
Abstract
Retinoblastoma is a malignant tumor arising from the embryonic neural retina and it is the most frequent intraocular tumor of childhood occurring in about 1 in 14,000–18,000 live births. Out of an estimated 8,000 children developing retinoblastoma each year worldwide, more than 5,000 [1] are diagnosed in developing countries and a high proportion of them would die of disease dissemination [2]. It is currently unknown if there is an increased incidence of retinoblastoma in some developing countries. Despite some studies reported incidence rates that are up to 3–7 times higher than the reported figures for Western Europe, there are no conclusive population-based studies supporting this probable increased incidence [3]. However, there is no doubt that pediatric oncologists practising in developing countries see more patients with retinoblastoma in their practice. The reason for this phenomenon is probably related to the higher frequency of disseminated retinoblastoma in that setting. There are wide differences in the prevalence of extraocular dissemination and survival in developing countries that are related to socioeconomical indicators [2]. In developed countries, survival rates over 90 % have been achieved decades ago, but as opposed to acute leukemias for example, this successful story is not dependant on highly intensive treatments but in early diagnosis, making intensive treatments not necessary. In middle-income countries, where most children present with advanced disease but still limited to the eye or microscopically disseminated, the cure rate is over 80 % but multimodality treatment is needed [4]. In most lower-income countries, where retinoblastoma presents with metastatic disease less than 30 % of the children survive [5].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Retinoblastoma is a malignant tumor arising from the embryonic neural retina and it is the most frequent intraocular tumor of childhood occurring in about 1 in 14,000–18,000 live births. Out of an estimated 8,000 children developing retinoblastoma each year worldwide, more than 5,000 [1] are diagnosed in developing countries and a high proportion of them would die of disease dissemination [2]. It is currently unknown if there is an increased incidence of retinoblastoma in some developing countries. Despite some studies reported incidence rates that are up to 3–7 times higher than the reported figures for Western Europe, there are no conclusive population-based studies supporting this probable increased incidence [3]. However, there is no doubt that pediatric oncologists practising in developing countries see more patients with retinoblastoma in their practice. The reason for this phenomenon is probably related to the higher frequency of disseminated retinoblastoma in that setting. There are wide differences in the prevalence of extraocular dissemination and survival in developing countries that are related to socioeconomical indicators [2]. In developed countries, survival rates over 90 % have been achieved decades ago, but as opposed to acute leukemias for example, this successful story is not dependant on highly intensive treatments but in early diagnosis, making intensive treatments not necessary. In middle-income countries, where most children present with advanced disease but still limited to the eye or microscopically disseminated, the cure rate is over 80 % but multimodality treatment is needed [4]. In most lower-income countries, where retinoblastoma presents with metastatic disease less than 30 % of the children survive [5].
Presenting Signs and Symptoms
Retinoblastoma presents in two distinct clinical forms:
-
1.
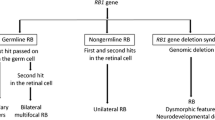
Heritable form (40 % of the total) which is bilateral in 90 % of the cases or unilateral usually with multifocal tumors in the remaining 10 %. In these cases there is a germline mutation of the RB1 gene, which has been identified within the chromosome 13q14 [6]. In developed countries, these are usually the result of a new germline mutation in the 75 % or it may be inherited from an affected parent in the remaining 25 % of the cases. However, in developing countries, especially in those where survival from retinoblastoma in the previous generation is unlikely, familial cases are much less common. Knudson proposed the “two-hit hypothesis,” in order to interpret mutational events in a developing retinal cell leading to the development of retinoblastoma [7]. The Rb1 gene product is crucial in regulating the transition of the cell through the G1 phase of the cell cycle and its inactivation seen in retinoblastoma cases result in deregulated cell proliferation. Mutations at this gene are responsible for retinoblastoma and they have been described in almost every exon of the gene without hot-spots.
-
2.
Sporadic form (60 % of the total): These cases are always unilateral and as such are not heritable. In this form, the RB1 mutation is only present in tumor cells in the affected retina.
Retinoblastoma is a tumor of the young child and occurs in a narrow age range [8]. The age at presentation correlates with laterality and also with socioeconomical factors that are influenced by late diagnosis. Patients with bilateral retinoblastoma present earlier, usually before 1 year of age, but in developing countries, it is not uncommon to see children with bilateral disease at 1 year or older. Those with unilateral disease often present in the second or third year of life. Familial cases are usually diagnosed by screening usually during the first months of life. However, in developing countries, screening of affected individuals is performed less frequently [9].
The typical presenting sign of retinoblastoma is leukocoria (abnormal white pupillary reflex) (Fig. 20.1a). Leukocoria is usually detected by the parents, who usually seek medical attention reporting this sign to the primary care physician. On rare occasions, the physicians are the ones that detect leukocoria in a physical examination. The detection of leukocoria through a flash photograph is becoming increasingly recognized in the medical practice. Leukocoria is a relatively specific sign with few differential diagnoses which always needs an expert evaluation by an ophthalmologist with a dilated examination of the retina under anesthesia. It is important to note that children with retinoblastoma presenting at this stage, usually look healthy, with no pain and adequate growth, so the pediatrician often tends to underestimate this complaint. Most children at the stage of leukocoria have poor or no vision in the affected eye, but since young children are not able to report decreased vision, this complaint is not common in retinoblastoma. Older children may complain of poor vision. Leukocoria may be difficult to detect in a regular examination done by the primary care physician because it may sometimes be visible when the child looks sideways or with low lighting conditions that lead to pupil dilation. Children presenting with leukocoria are still salvageable in most of the cases if diagnosed timely, so they should be promptly referred to experienced ophthalmologists. When children with leukocoria are not diagnosed timely, the disease invariably progresses to glaucoma leading to buphthalmia (increased eye size). These children usually complain of pain, irritability, or sometimes low-grade fever. Some of these cases are misdiagnosed as inflammatory eye conditions such as cellulitis, endophthalmitis or uveitis, and retinoblastoma may not be detected since leukocoria may not be easily noticeable in a severely swollen eye. If the diagnosis is not done, the disease progresses to rupture of the eye caused by tumoral invasion of the orbit leading to severe proptosis resulting in an orbital mass (Fig. 20.1b). In these situations, metastatic dissemination may have already occurred and the child usually looks severely sick and emaciated. Unfortunately, in developing countries, children are usually diagnosed at these later stages. In developed countries, parents usually seek medical attention after noticing leukocoria or because strabismus, which is an earlier presenting sign. Strabismus as a presenting sign of retinoblastoma is seldom considered in developing countries.

Clinical presentation of retinoblastoma (a) leukocoria (b) massive orbital dissemination
Biology and Patterns of Spread
Regardless of the specific mutation, the tumor grows from the nuclear layer of the retina either in an endophytic pattern seeding the vitreous or in an exophytic form into the subretinal space and to the choroid in more advanced states. Retinoblastoma usually produces a retinal detachment, and usually eyes with advanced disease show features of both exophytic and endophytic tumors. After filling the eye, retinoblastoma may disseminate to other organs. It usually does so after involving severely the eye structures, so metastatic dissemination is virtually not existent in cases with small tumors limited to the retina. Thus, extension inside the eye is usually a requisite before metastatic dissemination. The tumor can extend through the optic nerve and/or the subarachnoid space to the chiasm, the brain, and later to the meninges. Exophytic tumors may invade the choroid and later the sclera. The tumor usually remain into the eyeball for some period of time, but if left untreated, it eventually invades the orbit and beyond it to the surrounding structures. The metastatic pattern of retinoblastoma include the CNS and hematogenous metastasis involving the bone, bone marrow, or less frequently any other organ.
Histology
The diagnosis of retinoblastoma is usually made by an experienced ophthalmologist without needing pathological confirmation. However, after the eye is enucleated, it is extremely important that the eyeball is evaluated by an experienced pathologist in order to estimate with accuracy the degree and extent of tumoral dissemination in the eye structures. Retinoblastoma usually presents with small undifferentiated anaplastic cells with scanty cytoplasm and large nuclei, occasionally showing photoreceptor features. Calcification is a frequent finding. Retinoblastoma is a tumor of neuroepithelial origin and presents some similar characteristics to other neuroectodermic pediatric tumors. Thus tumor cells often express photoreceptor-differentiation antigens, neuron-specific enolase, and the ganglioside GD2. Retinoblastoma usually stains negative to CD99. However, immunohistochemistry is not vital to the diagnosis of retinoblastoma. Immunocytological evaluation is nevertheless important in cases of extraocular dissemination where tumoral cells should be readily identified by these techniques. The use of immunocytology for GD2 or for the transcription factor CRX has been reported effective for this goal [10]. The likelihood of extraocular relapse is directly correlated with microscopical invasion to critical eye structures such as the optic nerve in its orbital portion beyond the lamina cribrosa, the choroid when is massively invaded and the sclera that implies always a later stage of choroidal invasion [11].
Diagnostic and Extent of Disease Evaluation
When a child is diagnosed with retinoblastoma, it is important to assess the extent of the disease in two levels: (a) intraocular extension, which will predict eye salvage and (b) extraocular dissemination, which will predict patient survival.
Intraocular extension of retinoblastoma is assessed by the ophthalmologist using the indirect ophthalmoscope in a full examination with pupillary dilation under anesthesia. The extent of intraocular disease evaluation should consider the size of the tumors, their location especially their relationship to the macula and the presence of seeding to the vitreous or the subretinal space. On occasions, the tumor causes retinal detachment, which was previously regarded as a poor prognosis feature. Different systems were proposed to group these findings in order to predict eye salvage. The Reese-Ellsworth (R-E) grouping system was used for many years and proved to be effective in predicting eye salvage with radiotherapy treatment. More recently, an international system was proposed to predict eye salvage with modern chemotherapy treatments (Table 20.1) [12].
The evaluation of extraocular extension is usually done by the pediatric oncologist. For a proper extent of extraocular disease evaluation it is important to consider the prevalence of extraocular disease in a given patient population. Since extraocular retinoblastoma is very uncommon in developed countries, most authors recommend that no other staging procedure other than CNS imaging should be done for staging [13]. The low prevalence dissemination in the CSF or in the bone marrow makes evaluation of these sites of low yield and most authors in developed countries do not perform these evaluations routinely in their patients. However, in developing countries with high prevalence of these complications, a full extent of disease evaluation may be needed in most patients [14]. Because extraocular disease is so uncommon in developed countries, there has not been a consensus staging system for extraocular disease for years. However, in recent years, a group of international retinoblastoma experts developed a staging system by consensus that articulates with the intraocular grouping system proposed for the eye-conserving therapies with chemoreduction (Table 20.2) [15]. The TNM system has been recently updated in order to include patients with extraocular dissemination. So, each retinoblastoma program in developing countries should chose the staging system that better accommodates with their patient population, but it is important to be consistent in the use of a specific staging system over time in order to be able to compare the results.
Work-Up for Metastatic Disease
Regardless of the disease extension and the setting, all children with retinoblastoma should undergo a head and orbital, gadolinium-enhanced MRI. MRI is preferred to CT scan because it allows for a better estimation of the invasion to the optic nerve [16]. Guidelines for the evaluation of retinoblastoma with MRI have been recently published [16], but it is important to know that in order to obtain a detailed evaluation of the optic nerve or the choroid, it is necessary to use a high-resolution MRI with orbital coils, which are seldom available in developing countries. In addition, MRI is helpful to evaluate the pineal area where it avoids the exposure to radiation in these susceptible patients. However, many centers in developing countries would have only CT scan available for staging of their retinoblastoma patients. In these cases, CT may be useful to identify obvious optic nerve or orbital involvement and CNS extension. Thus, the first step in the initial evaluations in children with retinoblastoma includes a full ophthalmological evaluation and CNS imaging. After this first step, the treating group is able to determine if an eye salvage therapy is possible, if enucleation of the affected eye is needed or if the disease has already spread outside the eye and neoadjuvant therapy is required. At this point, clinical findings are also important to take a decision since children who can only be evaluated with low resolution CT scans and present with massive buphthalmia or glaucoma but imaging studies fail to disclose extension beyond the eyeball may be better treated with preoperative chemotherapy. In these children and in those in whom extraocular disease is present, further staging evaluations are necessary before starting neoadjuvant chemotherapy. These include bilateral bone marrow aspirates and biopsies and lumbar puncture with examination of the cytospin (in those without risks related to the procedure) [14]. A full bone marrow evaluation including bilateral biopsies and aspirates may improve the yield of these procedures, but they need to be done under general anesthesia, which is not always available or recommended in children with advanced disease in developing countries. In situations where children present with obvious extraocular disease, a single bone marrow aspiration could be done, and if the results are positive, no other bone marrow study would be needed. However, a more exhaustive bone marrow evaluation should be done when a single aspiration fails to show malignant cells, especially if no other metastatic site is present. Since retinoblastoma cells may adhere to the tube, the examination of the cytocentrifugate of the CSF specimen, should be always be done regardless of the cell count in order to improve the yield of this procedure. Bone scintigraphy is only recommended in children with confirmed metastatic disease or those with bone pain. This extensive work-up is only necessary at diagnosis in children with clinically advanced disease in whom preoperative enucleation is considered. In cases where enucleation of the affected eye was decided as initial therapy, staging procedures should be done only if high risk of metastatic disease is probably based on the pathological examination. Enucleation should not be delayed because of extent of disease evaluation other than CNS imaging. Children in whom conservative therapy is undertaken do not need any other staging procedure than CNS imaging.
Treatment
The treatment of retinoblastoma in developing countries poses significant and unique challenges for the treating physicians given the paucity of publications, the impact of socioeconomical factors and the influence of the availability of resources. It is important that children with retinoblastoma should be treated in centers with experience in the management of these tumors which on occasions would need the participation of eye hospitals in association with children or cancer centers in order to provide the best possible care. In order to provide tools for treating physicians in these settings, the SIOP-PODC (International Society of Pediatric Oncology-Pediatric Oncology in Developing Countries) published a consensus guideline for graduated-intensity therapy [17].
The Challenges in the Treatment of Unilateral Retinoblastoma: When Is Chemotherapy Indicated?
Initial enucleation of the affected eye is the treatment of choice for children with intraocular unilateral retinoblastoma [18]. It is the simplest and safest therapy for retinoblastoma; however, in some developing countries it is not widely accepted by the affected families. It is important to procure a prosthetic eyeball which can even be fitted in the operating room after the procedure to minimize cosmetic effects. Enucleation should be performed by an experienced pediatric ophthalmologist in order to obtain a long optic nerve stump and avoid eye rupture.
Eyes with secondary glaucoma, invasion of anterior segment (anterior chamber, iris), rubeosis iridis, hyphema, pseudohypopyon, and history of orbital cellulitis or those with affection of more than 2/3 of the retina or massive vitreous or subretinal seeding should be enucleated initially without delay. In some developing countries, as many as two-thirds of children present with enlarged eyeballs, that have a higher risk of microscopic extraocular dissemination [19–21]. These eyes may be difficult to enucleate and are at high risk of rupture [22], which would cause tumor seeding in the orbit. Occasionally, the tumor may be left behind in the resection margin of the optic nerve. These cases are at high risk of death if no further treatment is given and they need intensive chemotherapy and orbital radiotherapy to have a chance for cure. In many of these settings, radiotherapy and intensive chemotherapy are unfortunately not available. In these cases, pre-enucleation chemotherapy may theoretically reduce the tumoral volume in severely buphthalmic eyes, thereby reducing these risks [22]. If pre-enucleation chemotherapy is used, these children should be considered at higher risk for extraocular relapse and adjuvant chemotherapy should be given in all cases, regardless of the pathologic findings upon examination of the enucleated eye [23, 24]. It is important to enucleate these eyes no later than two or three chemotherapy cycles, because chemotherapy resistance may ensue, and the child may die of disseminated disease [25]. Enucleation is always needed in these cases, even when tumor response to neoadjuvant chemotherapy is spectacular, and no local therapy should be done.
After enucleation is done, the affected eyeball should be examined by an expert pathologist. Guidelines for uniform processing of enucleated eyes and for definition of involvement of the different eye structures have been published [26]. These guidelines are applicable in most centers since they do not imply any sophisticated procedure [26]. Centers managing cases where invasion to ocular coats is common should give priority to obtain a high-quality pathology examination. The pediatric oncologist uses this information to decide the need for adjuvant chemotherapy after enucleation. It is essential to identify invasion to the optic nerve, especially when it is beyond the lamina cribrosa paying particular attention to the status of the resection margin, the presence and extent of choroidal invasion, and any degree of scleral involvement. These children benefit from adjuvant chemotherapy but its use in children with other risk factors is controversial and it should be balanced with the potential toxicity of chemotherapy and the availability of second-line therapy in a given setting [11]. So, as a general rule, the use of adjuvant chemotherapy in a given setting should consider the following local scenarios (Table 20.3):
-
1.
The risk of toxicity-related death during a neutropenic episode or other toxic event. Some pathology risk factors such as isolated choroidal invasion or anterior segment invasion are associated to a relatively low risk of extraocular relapse if no adjuvant therapy is given (5–7 %). In centers with limited capacity for managing the side effects of chemotherapy or when patients with compliance problems are treated, the risk of a chemotherapy-associated death may outweigh the benefit of adjuvant chemotherapy in children with low-risk disease.
-
2.
The need of high-quality pathology assessments. This is essential, especially when withdrawal of adjuvant therapy is considered because unrecognized high-risk children may have an extraocular relapse that might have been avoided with adjuvant chemotherapy. Examples of this include omission of scleral invasion in cases with massive choroidal invasion, which is typically seen when an insufficient number of slides have been reviewed. Postlaminar optic nerve involvement may be missed when slides of insufficient quality are analyzed. When expert pathology examination is not available, better results may be obtained with the use of adjuvant chemotherapy in all patients when the prevalence of high-risk features is high.
-
3.
The availability of rescue therapy with high-dose chemotherapy and stem cell rescue. Extraocular relapse of retinoblastoma is seldom curable with conventional therapy (except of isolated orbital relapse) [27]. So, if this treatment modality is not available, all efforts should be done to prevent extraocular relapse, even when it is possible that some children would be over-treated with adjuvant chemotherapy.
-
4.
The choice of the chemotherapy regimen for adjuvant therapy. There is some evidence coming from non-randomized studies that intensive regimens including alkylating agents and occasionally anthracyclines are more effective in preventing extraocular relapse than moderate dose carboplatin-based regimens. However, this difference may be seen only in higher risk children and they are conceivably associated to higher risk of fatal toxicity and increased supportive care needs, so this small benefit will be lost in setting with less than optimal supportive care facilities. A list of published chemotherapy regimens is given in Table 20.3.
Bilateral Retinoblastoma and the Challenge of Conservative Therapy in Developing Countries
The indications for enucleation of affected eyes in bilateral retinoblastoma are essentially the same than for unilateral disease. Adjuvant therapy for enucleated eyes in cases of bilateral retinoblastoma should follow the same guidelines as those for cases of unilateral disease. However, there are specific challenges in developing countries. A variable proportion of children in developing countries present with one or both eyes with advanced intraocular disease, which are considered as Group D in the original International Classification. These cases present a dilemma in developing countries. It is necessary to remember that chemoreduction with systemic chemotherapy, the current standard conservative therapy of retinoblastoma originated from developed countries, where adequate supportive care measures and high technology resources operated by highly qualified teams. It is important to identify the local capabilities for conservative therapy in each setting. In low-income settings, usually conservative therapy is not available and since most children present with advanced disease, it is usually not a priority. It should be considered that enucleation would cure a high proportion of children with bilateral retinoblastoma in those settings; however, bilateral enucleation is seldom accepted by affected families. Hence, treating physicians often embark on conservative therapy using systemic chemotherapy when limited focal therapies are available. It is important that patients with intraocular disease not be exposed to treatments with conservative intent in a setting that has no facilities or experience in localized therapy. Conservative therapy of retinoblastoma is only feasible where an experienced ophthalmologist is available for evaluating these children under general anesthesia with adequate safety, focal therapies such as lasers and cryotherapy are available and patients are able to comply with frequent follow-up examinations over long periods of time. In some developing countries, centers of excellence for conservative treatment have been created. So, chemoreduction followed by focal therapy to avoid external-beam radiotherapy (EBRT) may not be feasible in some developing countries and may lead to poorer results when done in inadequate settings. This treatment is particularly dangerous in settings with a high rate of abandonment of follow-up, because children may die as a consequence of chemotherapy toxicity or partially treated tumors may reactivate and disseminate. As a general rule, conservative therapy of Group D eyes should not be considered routinely in centers with limited resources and enucleation is preferred, especially when the contralateral eye has less advanced disease. For eyes with less advanced disease, conservative therapies may achieve good results even in settings with limited resources. However, in developing countries, very few children present with less advanced disease and often EBRT is needed for tumor control. However, this modality is seldom available and it is not uncommon to see patients treated with several rounds of chemotherapy with a conservative intent that have progressive disease needing EBRT which is not available. In a proportion of these cases, especially in those with a single remaining eye, extraocular disease develops, as a consequence of family refusal of enucleation, but also because reluctance of treating physicians to consider enucleation when there is still time to save the child’s life. It should be always remembered that chemotherapy does not cure intraocular retinoblastoma [28]. Its only benefit is avoiding or delaying EBRT, which is associated with 6–17 % increased risk of mortality caused by radiation-induced second tumors during adulthood in developed countries. Avoidance of EBRT also results in improved cosmesis and lower probability of ocular side effects. However, there is no proven benefit in terms of ocular salvage. Therefore in developing countries with high prevalence of advanced disease, especially in those where compliance for follow-up examinations is not optimal, physicians leading a retinoblastoma program may consider developing EBRT facilities for conservative therapy. The advantages of such approach would be that children treated with EBRT need less-intensive follow-up and most those with Groups B and C may be cured with 2-month course of radiotherapy, whereas children treated with chemoreduction and local therapy usually need a more detailed and longer follow-up to consolidate tumor response and treat later relapses [29]. However, the limitation of such approach includes the need for qualified cataract surgery for repairing radiation-induced cataracts that occur in almost all irradiated patients, the need of safe anesthesia, and of course the availability of a linear accelerator for EBRT operated by qualified personnel.
The Challenge of Treating Overt Extraocular Disease
Children with overt extraocular retinoblastoma, regardless of the laterality, are at high risk of mortality. In developing countries, it is critical to differentiate those children with distant metastatic disease to those with only macroscopical dissemination to the orbit or regional lymph nodes since the latter are curable with moderately intense chemotherapy. Even though children with metastatic disease are not curable with conventional chemotherapy, they may benefit from it since it may help resolve severe pain caused by an orbital mass or emaciation caused by tumor dissemination [30]. Newly diagnosed retinoblastoma is a chemosensitive tumor that responds well to many chemotherapeutic agents, even at low doses. Thus, standard or low-dose chemotherapy with an intention of life prolongation should be given to children with metastatic disease. If high-dose chemotherapy followed by autologous stem cell rescue is available, it should be considered, especially for children with no CNS involvement.
Upfront surgery should not be attempted in children with massive orbital invasion and mutilating surgeries such as orbital exenteration should be avoided. Secondary surgery with a limited exenteration or resection of the residual mass is usually enough for tumor control if followed by adjuvant chemotherapy and radiotherapy [30].
The Challenge of Treatment of Patients Whose Parents Refuse Recommended Therapy
Enucleation of an affected eye may not be acceptable to some affected families and consent may not be given for this life-saving procedure [31]. This parental decision is influenced by many and complex socioeconomical, cultural, and religious factors that may be different in each setting. If left untreated, retinoblastoma is uniformly fatal; so every strategy to try to save these children should be employed. Centers where compliance is a substantial problem should establish a comprehensive program to approach these families [32]. Successful experiences have been reported in Central America [33], where multidisciplinary teams give support to high-risk families. There is no uniformly effective approach for this problem and each center should establish a culturally sensitive program to approach holistically this problem. Some centers use chemotherapy for the treatment of children whose families do not consent initial enucleation [34]. Even though this may give time to approach the families to reconsider their decision it should be done at the last resort, when other strategies have failed and definitive drop out is imminent. In these situations, the clinician must balance the risks and benefits of this approach. This strategy is associated with risks related to the possibility of chemotherapy-related toxicity, including death, in these high-risk families. In these situations, the clinicians should be convinced that enucleation is the only treatment with a curative intent and conservation of those eyes is not an option. If these eyes are not enucleated, extraocular relapse would be inevitable. These risks must be weighed against the fact that if no chemotherapy is given, the family will drop out and the child will die of disease dissemination. When pre-enucleation chemotherapy is used, and the eye is enucleated secondarily, the pathological evaluation of the enucleated eye may not be adequate for risk assignment. In these cases, adjuvant chemotherapy should be given.
The Challenge of Early Diagnosis: Are Media Campaigns Useful?
Theoretically, retinoblastoma would be the ideal tumor for screening since it occurs in a narrow age range, it is curable if diagnosed early, its natural history is well known from familial cases which prove the efficacy of early diagnosis in terms of ocular and patient survival [8]. However, retinoblastoma is a rare disease and the diagnosis can only be confirmed with a relatively invasive procedure such as ocular examination under anesthesia. Nevertheless many groups and organizations worldwide launched awareness campaigns directed to the public in order to disseminate information about leukocoria as a presenting sign of retinoblastoma [35]. The impact of these campaigns is difficult to estimate and to be more efficient they should also target doctors who are usually not aware of the possibility of cancer in children with leukocoria either [36]. These campaigns may be effective where disease presents with metastatic disease in a high proportion of children since they would not impact the possibility of ocular survival. Targeting treatment refusal is probably more cost-effective in these settings since usually the same countries that have high proportion of metastatic disease at presentation are the same that have compliance problems. There, the few children who are diagnosed timely may not be cured because their families would not consent enucleation.
Second Malignancies in Retinoblastoma Survivors
Children with the germline mutations of the RB1 gene are at increased risk of secondary non-retinoblastoma malignancies [37]. The occurrence of these malignancies is determined by the mutation but it is also influenced by treatment received [38]. The use, dose, and possibly the age when radiation therapy was administered increase the risk of secondary tumors in the irradiated field [39]. Typically the most common secondary tumors are sarcomas which may occur in the irradiation field or elsewhere in the body, including soft tissue tumors or osteogenic sarcoma, skin tumors such as melanoma and also epithelial tumors. It is probable that children with germline mutations for the Rb1gene do not have an increased susceptibility for secondary leukemias; so when this complication occurs, it is usually related to the use of chemotherapy [40]. Survival from a second tumor is higher when early diagnosis is achieved; so some authors recommend specific surveillance to detect these tumors earlier. Mortality for secondary acute leukemia is high. Overall, the prevalence of secondary malignancies is reported to increase steadily with age, leading to a mortality rate of 25–50 years of age [41], although with modern therapies, it may be much less [42]. Osteosarcoma and melanoma are more frequent in adolescents and young adults, whereas epithelial tumors are more frequent in older adults. Patients with non-hereditary retinoblastoma are not inherently at an increased risk of second malignancies [41].
Trilateral retinoblastoma differs to other secondary malignancies since it arises from the same putative progenitor cell, so the term refers to the association of bilateral retinoblastoma with an asynchronous intracranial primitive neuroectodermal tumor. The exact incidence of this malignancy is not known. Previous studies suggested that up to 3–9 % of patients with the genetic form would develop trilateral disease [43]. However, more recent series show a decreased prevalence and there is indirect evidence that it is less common in developing countries [44]. Occasionally a pineal mass is detected in a follow-up MRI in an asymptomatic child. In these cases, it is important to rule out pineal cysts which have been reported in children with retinoblastoma and always do not progress to malignant tumors [45]. Some authors suggested that the use of chemotherapy may reduce the incidence of trilateral retinoblastoma but these results were not based from population-based studies and included a low number of cases [46]. The prognosis has until recently been almost uniformly fatal, but when high-dose chemotherapy and autologous stem cell rescue is available, some children may be cured.
References
Kivela T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009;93(9):1129–31.
Canturk S, Qaddoumi I, Khetan V, Ma Z, Furmanchuk A, Antoneli CB, et al. Survival of retinoblastoma in less-developed countries impact of socioeconomic and health-related indicators. Br J Ophthalmol. 2010;94(11):1432–6.
BenEzra D, Chirambo MC. Incidence of retinoblastoma in Malawi. J Pediatr Ophthalmol. 1976;13(6):340–3.
Leal-Leal C, Flores-Rojo M, Medina-Sanson A, Cerecedo-Diaz F, Sanchez-Felix S, Gonzalez-Ramella O, et al. A multicentre report from the Mexican Retinoblastoma Group. Br J Ophthalmol. 2004;88(8):1074–7.
Kazadi Lukusa A, Aloni MN, Kadima-Tshimanga B, Mvitu-Muaka M, Gini Ehungu JL, Ngiyulu R, et al. Retinoblastoma in the democratic republic of congo: 20-year review from a tertiary hospital in kinshasa. J Cancer Epidemiol. 2012;2012:920468.
Dryja TP, Rapaport JM, Epstein J, Goorin AM, Weichselbaum R, Koufos A, et al. Chromosome 13 homozygosity in osteosarcoma without retinoblastoma. Am J Hum Genet. 1986;38(1):59–66.
Knudson Jr AG, Hethcote HW, Brown BW. Mutation and childhood cancer: a probabilistic model for the incidence of retinoblastoma. Proc Natl Acad Sci U S A. 1975;72(12):5116–20.
Abramson DH, Beaverson K, Sangani P, Vora RA, Lee TC, Hochberg HM, et al. Screening for retinoblastoma: presenting signs as prognosticators of patient and ocular survival. Pediatrics. 2003;112(6 Pt 1):1248–55.
Chantada GL, Dunkel IJ, Qaddoumi I, Antoneli CB, Totah A, Canturk S, et al. Familial retinoblastoma in developing countries. Pediatr Blood Cancer. 2009;53(3):338–42.
Terry J, Calicchio ML, Rodriguez-Galindo C, Perez-Atayde AR. Immunohistochemical expression of CRX in extracranial malignant small round cell tumors. Am J Surg Pathol. 2012;36(8):1165–9.
Chantada GL, Dunkel IJ, de Davila MT, Abramson DH. Retinoblastoma patients with high risk ocular pathological features: who needs adjuvant therapy? Br J Ophthalmol. 2004;88(8):1069–73.
Linn Murphree A. Intraocular retinoblastoma: the case for a new group classification. Ophthalmol Clin North Am. 2005;18(1):41–53, viii.
Azar D, Donaldson C, Dalla-Pozza L. Questioning the need for routine bone marrow aspiration and lumbar puncture in patients with retinoblastoma. Clin Experiment Ophthalmol. 2003;31(1):57–60.
Chantada GL, Rossi J, Casco F, Fandino A, Scopinaro M, de Davila MT, et al. An aggressive bone marrow evaluation including immunocytology with GD2 for advanced retinoblastoma. J Pediatr Hematol Oncol. 2006;28(6):369–73.
Chantada G, Doz F, Antoneli CB, Grundy R, Clare Stannard FF, Dunkel IJ, et al. A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer. 2006;47(6):801–5.
de Graaf P, Goricke S, Rodjan F, Galluzzi P, Maeder P, Castelijns JA, et al. Guidelines for imaging retinoblastoma: imaging principles and MRI standardization. Pediatr Radiol. 2012;42(1):2–14.
Chantada G, Luna-Fineman S, Sitorus RS, Kruger M, Israels T, Leal-Leal C, Bakhshi S, Qaddoumi I, Abramson DH, Doz F. SIOP-PODC recommendations for graduated-intensity treatment of retinoblastoma in developing countries. Pediatr Blood Cancer. 2013;60(5):719–27.
Sultan I, Wilson MW, Nawaiseh I, Mehyar M, Kharma S, Al-Qudimat M, et al. Enucleation for retinoblastoma: the experience of a single center in Jordan. Int Ophthalmol. 2010;30(4):407–14.
Carrim ZI, Kajaige J, Bowman RJ, Lavy TE, Scanlan P. First-year experience of chemotherapy for advanced retinoblastoma in Tanzania: disease profile, outcomes, and challenges in 2008. J Pediatr Ophthalmol Strabismus. 2012;49(3):176–83.
Bhurgri Y, Muzaffar S, Ahmed R, Ahmed N, Bhurgri H, Usman A, et al. Retinoblastoma in Karachi, Pakistan. Asian Pac J Cancer Prev. 2004;5(2):159–63.
Kashyap S, Meel R, Pushker N, Sen S, Bakhshi S, Sreenivas V, et al. Clinical predictors of high risk histopathology in retinoblastoma. Pediatr Blood Cancer. 2012;58(3):356–61.
Bellaton E, Bertozzi AI, Behar C, Chastagner P, Brisse H, Sainte-Rose C, et al. Neoadjuvant chemotherapy for extensive unilateral retinoblastoma. Br J Ophthalmol. 2003;87(3):327–9.
Zhao J, Dimaras H, Massey C, Xu X, Huang D, Li B, et al. Pre-enucleation chemotherapy for eyes severely affected by retinoblastoma masks risk of tumor extension and increases death from metastasis. J Clin Oncol. 2011;29(7):845–51.
Chantada G, Leal-Leal C, Brisse H, de Graaf P, Sitorus RS, Qaddoumi I, et al. Is it pre-enucleation chemotherapy or delayed enucleation of severely involved eyes with intraocular retinoblastoma that risks extraocular dissemination and death? J Clin Oncol. 2011;29(24):3333–4; author reply 5–6.
Bai S, Ren R, Li B, Xu X, Zhao B, Gao F, et al. Delay in the diagnosis of retinoblastoma in China. Acta Ophthalmol. 2011;89(1):e72–4.
Sastre X, Chantada GL, Doz F, Wilson MW, de Davila MT, Rodriguez-Galindo C, et al. Proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma. Arch Pathol Lab Med. 2009;133(8):1199–202.
Leal-Leal CA, Rivera-Luna R, Flores-Rojo M, Juarez-Echenique JC, Ordaz JC, Amador-Zarco J. Survival in extra-orbital metastatic retinoblastoma:treatment results. Clin Transl Oncol. 2006;8(1):39–44.
Shields CL, Mashayekhi A, Cater J, Shelil A, Meadows AT, Shields JA. Chemoreduction for retinoblastoma. Analysis of tumor control and risks for recurrence in 457 tumors. Am J Ophthalmol. 2004;138(3):329–37.
Wilson MW, Haik BG, Rodriguez-Galindo C. Socioeconomic impact of modern multidisciplinary management of retinoblastoma. Pediatrics. 2006;118(2):e331–6.
Ali MJ, Reddy VA, Honavar SG, Naik M. Orbital retinoblastoma: where do we go from here? J Cancer Res Ther. 2011;7(1):11–4.
Sitorus RS, Moll AC, Suhardjono S, Simangunsong LS, Riono P, Imhof S, et al. The effect of therapy refusal against medical advice in retinoblastoma patients in a setting where treatment delays are common. Ophthalmic Genet. 2009;30(1):31–6.
Mostert S, Arora RS, Arreola M, Bagai P, Friedrich P, Gupta S, et al. Abandonment of treatment for childhood cancer: position statement of a SIOP PODC Working Group. Lancet Oncol. 2011;12(8):719–20.
Luna-Fineman S, Barnoya M, Bonilla M, Fu L, Baez F, Rodriguez-Galindo C. Retinoblastoma in Central America: report from the Central American Association of Pediatric Hematology Oncology (AHOPCA). Pediatr Blood Cancer. 2012;58(4):545–50.
Luna-Fineman S, Alejos A, Amador G, Barnoya M, Benavides R, Castellanos M, et al. Pre-enucleation chemotherapy in advanced intraocular retinoblastoma. Central American experience: AHOPCA II. [Abstract]. Pediatr Blood Cancer. 2012;59(6):984.
Leander C, Fu LC, Pena A, Howard SC, Rodriguez-Galindo C, Wilimas JA, et al. Impact of an education program on late diagnosis of retinoblastoma in Honduras. Pediatr Blood Cancer. 2007;49(6):817–9.
Leal-Leal CA, Dilliz-Nava H, Flores-Rojo M, Robles-Castro J. First contact physicians and retinoblastoma in Mexico. Pediatr Blood Cancer. 2011;57(7):1109–12.
Eng C, Li FP, Abramson DH, Ellsworth RM, Wong FL, Goldman MB, et al. Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst. 1993;85(14):1121–8.
Marees T, Moll AC, Imhof SM, de Boer MR, Ringens PJ, van Leeuwen FE. Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst. 2008;100(24):1771–9.
Moll AC, Imhof SM, Schouten-Van Meeteren AY, Kuik DJ, Hofman P, Boers M. Second primary tumors in hereditary retinoblastoma: a register-based study, 1945–1997: is there an age effect on radiation-related risk? Ophthalmology. 2001;108(6):1109–14.
Gombos DS, Hungerford J, Abramson DH, Kingston J, Chantada G, Dunkel IJ, et al. Secondary acute myelogenous leukemia in patients with retinoblastoma: is chemotherapy a factor? Ophthalmology. 2007;114(7):1378–83.
Yu CL, Tucker MA, Abramson DH, Furukawa K, Seddon JM, Stovall M, et al. Cause-specific mortality in long-term survivors of retinoblastoma. J Natl Cancer Inst. 2009;101(8):581–91.
Turaka K, Shields CL, Meadows AT, Leahey A. Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer. 2012;59(1):121–5.
Kivela T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999;17(6):1829–37.
Antoneli CB, Ribeiro Kde C, Sakamoto LH, Chojniak MM, Novaes PE, Arias VE. Trilateral retinoblastoma. Pediatr Blood Cancer. 2007;48(3):306–10.
Beck Popovic M, Balmer A, Maeder P, Braganca T, Munier FL. Benign pineal cysts in children with bilateral retinoblastoma: a new variant of trilateral retinoblastoma? Pediatr Blood Cancer. 2006;46(7):755–61.
Shields CL, Meadows AT, Shields JA, Carvalho C, Smith AF. Chemoreduction for retinoblastoma may prevent intracranial neuroblastic malignancy (trilateral retinoblastoma). Arch Ophthalmol. 2001;119(9):1269–72.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Chantada, G.L., Leal, C.L. (2014). Retinoblastoma. In: Stefan, D., Rodriguez-Galindo, C. (eds) Pediatric Hematology-Oncology in Countries with Limited Resources. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-3891-5_20
Download citation
DOI: https://doi.org/10.1007/978-1-4614-3891-5_20
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-3890-8
Online ISBN: 978-1-4614-3891-5
eBook Packages: MedicineMedicine (R0)