers, and that they are relatively safe, since they do not integrate into the host cell genome. Importantly, the perceived unsuitability of Ad5 vectors as vaccine vehicles, due to preexistent or rapidly induced neutralizing antibody responses that would prohibit their use in prime/boost set-ups, can now be overcome by specific ablation of antibody binding sites in the hexon protein of the Ad capsid11.
In 1997, Wan et al. described the ex vivo transduction of DC with a replication deficient adenoviral vector encoding the polyoma middle T antigen12. A single injection of DC transduced with the Ad vector expressing polyoma middle T provided complete and specific protection against tumor cell challenge in 100% of vaccinated animals. A comparable study was performed by Song et al. with a replication deficient adenovirus expressing the reporter gene beta-galactosidase13. Using a murine metastatic lung tumor model with syngeneic colon carcinoma cells expressing beta-galactosidase, it was shown that immunization of mice with a genetically modified DC line or bone marrow-derived DC confers potent protection against a lethal tumor challenge, as well as suppression of pre-established tumors, resulting in a significant survival advantage. Since then, many similar tumor vaccination studies with Ad vectors encoding a myriad of TAA for various tumor types have been performed3 – 23. All these studies demonstrated that adenoviral transduction of DC resulted in high expression levels of the TAA of interest and efficient generation of immune responses directed against the tumor. A more general approach to treat different types of cancer is to transduce DC with Ad vectors encoding wild type p53. The tumor suppressor protein p53 is an attractive candidate for DC-based immunotherapy, because this protein is found abundantly in 50% of human malignancies but not in normal tissues. Several reports demonstrated that treatment with Ad-p53-transduced DC generated CTL directed to p53 and significantly slowed the growth of established tumors14 , 15. Thus, transducing DC with wild-type p53 may be a promising new tool for the immunotherapy of cancer.
In all the studies listed above, DC were ex vivo transduced with Ad vectors. Although more attractive, direct in vivo administration of Ad-based vaccines to patients is complicated by the fact that DC are relatively resistant to Ad infection. The infection of host cells by Ad5 is a two-step process. The first step is a high-affinity interaction of the knob domain of the Ad fiber with the cell surface receptor coxsackie and adenovirus receptor (CAR)16 , 17. Subsequent internalization, via receptor-mediated endocytosis, involves interactions between the Arg-Gly-Asp (RGD) sequences of the adenovirus penton base proteins with cellular αvβ3 and αvβ5 integrin receptors. Unfortunately, DC lack surface CAR expression, whereas CAR is abundantly expressed on many other cell types. In vitro the resistance to Ad infection of DC can be overcome by the use of high virus titers (at multiplicities of infection [MOI] exceeding 1,000). In vivo, however, this would lead to preferential bystander (non-DC) infection and unwanted cytopathic side effects. A logical approach to circumvent inefficient CAR-mediated Ad5 transduction is redirecting Ad5 entry (targeting) via alternative cell surface molecules abundantly expressed on DC. Several strategies have been explored in this regard (see Fig. 6.2). First of all, it proved possible to replace the tropism-determining fiber knob domain of Ad5 with that of a different Ad serotype. Replacement of the Ad5 fiber knob with the Ad35 fiber knob resulted in a dramatic increase in gene transfer efficiency to DC and their high-efficiency in situ transduction in human skin explants18 , 19. Similarly, it was recently demonstrated by Stone et al. that the use of the Ad11 led to an increased transduction efficiency of human immature DC as compared to Ad520. We have also shown that replacement of the Ad5 fiber knob with that of Ad3 resulted in increased transduction efficiencies of human DC and that this Ad5/3 was more specific for mature CD1a+CD83+ DC than Ad5/35, selectively targeting DC in the context of skin and melanoma-draining lymph nodes through binding to CD80/CD8621. Obviously, the utility of pseudotyping is limited by the natural diversity of Ad receptor recognition. More precise targeting of DC specific surface molecules requires synthetic design of targeted adenoviruses. For this, single- and two-component systems are being explored.

Schematic representation of different approaches to target adenoviruses to DC. During native infection, adenovirus serotype 5 (Ad5) enters the cell following high-affinity binding to the cellular receptor CAR, which is not expressed by DC. Replacing the tropism-determining fiber knob domain of Ad5 with that of a different adenovirus serotype results in a virus with a modified tropism, potentially leading to improved transduction of DC. Examples are Ad5/3 and Ad5/35 that can bind to CD80 and CD46 on the DC. Targeting Ad5 to DC has also been established by inserting peptide motifs, like the binding domain of CD40L, in the virus capsid that can bind to receptors on DC. Furthermore, bispecific targeting moieties have been used to target Ad5 to DC. These molecules can bind on one side to the adenoviral fiber and on the other side to a receptor on the DC. An example is the fusion protein sCAR-CD40L which can bind to the knob domain of Ad5 (thereby neutralizing its natural receptor binding) and to DC expressing CD40.
The design of single-component targeted Ad vectors by incorporating targeting ligands into adenovirus capsid proteins has been widely explored in the context of tumor targeting. For DC targeting, the RGD sequence has been incorporated in the fiber knob22 – 24. RGD targeting greatly enhanced DC transduction efficiency. This modification, however, does not abrogate binding to CAR and thus expands rather than targets Ad entry. However, it should be possible to combine such modifications with capsid protein mutations known to abolish native tropism25. Belousova et al. constructed a chimeric Ad containing the CAR-binding mutated wild-type fiber and a bacteriophage T4 fibritin fiber in which CD40L was incorporated26. Intradermal injection of this vector in human skin explants resulted in targeted, enhanced gene transfer to migrating DC, as well as in their phenotypic maturation27. Production of a virus containing only the CD40 targeted fibritin fiber unfortunately proved suboptimal, and thereby unsuitable for clinical application.
Complex binding ligands including antibodies have been successfully employed in two-component targeting strategies, where they were bound to the Ad fiber indirectly via a second protein moiety. We and others have demonstrated that using this approach to target Ad5 to CD40 expressing cells, the transduction efficiency increased to 95% at MOI 10010 , 28 – 30. Indeed, immune conjugate-mediated targeting of Ad5 vectors to CD40 resulted in the selective and enhanced transduction of DC in human skin explants and in lymph node suspensions and facilitated the efficient priming of high-avidity melanoma-reactive CTL9 , 10. Moreover, in vivo delivery of an Ad5 vector carrying tumor antigens and retargeted to CD40 through a CD40L-sCAR adapter protein resulted in efficient DC transduction in lymph nodes and resulted in superior tumor protection in the B16 melanoma model31. Indeed, we envisage the clinical use of such a recombinant CD40 adapter protein (consisting of the TNF-like domain of CD40L fused to soluble CAR), which represents a highly defined product that is clinically applicable with Ad5 as a highly flexible two-component DC-targeted Ad vector configuration, ultimately allowing vaccination with different TAA-encoding Ad vectors simultaneously, depending on the TAA expression profiles of the targeted primary or metastatic tumors.
5.2 Adeno-Associated Virus
Adeno-associated virus (AAV) are small, nonpathogenic parvoviruses that are dependent on larger helper viruses, such as adenoviruses, for their replication. AAV has established its position as one of the most popular gene delivery systems. This is mainly because of the long-term and efficient transgene expression in various cell types in many tissues including liver, muscle, retina, and the central nervous system32. However, there are some disadvantages associated with the application of AAV. The packaging capacity is relatively restricted and the large-scale production inefficient. Furthermore, the integration into the host genome is random, which can lead to unexpected activation or inhibition of endogenous gene expression (a major obstacle to in vivo clinical application). Different AAV serotypes have shown remarkably different expression patterns because of differences in cell entry and intracellular activities. For example, Ponnazhagan et al. demonstrated that at an MOI of 100 of AAV serotype 2, the efficiency of transduction among DC cultures derived from different normal blood donors, varied between 2 and 55%33. Nevertheless, transduction of DC with an AAV containing the cDNA encoding the HPV-16 E7 antigen generated CTL that showed MHC class I-restricted killing of cervical cancer cells34. Flow cytometric analysis of the DC populations revealed that AAV/E6 vector-pulsed DC had higher levels of CD80 and lower levels of CD86 than protein-pulsed DC35. Importantly, transducing DC with AAV encoding self-antigens resulted in the generation of functional CTL, thus suggesting that AAV-loading of DC is a good approach for generating CTL against TAA with low immunogenicity36 , 37.
5.3 Vaccinia Virus
Vaccinia virus is a double-stranded DNA virus of which the entire life cycle takes place within the cytoplasm of host cells. It has a wide host range and is capable of infecting almost all human cell types with high efficiency. This represents a clear disadvantage for DC targeting. An advantage of Vaccinia virus is its capacity for efficient infection and gene expression. A number of viral promoters can be chosen to control the timing and the level of transgene expression. Furthermore, the Vaccinia virus genome can accommodate at least 25 kb of foreign DNA sequence38 and its replication occurs exclusively in the cytoplasm, eliminating the possibility of chromosomal integration. An important potential disadvantage for clinical application of Vaccinia virus-based vaccines may be preexistent immunity in older patients vaccinated for smallpox. This may limit vaccination efficacy.
In 1998, Di Nicola et al. described that mature monocyte-derived DC were transducible with Vaccinia virus39. Since then, various studies have been performed demonstrating Vaccinia virus-mediated transduction of genes encoding the TAA EBNA-3A40, gp10041, MUC142 , 43, CEA43, or HPV16-E7. In general, all these studies showed the induction of antigen-specific Th and CTL responses, resulting in in vivo tumor rejection. However, it has been demonstrated that Vaccinia virus transduction hampers proper DC maturation44 , 45 making it necessary to induce DC maturation prior to transduction with this virus. Furthermore, transduction with Vaccinia virus inhibited expression of HLA-DR and reduced the secretion of cytokines important for DC migration, like RANTES, MIP-1α, and TNF-α46.
Two phase I clinical trials with Vaccinia virus-based melanoma vaccines have been performed47 , 48. In the first study, 6 patients were injected intravenously and subcutaneously with DC transduced in vitro with a modified Vaccinia virus encoding the human tyrosinase gene47. Treatment was well tolerated, except for low-grade fever (in 3/6 patients), mild erythema at the injection site (in 5/6 patients), and vitiligo (in 2/6 patients). A partial response, involving shrinkage of a subcutaneous nodule, later surgically removed, was observed in 1 patient, who then remained disease-free (>850 days). In 4 of 5 patients, significant and often long-lasting increases in frequency of T cells directed to tyrosinase were documented. In another study a comparable vaccine was directly injected in 20 patients three times at 4-week intervals (5 × 108 IU/injection)48. This did not elicit a measurable immune response to its transgene product in patients with stage II melanoma after repeated combined intradermal and subcutaneous vaccination, probably because DC maturation was hampered.
Another clinically tested Vaccinia virus-based vaccine is PROSTVAC-VF (Tricom). It consists of two vectors, both encoding the prostate tumor antigen prostate specific antigen (PSA) and three co-stimulatory molecules: ICAM-1, LFA-3, and B7.1 (CD80). A Vaccinia virus is used for the priming vaccination, followed by boost vaccinations with fowlpox vectors. The viruses are subcutaneously injected together with GM-CSF. In a randomized controlled phase II trial of 125 patients with castration resistant prostate cancer it was demonstrated that patients receiving PROSTVAC-VF had a longer median overall survival (25.1 months versus 16.6 months for patients receiving an empty control vector)49. A large phase III trial is now planned to confirm these promising results.
5.4 Herpes Simplex Virus
Herpes simplex virus (HSV) is a large DNA virus of which type I can infect DC with intermediate to high efficiency. DC infected with replication deficient HSV fail to become activated, downregulate a number of surface markers, and fail to produce a number of cytokines in response to activation stimuli, such that their T cell-activating capabilities are minimal50 , 51. To overcome this immune problem, the viral gene encoding virion host shut-off protein has been deleted52. This protein destabilizes mRNA in infected cells so that host protein synthesis is reduced in favor of translation from more rapidly produced viral mRNA. The resulting virus transduced DC as efficiently as the parental virus, but induced both expression of CD86 and an enhanced specific T cell-proliferative response. Transduction of DC with HSV–OVA (ovalbumin) or HSV–PSA and co-culture with CTL hybridomas resulted in specific activation of the CTL, indicating that transduced DC express these transgenes and process the tumor antigens for MHC-I mediated presentation to CTL. Mice immunized with HSV–PSA-transduced DC generated a specific CTL response that could be detected in vitro by a classic chromium release assay and these mice were protected from challenge with tumors that expressed PSA53. Thus far, HSV vectors have not been clinically used for in vivo vaccination or DC targeting.
6 RNA Viruses
6.1 Retrovirus
The idea of using retroviruses as gene delivery tools was introduced by Mann et al.54. The retrovirus family consists of single-stranded RNA viruses that measure 80–120 nm in diameter. These single-stranded RNA viruses replicate through a double-stranded DNA intermediate, which is integrated in the host genome. The most commonly used retroviral vectors are based on Moloney murine leukemia virus, in which the gag, pol, and env genes are replaced with an expression cassette. The major advantage of retroviral vectors is the lack of immunogenicity due to the removal of the genes encoding viral proteins. However, limitations of this vector are the instability of the viral particle, low viral titers, and the inability to transduce nondividing cells. DC residing in tissues and secondary lymphoid organs have lost their proliferative capacity, rendering retroviruses useless in terms of in vivo DC targeting.
6.2 Lentivirus
In contrast to oncoretroviral vectors, lentiviral vectors are capable of transducing nondividing cells, such as DC, at high transduction efficiencies. Importantly, like oncoretroviral vectors, lentiviral vectors do not encode viral proteins, thereby minimizing the potential for interfering with the function of the transduced DC55. Third-generation lentiviral vectors with enhanced safety profiles have been developed and used to transduce murine and human DC efficiently. These improved vectors contain a chimeric Rous sarcoma virus/HIV 5′ long terminal repeat (5′LTR) enhancer and promoter to initiate the transcription of genomic viral RNA56. The stronger chimeric promoter does not require HIV Tat protein, a transactivator of the transcription of HIV genomic RNA, to generate vector transcripts. In addition, the vectors have been made self-inactivating by deleting the majority of the U3 region in 3′LTR so that viral RNA cannot be produced in target cells57. These additional safety modifications further prevent the generation of replication-competent recombinants and should feasibilize clinical implementation.
It has been shown that immature DC are efficiently transduced with increasing doses of lentivirus without affecting cell viability. Transduction at low MOI did not result in phenotypical or functional maturation. Higher doses of lentivirus, however, resulted in upregulation of adhesion, costimulatory, and HLA molecules, as well as in increased allostimulatory capacity and secretion of interleukin (IL)-6, IL-8, and tumor necrosis factor-alpha57. Li et al. described that a single injection of murine bone marrow-derived DC transduced with a lentiviral vector encoding a truncated form of Neu protein stimulated the induction of CD4+ and CD8+ T cells in vivo and suppressed the growth of Her2/Neu overexpressing tumors58.
Recently, two papers were published describing the construction of lentiviral vectors in which expression of the transgene was targeted to DC. Lopes et al. used the mouse dectin-2 promoter to target expression of GFP to dectin-2 positive cells. This lentivector effected transgene expression in mouse bone marrow-derived DC and in human skin-derived Langerhans cells and dermal DC. In mice, transgene expression was detected in splenic dectin-2+ cells after intravenous injection and in CD11c+ DC in the draining lymph node after subcutaneous injection. A dectin-2 targeted lentivector encoding the human cancer antigen NY-ESO-1 primed an NY-ESO-1-specific CD8+ T cell response in HLA-A2 transgenic mice and stimulated a CD4+ T cell response59. Furthermore, a transcriptionally DC-targeted vector was constructed using the DC-STAMP promoter region to induce tolerance by transducing hematopoietic stem cells. This should result in tolerance because the DC that generated from these stem cells are not activated by the vaccine and thus remain in a steady-state immature condition. This vector induced long-term and cell-selective transgene expression in vivo. As expected, these transcriptionally targeted DC induced functional, antigen-specific CD4+ and CD8+ T cell tolerance in vivo, which could not be broken by subsequent immunization60.
Recently, a DC-SIGN targeted lentiviral vector was constructed by incorporation of an engineered glycoprotein derived from Sindbis virus. This targeted lentivector transduced DC in vitro with high specificity. Direct subcutaneous administration of the targeted lentivector in DC-SIGN transgenic mice induced a strong antigen-specific T cell and antibody response61.
Thus, there is a growing body of evidence to show that it is indeed possible to use lentiviral vectors for in vivo DC targeting applications.
7 Nonviral Gene Vehicles
7.1 Naked DNA and RNA
7.1.1 DNA
An elegant approach to circumvent (mostly safety-related) disadvantages associated with viral vectors is to transfect DC directly with plasmid DNA encoding full-length TAA. Advantages of DNA transfection include the easy construction and high stability of plasmid DNA and the possibility to include sequences that lead to better antigen presentation or DC activation. Furthermore, DNA vaccines are relatively safe, because there is no risk for recombination with wild-type viruses and the risk for insertional mutagenesis is low. Finally, it has been demonstrated that cutaneously applied plasmids can remain present in the skin for up to 5 months62. DNA-based vaccines thus direct antigen expression for extended periods, supporting persistent anti-tumor immune responses that could theoretically protect a patient from relapse. Major hurdles to the use of DNA as immunotherapeutic tools are the low efficiency with which DC are transfected and the general weakness of elicited immune responses. Possible ways of administration include the modification of target cells with the DNA ex vivo and the direct delivery of the DNA plasmid into the patient, for example, with the gene gun method, by tattooing into skin, or simply by intradermal or intramuscular injection. The gene gun methodology entails delivery of the DNA following its precipitation onto gold microparticles that are delivered to the skin under pressure by a ballistic delivery device63. This process does not induce traumatic injury and requires much less DNA to achieve comparable humoral immune responses as compared to intramuscular administration64. More recently, Bins et al. pioneered a tattoo approach whereby antigen-encoding DNA can be delivered to the epidermis65. In mice and primates this was shown to induce immunity to tumor and HIV antigens, respectively66. The resulting trauma to the epidermis also ensured DC activation and migration. The exact mechanism for induction of the immune response is still not entirely clear, but appears to involve processing of the antigen through both endogenous and exogenous pathways, leading to presentation of the antigen in the context of both MHC class I and II. The disadvantage of direct administration of DNA to patients is that any cell encountering the plasmid could be transfected with it. It was demonstrated by Raz et al. that after intradermal injection of plasmid DNA, cells resembling macrophages and DC, but also keratinocytes and fibroblasts, were transfected62. Other studies, however, demonstrated direct transfection only of skin DC following gene gun administration67 , 68. In contrast to this, Corr et al. published that the elicited immune response was the result of expression of the antigen by non-lymphoid tissues and transfer to APC69. After DNA vaccination DC appear to acquire antigen both by direct transfection and by cross priming. A study by Condon et al. furthermore revealed that gene gun immunization resulted in the migration of transfected skin-derived DC to the draining lymph nodes67. Recently, a way to target the expression of the gene to DC was described by Ni et al. who used a DC specific promoter based on a short sequence of the CD11c promoter to target expression of lacZ or EpCAM to DC in mice70.
A number of features can influence the nature and the potency of the DNA-elicited immune response. The composition of the DNA is a first important consideration for plasmid vaccines. For instance, hypomethylated CpG dinucleotide sequences that are relatively underrepresented in eukaryotic DNA serve as a PAMP and in man bind to TLR9 in plasmacytoid DC, increasing the immunogenicity of DNA vaccines71. Secondly, to increase the level of transgene expression, a strong promoter, like the CMV promoter, is required. Finally, the antigenicity of the encoded protein is of considerable importance in generating an effective immune response. Although DNA vaccines have shown promise in eliciting effective CTL responses to neoantigens, the fact that most TAA are self-antigens and thus weakly immunogenic requires DNA vaccines to be very potent to be clinically useful. Many studies have therefore focused on enhancing the immune response that is raised by DNA vaccines. Different approaches have tried to improve the delivery of the vaccine72, modification of the encoded antigen to increase its immunogenicity and DC targeting potential, for example, by fusion of the antigen to CD40L73 or FLT3L74, or modification of the microenvironment by addition of (DNA encoding) cytokines or chemokines75.
Only a few clinical studies with plasmid DNA in cancer patients have been published. In general, DNA vaccines were well tolerated, but had mixed results in raising cellular immunity. Tagawa et al. conducted a phase I trial in patients with stage IV melanoma76. Patients received intranodal injections of a DNA vaccine encoding tyrosinase epitopes. The vaccine was tolerated well, with only five patients demonstrating grade I–II toxicity. Immune responses by peptide-tetramer assay to tyrosinase were detected in 11 of 26 patients. However, no clinical responses were seen. In a study by Rosenberg et al., 22 patients with metastatic melanoma were injected intradermally or intramuscularly with plasmid DNA encoding gp100 melanoma–melanocyte differentiation antigen77. One patient exhibited a partial response of several subcentimeter cutaneous nodules, whereas all other patients had progressive disease. Of 13 patients with cells available before and after immunization, none exhibited evidence of the development of anti-gp100 T cell responses. A significant clinical or immunological response to plasmid DNA encoding the gp100 tumor antigen was thus not demonstrated. Recently, Miller et al. published the results of a phase I trial in which 6 patients with hormone-refractory prostate cancer were monitored for their ability to mount PSA-specific cellular responses with recombinant GM-CSF and IL-12 as immunoadjuvants after receiving a PSA DNA vaccine78. After vaccination, T cells recognized both PSA peptides and the naturally processed PSA protein. Several trials of DNA vaccines against human papillomavirus (HPV) related malignancies have been performed. HPV-related malignancies have the advantage that foreign HPV antigens are expressed and serve as TAA, rather than self antigens. Plasmid DNA encoding HLA-A2 epitopes from HPV16 E7 protein was incorporated in polymer microparticles and delivered intramuscularly. In a trial for anal dysplasia, increased T cell responses were reported in 10 of 12 patients79. In another study, the same plasmid was delivered to women with cervical intraepithelial neoplasia and most patients mounted a detectable immune response to HPV16-E7. More importantly, in 33% of the participating women, complete histological responses were documented.
Although definitive clinical evidence of the efficacy of DNA vaccines remains to be demonstrated, DNA vaccines do have several advantages and have proven to be safe in clinical applications. Further clinical investigations to improve their efficacy are therefore warranted.
7.1.2 RNA
Transfection of DC with specific or whole cell lysate-derived ribonucleic acid (RNA) has been demonstrated to be very effective in inducing potent TAA-specific cytotoxic T-lymphocytes80 , 81. Using whole cell lysates has the advantage that it does not require the definition of specific TAA and that it therefore might have broad clinical applicability. On the other hand, a potential drawback is the increased risk of inducing autoimmunity. The first preclinical data using RNA-loaded DC in vivo were presented by Boczkowski et al.80. This study showed that DC pulsed with mRNA from ovalbumin-expressing tumor cells were as effective in inducing CTL responses as DC pulsed with ovalbumin peptide. Since then, the effectiveness of this approach has been demonstrated in many in vitro studies using RNA coding for different TAA, like PSA82 , 83, the human papillomavirus proteins E6 and E784, human telomerase reverse transcriptase (hTERT)85 and human immunodeficiency virus capsid proteins86. Recently, Grunebach et al. demonstrated that transfection of monocyte derived DC with the RNA encoding Her-2/Neu and 4-IBBL resulted in an increased specific lysis of target cells by induced CTL lines87 compared to untransfected monocyte derived DC. More importantly, vaccination with mRNA loaded DC has been shown to induce protective and therapeutic anti-tumor responses in mice88 , 89. Several RNA delivery strategies have been explored, like electroporation90, lipofection85, or transfer through receptor-mediated endocytosis. Strobel et al. demonstrated that the use of liposomes was more effective than electroporation90 whereas Van Tendeloo et al. found that electroporation was more potent compared to lipofection or CD71 based endocytosis91.
RNA transfection thus represents a promising approach to engineer DC to present the whole and unique antigenic spectrum of a patient’s tumor and therefore several clinical trials have been performed to assess the efficacy of this approach in patients. The first vaccination study using RNA-transfected DC was a phase I trial designed to evaluate the safety, feasibility, and efficacy to induce T cell responses in patients with metastatic prostate cancer92. Immature monocyte-derived DC were transfected with in vitro transcribed PSA RNA. Increasing doses (1–5 × 107) of the modified DC were administered intravenously every 2 weeks and additionally 1 × 107 cells were administered intradermally at each vaccination. No major toxicity was observed and in general vaccination was well tolerated. After vaccination, all analyzed patients had PSA-reactive, IFN-γ secreting T cells, whereas in the pre-therapy samples no IFN-γ secreting T cells were detected. Furthermore, in a chromium release assay it was demonstrated that after vaccination there was a significant increase in PSA-specific killing of target cells90. In a phase II trial for the same disease, hTERT mRNA-transfected DC were administered to 20 patients93. Eleven of the patients received DC transfected with hTERT-encoding mRNA, whereas 9 patients received DC transfected with the mRNA encoding a chimeric lysosome-associated membrane (LAMP)-hTERT fusion protein to direct hTERT antigen processing into the class II pathway94. It was demonstrated that patients receiving the fusion protein mRNA construct exhibited more pronounced delayed type hypersensitivity reactions, enhanced CD4+ T cell responses, increased antigen-specific proliferative responses and improved CTL-mediated lytic activity when compared with immunization with the unmodified hTERT construct.
Several subsequent trials demonstrated safety, feasibility, immunogenicity, and moderate clinical efficacy of DC vaccines pulsed in vitro with TAA RNA95 – 97. It is, however, also possible to inject mRNA directly into skin. Intradermal application of naked mRNA in mice resulted in protein expression and the development of an immune response98. The same approach was used to vaccinate 15 melanoma patients99. For each patient a growing metastasis was removed and copy mRNA was produced. Autologous preparations were applied intradermally in combination with GM-CSF as adjuvant. This treatment proved to be feasible and safe. Furthermore, an increase in anti-tumor humoral immune responses was seen in some patients. However, a demonstration of clinical efficacy of direct injection of mRNA for anti-tumor immunotherapy was not shown in this study and must be evaluated in subsequent trials. Further strategies to stabilize naked RNA for in vivo applications should prove instrumental in this regard.
8 Nanoparticles and Liposomes
Nanotechnology is a relatively new focus of anti-cancer research and is used as a general term for the manufacture, manipulation and application of structures in the nanometer range. The ultimate goal of nanomedicine is to create medically useful nanodevices that can function inside the body. It is envisioned that nanodevices will be hybrids of biological molecules and synthetic polymers that can enter the cell and can interact with the DNA and proteins. In this regard, it should be possible to incorporate TAA into nanodevices and target them specifically to DC.
One of the first studies using this technology for immunotherapy described the construction of 100 nm cationic nanoparticles from warm oil-in-water microemulsion precursors. Plasmid DNA was coated on the surface of these cationic nanoparticles and the DC-targeting ligand mannan was incorporated in or deposited on the particles100. This approach significantly increased both IgG titer and Th1 cytokines upon immunization as compared to naked DNA transfection.
Fifis et al. coupled ovalbumin to smaller solid-core nanobeads of 40–50 nm, which, upon in vivo delivery, allowed them to localize to DC in the draining lymph nodes101. This resulted in the induction of two- to tenfold stronger immune responses as compared to larger bead sizes, indicating that the size of the nanodevice is important. A single dose of these beads protected the mice from tumors in two different models. Since then, many more studies were performed in mice, although it is difficult to compare all these, because all the nanoparticles used have different compositions and sizes and were tested in different mouse models.
In human skin, particles with a size of 40 nm were efficiently taken up by epidermal Langerhans cells after transcutaneous vaccination, whereas larger particles were only taken up by Langerhans cells around hair follicles102. The same group recently published a paper in which influenza protein-based nanoparticles were transcutaneously injected and compared to intramuscular vaccination in humans. In a study on 11 healthy volunteers, it was found that transcutaneous vaccination induced both effector CD4+ and CD8+ T cell responses, whereas intramuscular injection induced effector CD4+ T cells in the absence of CD8+ T cells103. An interesting paper by Prasad et al. was recently published that described the construction of nanoparticles containing whole tumor lysates from human solid tumors. Compared to conventional tumor lysates the nanoparticles containing tumor lysates were more efficient in inducing IFN-γ production in vitro while reducing the production of potentially immunosuppressive IL-10, although the amount of administered lysate was five times lower in the nanoparticle containing lysate as compared to conventional tumor lysate104. An explanation for this could be that nanoparticles can function as a Th1 adjuvant, because the particles can bind and activate TLR-2105, thereby inducing DC maturation.
The surface of nanoparticles can be modified to improve stability, but also to conjugate ligands to target the particles specifically to target cells (see Fig. 6.1). Ghotbi et al. described the construction of nanoparticles into which mannan was incorporated. These targeted nanoparticles were more efficiently taken up by murine bone marrow-derived DC than untargeted particles106. Cruz et al. recently reported on antibody-modified nanoparticles targeting DC through DC-SIGN and DEC-20595 , 107, resulting in T cell activation. Clearly such particles are attractive candidates for clinical translation as DC-targeted nanoparticle-based vaccines.
Liposomes can be regarded as a subtype of nanoparticles. Liposomes, bilayered phospholipid spheres of ≥100 nm, are excellent carriers of drugs or antigens that are currently used in a number of immunotherapeutic applications. They can accommodate almost any molecule of interest, whether it be peptides, proteins, or DNA, for the purpose of targeting, sustained release, and protection from degradation. The components of the lipids may vary, with neutral liposomes containing neutral lipids, and cationic and anionic liposomes containing lipids that are either positively or negatively charged. Depending on their lipid composition, liposomes can exhibit potent adjuvant-like properties96. Like nanoparticles, many liposomes with different composition and sizes have been tested for their capacity to target DC. The delivery of antigen-containing liposomes to DC can be facilitated by introduction of agents into the bilayer that bind selectively to molecular structures on the surface of the DC, such as antibodies, nanobodies or glycosylated motifs that can bind to specific CLR (see Fig. 6.1).
Targeting liposomes containing antigen or DNA to DC enhanced their capacity to induce humoral and CTL responses in vivo97 , 108 , 109. Moreover, specialized liposomes can be designed to enhance delivery of their payload to the cytosol, such as by using bilayer compositions which are pH-sensitive (pH-sensitive liposomes). This should facilitate subsequent processing for MHC-I mediated activation of specific CTL. Studies in mice have shown that the glycan modification of liposomes for APC targeting is a promising approach for the treatment of cancer. These glycoliposomes can also incorporate TLR-L motifs as well as DNA, since cationic lipids will spontaneously complex with DNA to form lamellar structures, so-called lipoplexes. A recent paper described the construction of antigen-containing liposomes that were engrafted with peptides from the TLR-5-ligand flagellin110. These DC targeted liposomes were efficiently taken up by murine bone marrow-derived DC and induced their maturation. Vaccination of mice with ovalbumin containing TLR-5 targeted liposomes increased the number of antigen specific CD8+ T cells, indicating that this is a promising approach to target liposomes to DC.
A novel subtype of nanoparticles are nano-engineered exosomes. Exosomes are small vesicles released by tumor cells and/or DC. Exosomes released by DC express high levels of costimulatory molecules including MHC class I and II, and because of this observation DC exosomes (dexosomes) are considered to represent an alternative pathway of antigen delivery and presentation. DC-derived exosomes can modulate immune responses by activating T cells. Exosomes can be engineered ex vivo and are an interesting new field in immunotherapy111. Artificial exosomes can be 30–100 nm in diameter and were developed by coating liposomes with peptide-MHC class I complexes. These artificial exosomes could activate and expand functional antigen specific T cells111.
9 Proteins, Antibodies, and Nanobodies
Antigenic proteins can be targeted to DC by coupling them to DC-specific antibodies. After uptake of the antigen-antibody complex by DC, the antigen will be presented and antigen-specific T cell responses can be raised. This has been successfully demonstrated using antigen targeted to DEC-205 and Langerin in murine models4 , 5 , 112 , 113. This approach also worked using DEC-205 or CD11c specific scFv antibodies coupled to gp100 and Her2-Neu, respectively114 , 115. Of note, antigen-antibody complexes were recently reported to be stored in specialized subcellular compartments in DC, ensuring prolonged presentation to, and activation of CTL116. Although these studies demonstrated that antigen targeting to DC induces specific immune responses, translation of these results to humans is difficult, because of different target expression patterns on DC in humans and mice, as is the case, for example, with many CLR. Kretz-Rommel et al. therefore used a mouse model with a humanized immune system to study DC-SIGN targeting. Targeting antigens to DC-SIGN in this model induced antigen-specific T cell responses117. Recently, Flacher et al. published a paper describing targeting of DC using antibodies in human skin118. Antibodies directed to DEC-205 or Langerin were injected intradermally and interestingly, efficient targeting of LC was observed, indicating that the antibodies can diffuse from the dermis towards the epidermis. The LC thus targeted through Langerin were not capable of priming T cell responses, however118.
Nanobodies are single-domain antigen-binding fragments of camelid (from camels or llamas)-specific heavy chain-only antibodies. These nanobodies bind antigen without requiring domain pairing and have a therapeutic advantage over classic antibody fragments because of their smaller size, robustness, and preference to target unique epitopes. Nanobodies have been successfully used to target toxic enzymes or to block specific molecular interactions. Cortez-Retamozo et al. used a nanobody directed to CEA to target the prodrug converting enzyme β-lactamase specifically to tumor cells, resulting in tumor specific toxicity after injection of the prodrug119. Another approach described the construction of nanobodies directed to EGFR to block the binding of the growth factor EGF specifically to its receptor120. Currently, two nanobodies for the treatment of thrombosis are being tested in clinical trials. Thus far, no literature is available describing the use of nanobodies to activate the immune system. In theory, however, coupling of TAA to a DC-specific nanobody should result in effective targeting. Indeed, a recent paper by de Groeve et al. does describe the construction of a DC-specific nanobody121.
Finally, synthetic long peptides should be mentioned as a DC-targeting strategy. Preclinical studies showed increased vaccination efficacy of synthetic long peptides over short peptides which was attributable to selective uptake by DC and prolonged antigen presentation to CD4+ T cells and CTL122. Several clinical trials with synthetic long peptides tumor vaccines emulsified in Montanide have since been carried out. In a phase II trial in which women with stage III vulval intraepithelial neoplasia received 3–4 vaccines of an HPV16 E6/E7 SLP vaccine in incomplete Freund’s adjuvant, clinical responses were observed in 15/19 patients, with complete regressions in 9 patients123. Responses were associated with the strength and breadth of induced CD4+ and CD8+ IFN-γ effector T cell responses. A next generation of synthetic long peptides vaccines is now under development, in which they are coupled to TLR ligands to ensure simultaneous DC targeting and activation124.
10 Conclusions and Future Developments
A wide variety of vehicles and targeting motifs are now identified and available for use in the design of DC-targeted vaccines. Although some targets are more DC-restricted than others, their selection is mostly predicated by the DC subset to be targeted and, importantly, the vaccine delivery route: some targets may be relatively specific for DC in one tissue microenvironment, but not necessarily in another. Further translational studies are urgently needed to explore the best (combination of) DC targeting motifs and the preferred DC subset(s) to be targeted, as well as the most optimal route of in vivo delivery to achieve efficacious DC-mediated immunization. When potentially immunosuppressive B cells, macrophages or other bystander APC are co-targeted as a collateral consequence of poor DC selectivity, this could lead to T cell tolerance rather than anti-tumor immunity. Also, care must be taken when selecting a DC-targeted vaccine formulation in terms of modulatory effects on the DC activation state and functionality. For tumor vaccination in particular it is of vital importance to ensure optimal DC activation upon vaccine delivery with preferential Th1 skewing and CTL activation, and to increase or decrease other arms of immunity as their relative importance in specific cancers becomes better known. In cancer patients where immunosuppressive conditions often prevail this may sometimes be a tall order, but it is nevertheless of the essence. Although DC-targeted vaccine approaches receive considerable attention and are becoming a major focus of attention in the tumor immunology field, clinical translation is seriously lagging with very few vaccines set to be tested in patients within the foreseeable future. Nevertheless, there is a general consensus in the field that in vivo DC targeting vaccines are a most promising way forward. Newly identified promising targeting motifs and advances in the fields of virology and nanotechnology should prove instrumental in developing effective new agents.
References
Mathers AR, Larregina AT (2006) Professional antigen-presenting cells of the skin. Immunol Res 36:127–136
Seya T, Shime H, Ebihara T et al (2010) Pattern recognition receptors of innate immunity and their application to tumor immunotherapy. Cancer Sci 101:313–320
Tacken PJ, Figdor CG (2011) Targeted antigen delivery and activation of dendritic cells in vivo: steps towards cost effective vaccines. Semin Immunol 23:12–20
Mahnke K, Qian Y, Fondel S et al (2005) Targeting of antigens to activated dendritic cells in vivo cures metastatic melanoma in mice. Cancer Res 65:7007–7012
Gurer C, Strowig T, Brilot F et al (2008) Targeting the nuclear antigen 1 of Epstein-Barr virus to the human endocytic receptor DEC-205 stimulates protective T-cell responses. Blood 112:1231–1239
Do Y, Park CG, Kang YS et al (2008) Broad T cell immunity to the LcrV virulence protein is induced by targeted delivery to DEC-205/CD205-positive mouse dendritic cells. Eur J Immunol 38:20–29
Idoyaga J, Lubkin A, Fiorese C et al (2011) Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc Natl Acad Sci USA 108:2384–2389
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327:291–295
Hangalapura BN, Oosterhoff D, Aggarwal S et al (2010) Selective transduction of dendritic cells in human lymph nodes and superior induction of high-avidity melanoma-reactive cytotoxic T cells by a CD40-targeted adenovirus. J Immunother 33:706–715
de Gruijl TD, Luykx-de Bakker SA, Tillman BW et al (2002) Prolonged maturation and enhanced transduction of dendritic cells migrated from human skin explants after in situ delivery of CD40-targeted adenoviral vectors. J Immunol 169:5322–5331
Abe S, Okuda K, Ura T et al (2009) Adenovirus type 5 with modified hexons induces robust transgene-specific immune responses in mice with pre-existing immunity against adenovirus type 5. J Gene Med 11:570–579
Wan Y, Bramson J, Carter R et al (1997) Dendritic cells transduced with an adenoviral vector encoding a model tumor-associated antigen for tumor vaccination. Hum Gene Ther 8:1355–1363
Song W, Kong HL, Carpenter H et al (1997) Dendritic cells genetically modified with an adenovirus vector encoding the cDNA for a model antigen induce protective and therapeutic antitumor immunity. J Exp Med 186:1247–1256
Nikitina EY, Chada S, Muro-Cacho C et al (2002) An effective immunization and cancer treatment with activated dendritic cells transduced with full-length wild-type p53. Gene Ther 9:345–352
Murakami T, Tokunaga N, Waku T et al (2004) Antitumor effect of intratumoral administration of bone marrow-derived dendritic cells transduced with wild-type p53 gene. Clin Cancer Res 10:3871–3880
Bergelson JM, Cunningham JA, Droguett G et al (1997) Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323
Tomko RP, Xu R, Philipson L (1997) HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci USA 94:3352–3356
Rea D, Havenga MJ, van Den Assem M et al (2001) Highly efficient transduction of human monocyte-derived dendritic cells with subgroup B fiber-modified adenovirus vectors enhances transgene-encoded antigen presentation to cytotoxic T cells. J Immunol 166:5236–5244
Ophorst OJ, Kostense S, Goudsmit J et al (2004) An adenoviral type 5 vector carrying a type 35 fiber as a vaccine vehicle: DC targeting, cross neutralization, and immunogenicity. Vaccine 22:3035–3044
Stone D, Ni S, Li ZY et al (2005) Development and assessment of human adenovirus type 11 as a gene transfer vector. J Virol 79:5090–5104
van de Ven R, Lindenberg JJ, Oosterhoff D et al (2009) Selective transduction of mature DC in human skin and lymph nodes by CD80/CD86-targeted fiber-modified adenovirus-5/3. J Immunother 32:895–906
Okada N, Saito T, Masunaga Y et al (2001) Efficient antigen gene transduction using Arg-Gly-Asp fiber-mutant adenovirus vectors can potentiate antitumor vaccine efficacy and maturation of murine dendritic cells. Cancer Res 61:7913–7919
Okada N, Tsukada Y, Nakagawa S et al (2001) Efficient gene delivery into dendritic cells by fiber-mutant adenovirus vectors. Biochem Biophys Res Commun 282:173–179
Worgall S, Busch A, Rivara M et al (2004) Modification to the capsid of the adenovirus vector that enhances dendritic cell infection and transgene-specific cellular immune responses. J Virol 78:2572–2580
Roelvink PW, Mi Lee G, Einfeld DA et al (1999) Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adenoviridae. Science 286:1568–1571
Belousova N, Korokhov N, Krendelshchikova V et al (2003) Genetically targeted adenovirus vector directed to CD40-expressing cells. J Virol 77:11367–11377
Korokhov N, Noureddini SC, Curiel DT et al (2005) A single-component CD40-targeted adenovirus vector displays highly efficient transduction and activation of dendritic cells in a human skin substrate system. Mol Pharm 2:218–223
Tillman BW, Hayes TL, DeGruijl TD et al (2000) Adenoviral vectors targeted to CD40 enhance the efficacy of dendritic cell-based vaccination against human papillomavirus 16-induced tumor cells in a murine model. Cancer Res 60:5456–5463
Pereboev AV, Asiedu CK, Kawakami Y et al (2002) Coxsackievirus-adenovirus receptor genetically fused to anti-human CD40 scFv enhances adenoviral transduction of dendritic cells. Gene Ther 9:1189–1193
Brandao JG, Scheper RJ, Lougheed SM et al (2003) CD40-targeted adenoviral gene transfer to dendritic cells through the use of a novel bispecific single-chain Fv antibody enhances cytotoxic T cell activation. Vaccine 21:2268–2272
Hangalapura BN, Oosterhoff D, de Groot J et al (2011) Potent anti-tumor immunity generated by a CD40-targeted adenoviral vaccine. Cancer Res 71:5827–5837
Rabinowitz JE, Samulski J (1998) Adeno-associated virus expression systems for gene transfer. Curr Opin Biotechnol 9:470–475
Ponnazhagan S, Mahendra G, Curiel DT et al (2001) Adeno-associated virus type 2-mediated transduction of human monocyte-derived dendritic cells: implications for ex vivo immunotherapy. J Virol 75:9493–9501
Chiriva-Internati M, Liu Y, Salati E et al (2002) Efficient generation of cytotoxic T lymphocytes against cervical cancer cells by adeno-associated virus/human papillomavirus type 16 E7 antigen gene transduction into dendritic cells. Eur J Immunol 32:30–38
Liu Y, Chiriva-Internati M, Grizzi F et al (2001) Rapid induction of cytotoxic T-cell response against cervical cancer cells by human papillomavirus type 16 E6 antigen gene delivery into human dendritic cells by an adeno-associated virus vector. Cancer Gene Ther 8:948–957
Chiriva-Internati M, Liu Y, Weidanz JA et al (2003) Testing recombinant adeno-associated virus-gene loading of dendritic cells for generating potent cytotoxic T lymphocytes against a prototype self-antigen, multiple myeloma HM1.24. Blood 102:3100–3107
Liu DW, Chang JL, Tsao YP et al (2005) Co-vaccination with adeno-associated virus vectors encoding human papillomavirus 16 L1 proteins and adenovirus encoding murine GM-CSF can elicit strong and prolonged neutralizing antibody. Int J Cancer 113:93–100
Merchlinsky M, Moss B (1992) Introduction of foreign DNA into the vaccinia virus genome by in vitro ligation: recombination-independent selectable cloning vectors. Virology 190:522–526
Di Nicola M, Siena S, Bregni M et al (1998) Gene transfer into human dendritic antigen-presenting cells by vaccinia virus and adenovirus vectors. Cancer Gene Ther 5:350–356
Subklewe M, Chahroudi A, Schmaljohn A et al (1999) Induction of Epstein-Barr virus-specific cytotoxic T-lymphocyte responses using dendritic cells pulsed with EBNA-3A peptides or UV-inactivated, recombinant EBNA-3A vaccinia virus. Blood 94:1372–1381
Yang S, Kittlesen D, Slingluff CL Jr et al (2000) Dendritic cells infected with a vaccinia vector carrying the human gp100 gene simultaneously present multiple specificities and elicit high-affinity T cells reactive to multiple epitopes and restricted by HLA-A2 and -A3. J Immunol 164:4204–4211
Trevor KT, Hersh EM, Brailey J et al (2001) Transduction of human dendritic cells with a recombinant modified vaccinia Ankara virus encoding MUC1 and IL-2. Cancer Immunol Immunother 50:397–407
Tsang KY, Palena C, Yokokawa J et al (2005) Analyses of recombinant vaccinia and fowlpox vaccine vectors expressing transgenes for two human tumor antigens and three human costimulatory molecules. Clin Cancer Res 11:1597–1607
Engelmayer J, Larsson M, Subklewe M et al (1999) Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J Immunol 163:6762–6768
Jenne L, Hauser C, Arrighi JF et al (2000) Poxvirus as a vector to transduce human dendritic cells for immunotherapy: abortive infection but reduced APC function. Gene Ther 7:1575–1583
Rehm KE, Connor RF, Jones GJ et al (2009) Vaccinia virus decreases major histocompatibility complex (MHC) class II antigen presentation, T-cell priming, and peptide association with MHC class II. Immunology 128:381–392
Di Nicola M, Carlo-Stella C, Mortarini R et al (2004) Boosting T cell-mediated immunity to tyrosinase by vaccinia virus-transduced, CD34(+)-derived dendritic cell vaccination: a phase I trial in metastatic melanoma. Clin Cancer Res 10:5381–5390
Meyer RG, Britten CM, Siepmann U et al (2005) A phase I vaccination study with tyrosinase in patients with stage II melanoma using recombinant modified vaccinia virus Ankara (MVA-hTyr). Cancer Immunol Immunother 54:453–467
Kantoff PW, Schuetz TJ, Blumenstein BA et al (2010) Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol 28:1099–1105
Salio M, Cella M, Suter M et al (1999) Inhibition of dendritic cell maturation by herpes simplex virus. Eur J Immunol 29:3245–3253
Kruse M, Rosorius O, Kratzer F et al (2000) Mature dendritic cells infected with herpes simplex virus type 1 exhibit inhibited T-cell stimulatory capacity. J Virol 74:7127–7136
Samady L, Costigliola E, MacCormac L et al (2003) Deletion of the virion host shutoff protein (vhs) from herpes simplex virus (HSV) relieves the viral block to dendritic cell activation: potential of vhs- HSV vectors for dendritic cell-mediated immunotherapy. J Virol 77:3768–3776
Willis RA, Bowers WJ, Turner MJ et al (2001) Dendritic cells transduced with HSV-1 amplicons expressing prostate-specific antigen generate antitumor immunity in mice. Hum Gene Ther 12:1867–1879
Mann R, Mulligan RC, Baltimore D (1983) Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 33:153–159
Breckpot K, Dullaers M, Bonehill A et al (2003) Lentivirally transduced dendritic cells as a tool for cancer immunotherapy. J Gene Med 5:654–667
Dull T, Zufferey R, Kelly M et al (1998) A third-generation lentivirus vector with a conditional packaging system. J Virol 72:8463–8471
Zufferey R, Nagy D, Mandel RJ et al (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15:871–875
Li GB, Lu GX (2010) A single low dose of dendritic cells modified with lentivirus containing a truncated neu gene can effectively suppress neu-overexpressing tumors. J Gene Med 12:604–612
Lopes L, Dewannieux M, Gileadi U et al (2008) Immunization with a lentivector that targets tumor antigen expression to dendritic cells induces potent CD8+ and CD4+ T-cell responses. J Virol 82:86–95
Dresch C, Edelmann SL, Marconi P et al (2008) Lentiviral-mediated transcriptional targeting of dendritic cells for induction of T cell tolerance in vivo. J Immunol 181:4495–4506
Yang L, Yang H, Rideout K et al (2008) Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat Biotechnol 26:326–334
Raz E, Carson DA, Parker SE et al (1994) Intradermal gene immunization: the possible role of DNA uptake in the induction of cellular immunity to viruses. Proc Natl Acad Sci USA 91:9519–9523
Williams RS, Johnston SA, Riedy M et al (1991) Introduction of foreign genes into tissues of living mice by DNA-coated microprojectiles. Proc Natl Acad Sci USA 88:2726–2730
Fynan EF, Webster RG, Fuller DH et al (1993) DNA vaccines: protective immunizations by parenteral, mucosal, and gene-gun inoculations. Proc Natl Acad Sci USA 90:11478–11482
Bins AD, Jorritsma A, Wolkers MC et al (2005) A rapid and potent DNA vaccination strategy defined by in vivo monitoring of antigen expression. Nat Med 11:899–904
Oosterhuis K, van den Berg JH, Schumacher TN et al (2012) DNA vaccines and intradermal vaccination by DNA tattooing. Curr Top Microbiol Immunol 351:221–250
Condon C, Watkins SC, Celluzzi CM et al (1996) DNA-based immunization by in vivo transfection of dendritic cells. Nat Med 2:1122–1128
Porgador A, Irvine KR, Iwasaki A et al (1998) Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J Exp Med 188:1075–1082
Corr M, von Damm A, Lee DJ et al (1999) In vivo priming by DNA injection occurs predominantly by antigen transfer. J Immunol 163:4721–4727
Ni J, Nolte B, Arnold A et al (2009) Targeting anti-tumor DNA vaccines to dendritic cells via a short CD11c promoter sequence. Vaccine 27:5480–5487
Klinman DM, Yi AK, Beaucage SL et al (1996) CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci USA 93:2879–2883
Gregoriadis G, Bacon A, Caparros-Wanderley W et al (2003) Plasmid DNA vaccines: entrapment into liposomes by dehydration-rehydration. Methods Enzymol 367:70–80
Xiang R, Primus FJ, Ruehlmann JM et al (2001) A dual-function DNA vaccine encoding carcinoembryonic antigen and CD40 ligand trimer induces T cell-mediated protective immunity against colon cancer in carcinoembryonic antigen-transgenic mice. J Immunol 167:4560–4565
Hung CF, Hsu KF, Cheng WF et al (2001) Enhancement of DNA vaccine potency by linkage of antigen gene to a gene encoding the extracellular domain of Fms-like tyrosine kinase 3-ligand. Cancer Res 61:1080–1088
Sin J, Kim JJ, Pachuk C et al (2000) DNA vaccines encoding interleukin-8 and RANTES enhance antigen-specific Th1-type CD4(+) T-cell-mediated protective immunity against herpes simplex virus type 2 in vivo. J Virol 74:11173–11180
Tagawa ST, Lee P, Snively J et al (2003) Phase I study of intranodal delivery of a plasmid DNA vaccine for patients with Stage IV melanoma. Cancer 98:144–154
Rosenberg SA, Yang JC, Sherry RM et al (2003) Inability to immunize patients with metastatic melanoma using plasmid DNA encoding the gp100 melanoma-melanocyte antigen. Hum Gene Ther 14:709–714
Miller AM, Ozenci V, Kiessling R et al (2005) Immune monitoring in a phase 1 trial of a PSA DNA vaccine in patients with hormone-refractory prostate cancer. J Immunother 28:389–395
Klencke B, Matijevic M, Urban RG et al (2002) Encapsulated plasmid DNA treatment for human papillomavirus 16-associated anal dysplasia: a Phase I study of ZYC101. Clin Cancer Res 8:1028–1037
Boczkowski D, Nair SK, Snyder D et al (1996) Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med 184:465–472
Nair SK, Boczkowski D, Morse M et al (1998) Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfected with RNA. Nat Biotechnol 16:364–369
Heiser A, Dahm P, Yancey DR et al (2000) Human dendritic cells transfected with RNA encoding prostate-specific antigen stimulate prostate-specific CTL responses in vitro. J Immunol 164:5508–5514
Heiser A, Maurice MA, Yancey DR et al (2001) Human dendritic cells transfected with renal tumor RNA stimulate polyclonal T-cell responses against antigens expressed by primary and metastatic tumors. Cancer Res 61:3388–3393
Thornburg C, Boczkowski D, Gilboa E et al (2000) Induction of cytotoxic T lymphocytes with dendritic cells transfected with human papillomavirus E6 and E7 RNA: implications for cervical cancer immunotherapy. J Immunother 23:412–418
Nair SK, Heiser A, Boczkowski D et al (2000) Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med 6:1011–1017
Weissman D, Ni H, Scales D et al (2000) HIV gag mRNA transfection of dendritic cells (DC) delivers encoded antigen to MHC class I and II molecules, causes DC maturation, and induces a potent human in vitro primary immune response. J Immunol 165:4710–4717
Grunebach F, Kayser K, Weck MM et al (2005) Cotransfection of dendritic cells with RNA coding for HER-2/neu and 4-1BBL increases the induction of tumor antigen specific cytotoxic T lymphocytes. Cancer Gene Ther 12:749–756
Ashley DM, Faiola B, Nair S et al (1997) Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med 186:1177–1182
Koido S, Kashiwaba M, Chen D et al (2000) Induction of antitumor immunity by vaccination of dendritic cells transfected with MUC1 RNA. J Immunol 165:5713–5719
Strobel I, Berchtold S, Gotze A et al (2000) Human dendritic cells transfected with either RNA or DNA encoding influenza matrix protein M1 differ in their ability to stimulate cytotoxic T lymphocytes. Gene Ther 7:2028–2035
Grunebach F, Muller MR, Nencioni A et al (2003) Delivery of tumor-derived RNA for the induction of cytotoxic T-lymphocytes. Gene Ther 10:367–374
Heiser A, Coleman D, Dannull J et al (2002) Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest 109:409–417
Su Z, Dannull J, Yang BK et al (2005) Telomerase mRNA-transfected dendritic cells stimulate antigen-specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J Immunol 174:3798–3807
Su Z, Vieweg J, Weizer AZ et al (2002) Enhanced induction of telomerase-specific CD4(+) T cells using dendritic cells transfected with RNA encoding a chimeric gene product. Cancer Res 62:5041–5048
Cruz LJ, Tacken PJ, Fokkink R et al (2011) The influence of PEG chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials 32:6791–6803
Sprott GD, Dicaire CJ, Gurnani K et al (2004) Liposome adjuvants prepared from the total polar lipids of Haloferax volcanii, Planococcus spp. and Bacillus firmus differ in ability to elicit and sustain immune responses. Vaccine 22:2154–2162
Fukasawa M, Shimizu Y, Shikata K et al (1998) Liposome oligomannose-coated with neoglycolipid, a new candidate for a safe adjuvant for induction of CD8+ cytotoxic T lymphocytes. FEBS Lett 441:353–356
Scheel B, Aulwurm S, Probst J et al (2006) Therapeutic anti-tumor immunity triggered by injections of immunostimulating single-stranded RNA. Eur J Immunol 36:2807–2816
Weide B, Carralot JP, Reese A et al (2008) Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J Immunother 31:180–188
Cui Z, Mumper RJ (2002) Genetic immunization using nanoparticles engineered from microemulsion precursors. Pharm Res 19:939–946
Fifis T, Gamvrellis A, Crimeen-Irwin B et al (2004) Size-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumors. J Immunol 173:3148–3154
Vogt A, Combadiere B, Hadam S et al (2006) 40 nm, but not 750 or 1,500 nm, nanoparticles enter epidermal CD1a+ cells after transcutaneous application on human skin. J Invest Dermatol 126:1316–1322
Vogt A, Mahe B, Costagliola D et al (2008) Transcutaneous anti-influenza vaccination promotes both CD4 and CD8 T cell immune responses in humans. J Immunol 180:1482–1489
Prasad S, Cody V, Saucier-Sawyer JK et al (2011) Polymer nanoparticles containing tumor lysates as antigen delivery vehicles for dendritic cell-based anti-tumor immunotherapy. Nanomedicine 7:1–10
Tamayo I, Irache JM, Mansilla C et al (2010) Poly(anhydride) nanoparticles act as active Th1 adjuvants through TLR exploitation. Clin Vaccine Immunol 17:1356–1362
Ghotbi Z, Haddadi A, Hamdy S et al (2011) Active targeting of dendritic cells with mannan-decorated PLGA nanoparticles. J Drug Target 19:281–292
Cruz LJ, Tacken PJ, Fokkink R et al (2010) Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J Control Release 144:118–126
Hattori Y, Kawakami S, Suzuki S et al (2004) Enhancement of immune responses by DNA vaccination through targeted gene delivery using mannosylated cationic liposome formulations following intravenous administration in mice. Biochem Biophys Res Commun 317:992–999
Arigita C, Bevaart L, Everse LA et al (2003) Liposomal meningococcal B vaccination: role of dendritic cell targeting in the development of a protective immune response. Infect Immun 71:5210–5218
Faham A, Altin JG (2010) Antigen-containing liposomes engrafted with flagellin-related peptides are effective vaccines that can induce potent antitumor immunity and immunotherapeutic effect. J Immunol 185:1744–1754
De La Pena H, Madrigal JA, Rusakiewicz S et al (2009) Artificial exosomes as tools for basic and clinical immunology. J Immunol Methods 344:121–132
Trumpfheller C, Caskey M, Nchinda G et al (2008) The microbial mimic poly IC induces durable and protective CD4+ T cell immunity together with a dendritic cell targeted vaccine. Proc Natl Acad Sci USA 105:2574–2579
Flacher V, Sparber F, Tripp CH et al (2009) Targeting of epidermal Langerhans cells with antigenic proteins: attempts to harness their properties for immunotherapy. Cancer Immunol Immunother 58:1137–1147
Johnson TS, Mahnke K, Storn V et al (2008) Inhibition of melanoma growth by targeting of antigen to dendritic cells via an anti-DEC-205 single-chain fragment variable molecule. Clin Cancer Res 14:8169–8177
Wei H, Wang S, Zhang D et al (2009) Targeted delivery of tumor antigens to activated dendritic cells via CD11c molecules induces potent antitumor immunity in mice. Clin Cancer Res 15:4612–4621
van Montfoort N, Camps MG, Khan S et al (2009) Antigen storage compartments in mature dendritic cells facilitate prolonged cytotoxic T lymphocyte cross-priming capacity. Proc Natl Acad Sci USA 106:6730–6735
Kretz-Rommel A, Qin F, Dakappagari N et al (2007) In vivo targeting of antigens to human dendritic cells through DC-SIGN elicits stimulatory immune responses and inhibits tumor growth in grafted mouse models. J Immunother 30:715–726
Flacher V, Tripp CH, Stoitzner P et al (2010) Epidermal Langerhans cells rapidly capture and present antigens from C-type lectin-targeting antibodies deposited in the dermis. J Invest Dermatol 130:755–762
Cortez-Retamozo V, Backmann N, Senter PD et al (2004) Efficient cancer therapy with a nanobody-based conjugate. Cancer Res 64:2853–2857
Roovers RC, Laeremans T, Huang L et al (2007) Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR Nanobodies. Cancer Immunol Immunother 56:303–317
De Groeve K, Deschacht N, De Koninck C et al (2010) Nanobodies as tools for in vivo imaging of specific immune cell types. J Nucl Med 51:782–789
Melief CJ, van der Burg SH (2008) Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 8:351–360
Kenter GG, Welters MJ, Valentijn AR et al (2009) Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Eng J Med 361:1838–1847
Khan S, Bijker MS, Weterings JJ et al (2007) Distinct uptake mechanisms but similar intracellular processing of two different toll-like receptor ligand-peptide conjugates in dendritic cells. J Biol Chem 282:21145–21159
Acknowledgements
This work was in part supported by the Netherlands Organization for Scientific Research (NWO-VIDI-grant 917-56-32) and by NIH/NIAID (grant 7R33 AI076096-05 & 04S1).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Oosterhoff, D., Curiel, D.T., de Gruijl, T.D. (2013). Antigen Targeting to Dendritic Cells for Cancer Immunotherapy. In: Curiel, T. (eds) Cancer Immunotherapy. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-4732-0_6
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4732-0_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-4731-3
Online ISBN: 978-1-4614-4732-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)