
Abstract
All life processes, whether plant, animal, or microbial, depend upon a complex network of enzyme-catalyzed chemical reactions for cellular growth and maintenance [1, 2]. As catalysts, enzymes facilitate reactions by enabling alternate reaction mechanisms with lower activation energy, but in no way modify the thermodynamic equilibrium constant or the free energy change of a chemical transformation. They generate enormous kinetic rate accelerations, often exceeding factors of 1012-fold relative to the rate of the uncatalyzed reaction. Enzymes are capable of performing many different chemistries, can be produced on a large scale, and typically operate at ambient temperatures and near neutral pH [3–5]. These attributes have captured the attention of generations of scientists and engineers alike and enabled the dramatic growth of the enzyme industry over the past century.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
All life processes, whether plant, animal, or microbial, depend upon a complex network of enzyme-catalyzed chemical reactions for cellular growth and maintenance [1, 2]. As catalysts, enzymes facilitate reactions by enabling alternate reaction mechanisms with lower activation energy, but in no way modify the thermodynamic equilibrium constant or the free energy change of a chemical transformation. They generate enormous kinetic rate accelerations, often exceeding factors of 1012-fold relative to the rate of the uncatalyzed reaction. Enzymes are capable of performing many different chemistries, can be produced on a large scale, and typically operate at ambient temperatures and near neutral pH [3–5]. These attributes have captured the attention of generations of scientists and engineers alike and enabled the dramatic growth of the enzyme industry over the past century.
All commercial enzymes are proteins, which are assembled in living cells from a combination of the 20 naturally occurring amino acids, according to specific sequences defined by genes within the cell’s DNA. Additional structural features are often present, such as carbohydrates, metal ions, and coenzymes which influence both the physical and catalytic properties of enzymes. The biosynthesis of a given enzyme is typically subject to several layers of cellular regulation and control; however, these controls can often be genetically or environmentally manipulated when overproduction of an enzyme is desired.
Enzymes have been used quite extensively as industrial catalysts and the current trend toward green, more sustainable processes is driving strong growth of this industry [5–9]. They possess the following attributes;
-
1.
They are specific in action, and thus minimize the occurrence of undesirable side reactions
-
2.
They are relatively inexpensive and can perform under mild conditions
-
3.
They are effective for chemical conversions (biocatalysis)
-
4.
They can be produced on a large scale by fermentation from common sugars and other renewable substrates
-
5.
They can be modified and improved to suit a particular application
-
6.
They are safe and acceptable for applications in food processes and medicinal treatments
-
7.
They are effective over a wide range of concentrations
Over 3,000 different enzymes have been isolated and characterized, although less than 100 have ever been used in industrial applications. Enzymes are often named according to the substrate they act upon, for example, cellulose degrading enzymes are called cellulases and those that degrade starch, a mixture of amylase and amylopectin, are referred to as amylases. Many individual enzymes also have common names such as subtilisin, a family of serine proteases, and the digestive proteases chymotrypsin, rennin, and pepsin. Another example is lysozyme, a glycosidase that hydrolyzes peptidoglycan, a key component of bacterial cell walls. Enzymes that act upon polymeric substrates are also described according to their site of action—polysaccharide-degrading enzymes that act within the polymer chain are referred to as endoglycosidases and those that remove sugar units from the termini of the substrate as exoglycosidases. A description of the system used to formally classify enzymes is given later in the chapter. Commonly used industrial enzyme classes, their substrates, and products are listed in Table 31.1.
The world market for industrial enzymes exceeded $US3.5 billion/year in 2010 and was growing at a rate of 5–8% per annum (Fig. 31.1) [10]. Over 50% of the sales come from proteolytic enzymes for use by the detergent, dairy, and leather industries [11]. The carbohydrases, mainly the amylases, isomerases, pectinases, cellulases, and hemicellulases, used in baking, brewing, fuel ethanol, starch, and textiles industries, represent nearly 40% of the total enzyme market [12]. Lipases, phytases, oxidoreductases, and other highly specialized enzymes make up the remainder of total enzyme sales. While markets have matured in North America and Western Europe, regions including Latin America and Asia are still experiencing strong growth driven by demand from the animal feed, textiles, detergent, and grain processing industries. The major worldwide enzyme producers are Novozymes A/S and Genencor, now part of DuPont.

Uses of industrial enzymes
A multitude of non-industrial uses for enzymes have also been developed [13]. Although the total volume of enzyme protein for specialty uses is relatively small, the global market was estimated to exceed $2.5 billion by 2011 [14]. Enzymes used for analytical, clinical, and research purposes include glucose oxidase, hexokinases, pyruvate kinase, uricase, glucose-6-phosphate dehydrogenase, amino acid oxidase, aminopeptidase, and others. Restriction enzymes, endonucleases, and oligonucleotide polymerases are essential tools in Molecular biology and Forensic analysis. Glycosyltransferases and other carbohydrate-modifying enzymes are also routinely employed as tools for carbohydrate research [15]. Another growing application for enzymes is in the field of industrial biocatalysis, whereby enzymes are used for chemical and pharmaceutical manufacture, often displacing traditional chemical catalysts [5, 13, 16–18].
The first section of this chapter outlines the history of the enzyme industry, the general properties of enzymes (classification, structure, catalysis, and kinetics), and the methods used for enzyme discovery and optimization. The second section of the chapter describes the production and applications of industrial enzymes, with a focus on those of greatest commercial importance. The application of biocatalysis in industry is covered in the third section, with an emphasis on the processes employed and the resulting products. The interested reader can find more detailed information in the books and reviews cited throughout the text.
Industrial Enzymes: General Properties
History of Enzyme Use
The history of enzyme use dates back to ancient times and many cultures applied enzymes from various natural sources for the production of beverages, foods, and textiles [2, 5]. It was not until the nineteenth century that the basic nature of enzymes was investigated and their specific actions characterized. Diastase, an amylase mixture prepared from germinating barley, was described by Anselme Payen and Jean-François Persoz in 1833. Soon after, in 1835, Theodor Schwann isolated the digestive enzyme pepsin. As early as 1836, the Swedish chemist Jöns Jacob Berzelius speculated that thousands of catalytic processes took place in every organism and hypothesized that “catalytic bodies” were responsible. A mixture of protease and carbohydrases derived from solid-phase cultures of the fungus Aspergillus oryzae was developed in 1894 by Jokichi Takamine and is still sold under the brand name Takadiastase.
The name “enzyme” itself was coined in 1878 by Wilhelm Kühne, derived from the Greek term meaning “in yeast.” Subsequent work by Emil Fischer beginning in 1894 led to a better understanding of enzymatic action, particularly the concept of substrate specificity. Further studies by Eduard Buchner demonstrated alcoholic fermentation in a cell-free system and he postulated that enzymes were responsible, although this theory was not widely accepted at first [19]. The protein nature of enzymes was demonstrated by James B. Sumner in 1926 through the crystallization of urease, a contribution for which he received the 1946 Nobel Prize [20]. The precise structure of enzymes and proteins in general remained elusive until 1965 when the 3D structure of lysozyme was finally deduced by Phillips and coworkers through X-ray crystallography [21].
Despite an incomplete understanding of enzymes, there was considerable use of crude enzyme preparations before 1950, primarily those derived from animal sources. Applications included cheese-making, leather production, brewing, cleaning, and the manufacture of malt extract. The primary limitation to the further development of enzymes was the inability to produce them on a large scale. This began to change after the mid-part of the twentieth century through the implementation of fermentation-based processes for the manufacture of bacterial amylases and proteases.
The development of large scale processes for the production of glucoamylase and proteases had significant impact within the starch processing and detergent industries, respectively. The ability to formulate enzymes and minimize sensitization of workers in enzyme production was another important development in the early 1970s, meeting a challenge which had significantly impeded the expansion of the enzyme industry. The advent of gene engineering in the late 1970s and early 1980s heralded an expansion of enzyme technologies that continues to this day. The first recombinant industrial enzyme product, a subtilisin protease variant, was marketed by Genencor International, Inc. in 1988 [22]. Additional engineered enzyme products were developed in the 1990s, as well as improved means to discover, engineer, and produce enzymes. In the last decade, the ability to rapidly sequence DNA and information technologies such as Bioinformatics have provided new tools for enzyme development. Powerful analytical methodologies including mass spectrometry, nuclear magnetic spectroscopy (NMR), and X-ray crystallography are now routinely used to solve enzyme structures and analyze the mechanisms underlying enzymatic catalysis [23]. The ability to design enzymes de novo is not yet routine, although some examples have been reported and further developments are highly likely.
The number of applications for enzymes has steadily expanded over the past 40 years and enzymes have gained wide use in the detergent, textiles, grain-processing, animal feed, pulp and paper, fuel ethanol, and chemical industries [5–10]. In addition to the economic benefits that enzymes offer, the need for clean and more sustainable industrial processes will inevitably stimulate new technologies for enzyme development and application.
Enzyme Structure and Properties
Enzymes are complex, three dimensional macromolecules that contain the chemical environment necessary to catalyze a particular reaction mechanism, in addition to possessing a template function that limits the set of possible substrates and the resulting products [3, 4]. All enzymes contain a defined region within their structure, known as the active site, where catalysis takes place. Each enzyme class catalyzes a specific reaction or a group of reactions with certain common characteristics. Many enzymes possess the ability to catalyze reactions with very high stereoselectivity in that they favor the production of only one of two possible stereoisomers. As macromolecules, enzymes can lose their catalytic properties when subjected to agents such as heat, strong acids or bases, organic solvents, or other conditions that break non-covalent bonds, a process called denaturation. Some enzymes can withstand quite extreme conditions, for example, temperatures exceeding 120°C and pH values of less than 1 [24]. The ability of an enzyme to turn over a substrate is referred to as enzyme activity. The activity of an enzyme is a function of the intrinsic nature of the enzyme itself, as well as environmental factors such as substrate concentration, temperature, and pH. All enzymes exhibit optima with regard to pH and temperature. Outside of these optimal regions, enzymes become less efficient and can undergo irreversible denaturation. Enzyme activity can be measured using an enzyme assay, where the concentrations of substrates and/or products are determined after a given period (end-point assays) or as a function of time (kinetic enzyme assays) [25].
All proteins, including enzymes, consist of a linear sequence of amino acids, referred to as the primary sequence. The N-terminal of the sequence possesses a free α-amino group, and the C-terminus contains a free α-carboxylic acid function. A typical enzyme is composed of several hundred amino acid residues with molecular weights in the 20–100 kDa range. The smallest enzyme yet described is 4-oxalocrotonate tautomerase, consisting of only 62 amino acids, whereas the large eukaryotic fatty acid synthase complex is composed of over 2,500 amino acid residues [26, 27]. Commercial enzymes such as proteases, amylases, cellulases, and lipases typically consist of 200–500 amino acids.
A key feature of enzymes and proteins in general is the ability to fold into more complex structures. It is the primary sequence of an enzyme that determines the 3-dimensional structure of an enzyme, although the protein folding process is complex and not yet fully understood [28]. Secondary structure refers to features such as alpha-helices, beta-sheets, and random coils which are defined by hydrogen bonding between different amino acid residues in the primary sequence. The identity and order of amino acids defines the secondary structural features. These secondary features fold further to form the tertiary structure of an enzyme, which is stabilized by a combination of both non-covalent (hydrogen, ionic, and hydrophobic bonds) and covalent bonds (disulfide bridges). The tertiary structure of an enzyme can be depicted in several ways, the most common being a ribbon diagram where the orientation of the amino acid backbone is represented, but amino acid side chains are omitted for clarity. A ribbon diagram of a protease is depicted in Fig. 31.2. Space filling models where all atoms are depicted are also common, particularly when visualizing the active site and other binding regions of an enzyme. Quaternary structure refers to complexes formed from two or more fully folded proteins. Such complexes can take the form of dimers, trimers, and higher order assemblies. These complexes can be formed from many sub-units of the same enzyme (e.g., homodimers and trimers) or several different molecules (e.g., heterodimers, trimers, tetramers, etc.). Many enzymes are catalytically inactive in monomer form and only express activity in quaternary complexes. Protein and enzyme structural hierarchy is depicted in Fig. 31.3.

Ribbon diagram of a protease

Protein and enzyme structural hierarchy
Many enzymes contain additional structural features beyond the amino acids that define the primary structure. These elements are called posttranslational modifications resulting from subsequent processing following the biosynthesis of the primary sequence. The two most common types of posttranslational modifications are phosphorylation and glycosylation with mono- and oligosaccharides. Glycosylation is of particular interest as the carbohydrate chains can account for a large proportion of an enzyme’s molecular weight. Glycosylation is described as either O-linked (serine and threonine residues) or N-linked (asparagine and glutamine residues) depending on whether the glycan chain is attached to the protein backbone through oxygen or nitrogen, respectively. Enzymes made in fungal hosts such as A. oryzae, A. niger and Trichoderma reesei are often glycosylated, in contrast to those produced in bacterial cells. Enzymes often contain additional structural elements including metal ions, phosphate and fatty acid esters, and organic cofactors (coenzymes) like heme, cobalamin, and flavin adenine nucleotide. In many instances, these modifications have regulatory or catalytic functions.
The primary structure of an enzyme can be determined by a process known as sequencing, with Edman degradation being the most widely used technique. Mass spectrometry is now used routinely to determine the amino acid sequence of an enzyme, as well as modifications to the amino acid backbone [29]. Several analytical techniques are used to determine enzyme 3-dimensonal structure, including circular dichroism for secondary structure and X-ray crystallography for tertiary and quaternary structure. NMR is also an invaluable tool in enzyme structure determination and can be used to observe enzyme dynamics [30].
Enzyme Classification
Enzyme nomenclature is defined according to the Enzyme Commission number (EC number), first developed in 1955 at the International Congress of Biochemistry in Brussels. The system is based upon the nature of the reaction that the enzymes catalyze, as opposed to structural or other characteristics. There are six major groupings within the EC system, each of which contains many subtypes. Enzymes that perform hydrolysis reactions (EC 3) such as proteases, amylases, lipases, and cellulases dominate the current markets for industrial enzymes, although examples from each of the other classes have also been commercialized, particularly in the area of biocatalysis (oxidoreductases, lyases, isomerases) and diagnostics (oxidoreductases, ligases). Table 31.2 summarizes the EC classes and the reactions they catalyze. The EC number is also referred to as the IUBMB (International Union of Biochemistry and Molecular Biology) number. The IUBMB periodically updates enzyme nomenclature and lists the corresponding CAS number for each IUBMB number. More detailed information can be found at the International Union of Biochemistry and Molecular Biology enzyme nomenclature website (www.chem.qmul.ac.uk/iubmb/enzyme) and the ExplorEnz database (www.enzyme-database.org).
Enzyme Catalysis and Kinetics
The basic principle by which enzymes and chemical catalysts in general operate is their ability to lower the activation energy (E a or ΔG ‡) for a chemical reaction by altering the reaction coordinate, thus facilitating the rate at which the conversion of substrates to products proceeds [31]. Lowering of the activation energy does not change the equilibrium position of a reaction and accelerates both the forward and reverse directions of a chemical reaction (Fig. 31.4).

A comparison of the reaction coordinates of a catalyzed vs. an uncatalyzed reaction. A catalyst provides an alternate reaction pathway with lower activation energy
The relation between activation energy, temperature, and reaction rate is defined by the Arrhenius equation (31.1) where R is the universal gas constant, T is temperature in Kelvin, k is the reaction rate coefficient, and A refers to the frequency factor. The equation demonstrates why catalysts can produce dramatic rate accelerations. For example, an enzyme that lowers the activation energy of a reaction by 10 kJ/mol at 25°C leads to a rate increase of around 50-fold, whereas a 40 kJ/mol reduction in E a gives a rate acceleration of 107 fold [32].
Reaction rates tend to increase with temperature and enzyme catalyzed reactions are no different, exhibiting a rate increase of approximately twofold for every 10°C rise in temperature. The physical stability of an enzyme is also related to temperature, a concept known as thermostability. Thus, enzymes display temperature optima with regard to their activity. Some enzymes have broad temperature optima, while others display maximal activity over narrower ranges of temperature. Enzymes from animal sources typically have optimal temperatures around 37°C, whereas those from microbial sources have widely varying optima, often correlating to the environment from which the source organism was isolated.
The action of enzymes can be divided into three distinct events—initial substrate recognition, followed by enzymatic catalysis and subsequent product release. The binding of a substrate to an enzyme is often highly specific and the nature of this event was first explained through the lock and key model, developed by Emil Fisher in 1894, which held that the structures of the substrate and enzyme were both rigid and complementary, such that similar but different substrates would not bind with the same affinity, relative to the preferred substrate. The induced fit model was put forward by Koshland in 1958 to overcome the shortcomings of the earlier theory and proposed that the conformations of both enzyme and substrate adapt upon binding in order to produce an enzyme/substrate complex that undergoes catalytic conversion to an enzyme/product complex. This model more readily explains how an enzyme is able to produce and stabilize the transition state that lies between a substrate and the resulting product during the catalytic process.
A number of theories have been proposed to explain how an enzyme lowers the activation energy for a chemical reaction and the topic is still the subject of much debate [33]. A common view is that enzymes stabilize the transition state of a reaction, thereby reducing the free energy of this state and promoting the rate of the reaction. Enzymes accomplish this through non-covalent interactions including hydrogen-bonds, Van der Waals, and electrostatic interactions. This hypothesis is supported by the observation that compounds designed to mimic transition states are often extremely potent enzyme inhibitors with far greater affinity for the enzyme relative to either the substrate or products of the enzyme. For example, Acarbose, a nitrogen-containing pseudo-tetrasaccharide, has a K d for glucoamylase of 10−12 M, far below that of maltotetraose, a substrate for this enzyme [34].
Enzymes also promote reactions through the assistance of chemical functionalities present within the active site that participate in the reaction by acting as acids, bases, nucleophiles, and counter ions. Such catalytic residues consist of amino acid side chains or elements within cofactors. Examples include carboxylic acid groups from glutamic and aspartic acid that participate in glycosidic bond cleavage by glycosidases, serine, histidine, and aspartic acid that comprise the catalytic triad of proteases and the nicotinamide groups from the soluble redox cofactors NADH and NADPH. Metal ions are also commonly employed by enzymes to perform some of the more demanding feats of chemical catalysis, for example, zinc-containing metalloproteases, iron-based oxygenases, and molybdenum nitrogenases [35].
Yet another means by which enzymes promote reactions is through conformational changes of the protein which increases strain on bonds within the substrate and bring catalytic residues into close proximity to the substrate [36]. Such conformational changes can be induced through substrate binding or from the different vibrational modes of the protein itself. The role of enzyme dynamics in catalysis is a topic of current interest, as is quantum tunneling, whereby a proton or electron transfer event occurs at rates faster than one would predict from the known activation energy by tunneling through the energy barrier instead [37]. The enzyme aromatic amine oxidase is thought to utilize quantum tunneling to oxidize tryptamine.
Enzyme Kinetics
The kinetic parameters for a free enzyme in solution are readily derived using the Michaelis–Menten approach describing pseudo-steady-state conversions [4, 38]. Consider (31.2) representing the conversion of a substrate S into a product P, catalyzed by an enzyme E. The rate of formation of an enzyme/substrate complex, ES, is denoted as k 1, the reverse reaction by k −1, and the rate of subsequent conversion to the free product by k 2 .
The Michaelis constant K m under steady-state conditions is defined as follows:
The term k cat can be substituted for k 2 and is referred to as the turnover number of an enzyme (units of s−1). The expression k cat /k m is widely used as a measure of the catalytic efficiency of an enzyme and is termed the specificity constant or turnover number. Where [S] ≫ K m, one can assume that all enzyme is bound to substrate (i.e., [E 0] = [ES]). Under these conditions, the maximal velocity of the reaction V max is a function of k 2, represented in (31.4), where E 0 is the initial enzyme concentration.
From this one can derive an expression for the velocity of an enzyme catalyzed reaction as a function of the substrate concentration [S], termed the Michaelis–Menten equation (31.5):
This relationship can be graphically represented by plotting reaction velocity vs. substrate concentration (Fig. 31.5) or alternately as a double reciprocal plot known as a Lineweaver–Burk plot (Fig. 31.6) where the x-intercept is −1/K m, the y-intercept is 1/v max, and the slope K m/V max.

Enzyme velocity vs. substrate concentration

A Lineweaver–Burke plot
Although the Michaelis-Menten approach provides a useful approximation of enzyme kinetics, there are many situations in which the relation of reaction velocity to substrate concentration is more complex and cannot be adequately modeled using the Michaelis-Menten approach. In many instances, the substrate is a heterogeneous material, such as starch or biomass. There may be other enzymes present, resulting in synergistic effects. Many enzymes possess additional sites other than the active site where molecules can bind and alter activity, a phenomenon known as allosteric regulation. Allosteric regulation can either enhance or decrease enzyme activity and is a common form of regulation observed in metabolic networks.
Enzyme Inhibition
Enzyme inhibitors may also modulate the activity of the enzyme, altering the observed kinetic behavior of the system. An enzyme inhibitor is defined as a substance that can bind to an enzyme and reduce the catalytic rate. There are two overall types of enzyme inhibition—reversible and irreversible inhibition. Irreversible enzyme inhibitors often react with an enzyme forming a covalent enzyme-inhibitor complex. Fluorophosphonate nerve agents (e.g., sarin) are examples of irreversible inhibitors that can react with serine proteases and acetylcholine esterases. Suicide inhibitors are a type of irreversible enzyme inhibitor that is converted into the reactive form through the action of the enzyme they act upon.
In the case of reversible inhibitors, the formation of an enzyme inhibitor complex [EI] is conceptually similar to the enzyme-substrate complex [ES]. The inhibition constant K i is used to describe the affinity of an inhibitor for an enzyme. Weak inhibitors have K i values in the millimolar range and above, whereas potent inhibitors can inhibit enzymes on a stoichiometric (1:1) basis and have subnanomolar K i values. Enzyme inhibitors can be both small and large molecules and can bind at the active site, substrate binding sites, or allosteric sites. Several types of enzyme inhibition are known and are summarized in Table 31.3. Examining the profile of Lineweaver-Burk plots at different inhibitor concentrations is a common means to determine the mode of inhibition.
Reversible enzyme inhibitors are often used to prevent unwanted enzymatic reactions, such as autoproteolysis in protease concentrates. Enzyme inhibitors can also be used to enhance the thermostability of enzyme formulations, provided the inhibitor is not so potent as to reduce the activity of the enzyme under use conditions in the presence of substrate [39].
Enzyme Discovery and Engineering
When a suitable enzyme for a desired application is not available, it becomes necessary to develop a new or improved enzyme. Traditionally, this has been done by screening living microorganisms from either environmental samples or culture collections. With the recent advances in molecular biology and the explosion of genomic sequences, it is possible to isolate novel enzymes through genetic engineering of existing molecules, environmental gene screening, or by genomic database mining [40–43].
The classic method for discovering new enzymes involves screening environmental isolates and culture collections. This process was used very successfully as far back as the 1950s for the discovery of antibiotics such as penicillins and streptomycin. The technique involves examination and screening of thousands of isolates from a diverse array of environments such as soil, plant, and aquatic samples. The limiting factor in this process is the throughput capabilities. A significant investment in time is required to cultivate and screen the organisms, of which many will not have the desired activity. However, one can significantly enhance this process by subjecting an environmental sample to some selective pressure that will enrich for the desired activity. An example of this would be to take a soil sample and then provide a particular compound as the sole carbon source. This would enrich for organisms that are capable of breaking down the compound and using it as a carbon source. Another approach would be to select environmental conditions that would mimic the conditions of the biotransformation. Environments such as the hot springs, mud pots, and fumaroles found in Yellowstone National Park reach temperatures over 80°C and pH values lower than 3. Organisms such as Sulfolobus solfataricus that grow optimally at 80°C and pH 3 will also express extracellular enzymes that work optimally at these conditions. In addition, one the most famous research enzymes, Taq Polymerase, came from an organism, Thermus aquaticus, isolated from Yellowstone National Park.
One of the limitations of screening environmental isolates is the cultivation of the organisms. In a single gram of soil, more than 108 microbes may be present, the majority of which cannot be cultivated. It is clear from molecular ecological studies that traditional cultivation techniques only capture a fraction, usually less than 1%, of the available diversity. To overcome this, direct cloning of DNA from environmental samples enables an unbiased representation of the microbial diversity. The collective genomes of all microorganisms in a given environment is known as the “metagenome” [44]. Briefly, this involves the isolation of DNA from environmental samples and subsequent cloning into plasmids or bacterial artificial chromosome (BAC). The plasmid libraries can then be screened for the desired activity. A comparison to the traditional screening method is shown in Fig. 31.7. For example, Knietsch et al. incubated environmental samples in the presence of glycerol or propanediol to enrich for organisms and activities that would oxidize short-chain polyols or reduce the corresponding carbonyls [45]. Total DNA was isolated from the samples and approximately 100,000 Escherichia coli strains were screened for the desired activity. In total, 16 strains were isolated that stably expressed the desired activity and a number of novel genes were identified that encoded alcohol dehydrogenases and putative oxidoreductases.

Processes for the isolation, screening, and expression of enzymes
A third approach to isolate novel enzymes is via genomic database mining. Genomic database mining is defined as the process of finding and extracting useful information from raw DNA sequence data sets. With the explosion of genomic sequencing, there is a wealth of information available to the researcher. The whole genomes of over 1,200 organisms can be found in databases such as ERGO (http://ergo.integratedgenomics.com) and NCBI (http://www.ncbi.nlm.nih.gov/genome). All three domains of life are represented, bacteria, archaea, and eukaryota, as well as viruses and various organelles (Table 31.4).
With the protein sequence of a molecule that has been characterized biochemically, one can easily identify a large number of sequence homologues in completed genomes. Genes of interest can be identified through BLAST analyses of completely sequenced genomes. In addition, putative biocatalysts can be analyzed for known motifs using ScanProsite (http://ca.expasy.org/tools/scanprosite), the presence of signal peptides using SignalP (http://www.cbs.dtu.dk/services/SignalP/), and genomic organization using the STRING analysis method (http://string.embl.de), for possible enzyme–gene relationships. An example of this was done with the identification of proteases in the hyperthermophiles [46]. Initial efforts to assess the extent and variety of proteases in hyperthermophiles by biochemical methods significantly underestimated this biocatalytic feature. With the benefit of genome sequence data, it is clear that the proteolytic genotypes of these organisms are more expansive than can be inferred from biochemical analyses with over 30 proteases/peptidases identified in each genome.
The last approach involves engineering of an existing enzyme [47]. Molecular biology, high-throughput screening, and other analytical methods have made it possible to redesign and improve wild-type enzymes through the generation and screening of enzyme variant libraries. Site-directed mutagenesis is the method of choice for assessing the role of individual amino acids in the enzymes’ catalytic mechanism or stability. A high-resolution crystal structure or a model based on the structure of a related protein is necessary to identify the residues to mutate. Using this technique, the amino acid of interest is either deleted or altered to another amino acid of choice. Subsequent biochemical analyses will yield information about the residues’ role in the protein. This method has been an important tool in determining protein structure–function relationships and elucidation of catalytic and protein stabilization mechanisms.
Whereas site-directed mutagenesis is a technique that focuses on changing very specific residues based on the protein structure, directed evolution and gene shuffling take a more global approach. An advantage of these methods is the limited knowledge of the proteins structure and mechanism that is required. The discovery of error-prone PCR was an important breakthrough in enzyme-variant generation. The mutation frequency of the error-prone PCR can be controlled at two levels, the reaction conditions and the choice of polymerase. An overview of this method is shown in Fig. 31.8.

Error-prone PCR for directed evolution of enzymes: (a) gene of interest; (b) error-prone PCR generating diversity; (c) expression of variants and characterization; (d) selection of variant with improved trait; (e) variant then serves as template for subsequent rounds of evolution
Another method for generating diversity is gene shuffling. Using this method, one can start with either a single gene or a family of related genes. In the first case, error-prone PCR is carried out and the diversity of the library can be further increased through gene shuffling (Fig. 31.9a). The other alternative is to use multiple genes from various sources with significant levels of identity, usually >70% (Fig. 31.9b). In both cases, the genes of interest are fragmented by a DNAse treatment and then allowed to reassemble via recombination thereby generating a library of variants.

Gene shuffling using a single gene (1) or a family of genes (2): (a) error-prone PCR generating diversity; (b) DNAse treatment; (c) homologous recombination of fragments and generation of diversity
Industrial Enzymes: Properties and Applications
The global market for industrial enzymes is dominated by hydrolases from the EC 3 family, namely α-amylases, glucoamylases, proteases, cellulases, and lipases. Altogether they account for over 85% of the total sales of enzymes. In recent years, additional enzymes have shown strong sales growth, including phytase, an animal feed additive for increasing the bioavailability of phosphorous, laccase for the biobleaching of denim, phospholipases for edible oil production, and xylanases for biomass degradation. Applications of the industrially important enzymes are listed in Table 31.5, along with their microbial sources and commercial applications [6].
Industrial enzymes are either extracted from natural sources (animal, plant, microbial) or manufactured though large-scale microbial fermentation processes. The latter method is responsible for the majority of the enzyme protein produced per year, accounting for over 85% of the total [48]. Submerged fermentation is the dominant fermentation technique, although solid state fermentation is still used for the production of some enzymes [49]. The advent of recombinant DNA technology has had a great influence on the production of enzymes for industrial purposes, both through improvements in the molecules themselves as well as the microbial hosts used in their production. The majority of technical enzymes are now produced in recombinant systems. This has offered many advantages to enzyme users. For instance, the price of enzymes has dropped significantly, while the purity of enzymes has increased overall. The improvement in purity was mainly achieved by the absence of enzymatic side activities, which has enabled much better control of enzyme activity in the industrial environment. General methods for the production, recovery, and formulation of enzymes by fermentation processes are covered in detail within the preceding chapter on Industrial Biotechnology (Chap. 30).
Despite the dominance of large-scale microbial fermentation processes for enzyme manufacture, many commercially relevant enzymes are still isolated from natural sources for both cost reasons, as well as consumer preference. The protease chymosin (rennin), used in cheese-making, is derived from rennet, a substance isolated from calf-stomach lining. While recombinant versions of chymosin expressed in microorganisms are available at lower cost, the demand for the animal-derived product remains strong, especially in Europe. Other animal-derived enzymes include lysozyme, derived from hen egg whites and the digestive aid Pancreatin, a mixture of amylase, lipase, and protease, manufactured from either cow or pig pancreatic extracts.
Plant-derived enzymes also have an important niche in the marketplace. For example, the cysteine proteases papain (papaya latex), ficin (fig latex), and bromelein (pineapple stalks) have been used as meat tenderizers, for contact lens cleaning, and as digestive aids. In the case of papain, latex from papaya trees is collected and dried, resulting in a crude extract. Further purification involves steps including extraction with water, reducing and chelating agents, salt precipitation, and solvent extraction. Highly pure material can be obtained through affinity purification using an immobilized substrate to isolate the enzyme from background components, although this form of purification is often rather costly. In recent years the FDA has restricted the use of papain for topical applications due to the risk of hypersensitivity and potential allergenicity. Malted barley is another important plant source of enzymes providing both α- and β-amylase, as well as β-glucanase, which are used on a very large scale for brewing and food applications. Lipoxygenase, an enzyme that oxidizes fatty acids, is isolated from soy beans. Production of industrial enzymes in recombinant plant systems has generated much interest, as well as controversy in recent years. While this mode of production has the potential to produce large volumes of enzymes directly from carbon dioxide and sunlight, there are concerns about contamination of non-GMO crops with enzyme-producing strains [50].
The major classes of industrial enzymes are discussed below, in addition to emerging enzymes of interest. Further information can be found in the reviews cited early in each section.
Proteases
Proteolytic enzymes (EC 3.4) are by far the most important of the commercially available industrial enzymes having found widespread use for laundry, automatic dishwashing, baking, diary, textiles manufacture, and other applications [11]. These enzymes, being essential parts of the metabolic system of most living organisms, can be isolated from innumerable sources. They can be divided into two overall categories—exoproteases (i.e., peptidases) that hydrolyze terminal peptide bonds releasing free amino acids, and endopeptidases that hydrolyze internal peptide bonds and convert proteins into smaller peptides. Proteases are also classified according to their catalytic mechanism, these subtypes are listed below;
-
Serine proteases (EC 3.4.21)
-
Cysteine proteases (EC 3.4.22)
-
Aspartic acid proteases (EC 3.4.23)
-
Metalloproteases (EC 3.4.24)
This classification is determined by amino acid sequence and through reactivity toward inhibitors that act on particular amino acid residues in the active site region of the enzyme. The serine proteases are widely distributed among microbes. The enzymes have a reactive serine residue in the active site and are generally inhibited by diisopropylfluorophosphate (DFP) or phenylmethylsulfonyl fluoride (PMSF). They are generally active at neutral and alkaline pH. The best-known serine proteases are the subtilisins, a large family of alkaline endoproteases which are the dominant enzymes used in cleaning applications owing to their wide temperature optima, broad substrate specificity, and resistance to denaturation by surfactants and other chemicals [51].
The occurrence of cysteine proteases is also widespread, occurring in both prokaryotes and eukaryotes. Examples include the plant-derived proteases papain and bromelein. These proteases are sensitive to sulfhydryl reagents, such as TLCK (tosyl lysyl chloromethyl ketone) and iodoacetic acid. Aspartic proteases are widely distributed among molds, including Aspergillus, Penicillium, and Rhizopus, but are less commonly found in bacteria and protozoa. They have their maximal activity at low pH, around 3–4. Many of the aspartic proteases are unstable above neutral pH and are not found in cultures growing at neutral or alkaline pH. Most aspartic proteases are sensitive to epoxy and diazoketone compounds. The pepsin-like aspartic proteases have been used commercially in processes such as soybean protein hydrolysis, and the rennin-like aspartic proteases have been used for clotting milk in a manner similar to animal rennins. The rennin-like proteases from Mucor and Endothia species have commercial applications in the manufacturing of cheese. The metalloproteases have pH optima between 5 and 9 and are sensitive to metal-chelating agents such as EDTA. Metalloproteases are widespread and most of the bacterial and fungal metalloproteases contain zinc. Calcium is required to stabilize their protein structure. The best-known metalloprotease is thermolysin produced by Bacillus thermoproteolyticus.
Protease Applications
Microbial proteases with widely different properties are produced commercially. Bacillus proteases, however, represent more than 95% of the sale of all proteases [52]. The most important use of Bacillus proteases is in detergents for both laundry and automatic dish (ADW) applications. Proteinaceous dirt often precipitates on clothes and it coagulates during the normal washing process. Similarly, protein soils can be difficult to remove from dishware and eating utensils through chemical-based detergents alone. The addition of proteolytic enzymes to the detergent cleaves such proteinaceous stains into soluble peptides, thus facilitating stain removal.
The alkaline serine protease of Bacillus licheniformis, also known as Subtilisin Carlsberg, (EC 3.4.21.62), a 274 amino acid protein, was the first protease to find widespread use in both nonionic and anionic detergents [53]. It readily dissolves proteins by cleavage of peptide bonds in a non-specific manner and is sold under the Trademark Alcalase® by Novozymes. It may be used at temperatures up to 65°C, and its pH optimum is close to 9.0, the pH normally used in washing fluids. Additional proteases have also been developed for solid and liquid detergents, including both wild-type and engineered enzymes, at both neutral and alkaline pH ranges. The serine protease Subtilisin Novo (BPN′) from Bacillus amyloliquefaciens has also found application as a detergent enzyme, crude versions of which contain a substantial content of α-amylase. An engineered version of BPN′ was developed by Genencor in 1984 by replacement of a methionine residue (Met 222) with amino acid residues less prone to oxidation by bleach (Ser, Ala and Leu) [22]. The subtilisins from other Bacillus species are also found in many commercial products, including B. lentus, B. clausii, B. halodurans and B. alcalophilus. Commercially available products include Savinase® (Novozymes), Purafect® (Genencor), and Properase® (Genencor) [6].
Sales of proteases were small and relatively unimportant until about 1965. Since then, the use of proteases in detergents has enabled rapid expansion of the enzyme industry as a whole. However, allergic symptoms were discovered in some workers handling enzymes in detergent factories in 1971. The public, particularly in the United States, was alarmed, and proteases were taken out of most detergents. It was later found that every risk could be eliminated with proper precautions in handling by using proteases in liquid form or by encapsulating the enzymes.
Proteases are also used in the leather industry for soaking, bating, and dehairing processes [54]. Their use has enabled reductions in the amounts of hazardous chemicals including sodium sulfide used by the industry. The alkaline protease from B. amyloliquefaciens is used alone or in combination with sulfide for hide treatment and dewooling. A protease from an alkalophilic Bacillus sp. is successfully used for dehairing of ox hides in combination with lime, apparently because the enzyme is stable at pH as high as 12. Serine proteases are also used for wool scouring, a process which involves the removal of the scales naturally present on wool fibers, thus reducing the tendency of wool garments to shrink upon washing.
In the brewing industry, there is a development toward substitution of malted grain with unmalted barley and exogenous enzymes, including α-amylase, β-glucanase, and proteases of microbial and plant origin. The neutral protease from B. amyloliquefaciens and the thermostable neutral protease Bacillus subtilis var. thermoproteolyticus have been used by brewers successfully to hydrolyze barley proteins into amino acids and peptides in order to reduce foaming and haze in the finished product. The choice of enzyme and the condition applied are important as the production of certain peptides can impart undesirable flavors that should be avoided. The use of carbohydrases in brewing is covered in the “Amylases” section below.
Proteases are used in many food applications, primarily baking and cheese manufacture, as digestive aids and in the preparation of protein hydrolyzates, such as those derived from soybean protein. These proteases are derived from a range of different organisms, including Bacillus subtilus, Streptomyces griseus, Aspergillus niger, Aspergillus flavusoryzae, Mucor miehei, and Mucor pusillus.
Rennin, an aspartyl protease mentioned earlier, is found in the fourth stomach of nursing calves. Because of a decline in veal consumption and an increased demand for cheese, the dairy industry found it difficult to obtain sufficient quantities of this enzyme from natural sources. The increasing market for rennin (around $100 million/year) provided the necessary incentive for commercializing a recombinant DNA-derived chymosin. Prochymosin, the self-processing precursor of chymosin, was expressed in E. coli as an insoluble, refractile protein aggregate. The cells were lysed, and the insoluble prochymosin mass was isolated by centrifugation. The prochymosin was solubilized by 8 M urea with high pH treatment, renatured, and activated, and then chymosin was purified by ion exchange chromatography. The recombinant E. coli chymosin is no longer commercially available. Today recombinant chymosin is produced in the microorganisms Kluyveromyces lactis, A. niger and T. reesei. These organisms secrete prochymosin. After prochymosin is separated from the cell broth, a pH drop induces the autocatalytic cleavage of prochymosin, which yields the active chymosin. This process is much cheaper than the former downstream processing from E. coli fermentations.
In addition to the recombinant chymosin, several coagulants for cheese-making are available from microbial origin. These are endoproteases from Rhizomucor miehei, Rhizomucor pussilus, and incidentally the plant-derived endothiapepsin from Cryphonectria parasitica.
Protease Production
The protease fermentation of Bacillus bacteria takes place under strictly contained conditions in conventional equipment for submerged fermentations. The aeration rate is about 1 vvm (volume of air per volume of medium per minute). Vigorous agitation is used to improve air distribution and oxygen transfer. The fermentation temperature is around 37°C, and the time cycle is 2–4 days. The composition of the fermentation medium is important to the yields of protease. Proteins of many different sources are used in commercial media. Carbohydrates are used as an energy source. The C/N ratio is important to the success of the process. Protein should be present in high concentration, and carbohydrate must not be in excess. A convenient way of obtaining this is to conduct fed-batch fermentation, feeding carbohydrate during the run and maintaining the carbohydrate concentration below 1%. Continuous fermentation of protease on a commercial scale is currently not practiced. The recovery and finishing of Bacillus protease involves the following steps.
-
1.
Cooling to about 4°C to prevent microbial spoilage.
-
2.
Precipitating undesirable salts using flocculants or filter aids.
-
3.
Removing particles by centrifugation or filtration.
-
4.
Removing pigments and odors with activated carbon treatment.
-
5.
Concentration by ultrafiltration to product an ultrafiltered concentrate (UFC).
-
6.
Sterile filtration to reduce microbial contamination.
-
7.
Addition of formulation chemicals in order to produce a stabilized concentrate.
-
8.
If a solid protease product is desired then granulation technologies are used.
Most, if not all, proteases are now produced on an industrial scale using genetically engineered organisms that overexpress either the wild-type enzymes (through homologous expression or self-cloning) or variants that have been obtained through protein engineering. The major reason for using the genetically engineered organisms is to reduce production cost. The overexpressing Bacilli can secrete proteases into the fermentation broth in very high yields. The secreted protease becomes the dominant protein in the broth. This greatly facilitates and reduces the cost of downstream processing and purification.
Amylases
The commercial importance of amylolytic enzymes is now well established and has driven significant expansion in the grain processing and fuel ethanol industries [55–58]. Amylases are also used in combination with proteases for cleaning applications. These enzymes catalyze the hydrolytic reactions of amylose (unbranched starch), amylopectin (branched starch), glycogen (animal starch), and related glucose polymers. Amylases, according to their difference in modes of action, can be divided into;
-
1.
α-Amylase (EC 3.2.2.1), an endoglycosidase, hydrolyzes α-1,4-linkages randomly to yield a mixture of oligosaccharides, maltose, and glucose
-
2.
β-Amylase (EC 3.2.1.2), an exoglycosidase, cleaves successive maltose units from the nonreducing end of starch
-
3.
G4-amylase (EC 3.2.1.60), an exoglycosidase, hydrolyzes maltotetraose (G4) units from the non-reducing ends of starch
-
4.
Glucoamylase (or amyloglucosidase) (EC 3.2.1.3) sequentially liberates glucose units from the non-reducing termini of amylose and amylopectin
-
5.
The debranching enzymes such as pullulanase (EC 3.2.1.41) attacks the α-1,6-linkages at the branching point of amylopectin
The properties of amylases listed above have led to their use in many applications, including;
-
1.
To produce grain syrup, glucose syrup, liquid glucose, and crystalline glucose
-
2.
To produce high-fructose corn syrup (HFCS) in combination with glucose isomerase
-
3.
To solubilize and saccharify starch for ethanol production in brewing, distilling, and fuel industries
-
4.
To modify the viscosity of starch used in coating printing papers
-
5.
To remove starch sizes applied to cotton thread before weaving in the textiles industry
-
6.
To produce maltose-containing syrups in brewing and baking industries
-
7.
To reduce the viscosity of sugar syrups used in various food and sugar products
-
8.
As a component in digestive aids
-
9.
To remove starch stains on clothes/dishes
α-Amylase is produced commercially by using both fungal and bacterial species. Fungal amylase has relatively low heat stability and its major application is in the baking industry to supplement the variable activity of amylase present in wheat flour. Bacterial amylases are much more heat stable and it is used in brewing, starch degradation, alcohol, textiles, and detergent industries. The organisms commonly used for the commercial production of α-amylase include:
-
Fungi
-
Aspergillus oryzae
-
Trichoderma reesei
-
-
Bacteria
-
Bacillus subtilis
-
Bacillus licheniformis
-
Bacillus amyloliquefaciens
-
A. oryzae can be grown in either semisolid or submerged culture. In semisolid culture, it produces several enzymes, primarily α-amylase, glucoamylase, lactase, and protease. In submerged culture, the production of α-amylase is increased and the formation of other enzymes becomes minimal. The use of this fungal amylase in the baking industry speeds up the yeast (Saccharomyces carlsbergenis) fermentation; produces stiffer, more stable dough; and improves the texture, porosity, digestibility, and shelf life of bread. The fungal α-amylase delivers its optimal activity at pH 5–7 and at 50–55°C.
In the last decade, both protein engineering and recombinant DNA technology have been used to improve the thermostability of amylases by increasing the negative charges at its calcium-binding site and to improve the enzyme’s resistance to oxidative compounds present in bleach containing detergents by substituting the oxidation-sensitive methionine residue with leucine. Genencor and Novozymes market their improved enzymes under the trade names of PuraStar® OxAm and Duramyl®, respectively.
Amylase Production
Different amylase-producing organisms may require different fermentation conditions for optimal enzyme production. When B. subtilis is used, the fermentation medium may contain starch, cornsteep liquor, yeast, phosphate, and some mineral salts. The amylase treatment on starch is often short to prevent the significant accumulation of glucose, which is inhibitory to the Bacillus amylase fermentation. The fermentation is run at neutral pH and at around 35°C. Care must be taken to prevent contamination by unwanted microorganisms. The time cycle is about 48 h. Whole mash may be used directly for starch liquefaction and saccharification or the mash may be processed to produce liquid or crystal enzyme preparation with high purity. The processing, involving filtration or centrifugation of the bacterial fermentation broth, presents a challenge to the recovery plant. Pretreatment with coagulating or flocculating agents is often needed. The amylase produced by this Bacillus strain is relatively unstable, but the addition of calcium chloride improves the stability.
Fungal glucoamylase, an exoglucosidase, drives the further degradation of starch following initial solubilization by α-amylase (often of a bacterial origin). Both A. niger and increasingly T. reesei are used for the production of glucoamylase. The fungal fermentation starts with a medium containing 25–30% starch and around 10% cornsteep liquor. Incremental or continuous feeding of concentrated nutrients may be used to circumvent the problems caused by a concentrated initial medium. The fermentation pH is about 4 and temperature around 28°C. The fermentation has a high oxygen demand. High oxygen tension, however, inhibits enzyme production. Levels of dissolved oxygen (DO) that approach zero are not atypical in this fermentation. After the completion of the fermentation in 4–5 days, the fermentor mash is cooled and filtered to remove cells and insoluble matters. Trans-glucosidase may be removed using clay, destroyed preferentially using proteases at certain pHs and temperatures, or inactivated by magnesium oxide. Contamination of glucoamylase by the transglucosidase activity may result in the loss of 5–10% of glucose to isomaltose and panose by a reversion process. These reversion products also impede the crystallization of glucose.
Amylases in Wet and Dry Milling Processes
The enzymatic hydrolysis of starch to glucose has long since superseded the acid hydrolysis route using hydrochloric acid. The enzymatic process produces fewer side products, does not involve a corrosive acid, and allows the use of less pure starch products whose protein contaminants would, upon acid hydrolysis, give amino acids and browning reactions. Fungal glucoamylase in combination with bacterial α-amylase makes a complete enzymatic mixture for hydrolysis of starch to glucose. Corn is the dominant source of starch in the United States and is processed by either wet or dry milling processes to produce ethanol, starch, dextrins, corn syrup, glucose, and fructose syrups, in addition to byproducts of significant value such as corn oil and dry distillers grains and solubles (DDGS), a protein-rich animal feed [59].
Corn wet milling involves initial separation of the grain kernel into the germ, fiber, protein, and starch components prior to fermentation. The corn wet milling process is depicted in Fig. 31.10. Liquefaction of starch to form maltodextrins is aided by the action of bacterial α-amylase, whereas fungal glucoamylases are used in order to saccharify maltodextrins to glucose. Prior to liquefaction, granular starch is gelatinized by heat treatment at temperatures of up to 108°C for 5 min in the presence of a thermostable amylase. The α-amylase from B. amyloliquefaciens functions at pH 5.5–7 and 90°C and α-amylase from B. licheniformis functions at pH 5.5–9 and temperatures as high as 110°C. In the subsequent saccharification process, an appropriate amount of fungal glucoamylase is added to the thinned starch (30–50% dry substance) with stirring at 55–60°C and pH 4–5 for 48–72 h. This achieves a final DE (dextrose equivalents) of about 97, with about 94% of the dry weight being glucose. The equilibrium concentrations of the saccharides formed by resynthesis limit the maximum degree of hydrolysis obtainable. Because the activity of glucoamylase toward the branching points (the α-1,6-linkages) is low, it may be advantageous to use a debranching enzyme such as pullulanase early in the hydrolysis process.

Flowsheet of wet corn milling process (Copyright FLSmidth Co. and reproduced with permission)
Dry milling of corn is the dominant means by which corn is converted into bioethanol (Fig. 31.11) and delivers improved process economics relative to corn wet milling. The process begins with the grinding of whole corn into flour in a hammer mill. This flour is slurried, the pH adjusted to around 5.8, and treated at 80–90°C with α-amylase for 30–45 min in order to reduce viscosity. This slurry is then gelatinized and liquefied in a similar manner to the corn wet milling process. Simultaneous saccharification and fermentation (SSF) combines the conversion of maltodextrins to glucose by glucoamylase and fermentation to ethanol. The yeast Saccharomyces cerevisiae is typically used, the fermentation period lasting from 48 to 72 h. Following fermentation, the spent mash (referred to as “beer”) is distilled to produce 95% (190 proof) ethanol which is further dried and ultimately blended into gasoline.

Overview of a corn dry-milling process
Enzymes for the processing of granular starch directly to glucose at lower temperatures (<50°C) have been developed, for example, Stargen™ (Genencor) and BPX (Novozymes). The Granular starch hydrolysis (GSH) process eliminates the cooking step and combines the liquefaction, SSF steps, further streamlining the overall economics of corn drymilling.
Amylases are widely used in cleaning products, primarily laundry and automatic dish-washing detergents. The ability to hydrolyze starch in food soils greatly boosts the ability of the detergent to remove stains. Many amylases are active at the high pH values and temperatures that some applications demand. In addition, detergent amylases must be resistant to proteolysis and bleaches. Typical dosing levels in the wash are in the 0.1–5 ppm range. In recent years, much effort has gone into developing amylases and other detergent enzymes that operate with improved efficiency at lower temperatures, allowing energy savings.
Glucose Isomerase
Starch degradation using α-amylase and glucoamylase produces corn syrup with glucose concentrations of up to 94% on a dry weight basis. The glucose can then be isomerized to a mixture of glucose and fructose by glucose isomerase (GI), an enzyme present in many microorganisms [60]. The product, known as HFCS, has become the dominant sweetener used in food and beverages, displacing fructose syrups derived from the action of invertase on sucrose. Industrial production of HFCS represents one of the largest biocatalytic processes in current practice and is discussed further in the third section of this chapter.
Many organisms are glucose isomerase producers. Most of them produce xylose isomerase with low specificity and glucose can be an alternative substrate for the enzyme. Table 31.6 lists microorganisms used industrially for glucose isomerase production, along with their reported yields. The desirable application conditions of glucose isomerase are pH 7.0–8.0 and 65°C. Glucose isomerase fermentation typically has a cycle time of about 2 days. The fermentation conditions vary from producer to producer. Many glucose isomerase-producing organisms require xylose and cobalt for full enzyme induction. Xylose is too rare and expensive to be used in commercial fermentation processes and cobalt ions remaining in the spent fermentation medium constitute a serious environmental hazard. However, mutants that do not require xylose and cobalt have been obtained for commercial production of glucose isomerase.
Almost every known glucose isomerase is an intracellular enzyme and can only be extracted from the cells in relatively low concentrations, so it is expensive to use this enzyme as a soluble and “once only” enzyme. Immobilized enzyme/cell technology is the key scientific advance that has made the use of glucose isomerase for HFCS production a commercial success. Improved mutants of GI are still actively sought, particularly those with altered pH optima and increased temperature resistance.
Cellulases
Cellulases (EC 3.2.1) are a large group of enzymes that find use in many industrial applications [61]. Cellulose is a homopolymer of glucose monomers, which are linked by β-1,4 bonds. The difference of the linkages distinguishes cellulose from starch and determines the difference in macroscopic properties of these two natural polymers. Starch functions as energy storage in the form of glucose and is consequently used by humans as a food ingredient, whereas cellulose is a structural polymer, which is used for production of paper and textiles. Cellulases can be separated into three distinct groups: the endo-1,4-β-glucanases (EC 3.1.2.4), which catalyze the hydrolytic cleavage within the cellulose polymers; the exo-1,4-β-glucanases (cellobiohydrolases, EC 3.2.1.91), which attack the polymer from the ends releasing cellobiose; and the β-glucanases (EC 3.2.1.21), which catalyze the hydrolysis of cellobiose into glucose monomers.
Cellulases are found in fungi and bacteria. Of commercial interest are fungal enzymes from Aspergillus sp. or T. reesei and a few bacterial enzymes. They are either used as a multicomponent, which contain all enzyme types and are found in T. reesei (Hypocrea jecorina), or as a monocomponent enzyme product, which consists of only one of the three types of enzymes. The multicomponent enzyme preparations can be produced from a selected cellulose overproducing strain of the wild-type organism, whereas the monocomponent cellulases are mainly produced in recombinant production systems.
Currently, the main use of cellulases is in the textiles, paper, and laundry detergent industries. In textiles applications, cellulases are used to change the appearance of the fabric by enzymatic removal of cellulose fibrils at the surface and in the so-called stone-washing process of denim jeans, which gives them the aged look. In the paper industry, cellulases are used as process aids, where they increase the efficiency of the papermaking process by saving energy or allowing higher machine speed. The properties of the paper can also be influenced by cellulose usage. As detergent ingredients, cellulases assist in the cleaning process and can maintain the new look of the washed textiles by removing pills or micro fibrils from the surface of cellulose-based garments.
Hemicellulases
Hemicellulases are a diverse set of enzymes that degrade mixed polysaccharides present in plant derived biomass, known collectively as hemicelluloses [62, 63]. Hemicellulose polymers comprise a variety of different sugars, principally 5-carbon sugars (xylose, arabinose), 6-carbon sugars (mannose, galactose), and uronic acids (galacturonic and glucuronic acids). They also contain ester linkages with organic acids such as acetic and ferulic acids, or with alcohols, for example, methyl esters of galacturonic acid. Accordingly, the hemicellulases span a range of different enzyme activities capable of cleaving both glycosidic and ester bonds. Hemicellulases are widely distributed in the microbial world and are found in bacteria, fungi, and yeasts from many environments. Some more common hemicellulases are listed below. A more comprehensive account on hemicellulases can be found in Chap. 33 on Biomass Conversion in this volume and the references cited within.
-
α-l-Arabinofuranosidase (EC 3.2.1.55)
-
Endo-α-1,5-arabinase (EC 3.2.1.99)
-
α-Galactosidase (EC 3.2.1.22)
-
Endo-galactanase (3.2.1.89)
-
α-Glucuronidase (EC 3.2.1.139)
-
Endo-β-1,4-mannanases (EC 3.2.1.78)
-
Exo-β-1,4-mannanases (EC 3.2.1.25)
-
Endo-β-1,4-xylanases (EC 3.2.1.8)
-
Exo-β-1,4-xylanases (EC 3.2.1.37)
-
Acetyl xylan esterase (EC 3.1.1.72)
-
Ferulic acid esterase (EC 3.1.1.73)
Hemicelluloses, like xylan, arabinogalactan, or glucomannan, are part of many natural materials from plant origin and have a variety of uses. Consequently, xylanases, glucomannanases, and mannanases find use in many technical applications, for example, as detergent additives, for flour processing in baking, as animal feed additives, as well as in the textiles and pulp and paper industry.
The pectins are a related but distinct class of plant polysaccharide composed of poly-β-1,4-galactouronic acid chains with varying degrees of methyl ester functionality. Four different enzyme classes help degrade pectin and are referred to collectively as “pectinases” [64]. They are pectate lyase (4.2.2.2), pectin lyase (EC 4.2.2.10), polygalacturonase (EC 3.2.1.15), and pectin methylesterases (EC 3.1.1.11). The predominant use of pectinases is for the clarification of fruit juices by hydrolysis of haze-forming pectins. Pectate lyase is also used for cotton scouring by modifying pectin within the cotton structure and enhancing the subsequent washing steps. Another textiles application for which hemicellulases are used is for desizing of fabric.
Lignocellulose Degradation
The use of cellulase/hemicellulase blends for conversion of lignocellulosic biomass into fermentable sugars for the production of ethanol and other chemicals promises to greatly expand the market for these enzymes [65]. The quest for economic processes for the utilization of cellulosic biomass has faced many challenges, for example, the cost of and amount of the enzymes required to carry out the process. As much as 100-fold more native cellulase protein (as compared to amylase protein for breakdown of starch) is required for conversion of pretreated substrate (e.g., corn stover) to fermentable sugars. In addition, chemical or thermal pretreatment of cellulosic biomass is needed in order to enable subsequent enzymatic hydrolysis of polysaccharides. Despite these hurdles, much progress has been made in recent years through intensive research programs conducted by government, industry, and academic institutions and have led to the construction of several pilot and demonstration plants, primarily for ethanol production. These efforts have led to identification of new enzymes for biomass degradation, enzymes with greater thermostability, and the optimization of the ratios of the components in enzyme blends.
One strategy to minimize ethanol production cost is to run simultaneous SSF, which utilizes ethanologens engineered to operate in high-temperature environments. Ideally, the fermentation organism has ability to utilize C5 sugars derived from the hemicellulose component, with acceptable productivities in the presence of numerous byproducts of the biomass pretreatment process, leading to lower overall production costs [66]. Several enzyme products designed specifically for biomass processing are now commercially available, for example, the Accellerase™ product line from Genencor.
Lipases and Esterases
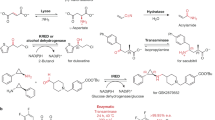
Lipases (EC 3.1.1.3) and esterases (EC 3.1.1.1) are enzymes that catalyze the hydrolytic cleavage of ester bonds (Fig. 31.12), but differ in their substrate spectrum. Lipases have the special capability to catalyze the hydrolysis of water-insoluble substrates such as fats and lipids. Like many other enzyme-catalyzed reactions, the ester hydrolysis is a reversible process, which allows using lipases and other esterases for the synthesis of esters. The use of lipases and esterases as biocatalysts is described in the Industrial Biocatalysis section of this chapter.

Lipases and esterases catalyze the hydrolysis of ester bonds
True lipases show the interfacial activation phenomenon in their catalytic activity pattern. At low concentration of water-insoluble substrates, lipases are almost inactive, and the hydrolytic activity does not increase linearly. At a certain substrate concentration, however, the hydrolytic activity of lipases increases rapidly and the lipase kinetics resembles “normal” enzyme kinetics. This boost in activity is related to the formation of water-insoluble substrate aggregates such as micelles or another second phase. Only when this second phase is present, do lipases become fully active. This interfacial activation is caused by a large conformational change in the 3D structure of the lipases. In their water-soluble form, the active site is covered by a lid, which prevents the substrates from reaching it. At the lipid/water interface, the lid is opened and the active site is accessible to the substrates. In addition, the now accessible area is mainly hydrophobic, which gives the open-form lipase the shape and behavior of conventional surfactant molecules with a hydrophilic and a hydrophobic moiety in one single molecule.
Lipases are found in a wide variety of bacteria, yeasts, and filamentous fungi. Due to their unique properties, lipases are used in all applications where lipids are modified or need to be removed [67]. In the detergent industry, lipases are added to laundry detergents for the removal of fat-containing stains. In the paper industry, lipases remove fatty pitches from paper in the manufacturing process. These pitches stem from remaining resin of the wood chips.
In the food industry, lipases are used in lipid modification processes. In these processes, the texture, digestibility, or physical properties of natural lipids are modified by lipase-catalyzed transesterification reactions with lipids other than the original fatty acids. In the baking industry, lipases are used to influence the quality of bread through modification of the wheat flour lipids. Finally, lipases are used for flavor enhancement of cheese in the dairy industry. The phospholipases (EC 3.1.1.4) related class of enzymes which act upon phospholipids such as lecithin [68]. The major use of these enzymes is for degumming of vegetable oils, for egg processing, and in the baking industry.
A new industrial application for lipases has been developed in the field of renewable energy. Lipases are used for methylation of plant oils such as rapeseed oil. The resulting methylated oils (biodiesel) are used as a replacement or supplement for fossil fuel-derived diesel. As of today, the costs of the biocatalyst, however, prevent its commercial application and chemical processes such as saponification of fats with sodium methoxide is currently preferred.
Phytase
Phytases are a class of phosphate ester hydrolase (a phosphatase) that act upon phytic acid (phytate, myo-inositol-1,2,3,4,5,6-hexakisphosphate), the dominant form of phosphate in plant feedstuffs, including cereal grains and oil seeds. The use of phytases as additives in feed for monogastric animals has expanded greatly in recent years. They increase the bioavailability of phosphate, thus enhancing animal growth while reducing the amount of phosphate that would otherwise end up in the environment [69].

Most commonly used in swine and poultry feed, phytases also increase the bioavailability of other minerals (calcium, magnesium, iron, and zinc) and nutrients (amino acids, proteins) by hydrolysis of complexes with phytate (phytin) that tend to sequester these nutrients.
Phytases are found in many organisms where their functions include phosphate metabolism and the modulation of inositol phosphate regulatory metabolites. Phytases are characterized according to the position at which they initially remove phosphate from phytic acid. Plant phytases (EC 3.1.3.26) remove phosphate from the 4- and 6-positions of phytic acid, whereas those from microbial sources (EC 3.1.3.8) such as Aspergillus species tend to remove phosphate from the 3-position. There are exceptions to these rules, for example, phytases from E. coli, Buttiauxella and Peniophora sp. act at the 6-position of phytate. Both acid- and alkaline-phytases have been described. Those from microbial sources usually belong in the former category with pH optima in the 4.5–6.5 range. Phytases expressed in eukaryotic organisms are typically glycosylated, a feature than has been found to confer stability by reducing the protein aggregation at elevated temperatures. There are four different types of phytase, listed in Table 31.7.
Bacterial and fungal phytases are most readily produced at scale and possess the activity and stabilities required for an industrial product. Nonetheless, most wild-type phytases have temperature optima in the 40–60°C range and considerable activity is lost at the elevated temperatures (>90°C) attained in the steam pelleting process used to manufacture feed pellets. Ideally, a phytase product must retain at least 80% of the initial activity upon transient exposure to temperatures of 90°C or greater. For this reason, both protective enzyme granulation technologies and phytases engineered for greater thermostability have been developed to minimize loss of activity during the feed pelleting process. Commercial Phytase products include Phyzyme® XP (Danisco), Ronozyme® (DSM), and Natuphos™ (BASF).
Phytases have also found application in the starch processing industry where they are used to reduce the levels of phytate derived from corn and other feedstocks. Enzyme blends containing both α-amylase and phytase are commercially available. Reduction of phytate in the starch liquefaction process has numerous benefits including;
-
Ethanol yield improvement
-
Removal of phytate-mediated inhibition of α-amylases
-
Viscosity reduction during liquefaction
-
Release of inositol, a yeast nutrient
-
Production of low-phytate, high phosphate DDGS for animal feed
Overall the market for phytase is poised for further growth, particularly in Asia and other developing regions, driven by demand for meat and need to minimize the associated effects on the environment.
Oxidoreductases
Oxidoreductases (EC family 1) are enzymes that add or remove electrons from a substrate. Cofactors are usually required to assist electron transfer and a stoichiometric electron donor or acceptor is always needed. Applications for oxidoreductases have been limited in comparison to the hydrolases, but have grown substantially in recent years [70]. Oxidases using oxygen as a terminal electron acceptor have found the greatest application, whereas those that require an electron source are still restricted in their practical application. Oxidoreductases of particular prominence are discussed below.
Glucose oxidase (EC 1.1.3.4) converts d-glucose into d-gluconolactone utilizing molecular oxygen (O2) as the ultimate electron acceptor and producing hydrogen peroxide in the process [71]. This enzyme is used for scavenging traces of oxygen in food products, generation of hydrogen peroxide as a preservative, and for the detection of glucose in body fluids. While the later application does not consume a large amount of enzyme by weight, it forms the basis of modern glucose meters used by diabetics and others to monitor blood glucose levels. Glucose oxidase is commonly used in conjunction with catalase (EC 1.11.1.6), an oxidoreductase that converts hydrogen peroxide into oxygen and water [72]. Catalase is also used to decompose excess hydrogen peroxide in textiles bleaching applications.
Laccases (EC 1.10.3.2) are a class of copper-containing oxidases that transfer electrons from phenolic substrates to oxygen, producing water in the process [73]. The phenolic substrates are also known as mediators and can remove electrons from a wide array of substrates including lignin and dyes. Laccases occur in plants, bacteria, and fungi and have a characteristic blue color in their pure form. Commercial applications include bleaching of indigo in denim manufacture, decolorization of dyes in wastewater, and for degradation of lignocellulosic biomass [74].
Emerging Industrial Enzymes
New applications for established classes of industrial enzymes such as proteases and amylases are developed every year [6]. By contrast, products based upon new classes of enzyme are less common, although there is great potential for new enzymes that enable chemistries which cannot be performed under mild conditions. Some examples are discussed below.
Organophosphate hydrolase (OPH) (EC 3.1.8.1) and organophosphorous acid anhydrolases (OPAA) (EC 3.1.8.2) are enzymes that detoxify organophosphorous compounds including pesticides (e.g. paraxon, parathion, malathion) and chemical warfare agents (e.g., sarin, soman, tabun) [75]. These enzymes are being assessed for use by primary responders to counter chemical agents, although even larger potential lies in dealing with organophosphate pesticide contamination in agriculture.
Asparaginase (EC 3.5.1.1) is an enzyme that converts the amino acid asparagine to aspartic acid and ammonia by hydrolysis of the side-chain amide functionality. This class of enzyme is used to reduce the formation of acrylamide, a neurotoxic compound, in starchy foods such as French fries, by treatment of the foodstuff prior to cooking [76]. Products containing asparaginase are sold under the tradenames Acrylaway® and PreventASe™.
A bacterial arylesterase capable of peracid formation under aqueous conditions was developed by Genencor for biobleaching applications and is currently sold under the tradename PrimaGreen® EcoWhite. This enzyme has a unique active site configuration which disfavors hydrolysis of esters and readily performs both perhydrolysis and acyltransfer reactions [77]. The enzyme is formulated with propylene glycol diacetate and catalyzes the formation of peracetic acid in the presence of dilute aqueous hydrogen peroxide.
Section III: Industrial Biocatalysis
The use of enzymes for the synthesis of bulk and fine chemicals represents a somewhat specialized application relative to their wider industrial uses. Nonetheless, biocatalysis is becoming increasingly relevant within the chemical industry for the production of a wide range of materials (see Table 31.8) [5, 16–18]. Broadly defined, a biocatalytic process involves the acceleration of a chemical reaction by a biologically derived catalyst. In practice, the biocatalysts concerned are invariably enzymes and are used in a variety of forms. These include whole cell preparations, crude protein extracts, enzyme mixtures, and highly purified enzymes—both soluble and immobilized. The products of biocatalytic processes are as varied, ranging from the synthesis of fine chemicals and pharmaceuticals to polymer precursors, flavor, fragrances, foodstuffs, and fuels. The common theme is that the use of a biocatalyst has either lowered the cost of production or facilitated the development of a new product.
Biocatalysts often enable one to perform chemical transformations with greater selectivity or under milder conditions than might be achieved through conventional chemical means [78]. Large reductions in waste streams have been achieved in many instances. The term “bioconversion” has also been used in order to distinguish biocatalytic processes involving the transformation of a defined starting material into specific products using either isolated enzymes or resting cells. Despite the widespread recognition of the potential advantages of biocatalysis, the number of large-scale industrial processes is still relatively limited. A summary of the products of these processes is listed in Table 31.9.
The use of biocatalysis for the production of chemicals started to receive serious interest in the 1960s with the development of immobilized aminoacylases for the production of chirally pure amino acids by Tanabe Seigaku of Japan, as well as the application of penicillin acylase for the production of 6-aminopenicillanic acid (6-APA), a key intermediate in the production of semi-synthetic antibiotics. The production of HFCS began in 1968 with implementation of a batch process using whole cells originally developed by Takasaki et al. [79]. The industrial production of HFCS remains as the largest bioconversion worldwide. These applications coincided with the growing use of proteases for cleaning and significant improvements in the production and isolation of industrial enzymes. The advent of recombinant DNA technology in the 1970s was followed by the development of the powerful PCR reaction for DNA amplification in 1984 by Kary Mullis while working at Cetus Corporation. Aside from revolutionizing biomedicine, these and other technologies have led to the ability to alter and improve many wild-type enzymes to the point that new bioconversions are now economically feasible.
The majority of industrial biocatalytic processes involve the use of hydrolytic enzymes including proteases, transaminases, glycosidases, aminoacylases, and lipases as well as several additional enzyme classes (Table 31.10) [80]. In some instances, hydrolytic enzymes can be used to drive the reverse reaction as in the formation of ester bonds through the use of lipases. In contrast, oxidases and other enzymes requiring movement of electrons commonly rely on the supply of a cofactor that may need to be recycled for practical application. These processes are typically limited to those involving respiring cells capable of regenerating such cofactors, or through the supply of a stoichiometric electron donor/acceptor, for example, hydrogen peroxide or oxygen. An emerging application of redox enzymes is in diagnostics and other sensitive analytical devices where direct electronic detection of a chemical reaction is possible.
One of the greatest hurdles for the application of biocatalysis is the need to operate processes under conditions that can differ dramatically from those in which the enzymes evolved. Many techniques are used in order to preserve catalytic activity and minimize the costs associated with the biocatalyst. In cases where the cost of the biocatalyst is a concern, an enzyme might be immobilized and used in a packed column or a fluidized bed reactor so as to enable reuse. Here also the enzyme must be stable for extended periods and may even be used under nonaqueous conditions and elevated temperatures. Recombinant technology has revolutionized the applications of biocatalysts through the ability to modify existing proteins. When combined with high-throughput screening and other analytical methods, it is now possible to redesign and improve wild-type enzymes such that they can be used in areas not previously possible. Coupled with these advances are those in the large-scale production and recovery of industrial enzymes, in addition to improvements in formulation and stabilization of enzymes.
The following sections focus on the elements of industrial biocatalysis, including the definition of a biocatalytic process, rendering, biocatalysts into immobilized forms, and performing bioconversions using whole cells. Key issues relating to the design of bioreactors are highlighted and several larger-scale bioconversions are discussed in further detail, followed by some emerging trends in biocatalysis.
Biocatalytic Processes
The nature of a biocatalyst often defines its utility for industrial bioconversions and determines the conditions under which it can be used. For example, the biocatalyst must be stable enough for practical application and of sufficiently low cost so as not to affect the overall economics of the process. Biocatalysts come in many forms including highly purified enzymes, enzyme mixtures, and whole cells. The form used is often related to cost and performance considerations. Although a crude enzyme preparation might be relatively cheap, the presence of additional enzyme activities might affect the purity and yield of the product. Similarly, the need for cofactor recycling might weigh against the use of a purified enzyme in favor of a respiring whole cell. Another important factor relates to the exquisite selectivity of many enzymes. This has both advantages and disadvantages in that a particular enzyme might be too narrow in its substrate preference to allow the use of unnatural substrates, thus limiting the potential application of that enzyme. Interestingly, some enzymes demonstrate broad substrate selectivity while retaining a high degree of stereoselectivity. A case in point is the lipase B isolated from Candida antarctica [81]. Despite the fact that this enzyme was isolated from a psychrophilic organism (cold-adapted), it demonstrates a high degree of thermostability, being functional at over 80°C, especially in an immobilized form. Another enzyme displaying broad specificity is rabbit muscle aldolase, useful for the formation of C–C bonds [82]. Selection and development of the right biocatalyst for a given transformation is often the key for successful commercialization of a biocatalytic conversion.
Immobilized Enzymes
There has been considerable effort directed toward the immobilization of both enzymes and whole cells in a wide array of formats [83, 84]. Initial attempts to immobilize enzymes on naturally derived supports such as charcoal were conducted early in the twentieth century and eventually led to the development of more robust biocatalysts immobilized on synthetic resins by the mid-1950s. Immobilization often confers a number of advantages relative to the free biocatalyst including ease of removal from the process stream, potential for reuse, improvements in stability, favorable alterations in kinetic parameters, suitability for continuous production, and in some cases the ability to operate in organic solvents. The focus of this section is on the immobilization of enzymes; however, many of the same principles apply to whole cells, the primary difference being the fact that immobilized cells are often less stable than individual enzymes and may contain additional undesired enzyme activities.
Immobilization needs to improve the performance of an enzyme enough to offset the costs associated with the procedure. Such gains can be measured in terms of improvement in the total amount of product produced per unit of enzyme, an increase in the rates and volumetric efficiency of a bioconversion, improvement in the ease of removing the biocatalyst, or through the enabling of new applications for a given enzyme. It is often the case, however, that the cheapest immobilization methods suffer from a number of drawbacks including lack of both enzymatic and mechanical stability, leaching of the enzyme, fouling of the support, and limited enzymatic activity. The many methods for producing immobilized enzymes can be divided into these subcategories:
-
1.
Adsorption to a matrix such as carbon, chitin, diatomaceous earth, and ion exchange resins
-
2.
Crosslinking enzyme crystals and whole cells with gluteraldehyde and other agents
-
3.
Gel entrapment in silica sol-gels, alginate, and protein matrices
-
4.
Covalent attachment to resins and other carriers
-
5.
Encapsulation within a membrane or liposome
Many factors influence the catalytic efficiency and kinetics of an immobilized enzyme. The immobilization process itself can lead to loss of enzymatic activity through incomplete capture of the enzyme or through denaturation of the enzyme protein. Such denaturation may arise from the action of reagents used in the immobilization process, for example, the use of carbodiimides during covalent immobilization methods, or through partial unfolding of the protein upon contact with hydrophobic surfaces such as those encountered in the adsorption of enzymes onto polymeric resins. Immobilized enzymes often demonstrate dramatic improvements in stability over free enzymes, especially in organic solvents or at elevated temperatures. This is particularly the case where enzymes have been attached to a solid support through multipoint covalent attachment or entrapped within a rigid matrix. In such instances, the potential for loss of activity through unfolding of the enzyme is reduced.
Kinetics of immobilized enzymes. Another major factor in the performance of immobilized enzymes is the effect of the matrix on mass transport of substrates and products [85]. Hindered access to the active site of an immobilized enzyme can affect the kinetic parameters in several ways. The effective concentration of substrates and products is also affected by the chemistry of the matrix, especially with regard to the respective partition coefficients between the bulk solution and the matrix. Immobilization of an enzyme typically reduces the rate of diffusion of a substrate to the active site of an enzyme and increases the apparent K m while reducing V max. The nature of the mass transfer effect depends on the fashion in which the enzyme is immobilized. Enzymes immobilized on the surface of a carrier will experience external mass transfer limitations between the bulk solution and the surface, whereas those entrapped within a porous matrix are also affected by internal mass transfer limitations due to the reduction in the rate of diffusion of substrate and products through the matrix.
The effect of diffusional limitations is given by the Damköhler number D a, defined as the maximal rate of reaction divided by the maximal rate of diffusion:
where D a ≫ 1 diffusion limits the observed rate, whereas when D a ≪ 1 the reaction rate is limiting. The effectiveness factor η is commonly used to assess the effect of immobilization on a given enzyme and is defined as the ratio of the rate with diffusional limitation vs. the rate without diffusional limitation. The relationship is a function of bulk substrate concentration β, defined as [S]/K M in addition to the ratio of the bulk substrate concentration [S R] to that within the matrix [S r]. Thus, the rate of reaction within a matrix r s is given by the modified Michaelis–Menten equation:
Current methods for large-scale immobilization. Although there are numerous methods for enzyme immobilization described in the scientific literature, relatively few methods have been successfully applied on an industrial scale. By volume the largest process is the immobilization of glucose isomerase for the production of HFCS. Several methods have been used, summarized below:
-
Adsorption to polyethylenimine (PEI) treated alumina, followed by gluteraldehyde cross-linking (UOP, 1981)
-
Adsorption onto anion exchange resins (e.g., DEAE-cellulose/TiO2 based resin)
-
Cross-linking of lysed cells with gluteraldehyde followed by extrusion
-
Incorporation into clay/PEI/gluteraldehyde cross-linked matrix, mechanical formation of granules
Over 500 t of immobilized GI are produced per annum, the majority through the incorporation of GI into a porous clay-based composite. The largest producer is Genencor. The process begins with a whole-cell lysate derived from GI fermentation to which is added PEI, bentonite clay, and diatomaceous earth, followed by cross-linking with gluteraldehyde. The resulting flocculant solid is filtered and pressed to remove excess water. Particles are fashioned mechanically by extrusion and dried in a fluidized bed dryer. The resulting immobilized GI is extremely stable, with a half-life of over 1 year when used in a packed bed reactor at temperatures of over 60°C. One kilogram of immobilized GI is sufficient to produce 20,000 kg of HFCS.
Another method for large-scale immobilization of lipases involves the formation of silica granulates, a technology developed by Novozymes A/S (Denmark) with the aim of producing a low-cost immobilized lipase for transesterification of food oils [86]. This process involves initial coating of a silicate powder with a crude lipase solution, followed by addition of a binding agent and mechanical granulation. The resulting granulates are typically 100–300 μm in diameter and are best suited to nonaqueous biocatalysis. A process for the transesterification of food oils based on a silica granulate containing Thermomyces lanuginosa lipase, Lipozyme TL-IM, was developed in a joint venture between Novozymes and ADM and has produced transesterified oils in the United States since 2002. The process itself uses a portable packed bed reactor operating at temperatures well below that required for chemical transesterification. Another advantage of the bioprocess is the fact that that product does not need to be purified by extraction with water as is necessary in the traditional chemical process.
A considerable number of industrial bioconversions utilize covalently immobilized biocatalysts. Examples include Penicillin acylases V and G, aminoacylases, and aspartase. In some cases the biocatalyst is immobilized through cross-linking and in others the catalyst is captured by a reactive resin [87]. Covalent immobilization often leads to extremely stable catalysts with high potential for reuse and exhibiting minimal leaching of enzyme during operating conditions. A variety of both organic and inorganic supports has been used, typically modified with a reactive functional group. The oxirane-based acrylic resins Eupergit C® (Röhm GmbH, Germany) and more recently AmberzymeTM (Rohm and Haas, USA) have proven to be versatile carriers for many enzymes. Attachment of an enzyme to an oxirane resin involves agitating a solution of the enzyme with the resin at approximately pH 8 for 12–24 h, followed by washing to remove non-covalently bound material (Fig. 31.13). The absence of amine-containing buffers (e.g., TRIS) or impurities is an important requirement for achieving high enzyme loading on the resin. The total activity of enzyme-functional beads is proportional to the amount of enzyme bound up to a certain point, past which the specific activity falls, most likely due to mass transport limitations. This effect is depicted in Fig. 31.14.

Covalent attachment of enzymes to oxirane-functional polymeric beads (Courtesy of M. Elizabeth Miller and Rohm and Haas)

Relationship between Penicillin G Acylase loading level and on-bead enzyme activity using Amberzyme™ Oxirane resin: loadings at higher challenge levels (Courtesy of M. Elizabeth Miller and Rohm and Haas)
Adsorption of enzymes to various polymeric resins is a straightforward means for immobilization. Zwitterionic molecules such as proteins can bind to both anionic and cationic ion exchange resins. Hydrophobic macroporous resins are also useful for immobilizing many enzymes, particularly lipases. For example, an immobilized form of Candida antarctica lipase B (CAL-B) on acrylic resin has been sold for many years under the name, Novozyme® 435 (N435). The enzyme is produced in a modified Aspergillus organism by submerged fermentation and is subsequently adsorbed onto a macroporous acrylic resin. Once immobilized, the enzyme is very robust and ideally suited to reactions in organic media where it has been widely used by the research community. Several pilot-scale processes have been developed based upon this catalyst including the production of sugar-based surfactants through the solvent-free esterification of ethyl glucoside with long-chain fatty acids. N435 has also been used for the synthesis of chiral alcohols, amines, and carboxylic acids. The use of this biocatalyst for the synthesis of biodegradable polyesters was developed by Baxenden (United Kingdom) and scaled to 2 t per annum. The cost and limited availability of this catalyst is one of the limiting factors in the wider use of CAL-B in polymer synthesis.
The last decade has seen great progress in materials science, an example being the development of practical methods for the synthesis of a great variety of mesoporous solids and nanoparticles. A number of researchers have used these materials as substrates for enzyme immobilization [88]. Advantages of nanomaterials for enzyme immobilization include very high surface areas, narrow size distributions, and the ability to tailor surface chemistries for optimal enzyme activity. Nanomaterials have also been designed to facilitate electron transport between electrodes and redox active proteins.
Enzymes can also be immobilized using techniques that mimic natural processes for the biosynthesis of inorganic materials [89]. One strategy employs a templating molecule capable of precipitating an inorganic solid like silica (SiO2) from a soluble precursor—sodium silicate being one example. An enzyme can be premixed with the templating molecule, or even covalently attached to a templating agent. Addition of the enzyme/template solution to an inorganic precursor results in the rapid formation of a colloidal coprecipitate consisting of the enzyme, the template, and the inorganic matrix (Fig. 31.15). A portion of the enzyme becomes entrapped within the composite matrix, often retaining high catalytic activity. The solid can be filtered and dried, or spray-dried directly. An application of this technology has been to formulate improved anti-fouling paints for ship hulls as an alternative to toxic tin-based coatings [90].

A template-driven silicate coprecipitation process results in the formation of a active enzyme/template/silica coprecipitate
Whole-Cell Biocatalysis
Whole-cell biocatalysis is a productive and practical style of conducting biocatalytic reactions [91]. Such reactions, as implied by the term, are done with structurally intact cells. Usually, viable respiring cells are used, but not exclusively. There are many reasons why a whole-cell reaction might be preferred to a cell-free reaction using crude or purified enzymes. Often, whole-cell biotransformations use enzymes, which are not normally excreted into the growth medium. Quite often these enzymes, which are normally intracellular, are unstable outside of the cell and quickly lose activity making them unsuitable for reactions without further stabilization work. Thus, the specific productivity or total turnovers per mole of catalyst may be severely compromised compared to that seen when used in the whole cell due to inactivation of the enzyme in the reaction conditions.
Whole-cell biocatalytic reactions are most often used when the biotransformation to be conducted requires the input of energy. In biological systems this usually takes the form of reduced pyrimidine nucleotides or ATP, but can be many of a number of reduced cofactors or modified reaction components. Using the whole cell allows the technologist to take advantage of the intact, preformed cellular machinery to efficiently provide the required cofactors or components. In order to provide the energy to catalyze these reactions, a source of reducing power is usually required. The co-oxidation of an oxidizable substrate such as glucose or ethanol can provide this energy. The partitioning of the enzymes inside the cells from the bulk medium of the reaction also can be advantageous to let the cellular machinery keep the reaction medium around the enzymes constant and allow for the accumulation, removal, or in other respects processing of the reaction products (see Fig. 31.16). Many techniques exist for the in situ removal of reaction products or side products using whole cells [92]. This task is often more difficult with cell-free reactions unless the catalyst is immobilized or in some other way partitioned from the bulk reaction medium.

Oxidized and reduced forms of nicotinamide adenine dinucleotide
Another major factor when considering whole-cell vs. cell-free reactions is the overall reaction kinetics. Some enzymatic reactions utilize a complex multicomponent enzyme system. Reconstitution of the crude or purified enzyme components are not usually as effective in vitro as they are when they remain in the intracellular milieu. Whole cells have often been called “little bags of enzymes.” Although this is an oversimplification, it is a useful concept to consider. Whole cells sequester the enzyme components in a small but concentrated form, which is usually optimal for high efficiency. Whole cells also contain co-factors, including the systems that recycle them and control pH and ionic strength. Altogether these factors combine to make whole cells a very useful form for the presentation and use of sensitive enzyme catalysts.
A practical point of using whole-cell biocatalysts is their inherent ease of preparation, use, and removal. There is a variety of growth considerations, as with any fermentation, but the major ones with respect to the use in biotransformations surround the induction of the enzyme(s) of interest and the repression of enzymes which might compete with the desired process or degrade the desired catalyst. Generally, a medium can be used in which the cells can be used directly after growth for the biotransformation. Some consideration needs to be given to the extraction of the product with respect to the growth medium. Often medium components from rich media used for fermentations can interfere with recovery or extraction procedures. The effort required to reduce the complexity of the medium, to one as defined as possible, is usually worth the trouble. Having the resulting growth medium as much like water as possible simplifies downstream work. If the cells must be removed from the growth medium and resuspended in a different biotransformation medium, they can be removed by centrifugation or the medium exchanged by microfiltration. Once the biotransformation is complete, the catalyst can be removed by centrifugation or filtration. Concentrating the medium after the biotransformation by removal of water will enhance many recovery processes. The concentration of the medium salts in this process often makes extraction much more efficient.
Although there are many advantages to the use of whole cells for biotransformations, there are certain limitations that must be considered. One consideration is the transport of substrates and products across the cell membrane. In life, the cell membrane is a proton-tight barrier to the rest of the world. It is generally impermeable to charged molecules and to water, but may have permeability to hydrophobic molecules. Often cells have specific transport systems that move compounds in and out as the cell needs them. In order for a whole-cell biotransformation to proceed, the substrate of interest must be transported across the cell membrane to the active enzyme or enzyme system. The same issues exist, of course, for the reaction product. Although it is not generally an issue, for biotransformations where the substrate is not transported but enters the cell by passive diffusion, mass transport must be considered. A simple dilution of the cell density can demonstrate if this is a problem if the rate is not proportional with cell density. Another limitation, which may be observed, is the nonspecific metabolism of the substrate or product by other enzymes in the cell. It is not uncommon to find a small percentage of the substrate or product oxidized or reduced by enzymes not in the desired pathway or reaction. This can sometimes be addressed by genetic techniques or by growth conditions. The commercial-scale use of whole-cell biocatalysts is typically limited to cells produced on-site and transferred under containment to the process vessel given the biosafety and economic issues inherent to transporting live microbes in large quantities.
A variation on the whole-cell biotransformation theme is the use of permeabilized cells. Whole cells can be rendered permeable to small molecules yet remain essentially intact by contacting them for a short time with low concentrations of solvents. This process has the effect of making “holes” in the cellular membrane while leaving enough of the cell membrane and cell wall intact to still contain the enzymes and other macromolecules. The permeabilized cells can then be treated much like immobilized enzymes. This technique is especially useful when transport issues are found to be limiting the reaction.
A related topic appropriate to this discussion is the use of solvents in whole-cell or permeabilized cell biotransformations. Solvents can be added to increase the solubility of substrates in the reaction medium. The specific solvent and concentration must be empirically determined and can be highly variable. Solvents, in low concentration, can serve as a source of energy for reactions requiring reducing equivalents. In such cases the solvents may serve to both supply energy and help solubilize the substrate. Although not generally considered a solvent, the use of a second nonmiscible liquid such as vegetable oil or hexadecane can be used in many whole-cell biotransformations to “buffer” the concentration of substrates and products with respect to the aqueous phase. Two liquid phase reactions can dramatically increase the yield obtainable from a specific biotransformation.
Bioreactor Configurations
The manner in which a bioconversion is performed is dictated by the nature of the biocatalyst, the chemistry involved, and process economics [93]. The overall aims of a bioconversion are the same as for any process, to maximize the production of a given material at the lowest overall cost. In some cases, this might mean maximizing the volumetric productivity (Qp in units of mol m3/s) of the reactor. Alternately, it might be most important to enable the more efficient recovery through maximizing the ratio of desired to undesired products. If the cost of the biocatalyst is limiting then the catalyst productivity (P cat) must be maximized, a function of the intrinsic activity of the catalyst itself and the fashion in which it is used.
A variety of reactor configurations has been developed for both batch and plug-flow modes of operation. Bioconversions, involving the transformation of a defined substrate into a product, are typically less demanding than fermentations involving growing cells where many additional factors such as oxygenation, feed rate, and the supply of trace nutrients may need to be tightly monitored. Nonetheless, the implementation of bioconversions, particularly larger scale operations, requires a considerable degree of engineering. Important factors that must be considered when selecting a reactor configuration for performing a given bioconversion include:
-
Nature and solubility of substrates and products
-
Form of the biocatalyst (whole cells, soluble or immobilized enzymes)
-
Cost of biocatalyst relative to the overall process
-
Kinetic parameters of the biocatalyst
-
Substrate/product equilibrium
-
Degree of product or substrate inhibition of the biocatalyst
-
Operational stability of biocatalyst
-
Product recovery
The bioreactor configurations in common use are illustrated in Fig. 31.17.

Bioreactor configurations
Batch reactors. The majority of smaller-scale bioprocesses are carried out in batch mode. A key advantage of many batch reactors is the ability to run a range of different bioconversions with the one piece of equipment. The main disadvantage is the downtime required for emptying and recycling the reactor and cases where product inhibition of the biocatalyst is significant. The simplest configuration involves the use of a stirred vessel containing the biocatalyst to which one or more substrates (S t concentration) are added resulting in conversion to the desired product(s) over a given time. The required batch time (T b) of such a system is a function of the kinetic parameters of the biocatalyst (K m, V max), the catalytic density (maximum enzyme activity per volume), and the initial substrate concentration (S o), represented in the equation below.
In some cases, continuous extraction of products allows extended operation and high volumetric efficiency. Reactors run in this mode are referred to as continuous-stirred batch reactors (CSBR). In this case, the bioconversion is run under approximately steady-state conditions where the position of reaction equilibrium lies toward the products of the conversion. In this case the concentration of product (proportional to S i − S o) at a given reactor residence time becomes a function of both the flow rate (Q) into the reactor and reactor volume, in addition to the factors discussed above for batch mode reactors (i.e., catalyst parameters and density, inlet substrate concentration S i, and outlet substrate concentration S o).
Another favorable aspect of stirred batch reactors is the fact that they are compatible with most forms of a biocatalyst. The biocatalyst may be soluble, immobilized, or a whole-cell preparation; in the latter case a bioconversion might be performed in the same vessel used to culture the organism. Recovery of the biocatalyst is sometimes possible, typically when the enzyme is immobilized or confined within a semi-permeable membrane. The latter configuration is often referred to as a membrane reactor. An example is the hollow fiber reactor where enzymes or whole cells are partitioned within permeable fibers that allow the passage of substrates and products but retain the catalyst. A hollow-fiber reactor can be operated in conjunction with the stirred tank and operated in batch or continuous mode. A tubular reactor configuration consisting of hollow fibers is also useful for continuous plug-flow mode operation.
Plug-flow bioreactors. Continuous bioconversions can also be carried out using so-called “plug-flow” reactors where a substrate is converted to a product by passage through a biocatalyst in immobilized form. The packed-bed bioreactor is the most common, although fluidized-bed bioreactors are also used. The principle advantages of packed-bed reactors are the greatly increased catalytic density relative to batch reactors allowing for compact designs and suitability for continuous production. There are several technical issues that need to be addressed before a given bioprocess can be performed in this fashion. The catalyst must be immobilized in a form compatible with a packed-bed format, usually immobilized on rigid particles that enable optimal fluid flow through the bed. The particles must be strong enough to support their own weight and also resist fouling by impurities such as particulates in the substrate. The rate of the conversion dictates the required bed thickness, or alternatively, the need for recycling in order to drive a conversion to completion. In this case, the concentration of product (proportional to S i − S o) at a given reactor residence time (T p) becomes a function of both the flow rate (Q) into the reactor and reactor volume in addition to the factors discussed above for batch mode reactors (i.e., catalyst parameters and density, inlet substrate concentration S i, and outlet substrate concentration S o).
Continuous bioconversions using packed-bed reactors are particularly well suited for high-volume bioprocesses including HFCS and enzymatic transesterification of fats and oils. As shown in Table 31.11 for an HFCS production reactor using Glucose isomerase, PFR is advantageous in terms of short residence time critical for minimum color formation, high productivity, and low cost. An overall summary of the relative advantages and disadvantages of different bioreactor configuration is listed in Table 31.12.
Recovery of products often dictates the fashion in which a bioconversion is carried out. Liquid/liquid two-phase systems have proven useful, particularly when substrates of low water solubility are used. In these cases it is often possible to operate the reactor in continuous mode by removing and recycling the phase containing the products. The mode of product recovery depends on the nature of the product and the medium in which it is dissolved, in most cases water. Distillation can be used for product recovery in some instances, whereas extraction using a solvent might be used for nonpolar products. In many cases the removal of water from dilute solutions of product can add considerable cost to a bioprocess. For this reason there has been widespread interest in the use of solvents other than water for biocatalysis, discussed below.
Nonaqueous Biocatalysis
Enzymes are, for the most part, soluble in water and not obviously suited for use in organic solvents. Many enzymes are denatured by exposure to solvents and still others require water as part of their catalytic action. Prior to 1980, there were several reports of biocatalysis in solvents; however, it was Klibanov and co-workers in the 1980s who first clearly demonstrated the potential of biocatalysis in organic media [94–96]. The use of such media, typically organic solvents, has greatly expanded the scope of biocatalysis for several reasons:
-
Ability to use substrates with low water solubility
-
Modulation of enzyme regio- and enantioselectivity
-
Improved product yields in some cases
-
Improved product recovery
-
Enhanced biocatalyst stability
-
Prevention of microbial contamination
Hydrolytic enzymes such as lipases and proteases catalyze readily reversible reactions and will often promote reverse hydrolysis at reduced water activities. Water can be removed with desiccants, as an azeotrope with a solvent or through application of a vacuum. Lipases have proven particularly useful in this regard, allowing the formation of esters from alcohols and either free carboxylic acids or esters.
An example is the synthesis of fatty acid esters in hexane from fatty acids and alcohols, a reaction catalyzed by many lipases including Porcine pancreatic lipase (PPL), P. alcaligenes lipase (LIPOMAXTM, Genencor), CAL-B (N435, Novozymes), and Candida rugosa lipase (Amano AYS, Amano) among others (Fig. 31.18). Many other classes of enzyme have also been used in organic solvents such as acyltransferases, glycosidases, dehydrogenases, oxidases, and dehalogenases, for the most part on the laboratory scale. Another major advantage of nonaqueous biocatalysis is the greatly expanded set of possible substrates, particularly those of low or negligible solubility in water. Examples of such compounds include long-chain fatty acids and ester derivatives, steroids, aromatic compounds, and many pharmaceutical intermediates. Several texts have been dedicated to the topic of synthetic biocatalysis, for example, the comprehensive handbook edited by Drauz and Waldmann [97].

Lipase-mediated ester formation
A wide variety of organic solvents have been used to conduct bioconversions including nonpolar solvents such as isooctane, n-hexane, and toluene, in addition to methanol, acetone, and other water-miscible solvents. Dipolar aprotic solvents dimethylformamide (DMF) and dimethylsulfoxide (DMSO) are also compatible with many enzymes and are often used to enhance the solubility of substrates in combination with a nonpolar solvent. Tertiary alcohols such as t-butanol and t-amyl alcohol have been used for many lipase-mediated esterifications as sterically hindered tertiary alcohols are not typically good substrates for most enzymes. It should be noted that the presence of small amounts of water is essential for the effective use of most biocatalysts in organic solvents. In some cases an enzyme may only require a monolayer of water molecules on its surface in order to operate. In other cases there may need to be enough water to form reverse micelles where the biocatalyst is contained within a predominantly aqueous environment. The amount of water needed for maximal enzyme performance is usually cited in terms of water activity (A w). Some enzymes (class “A”) operate efficiently at low water activities and become less efficient at catalyzing certain reactions such as ester formation at high A w. Other enzymes will operate most efficiently at high water activities (class “B”). The enzyme activity as a function of water activity for these two classes of enzyme is represented graphically in Fig. 31.19.

Enzyme activity vs. water activity plot
Few industrially important bioprocesses are carried out exclusively in organic solvents. Two-phase systems are more common, however, consisting of a water-immiscible solvent that allows adequate dissolution of substrates and products and an aqueous phase containing the biocatalyst [98].
Products of Biocatalysis
Although numerous chemicals have been produced at a pilot scale using biocatalysis, there are only a modest number of materials produced at the ton scale or greater [99]. Some materials such as fructose syrup, acrylamide, and aspartame are produced on a large scale (>1,000 t/year), whereas others, including most pharmaceutical intermediates, are manufactured at considerably smaller volumes. A summary of some current products produced through biocatalytic processes is given in Table 31.13.
High-fructose corn syrup. Glucose isomerase (GI) is used for the large-scale isomerization of glucose syrups produced from the enzymatic hydrolysis of corn starch (Fig. 31.20). The product, commonly known as HFCS, typically contains 42% fructose, 50% glucose, 6% maltose, and 2% maltotriose. Fructose, the monosaccharide commonly called fruit sugar, is about 50% sweeter than sucrose, the disaccharide familiarly known as table sugar.

Isomerization of d-glucose to d-fructose catalyzed by glucose isomerase
Annual production of HFCS amounts to over ten million tons, selling at around 30 cents/lb for the 42% grade (HFCS-42). HFCS is also available in the form of 55% syrup (HFCS-55), 90% syrup (HFCS-90), or as 99% pure crystalline fructose. The lowest grade, HFCS-42, is 15–20% cheaper than liquid invert sugar on a dry weight basis and is predominantly used in soft drinks. The baking industry ranks as the second largest user. Combining HFCS-42 and HFCS-90 gives HFCS-55, which has about the same degree of sweetness as sucrose. It is used as a sweetener and flavor enhancer in fruit-flavored soft drinks. Fructose enhances flavors, whereas sucrose masks them. Because the molecular weight of fructose is approximately half that of sucrose, a smaller amount of it is needed to sweeten a product to desired levels, and sweetener calories in the product can be reduced by about one third. The major use of HFCS-90 is in dietetic foods and drinks. Crystalline fructose of over 99% purity is obtained by drying the 90% pure HFCS. Pure fructose is about 70% sweeter than sucrose, and as an essentially pure sweetener, fructose allows the full taste of product flavors to develop. It has also found uses in low calorie foods and drinks. The amount of HFCS consumed per capita has risen dramatically in the last 30 years and there is some controversy about the possible health effects of high level consumption [100].
In many industrial cases, whole microbial cells containing GI are immobilized by physical means such as entrapment or encapsulation in polymeric materials or by chemical methods such as intermolecular cross-linking with gluteraldehyde or covalent binding with diazotized diamino compounds. Commercially, soluble glucose isomerase is also immobilized on DEAE-cellulose. The immobilized glucose isomerase can usually be used for over 1,000 h at a temperature of around 65°C. When the column enzyme activity decreases, the flow rate of the incoming glucose syrup can be adjusted so that the conversion of glucose to fructose is maintained constant.
Acrylamide. One of the largest scale industrial bioconversions is the process for the bioconversion of acrylonitrile to acrylamide, which is used in coagulants for water treatment, soil conditioners, paper treatment, adhesives, paints, and secondary oil recovery [101]. The worldwide production volume for the bioconversion process is in the tens of thousands of tons per year range. The reaction is a hydration of a nitrile to an amide and has traditionally been done by either a sulfuric acid or a copper-catalyzed hydration process. These processes are rapidly being phased out and replaced with a bioconversion process that has increased volumetric productivity, higher energy efficiency, decreased costs, and lower environmental impact.
The commercial bioconversion process employs the enzyme nitrile hydratase, which catalyzes the same reaction as the chemical process (Fig. 31.21). The bioconversion process was introduced using wild-type cells of Rhodococcus or Pseudomonas, which were grown under selective conditions for optimal enzyme induction and repression of unwanted side activities. These biocatalysts have since been replaced with recombinant cells expressing nitrile hydratase. The process consists of growing and immobilizing the whole-cell biocatalyst and then reacting them with aqueous acrylonitrile, which is fed incrementally. When the reaction is complete, the biocatalyst is recovered and the acrylamide solution is used as is. The bioconversion process runs at 10°C compared to 70°C for the copper-catalyzed process, is able to convert 100% of the acrylonitrile fed compared to 80%, and achieves 50% concentration of product compared to 30% concentration. Thus, the bioconversion process does not have to recycle the reaction to complete the hydration and the final liquor does not have to be concentrated as most of the commercial acrylamide is sold as a 50% solution.

Enzymatic conversion of acrylonitrile to acrylamide
It should be noted that this elegant example of replacing a chemical catalyst with a biocatalyst that has higher efficiency, lower costs, and is environmentally friendly did not happen overnight. It was first introduced in 1985 and has become progressively improved to the currently described state. That caveat being said, the current availability of biotechnological tools would today probably be fivefold shorter. The ability to identify and manipulate the enzyme, select the improved catalyst, and express it in a variety of host organisms has improved significantly in the last 20 years.
Semi-synthetic antibiotics. In 1959, Batchelor and coworkers in the Beecham Research Laboratories in England discovered that the penicillin nucleus, 6-APA, accumulated during fermentation when side chain precursors were omitted. This 6-APA could be used for the chemical synthesis of entirely new types of penicillin by coupling with new side chains. Shortly thereafter, several sources of penicillin amidase were found that would cleave the phenylacetyl side chain from penicillin G, thus producing a more economical source of 6-APA. A vast number of “synthetic penicillins” have been generated, and a few have achieved clinical importance [102]. Several objectives were sought:
-
1.
To broaden the inherent utility of penicillin to include gram-negative pathogens not inhibited by the natural penicillins
-
2.
To improve its stability and absorption
-
3.
To increase resistance to penicillinase producing pathogens
-
4.
To decrease allergenicity
-
5.
To improve other factors pertinent to clinical use
The broad objectives have been achieved with varying degrees of success. Table 31.14 shows the structures of some of the semisynthetic penicillin that have become important chemotherapeutics.
The natural penicillins, primarily G and V, have a relatively narrow spectrum. They act mostly on gram-positive organisms. The fact that proper selection of precursors could lead to new variations in the penicillin side chain offered the first source of synthetic penicillins. Penicillin V, derived from a phenoxyacetic acid precursor, attracted clinical use because of its greater acid tolerance, which made it more useful in oral administration. Also, the widespread use of penicillin eventually led to a clinical problem of penicillin-resistant staphylococci and streptococci. Resistance for the most part involved the penicillin-destroying enzyme, penicillinase, which attacked the beta-lactam structure of the 6-APA nucleus. Semisynthetic penicillins such as ampicillin and carbenicillin have a broader spectrum. Some, such as methicillin, oraficillin, and oxacillin, are resistant to penicillinase. In 1984, Beecham introduced Augmentin, which was the first combination formulation of a penicillin (amoxicillin) and a penicillinase inhibitor (clavulanic acid). Worldwide production of semisynthetic penicillins is currently around 10,000 t/year, the major producers are Smith Kline Beecham, DSM, Pfizer, and Toyo Jozo.
As in the penicillin studies, the chemotherapeutic properties of cephalosporin C can also be improved through synthetic modification of the 7-aminocephalosporanic acid (7-ACA) nucleus. Several semi-synthetic cephalosporins have been produced and are used clinically. The leading agents in this category include cefaclor (first-generation cephalosporin, developed by Lilly), cefoxitin (second-generation, by Merck), cefuroxime axetil (oral form, second-generation, by Glaxo), and ceftriaxone (third-generation, by Roche). Large-scale enzymatic removal of the 6-acyl side chain of penicillins is widely used in the commercial production of both 6-APA and the ring-expanded analogue, 7-aminodesacetoxy-cephalosporanic acid (7-ADCA). Two enzymes are used commercially, Penicillin acylases G and V. Penicillin G acylase is used in most cases, originally isolated from various sources including E. coli, Bacillus megaterium, and Streptomyces lavendulae. Penicillium acylase V, used to a lesser extent, is derived from organisms including Beijerinckia indica var. Penicillium, Fusarium sp., and Pseudomonas acidovorans. Bacterial acylases have also been found that cleave the 7-acyl side chain of cephalosporin C to form 7-ACA.

Immobilized forms of penicillin amidases and acylases have replaced whole-cell biocatalysts for the production of 6-APA and 7-ACA as they can be reused many times, in some cases for over 1,000 cycles. Another major advantage is the purity of the enzyme, lacking the β-lactamase contaminants often present in whole cells. The productivity of these biocatalysts exceeds 2,000 kg product/kg catalyst. A typical process for the production of 6-APA employs immobilized penicillin G acylase covalently attached to a macroporous resin. The process can be run in either batch or continuous modes. The pH of the reaction must be maintained at a value between 7.5 and 8 and requires continuous adjustment to compensate for the drop caused by the phenylacetic acid generated during the course of the reaction. Recycle reactors have been used, as they allow both pH control and the use of packed bed reactors containing the immobilized catalyst. The enzymatic process is cheaper, although not greatly so, than the older chemical route to 6-APA, although it produces far less waste and doesn’t require hazardous chemicals such as trimethylsilyl chloride. The biological production of 7-ACA, developed by Hoechst and currently practiced by Biochemie, is another excellent example of the waste reductions possible through implementation of a bioprocess, in this case from 31 to 0.3 kg per kg of product. Overall, the discovery and the development of penicillin and cephalosporin acylases greatly accelerated the commercialization of many semi-synthetic penicillins and cephalosporins and highlight the utility of biocatalysis for the selective modification of complex molecules.
Amino acids. Amino acids are produced by both fermentation and biocatalysis for use in animal feed, fertilizer, as flavor enhancers, dietary supplements, and in pharmaceutical manufacture [103]. By volume, the most important products are l-lysine, l-methionine, l-threonine, and l-tryptophan, most of which are produced by fermentation. Biocatalysis still plays an important role in amino acid supply and has been used to synthesize both l- and d-amino acids, including a variety of nonnatural analogues. The major producers are Degussa, Tanabe Seiyaku, and Kyowa Hakko.
Several enzyme classes have been used to synthesize amino acids, listed below;
-
Aminoacylases
-
Hydantoinases
-
Amidases
-
Dehydrogenases
-
Racemases
-
Lactamases
-
Lyases
Aminoacylases (EC 3.5.1.14) were first used for the production of both d- and l-amino acids in the mid-1950s in batch mode processes to effect the selective deacetylation of chemically synthesized racemic N-acetyl-dl-amino acids. The most widely used enzyme was derived from A. oryzae, selective for the l-isomer. The technology was further developed by Chibata and coworkers at Tanabe to the point where the aminoacylase was immobilized to an ion-exchange resin and used in a packed-bed reactor allowing continuous production [104]. More recently, d-aminoacylases have been isolated and used for the synthesis of d-amino acids. In most schemes, the unwanted isomers are racemized chemically under forcing conditions (pH <3, >80°C) or, alternatively, with a racemase enzyme and subsequently recycled. Examples of amino acids produced through this process include l-methionine, l-valine, l-phenylalanine, and l-tryptophan. The immobilized enzyme is quite stable with a half-life of over 1 month at 50°C. When the total activity of the reactor drops, more of the enzyme can be added to the carrier. Productivity of the continuous process ranges from 70 to over 200 t/year for a 1,000-L column and is approximately 40% less costly than the older batch process. Figure 31.22 depicts the chiral resolution of racemic N-acetyl d, l-methionine.

Chiral resolution of racemic N-acetyl-dl-methionine
Another widely adopted route to chirally pure amino acids is the hydantoin route. The enzyme hydantoinase allows the selective hydrolysis of l-hydantoins to form a carbamoyl amino acid that is subsequently converted to the free-l-amino acid by carbamoylase (Fig. 31.23). An advantage of this process is the fact that racemization of hydantoins is far more facile than for N-acetyl amino acids. This process has been used for many years for the large-scale production (>1,000 t/year) of both d-phenylglycine and 4-hydroxy-d-phenylglycine, used in the synthesis of semi-synthetic antibiotics.

Hydantoinase/carbamoylase process for the production of d-amino acids
Dehydrogenases are also used for amino acid production, albeit on a far smaller scale. An example is the unnatural amino acid l-tert-leucine, manufactured by Evonik (formerly Degussa), used as an intermediate for the manufacture of peptidomimetic drugs such as HIV protease inhibitors. The route used to synthesize this material is noteworthy in that it employs Leucine dehydrogenase, a redox enzyme with cofactor regeneration driven by the decomposition of ammonium formate, in turn catalyzed by formate dehydrogenase (Fig. 31.24). The cofactor in this case is NADH modified with polyethyleneglycol (NADH-PEG) so as to increase its molecular weight and allow retention within a membrane reactor. Total cofactor recycle efficiency is very high at over 125,000 cycles. This elegant approach has also been used to produce other unusual amino acids including β-amino acids.

Synthesis of l-tert-leucine is carried out using a cofactor recycle process using formic acid as the stoichiometric hydrogen donor
l-Aspartic acid is used in production of aspartame, in pharmaceuticals, and as a food additive. A bioprocess for the production of this amino acid was first commercialized in 1973 by Tanabe Seiyaku Co. (Japan) and involves the stereoselective addition of ammonia to fumarate catalyzed by l-aspartate ammonia lyase (Aspartase, EC 4.3.1.1). l-Aspartic acid was originally produced by fermentation; however, more recent processes use whole E. coli cells immobilized within either an acrylamide or cross-linked polysaccharide matrix. In this case the immobilized cell-based catalyst is far more stable than immobilized forms of the isolated enzyme, with an operational lifetime of over 120 days. The process is typically run in a continuous fashion with recovery of the l-aspartic acid readily achieved through crystallization. The major producers are Tanabe Seiyaku Co. and Kyowa Hakko Co. with a combined production of over 10,000 t per annum.
A similar process is also used for the production of l-malic acid from fumarate, in this case using a hydratase enzyme derived from Brevibacterium ammoniagenes. Another variation of the Tanabe technology involves the synthesis of l-alanine from l-aspartic acid through the use of immobilized whole cells (Pastilla dacunae) containing aspartate-decarboxylase.
l-Lysine is produced on an enormous scale, over 500,000 t/year, mostly through fermentation using genetically modified organisms. A biocatalytic route was also used to produce l-lysine, the most popular being the Toray process where chiral resolution of dl-α-amino-ε-caprolactam was achieved using a combination of a lactamase selective for the l-lactam and a dl-racemase (Fig. 31.25). A blend of whole-cell biocatalyst strains was used, consisting of Cryptococcus laurentii (l-ACL lactamase) and Achromobacter obae (ACL racemase). Yields of close to 100% are realized with productivities of over 100 g/L/day. At its peak, this process produced over 10,000 t of l-lysine per year.

Synthesis of l-lysine from dl-α-amino-ε-caprolactam
Aspartame. Aspartame is a high-intensity sweetener (200-fold that of sucrose) consisting of l-aspartyl-l-phenylalanine methyl ester (l-Asp-l-Phe-OMe). Around 10,000 t are produced per annum, mostly by a chemical process owned by Nutrasweet Corp. Holland Sweetener Company (a DSM/Tosoh joint venture) uses the protease thermolysin (EC 3.4.24.27) to synthesize aspartame from a chemically protected l-aspartic acid derivative (Z-l-Asp) and racemic phenylalanine methyl ester (dl-Phe-OMe). The enzyme only accepts l-phenylalanine-OMe and the unused d-isomer can be isolated, racemized, and recycled. This process is depicted in Fig. 31.26.

Thermolysin-mediated synthesis of aspartame
Chiral pharmaceutical intermediates. The use of biocatalysis for producing chirally pure pharmaceuticals is now an accepted technology that complements alternative chemical and physical methods [105, 106]. The size of the world market for single enantiomer drugs, over $225 billion in 2005, offers plenty of incentive to develop competitive biocatalytic routes for key intermediates [107]. The market for chiral intermediates themselves, although far smaller, is still considerable and was projected to rise from over $7 billion in 2002 to over $15 billion by 2012 [108]. The majority of these intermediates are generated through traditional chiral pool resolution, 35% by asymmetric chemical synthesis and 15% through the application of biocatalysis (Fig. 31.27) [109]. The number of biocatalytic processes is increasing as more traditional chemical companies begin to view enzymes more as reagents than exotic materials. In most cases biocatalysis is one of many options for the synthesis of given material and must demonstrate economic benefits in order to be adopted.

Examples of pharmaceuticals synthesized using biocatalysis
One of the inherent advantages of enzymes is the ability to discriminate between stereoisomers, often generating products with enatiomeric excesses (i.e., of over 98%). Judicious application of biocatalysis can also reduce the number of chemical steps needed to synthesize certain drugs, leading to hybrid chemoenzymatic processes with lower costs and less waste. The range of enzymes used in the synthesis of chiral intermediates has expanded beyond esterases and acylases and now includes oxidoreductases, glycosyltransferases, and C–C bond-forming enzymes (e.g., aldolases). A number of excellent reviews and books have been dedicated to the application of biocatalysis in the production of pharmaceuticals and should be consulted for detailed information [105–111]. A few examples of the use of biocatalysis are discussed below.
Naproxen and ibuprofen are nonsteroidal antiinflammatory drugs (NSAIDs) widely available as OTC medications. In both cases the active (S)-enantiomer is far more potent. Selective hydrolysis of racemic esters of these drugs enables the production of both (S)-naproxen and (S)-ibuprofen. A number of bioroutes have been developed for the synthesis of these drugs. Most commercial processes are based on the selective hydrolysis of racemic esters performed by lipases and esterases, although the selective hydrolysis of nitriles with nitrilases has also been used. One such process for (S)-naproxen, was developed by Chirotech using a recombinant esterase, generates titers of around 150 g/L in a batch-mode process. Recovery of the (R)-ester is readily achieved by centrifugation and is subsequently racemized by base treatment and returned to the reactor (Fig. 31.28).

Dynamic chiral resolution of naproxen ethyl ester
The blockbuster anti-cholesterol agents Lipitor® (atorvastatin) and Crestor® (rosuvastatin) generated combined sales of over $15 billion in 2010. Several companies have developed improved biocatalytic methods for the synthesis of ethyl (S)-4-chloro-3-hydroxybutyrate (ECHB), a key intermediate for these drugs [112]. One popular route to ECHB involves selective reduction of ethyl 4-chloroacetoacetate with a whole-cell biocatalyst (Fig. 31.29). Daicel Chemical Industries of Japan produces over 100 t/year of ECHB using two enzymes coexpressed in E. coli to carry out this reduction. An NADH-dependant carbonyl reductase, originally isolated from Kluyveromyces aestuarii, and an engineered formate dehydrogenase dehydrogenose from Mycobacterium vaccae resulted in ECHB yields of nearly 50 g/L/h. Alternative biocatalytic routes have been developed by DowPharma in collaboration with Verenium (formerly Diversa), and also by Codexis.

Synthesis of ethyl (3S)-4-chloro-3-hydroxybutanoate (ECHB) from ethyl-4-chloroacetoacetate through whole-cell microbial reduction
Diltiazem, a benzothiazepin used widely for the treatment of high blood pressure, is derived from a p-methoxyphenyl-substituted glycidyl ester with two stereocenters. Lipase-mediated resolution is used to obtain the desired (2R, 3S)-ester. Both DSM and Sepracor have commercialized processes for this intermediate using lipases including those from C. rugosa and Serratia marcesens. A typical process is run in a membrane reactor using a two-phase solvent/water mixture. The unwanted isomer is hydrolyzed to a carboxylic acid that subsequently decomposes to an aldehyde and CO2 (Fig. 31.30). The addition of bisulfite to the reactor prevents inhibition of the lipase by complexing with the aldehyde byproduct and allowing extraction into the aqueous phase.

Lipase-mediated synthesis of the key intermediate for Diltiazem
A number of other important drug intermediates are produced at scale (>1,000 kg/year) by biocatalysis including d-phenylglycine and d-(p-hydroxyphenyl)glycine for antibiotics, nicotinamide, 6-hydoxynicotinic acid, (R)-glycidol, and d-pantothenic acid. Many fine chemical companies have developed a biocatalysis capability such as Avecia (now a part of Nitto Denko Corp.) and Lonza to address the growing market for chiral intermediates. Others specialize in the development of novel biocatalysts of use for fine chemical synthesis.
Herbicides. Herbicides are used in great quantities globally, accounting for over $18 billion in sales in 2009. A number of herbicides are produced as racemates and, as is the case for many pharmaceuticals, one enantiomer is often more potent than the other. Examples include Frontier, produced by BASF, and Metolochlor, a Syngenta product. The active enantiomer, (S)-metolachlor, was introduced in 1999. BASF introduced OutlookTM, the S-enantiomer of Frontier, in 2001.

Whereas (S)-metolochlor is produced chemically by asymmetric hydrogenation, a biocatalytic route is employed by BASF for the production of (S)-methoxyisopropylamine on the multithousand-ton scale per annum. Resolution of racemic methoxyisopropylamine is achieved through selective enzymatic acylation of the unwanted (R)-isomer. This amide is then separated, racemized, and recycled (Fig. 31.31). A similar approach is used by BASF to produce a range of additional chiral amines as part of the ChiPros® portfolio.

Chiral resolution of 2-methoxyisopropylamine
Another chiral small molecule, (S)-2-chloropropionic acid (S-CPA), is a key intermediate in the synthesis of the 2-phenoxypropionic acid class of herbicides, including Fusilade (Avecia) and Mecoprop (BASF). Kinetic resolution of racemic CPA is performed with a whole-cell biocatalyst expressing a dehalogenase enzyme selective for the (R)-enantiomer and producing (S)-lactic acid, itself a useful byproduct. The S-CPA is extracted with a solvent following removal of the biocatalyst by filtration. Removal of the solvent by distillation gives crude S-CPA, which can be further purified. The process was developed by Avecia (purchased by Dr. Reddy's Laboratories in 2005) and produces over 2,000 t per annum.
Carbohydrates. Aside from starch processing and the production of HFCS, there are few biocatalytic processes currently employed for the large-scale synthesis of carbohydrates [113, 114]. A number of carbohydrates are, however, produced on the smaller scale. They include monosaccharides such as xylitol, l-sorbose, and sialic acid, alkyl glycosides, sugar fatty acid esters, cyclodextrins, and oligosaccharides including gluco-, galacto-, and fructooligosaccharides. Many of these processes involve the formation of a glycosidic bond, the carbohydrate version of an acetal or ketal. Three types of enzyme are used for glycoside formation, depicted in Fig. 31.32.

Formation of glycosides using (a) a glycosidase and a free sugar; (b) a transglycosidase and a glycoside; (c) a glycosyltransferase and a sugar nucleotide
Glycosidases typically hydrolyze glycosidic bonds, but can be made to work in reverse under certain conditions. An example is the β-glucosidase from sweet almonds, which can form glycosides from free (unactivated) sugars and aliphatic alcohols. Transglycosidases are used to interconvert glycosides. Many glycosidases also have transglycosidase activity. The industrial production of butyl-α-d-glucoside involves the transglycosylation of maltose with butyl alcohol in a two-phase system employing the α-transglucosidase from A. niger [115].
Glycosyltransferases are the most specific of the enzymes used to form glycosidic bonds and have also been used to synthesize complex oligosaccharides in a far more direct fashion than is possible by chemical means alone. One disadvantage of glycosyltransferases is the need for activated sugar nucleotide donors, such as UDP-glucose (UDP = uridine diphosphate). Recent progress in the practical application of glycosyltransferases has been made possible through the commercial availability of recombinant enzymes and elegant schemes for the in situ regeneration of sugar nucleotide donors using cheaper energy sources such as phosphenolpyruvate.

For example, production of a complex pentasaccharide, Sialyl Lewis X (SLex), was achieved on the multikilogram scale by Cytel Corporation using technology developed by Wong and Whitesides whereby all of the required sugar nucleotides were regenerated in a multienzyme scheme [116]. This molecule was originally identified as a possible therapeutic agent for the prevention of reperfusion injury following heart attacks. Although this drug ultimately failed in clinical trials, the technology has since been used in the production of carbohydrates for infant formula, as anti-infectives, and for the production of glycosylated therapeutic proteins.
Future Trends in Biocatalysis
Biocatalysts. Continued discovery of new enzymes and microorganisms will further drive the development of biocatalysis by both enabling new chemistries and lowering the cost of existing technologies. It has been estimated that over 98% of the microbial diversity in Earth remains untapped. The potential of this resource cannot be underestimated. For example, the discovery of extremophilic bacteria existing at temperatures exceeding 120°C and extreme pH values has already led to the development of thermostable enzymes for a variety of applications, of particular note the PCR reaction conducted with DNA polymerases. Libraries of enzymes derived from extremophiles are now commercially available. Table 31.15 lists different classes of extremophiles from which useful enzymes have been derived. Further information on extremophiles and their potential utility for biocatalysis can be found in the reviews by Kumar et al. [117] and Gomes and Steiner [118].
Enzyme engineering will increasingly drive the development of robust enzymes tailored to specific bioprocesses. The strategies used for improving the stability of enzymes will continue to focus on the generation and screening of libraries of mutants produced by random or targeted amino acid substitutions; however, alternative approaches also include the rational design of stabilizing protein motifs (e.g., salt bridges) and even the incorporation of unnatural amino acid analogues [119]. A case in point is the passive incorporation of fluorinated analogues such as hexafluo-roisoleucine into proteins by Tirrel and coworkers combined with the observation that fluorinated amino acids can stabilize protein folds in some instances [120]. The ability to design enzymes from unrelated proteins is another long-term goal. Similarly, computational design of enzymes from first principles has great promise, although the field is still in its infancy [121, 122]. Improved production methods for enzymes useful for biocatalysis will also enable the commercialization of many currently uneconomical processes. Gains are also to be expected from refinement of methods used for enzyme immobilization and bioreactor design. The development of improved supports for enzyme immobilization through the application of nanotechnology has particular promise through the enhancement of stability and productivity of immobilized enzymes.
Novel media for biocatalysis. The development of novel media in which to conduct biocatalytic transformations has been subject to investigation for almost as long as biocatalysis itself, with greatest focus on the use of organic solvents. In recent years several new directions have gained attention. In particular, the use of ionic liquids (Fig. 31.33) and supercritical carbon dioxide (scCO2) have extended the range of conditions under which biocatalytic processes can be applied, primarily through allowing the solubilization of substrates under conditions amenable to enzymatic catalysis and/or facilitating the isolation of products from the reaction medium [123–126]. The desire to reduce waste generation and eliminate the need for volatile and potentially flammable solvents is also spurring the development of novel media for biocatalysis.

Structures of some ionic liquids
Ionic liquids based on salts of alkylimidazoles and pyridines have been used for the synthesis of esters of ascorbic acid and other sugars on the laboratory scale and show particular advantage in their ability to dissolve otherwise incompatible substrates. One of the key features of these solvents is their extremely low vapor pressures and excellent ability to dissolve both polar and nonpolar materials. Reactions can be run under vacuum allowing the removal of volatile byproducts such as water and enable one to shift the equilibrium of a reaction towards product formation, as in the case of reverse hydrolysis used for the formation of esters, glycosides, and peptide bonds. Product recovery can be facilitated through continuous extraction or phase separation allowing recycling of the solvent. The ability to alter the nature of both the cation and anion of ionic liquids independently affords a degree of tunability, a useful property when attempting to optimize a bioprocess. To date, bioprocesses in ionic liquids have predominantly employed lipases; however, other classes of enzyme such as proteases and glycosidases can also be used. Baker’s yeast and other whole-cell biocatalysts have also been used successfully for enantioselective reductions in ionic liquids.
Supercritical fluids (SCFs) and in particular scCO2 have also been shown to provide a unique medium in which to perform biocatalytic transformations. The ability to perform biocatalysis in a SCF was first demonstrated in 1985 by several groups. There are several advantages to the use of SCFs, as well as some drawbacks that need to be addressed if large-scale processes are to be developed.
Advantages:
-
Improved solvation of nonpolar materials
-
Improved mass transfer relative to water and other liquids
-
Ability to modulate solvent properties through pressure changes
-
Ability to remove solvent by depressurization
-
Nontoxic and environmentally friendly
-
Ease of product recovery
Disadvantages:
-
Not compatible with all enzymes
-
Need for specialized equipment
-
Cosolvents often needed
-
Water activity often important
-
No large-scale processes developed to date
The conditions under which CO2 becomes supercritical are mild (Table 31.16) and compatible with many, but not all enzymes. Other SCFs aside from scCO2 have also been used to conduct bioconversions, for example, trifluoromethane, ethane, and propane.
In some cases an enzyme that performs poorly in scCO2 will exhibit greatly improved activity in an alternative SCF. An example is the increased activity of subtilisin in supercritical propane and propane/CO2 mixtures as compared to scCO2 alone. A particularly interesting property of SCFs is the ability to dramatically alter the solvent properties (e.g., dielectric constant) through changes in temperature and pressure. Increases in pressure can enhance enzyme activity in some cases. Enzyme activity in SCFs is also markedly affected by water activity, as is the case in organic media, and the presence of small amounts of water is often essential for effective catalysis. Lipases are particularly suitable for supercritical biocatalysis and are often used in immobilized form owing to increased stability and ease of handling. Other enzymes have also been employed including proteases, oxidases, and glycosidases. The use of whole cells in supercritical CO2 has also been reported. Matsuda et al. in 2001 employed the bacterium B. megaterium to catalyze the carboxylation of pyrrole at rates exceeding those possible in water [127].
Often a cosolvent is used in order to solubilize particularly polar substrates such as sugars and amino acids. Surfactants or additional solvents may also allow adequate solvation of enzymes. In some cases two-phase systems can be used to conduct bioconversion. For example, Reetz et al. employed both SCFs and ionic liquids in a semi-continuous process for the esterification of alcohols where the enzyme resided in the IL phase and scCO2 enabled substrate solvation and product removal [128].
Products. Biocatalysis will increasingly be used for the production of bulk chemicals including fuels, polymers, and other large-volume materials. Much of this development will be driven by large chemical manufacturers, many of whom have invested heavily in the development of in-house biocatalysis capabilities. Examples include BASF, Dupont, Celanese, DSM, Dow Corning, Lonza, and Evonik. The major factor driving this investment is the realization that in many instances bioprocesses can improve productivity, lower costs, and reduce waste streams. The production and modification of bulk polymers holds particular promise as enzymes may enable the production of polymeric architectures that are not readily made through purely chemical routes, for example, polyesters derived from sugars and other polyols [129].
Production of fine chemicals and active pharmaceutical ingredients (APIs) will also continue to benefit from the judicious application of biocatalysis, in many cases as part of multistep synthetic schemes. Of particular relevance is the increasing demand for chirally pure pharmaceuticals, driven by concerns about the unwanted side-effects often associated with racemic drugs. Another growth area is likely to be the production of biologically active carbohydrates, traditionally requiring complex and expensive chemistries for production, to be used as pharmaceuticals, in infant formula, and as nutritional supplements.
Continued development of the biorefinery concept should eventually lead to sources of renewable materials at costs competitive with those from petrochemical sources through the effective utilization of biomass from various sources (cellulosic, agricultural waste, garbage, vegetable oils). Many of these materials will be amenable to further modification through biocatalytic processes, for example, the production of biodiesel using lipases and fatty acids. Industrial enzymes overall should also see increased use in medicine as diagnostics and as components in biosensors and other devices.
Summary and Conclusion
Enzymes are currently applied across many industries and are used as processing aids, incorporated into consumer products, and used in biocatalysis for chemical production. The global market for industrial enzymes will continue to expand as enzymes find their way into new markets and attract greater interest from the chemical industry at large. Several factors will contribute to this growth:
-
Improved knowledge of enzyme mechanisms
-
Reduction in the production costs of industrial enzymes
-
Improved means for enzyme immobilization and bioprocess engineering
-
Novel schemes for cofactor recycling
-
Advances in metabolic engineering of whole microorganisms
-
Greater awareness of the environmental benefits of enzyme technology
Ultimately, the distinction between biological and chemical catalysis may become less distinct as technologies for modified enzymes, enzyme mimetics, and chemoenzymatic catalysis advance [130]. In the meantime, however, it is clear that further application of enzyme technology within industry will greatly benefit society in the twenty-first century.
References
Goodsell DS (2009) The machinery of life. Springer, New York
Renneberg R (2008) Biotechnology for beginners. Academic, Amsterdam
Jencks WP (1987) Catalysis in chemistry and enzymology. McGraw-Hill, New York
Cook PF, Cleland WW (2007) Enzyme kinetics and mechanism. Garland Science, New York
Buchholz K, Kasche V, Bornscheuer UT (2005) Biocatalysts and enzyme technology. Wiley-VCH, Weinheim
Aehle W (ed) (2007) Enzymes in industry: production and applications, 3rd edn. Wiley-VCH, Weinheim
Polaina J, MacCabe AP (eds) (2007) Industrial enzymes, structure, functions and applications. Springer, Dordrecht
Whitehurst RJ, Van Oort M (eds) (2010) Enzymes in food technology, 2nd edn. Wiley-Blackwell, Oxford
Olempska-Beer ZS et al (2006) Food-processing enzymes from recombinant microorganisms—a review. Reg Toxicol Pharm 45:144–158
Dewan SS, Enzymes in industrial applications: global markets (2011) Report BIO030F, BCC Research, Wellesley, MD
Sandhya C, Nampoothiri KM Pandey A (2005) Microbial proteases. In: Barredo JL (ed) Microbial enzymes and biotransformations, Methods in biotechnology, vol 17. Humana Press, Totawa, pp 165–179
Eggleston G (2007) Advances in the industrial application of enzymes on carbohydrate-based materials. In: Eggleston G, Vercellotti JR (eds) Industrial application of enzymes on carbohydrate-based materials, ACS symposium series, vol 972. American Chemical Society, New York, pp 1–16
Aehle W (ed) (2004) Nonindustrial enzyme usage. In: Enzymes in industry: production and applications (Ch. 6), 2nd edn. Wiley-VCH, Weinheim
World enzymes (2007) Study #2229, The Freedonia Group, Cleveland, OH
Lairson LL et al (2008) Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem 77:521–555
Straathof AJJ, Aldercreutz P (eds) (2001) Applied biocatalysis, 2nd edn. Harwood, Amsterdam
Bommarius AS, Riebel BR (2004) Biocatalysis—fundamentals and applications. Wiley-VCH, Weinheim
Liese A (2000) Industrial biotransformations. Wiley-VCH, Weinheim
Kohler R (1972) The reception of Eduard Buchner’s discovery of cell-free fermentation. J Hist Biol 5:327–353
Sumner JB (1946) The chemical nature of enzymes. Nobel Lecture, 12 Dec 1946
Phillips DC (1967) The hen egg-white lysozyme molecule. Proc Natl Acad Sci U S A 57:484–495
Estell DA, Graycar TP, Wells JA (1985) Engineering an enzyme by site-directed mutagenesis to be resistant to chemical oxidation. J Biol Chem 260:6518–6521
Manz A, Pamme N, Lossifidis D (2004) Bioanalytical chemistry. World Scientific Publishing Company, Singapore
Houg DW, Danson MJ (1999) Extremozymes. Curr Opin Mol Biol 3:39–46
Reymond J-L, Fluxa VS, Maillard N (2009) Enzyme assays. Chem Commun 1:34–46
Chen LH et al (1992) 4-Oxalocrotonate tautomerase, an enzyme composed of 62 amino acid residues per monomer. J Biol Chem 267:17716–17721
Smith S, Witkowski A, Joshi AK (2003) Structural and functional organization of the animal fatty acid synthase. Prog Lipid Res 42:289–317
Dill KA, Ozkan SB, Shell MS, Weiki TR (2008) The protein folding problem. Annu Rev Biophys 37:289–316
Gevaert K, Vandekerkhove J (2000) Protein identification methods in proteomics. Electrophoresis 21:1145–1154
Mittermaier A, Kay LE (2006) New tools provide new insights in NMR studies of protein dynamics. Science 312:224–228
Masel RI (2001) Chemical kinetics and catalysis. Wiley-Interscience, Oxford
Bugg T (2004) Introduction to enzyme and coenzyme chemistry, 2nd edn. Wiley-Blackwell, Oxford, p 31
Benkovic SJ, Hammes GG, Hammes-Schiffer S (2008) Free energy landscape of enzyme catalysis. Biochemistry 47:3317–3321
Sauer J et al (2000) Glucoamylase: structure/function relationships and protein engineering. Biochim Biophys Acta 1543:275–293
Messerschmidt A (ed) (2011) Handbook of metalloproteins. Wiley-Blackwell, Oxford
Eisenmesser EZ et al (2005) Intrinsic dynamics of an enzyme underlies catalysis. Nature 438:117–121
Sutcliffe MJ, Scrutton NS (2002) A new conceptual framework for enzyme catalysis. Eur J Biochem 269:3096–3102
Menten L, Michaelis MI (1913) Die kinetik der invertinwirkung. Biochem Z 49:333–369
Estell DA (1991) Liquid detergent with stabilized enzyme. US Patent 5,039,446
Yeh W-K, Yang H-C, McCarthy JR (eds) (2010) Enzyme technologies: metagenomics, evolution, biocatalysis and biosynthesis, Part A. Wiley, New York
Martin CH et al (2009) Synthetic metabolism: engineering biology at the protein and pathway scales. Chem Biol 16:277–286
Warnecke F, Hess MA (2009) A perspective: metatranscriptomics as a tool for the discovery of novel biocatalysts. J Biotechnol 142:91–95
Eggert T (2006) Optimization of industrial enzymes by molecular engineering. In: Liese A, Seelbach K, Wandrey C (eds) Industrial biotransformations (Ch. 4), 2nd edn. Wiley-VCH, Weinheim
Rondon MR et al (2000) Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol 66:2541–2547
Knietsch A et al (2003) Construction and screening of metagenomic libraries derived from enrichment cultures: generation of a gene bank for genes conferring alcohol oxidoreductase activity on Escherichia coli. Appl Environ Microbiol 69:1408–1416
Ward DE et al (2002) Proteolysis in hyperthermophilic microorganisms. Archaea 1:63–74
Böttcher D, Bornscheuer UT (2010) Protein engineering of microbial enzymes. Curr Opin Microbiol 13:274–282
Dodge T (2009) Production of industrial enzymes. In: Whitehurst RJ, van Oort M (eds) Enzymes in food technology, 2nd edn. Wiley-Blackwell, Oxford, UK
Krishna C (2005) Solid-state fermentation system—an overview. Crit Rev Biotechnol 25:1–30
Hood EE (2002) From green plants to industrial enzymes. Enzyme Microb Technol 30:279–283
Kumar D et al (2008) Microbial proteases and application as laundry detergent additive. Res J Microbiol 3:661–672
Outtrup H, Jørgensen ST (2008) The importance of bacillus species in the production of industrial enzymes. In: Berkeley R, Heyndrickx M, Logan N, De Vos P (eds) Applications and systematics of Bacillus and relatives. Blackwell Science, Oxford
Maurer K-H (2004) Detergent proteases. Curr Opin Biotechnol 15:330–334
Choudhary RB, Jana AK, Jha MK (2004) Enzyme technology applications in leather processing. Indian J Chem Technol 11:659–671
De Souza PM, Magalhães PO (2010) Application of microbial α-amylase in industry—a review. Braz J Microbiol 41:850–861
Gupta R et al (2003) Microbial-amylases: a biotechnological perspective. Process Biochem 38:1599–1616
Kumar P, Satyanarayana T (2009) Microbial glucoamylases: characteristics and applications. Crit Rev Biotechnol 29:225–255
Ito S et al (2003) Carbohydrate-active enzymes from alkaliphiles. J Appl Glycosci 50:257–262
Rausch KD, Belyea RL (2006) The future of co-products from corn processing. Appl Biochem Biotechnol 128:47–86
Bhosale SH, Rao MB, Deshpande VV (1996) Molecular and industrial aspects of glucose isomerase. Microbiol Rev 60:280–300
Sukumaran RK, Singhania RR, Pandey A (2005) Microbial cellulases—production, applications and challenges. J Sci Ind Res 64:832–844
Shallom D, Shoham Y (2003) Microbial hemicellulases. Curr Opin Microbiol 6:219–228
Soundari SG, Sashi V (2009) Bacterial xylanases. Asian J Microbiol Biotechnol Environ Sci 11:677–682
Hoondal GS et al (2002) Microbial alkaline pectinases and their industrial applications: a review. Appl Microbiol Biotechnol 59:409–418
Sun Y, Cheng J (2002) Hydrolysis of lignocellulosic materials for ethanol production: a review. Bioresour Technol 83:1–11
Shanmugam KT, Ingram LO (2008) Engineering biocatalysts for production of commodity chemicals. J Mol Microbiol Biotechnol 15:8–15
Anuradha P et al (2009) Microbial lipases: a potential tool for industrial applications. J Pure Appl Microbiol 3:301–306
De Maria L et al (2007) Phospholipases and their industrial applications. Appl Microbiol Biotechnol 74:290–300
Singh B, Kunze G, Satyanarayana T (2011) Developments in biochemical aspects and biotechnological applications of microbial phytases. Biotechnol Mol Biol Rev 6:69–87
Xu F (2005) Applications of oxidoreductases: recent progress. Ind Biotechnol 1:38–50
Bankar SB, Bule MV, Singhal RS, Ananthanarayan L (2009) Glucose oxidase—an overview. Biotechnol Adv 27:489–501
Chelikani P, Fita I, Loewen PC (2004) Diversity of structures and properties among catalases. Cell Mol Life Sci 61:19–208
Riva S (2006) Laccases: blue enzymes for green chemistry. Trends Biotechnol 24:219–226
Majeau J-A, Brar SK, Tyagi RD (2010) Laccases for removal of recalcitrant and emerging pollutants. Bioresour Technol 101:2331–2350
Theriot CM, Grunden AM (2010) Hydrolysis of organophosphorus compounds by microbial enzymes. Appl Microbiol Biotechnol 89:35–43
Pedreschi F, Kaack K, Granby K (2008) The effect of asparaginase in the formation of acrylamide in French fries. Food Chem 109:386–392
Mathews I et al (2007) Structure of a novel enzyme that catalyzes acyl transfer to alcohols in aqueous conditions. Biochemistry 46:8969–8979
Ager DJ, May O (2008) Pharmaceutical industry: biocatalysts and chemocatalysts. In: Begley TP (ed) Wiley encyclopedia of chemical biology. Wiley, New York, pp 1–15
Takasaki Y, Tanabe O. Enzyme method for converting glucose in glucose syrups to fructose. US Patent 3,616,221
Davis BG, Boyer V (2001) Biocatalysis and enzymes in organic synthesis. Nat Prod Rep 18:618–640
Kirk O, Christensen MW (2002) Lipases from Candida antarctica—unique biocatalysts from a unique origin. Org Proc Res Dev 6:446–451
Greenberg WA et al (2009) Aldolase enzymes for complex synthesis. In: Whittall J, Sutton P (eds) Practical methods for biocatalysis and biotransformations (Ch. 6). Wiley, Chichester
Guisan JM (ed) (2006) Immobilization of enzymes and cells (Methods in biotechnology), 2nd edn. Humana Press, Totowa
Přenosil JE, Kut ÖM, Dunn IJ, Heinzle E (2009) Biocatalysis, 2. Immobilized biocatalysts. In: Bellussi G et al (eds) Ullmann’s encyclopedia of industrial chemistry. Wiley-VCH, Weinheim
Berendsen WR, Lapin A, Reuss M (2006) Investigations of reaction kinetics for immobilized enzymes—identification of parameters in the presence of diffusion limitation. Biotechnol Prog 22:1305–1312
Fernandez-Lafuente R (2010) Lipase from Thermomyces lanuginosus: uses and prospects as an industrial biocatalyst. J Mol Catal B: Enzym 62:197–212
Mateo C et al (2007) Advances in the design of new epoxy supports for enzyme immobilization-stabilization. Biochem Soc Trans 35:1593–1601
Kim J, Grate JW, Wang P (2008) Nanobiocatalysis and its potential applications. Trends Biotechnol 26:639–646
Betancor L, Luckarift HR (2008) Bioinspired enzyme encapsulation for biocatalysis. Trends Biotechnol 26:566–572
Kristenen JB et al (2010) Biomimetic silica encapsulation of enzymes for replacement of biocides in antifouling coatings. Green Chem 12:387–394
Becker U et al (2008) Industrial applications of whole-cell biocatalysis. Pharm Technol 41(S6):S8–S11
Bechtold M, Panke S (2009) In situ product recovery integrated with biotransformations. Chimia 63:345–348
Shuler ML, Kargi F (2002) Bioprocess engineering: basic concepts, 2nd edn. Prentice-Hall, Upper Saddle River
Koskinen AMP, Klibanov AM (eds) (1995) Enzymatic reactions in organic media. Springer, Berlin
Klibanov AM (2001) Improving enzymes by using them in organic solvents. Nature 409:241–246
Adlercreutz P (2008) Fundamentals of biocatalysis in neat organic solvents. In: Carrea G, Riva S (eds) Organic synthesis with enzymes in non-aqueous media (Ch. 1). Wiley-VCH, Weinheim
Drauz K, Waldmann H (eds) (2002) Enzyme catalysis in organic synthesis, 2nd edn. Wiley-VCH, Weinheim
Krieger N et al (2004) Non-aqueous biocatalysis in heterogeneous solvent systems. Food Technol Biotechnol 42:279–286
Tufvesson P et al (2010) Process considerations for the scale-up and implementation of biocatalysis. Food Bioprod Process 88:3–11
Parker K, Salas N, Nwosu VC (2010) High fructose corn syrup: production, uses and public health concerns. Biotechnol Mol Biol Rev 5:71–78
Yamada H, Kobayashi M (1996) Nitrile hydratase and its application to industrial production of acrylamide. Biosci Biotechnol Biochem 60:391–1400
Bruggink A (ed) (2001) Synthesis of β-lactam antibiotics: chemistry, biocatalysis & process integration. Kluwer Academic, Dordrecht
Sonke T, Kaptein B, Schoemaker HE (2009) Use of enzymes in the synthesis of amino acids. In: Hughes AB (ed) Amino acids, peptides and proteins in organic chemistry: origins and synthesis of amino acids (Ch. 3), vol 1. Wiley-VCH, Weinheim
Tosa T, Mori T, Fuse N, Chibata I (1969) Studies on continuous enzyme reactions 6: enzymatic properties of DEAE-sepharose aminoacylase complex. Agr Biol Chem 33:1047–1056
Patel R (ed) (2006) Biocatalysis in the pharmaceutical and biotechnology industries. CRC Press, Florida
Woodley JM (2008) New opportunities for biocatalysis: making pharmaceutical processes greener. Trends Biotechnol 26:321–327
Erb S (2006) Single-enantiomer drugs poised for further market growth. Pharm Technol 30(suppl):s14–s18
Meyer H-P, Turner NJ (2009) Biotechnological manufacturing options for organic chemistry. Mini-Rev Org Chem 6:300–306
Stinson SC (2001) Chiral pharmaceuticals. Chem Eng News 79:79–97
Tao J, Xu J-H (2009) Biocatalysis in development of green pharmaceutical processes. Curr Opin Chem Biol 13:43–50
Pollard DJ, Woodley JM (2007) Biocatalysis for pharmaceutical intermediates: the future is now. Trends Biotechnol 25:66–73
Liljeblad A, Kallinen A, Kanerva LT (2009) Biocatalysis in the preparation of the statin side chain. Curr Org Synth 6:362–379
Chen X, Kowal P, Wang PG (2000) Large-scale enzymatic synthesis of oligosaccharides. Curr Opin Drug Discov Devel 3:756–763
Taniguchi H (2008) Biocatalysis-based development of oligosaccharides in Japan. In: Hou CT, Shaw J-F (eds) Biocatalysis and bioenergy (Ch. 17). Wiley, New York
Monsan P, Paul F, Pelenc V, Boures E (1996) Enzymatic production of α-butylglucoside and its fatty acid esters. Ann New York Acad Sci 799:633–641
Wong C-H, Whitesides G (1994) Enzymes in synthetic organic chemistry. In: Wong C-H et al (eds) Tetrahedron organic chemistry, vol 12. Pergamon, Oxford
Kumar L, Awasthi G, Singh B (2011) Extremophiles: a novel source of industrially important enzymes. Biotechnology 2011:1–15
Gomes J, Steiner W (2004) The biocatalytic potential of extremophiles and extremozymes. Food Technol Biotechnol 42:279–286
Arnold U (2009) Incorporation of non-natural modules into proteins: structural features beyond the genetic code. Biotechnol Lett 31:1129–1139
Tang Y, Tirrell DA (2001) Biosynthesis of a highly stable coiled-coil protein containing hexafluoroleucine in an engineered bacterial host. J Am Chem Soc 123:11089–11090
Marti S et al (2008) Computational design of biological catalysts. Chem Soc Rev 37:2634–2643
Vieceli J, Muellegger J, Tehrani A (2006) Computer-assisted design of industrial enzymes. The resurgence of rational design and in silico mutagenesis. Ind Biotechnol 2:303–308
Gorke J, Srienc F, Kazlauskas R (2010) Toward advanced ionic liquids. Polar, enzyme-friendly solvents for biocatalysis. Biotechnol Bioprocess Eng 15:40–53
van Rantwijk F, Madeira Lau R, Sheldon RA (2003) Biocatalytic transformations in ionic liquids. Trends Biotechnol 21:131
Cantone S, Hanefeld U, Basso A (2007) Biocatalysis in non-conventional media—ionic liquids, supercritical fluids and the gas phase. Green Chem 9:954–971
Matsuda T, Harada T, Nakamura K (2005) Biocatalysis in supercritical CO2. Curr Org Chem 9:299
Matsuda T, Ohashi Y, Harada T, Yanagihara R, Nagasawa T, Nakamura K (2001) Conversion of pyrrole to pyrrole-2-carboxylate by cells of Bacillus megaterium in supercritical CO2. Chem Commun 21:2194–2195
Reetz MT, Wiesenhoefer W, Francio G, Leitner W (2002) Biocatalysis in ionic liquids: batchwise and continuous flow processes using supercritical carbon dioxide as the mobile phase. Chem Commun 9:992–993
Cheng HN, Gross RA (2008) Polymer biocatalysis and biomaterials: current trends and developments. In: Cheng HN, Gross RA (eds) Polymer biocatalysis and biomaterials II, ACS symposium series, vol 999, pp 1–20
Wohlgemuth R (2007) Interfacing biocatalysis and organic synthesis. J Chem Technol Biotechnol 82:1055–1062
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media New York
About this chapter
Cite this chapter
McAuliffe, J.C. (2012). Industrial Enzymes and Biocatalysis. In: Kent, J. (eds) Handbook of Industrial Chemistry and Biotechnology. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-4259-2_31
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4259-2_31
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-4258-5
Online ISBN: 978-1-4614-4259-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)