
Abstract
In contrast to a classic view that the brain was a passive receiver of the pain message, Melzack and Wall proposed in 1965 that the brain exerted descending control upon spinal nociceptive transmission. This chapter reviews the substrates mediating endogenous pain-modulation. The respective roles of the ventrolateral periaqueductal gray (vlPAG) and rostroventromedial medualla (RVM) in the mediation of not only pain-inhibitory, but also pain-facilitatory responses are described behaviorally, neuroanatomically, neurochemically and neurophysiologically. How this intrinsic pain-modulatory system is activated by other brain nuclei (limbic system and cortex) and by exogeneous environmental factors is described. The emergence of sex differences and neurohormonal factors in mediating pain modulation is also considered. The neurophysiological underpinnings of RVM "ON-" and "OFF-cells" and their respective roles in endogenous pain-facilitatory and pain-inhibitory responses are discussed in detail. Finally, how these pain-modulatory responses relate to other neurobehavioral adapatations are considered, including how animal studies can inform human pain conditions and their detection through functional imaging.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Brief History
The brain was classically viewed as a passive receiver of the “pain message,” information in response to tissue damage that was transmitted by somatosensory neurons and relayed through the spinal cord dorsal horn and thalamus to the cortex. In some ways, this concept was only minimally advanced over the seventeenth-century model put forth by the philosopher and mathematician, Rene Descartes, who envisioned particles traveling from the periphery, through the nerves, and up to the brain, where they would ring a figurative alarm bell, signaling pain. The problem with this model is that pain is not a direct function of tissue damage. Pain experienced as the result of a given injury varies from person to person and depends on the behavioral context and competing sensory inputs. The eminent anesthesiologist Henry Beecher considered the subjective nature of pain in the middle of the last century and concluded that “There was no dependable relationship between the extent of a pathological wound and the pain experienced ….” This led many to conclude that any pain that could not be tied to an injury or disease processes was not “real” and, at worst, a manifestation of psychological and psychosomatic aberration. This view was difficult to reconcile with the subjective nature of pain, the influence of cognitive and emotional variables on pain, and clinical reports of pain, sometimes extraordinarily intense pain, in the absence of ongoing, observable tissue damage.
Ronald Melzack and Patrick Wall in their 1965 “gate control theory” posited descending control of spinal nociceptive transmission by the brain, but a concrete neural basis for descending control was not provided until 1969, with the discovery of “stimulation-produced analgesia” from the midbrain periaqueductal gray (PAG). Work from a number of laboratories led to the definition of a brain stem pain-modulating system with key links in the PAG and rostral ventromedial medulla (RVM). Interest in this system was further heightened when Tony Yaksh and others using a microinjection mapping approach showed that this PAG-RVM system was an important substrate for opioid analgesic drugs.
Over the last two decades, this system has been recognized to exert bidirectional control, facilitating as well as inhibiting pain and contributing to a range of pathological and persistent pain states. Part of the neural basis for bidirectional control is two distinct populations of neurons in the RVM, “ON-cells” (which facilitate pain) and “OFF-cells” (which suppress pain processing). Sex differences in pain and analgesia can also be explained, at least in part, on the basis of differences in this pain-modulating system. Top-down recruitment of this system, for example, from the amygdala or hypothalamus, is now understood to provide a basis for cognitive and emotional influences on pain.
Central Pain-Modulatory Systems
Activity in Pathways that Transmit Signals Related to Pain is Regulated by Brain Modulatory Systems that Can Inhibit Pain (Producing Analgesia, Hypoalgesia, Antinociception, Anti-hyperalgesia) or Facilitate Pain (Producing Hyperalgesia, Pronociception, Allodynia) Based Upon Prior Experience, other Behavioral Priorities, Contingencies in the Physical or Social Environment, or Stimulus History (e.g., Prior Injury Leading to Inflammation)
This chapter focuses on intrinsic pain-modulating systems, considering the neuroanatomical, neurochemical, and neurophysiological substrates by which the brain regulates transmission of pain-related sensory information from the periphery to the brain. This chapter describes pain-modulating processes as part of dynamic interacting systems that promote adaptive behavior in response to actual or potential tissue damage by suppressing or facilitating pain. Inhibition of pain allows an organism to focus on other behavioral priorities. By contrast, pain-facilitating processes enhance vigilance in situations in which injury is likely, and after an injury has occurred, promote recuperative behaviors that protect injured body parts. Thus, for example, it is important not to be distracted by injuries when trying to escape from a predator, but once safely in the burrow, it is equally important to protect injured body parts. Unfortunately, pain-facilitating processes can contribute to pathological pain states by supporting overactivity and dysfunction in pain-transmission pathways. Activation of pain-modulatory systems can occur as part of feedback loops triggered by noxious stimuli, but also through top-down mechanisms, which mediate the influences of cognitive and emotional factors on pain.
Important terms related to pain modulation should be defined at the outset. Analgesia is operationally defined as a complete suppression of the sensory experience of pain through a complex process that interferes with the sensory-discriminative, motivational-affective, and/or cognitive components of pain. Hypoalgesia refers to a reduction in pain sensitivity, or elevation of threshold, without complete inhibition. Hyperalgesia refers to enhanced pain sensitivity, including lowered threshold and/or increased response to suprathreshold stimuli. Allodynia refers to pain evoked by innocuous stimuli, that is, by stimuli that are below the normal pain threshold. Anti-hyperalgesia refers to reversal of potentiated pain states without suppression of normal nociceptive transmission. Antinociception is defined as an inhibition of neural processes related to tissue damage and pronociception as facilitation of those processes.
Strictly speaking, analgesia and hyperalgesia should be used only in the context of pain reports from human experimental subjects and patients because only humans can verbally report sensory experience. Antinociception and pronociception, rather than analgesia and hyperalgesia, are therefore often used in referring to alterations of nociceptive behaviors in experimental animals. However, this distinction is not always observed, and the present chapter will use analgesia and hyperalgesia, as well as antinociception and pronociception, when discussing experimental findings in animals.
To place the importance of the central mechanisms of pain modulation in proper perspective, it is important to refer to related chapters in the basic section (Sensory Systems: Somatosensation; Neurochemistry; Neuropharmacology: Opioids) and in the clinical section (Peripheral Receptors; Spinal “Wind-up”; Somatic Pain; Headache; Dental Pain).
Variability of Pain and the Influence of Cognitive and Emotional Variables on the Pain Response Are Explained by Intrinsic Brain Pain-Modulating Systems
Descending control of spinal sensory systems by the brain had been described by the mid-1950s, but a paradigm shift from the view of the pain system as passively transmitting information related to tissue damage occurred with the “gate control theory” proposed by Ronald Melzack and Patrick Wall in 1965. Their model is widely known for proposing interactions between large- and small-diameter afferent input within the dorsal horn, opening and closing a “gate” for pain. However, they also postulated that there were descending influences from the brain, influences that could determine whether a particular primary afferent input was transmitted rostrally from the spinal cord to the brain.
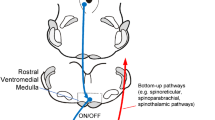
The Melzack-Wall gate control theory was highly influential and provided a valuable heuristic framework for thinking about pain, but a concrete neurobiological framework for their postulated descending control system remained to be identified. This changed in 1969, when electrical stimulation of the midbrain periaqueductal gray (PAG) in the rat was shown to dramatically suppress behavioral responses to stimuli that would normally be considered painful. Although those initial experiments were relatively crude in their pain measures, subsequent work, particularly by John Liebeskind and colleagues at UCLA using a variety of pain tests, and the extension to other species, including humans, confirmed the initial observations. Electrical stimulation effects were also shown to be specific for pain. Locomotion and other sensory systems were unaffected. The appreciation of pain modulation as a specific function of defined brain circuits was a watershed in the scientific approach to pain, because it indicated that the brain had the capacity to control its own pain-related inputs. As outlined in more detail below, it is now understood that defined brain stem circuits with essential links in the PAG and RVM, in the pontine noradrenergic cell groups, and in the caudal lateral medulla, allow top-down and feedback control of pain transmission by regulating nociceptive transmission at the level of the dorsal horn (Fig. 89.1). Although originally defined by the analgesia observed following electrical stimulation, these systems are now recognized to exert bidirectional control, facilitating as well as suppressing pain.

Principal brain stem pain-modulating systems are the PAG-RVM system and norepinephrine-containing cell groups (A5, locus coeruleus, and A7) in the lateral pons. The PAG influences the spinal cord dorsal horn indirectly, through a relay in the RVM. RVM ON-cells and OFF-cells project to the dorsal horn, having net pronociceptive (ON-cells) and antinociceptive (OFF-cells) actions. The PAG-RVM system also exerts some of its effects via the pontine norepinephrine-containing cell groups. NE cell groups norepinephrine-containing cell groups, PAG periaqueductal gray, RVM rostral ventromedial medulla
The PAG-RVM System Emerged as a Pain-Modulating Circuit
The best-characterized pain-modulating system has critical relays in the PAG and RVM (Fig. 89.1). The PAG is a cell-rich region surrounding the cerebral aqueduct in the midbrain. The RVM is defined functionally, rather than cytoarchitecturally, as that area in which electrical stimulation with low electrical currents (10 nA or less) produces antinociception. The RVM includes the nucleus raphe magnus and adjacent ventral aspect of the nucleus reticularis gigantocellularis (referred to as the n. reticularis gigantocellularis pars alpha or sometimes as the n. reticularis paragigantocellularis). Numerous behavioral studies have demonstrated that nonselective activation of neurons within the PAG or RVM, using electrical stimulation or nonspecific neuroexcitants, has a potent antinociceptive effect.
Electrical stimulation at either site elicits behavioral antinociception and suppresses dorsal horn nociceptive activity. Based on these observations, and on anatomical connections from the PAG to the RVM and then from the RVM to the dorsal horn of the spinal cord, Alan Basbaum and Howard Fields in the 1970s proposed a descending pain-inhibitory system consisting of three tiers: the PAG, the RVM, and the dorsal horn. The PAG-RVM connection most relevant for pain modulation arises in the ventrolateral PAG (vlPAG) and is distinct from lateral/dorsolateral projections thought to be important in other aspects of homeostatic behavior in which the PAG has an important role, including aggression and active defense. The functional significance of the PAG-RVM connection is demonstrated by the fact that inactivation of the RVM attenuates or blocks descending inhibition from the PAG. This indicates that the influence of the PAG on the spinal dorsal horn is indirect, through its connections to the RVM. The major descending projection of the RVM travels through the dorsolateral funiculus, terminating at all levels of the dorsal horn. This system influences spinal nociceptive processes important for local nocifensor reflexes, as well as ascending messages important for the affective and sensory-discriminative aspects of pain.
The PAG-RVM system employs a number of neurotransmitters and neuropeptides. Serotonin, enkephalins, substance P, neurotensin, and most prominently GABA have all been identified in neurons projecting from the vlPAG to the RVM. Neuromodulators found in RVM neurons include substance P, enkephalin, thyrotropin-releasing hormone, galanin, somatostatin, cholecystokinin, and acetylcholine. As with the PAG-to-RVM connection, GABA is a prominent component of the RVM projection to the spinal cord. The projection from the RVM to the spinal cord also includes axons of the B3 serotonin-containing cell group (although it should be emphasized these are a minority of RVM neurons, only about 20% in rat, even in n. raphe magnus). Early views of the RVM emphasized serotonin as the mediator of descending inhibition from this region. That perspective proved to be an oversimplicification, as elimination of serotonin or antagonism of spinal serotonin receptors can attenuate facilitation, as well as inhibition, from the RVM. The exact role of serotonin in descending pain modulation therefore remains an interesting open question.
Both the PAG and RVM can also influence the dorsal horn indirectly, through interactions with pontine noradrenergic systems. The role of descending noradrenergic pathways in nociceptive modulation is considered in the next section.
At this point, it is important to note that neither the PAG nor the RVM is a pain-modulating “center,” dedicated exclusively to pain modulation and having no other functions. These regions are part of the brainstem core and, as emphasized by Thelma Lovick, Richard Bandler, Michael Fanselow, and more recently Peggy Mason, coordinate behavioral and physiological aspects of defense, bodily homeostasis, and reproduction. However, there is evidence for specific populations of pain-modulating neurons in each region (these have been studied most intensively in the RVM, see section “Two RVM Cell Populations, “ON-Cells” and “OFF-Cells,” Are the Basis for Bidirectional Pain Control from this Region” below). Presumably, these pain-modulating neurons interact with local circuits to implement integrated responses to internal and external challenges.
Pontine Noradrenergic Systems Also Contribute to Pain Modulation
It had been known since early in the twentieth century that direct spinal application of catecholamines suppressed behavioral responses to noxious stimulation. However, it was only with the delineation of brain monaminergic cell groups by Annica Dahlström and Kjell Fuxe in the mid-1960s and the characterization of the PAG-RVM system that the ponto-spinal noradrenergic pathways began to be considered as a second major brain stem pain-modulating system (Fig. 89.1). Noradrenergic pathways arising in lateral pontine noradrenergic cell groups (A5, A6 [also known as the locus coeruleus], and A7) project to the dorsal horn, and electrical stimulation in the region of these cell groups produces behavioral antinociception that is blocked by spinal administration of α2-adrenergic receptor antagonists. The analgesic effect of pontine stimulation is mimicked by spinal administration of α2-receptor agonists, which in fact provides clinically useful analgesia. However, as with the PAG-RVM system, the influence of noradrenergic systems is bidirectional, and activation of spinal α1-receptors is pronociceptive.
Descending noradrenergic pathways can act independently of the PAG-RVM system, but are also engaged by that system, and contribute to its effects on spinal nociceptive transmission. Thus, the antinociceptive actions of manipulating the PAG or RVM can be attenuated, although not blocked completely, by antagonizing α2-adrenergic receptors at the spinal level. This observation indicates that spinal noradrenergic transmission is required for the PAG-RVM system to exert a potent antinociceptive effect. The PAG has projections to noradrenergic neurons in the A7 cell group, which is also reciprocally connected with the RVM (Fig. 89.1), consistent with activation of noradrenergic pathways by the PAG and RVM. Whether noradrenergic neurons regulate basal nociceptive tone remains unclear.
It is important to recognize that pontine noradrenergic neurons, like the PAG and RVM, engage in numerous functions such as attention, arousal, and learning and memory in addition to pain modulation and have ascending, as well as descending, projections. Moreover, noradrenergic neurons, particularly in A5 and A7, are intermingled among non-catecholaminergic neurons in the same region, and thus, effects of electrical stimulation in these areas are not necessarily mediated entirely by activation of noradrenergic neurons.
The PAG-RVM System Serves as a Critical Target for Opioid Analgesic Drugs
A central site of action for opiate drugs was discovered by Kang Tsou and C.S. Jang in 1964 with their demonstration that morphine administration into the ventrolateral PAG (vlPAG) led to profound analgesia. With the discovery of stimulation-produced analgesia from the PAG and delineation of the PAG-RVM system, it became apparent that opioid analgesic drugs take advantage of this endogenous pain-modulating system to produce their effects. Subsequent extensive brain mapping studies by Tony Yaksh and others demonstrated that direct focal application of small quantities of morphine or other opioids in the PAG or RVM, like electrical stimulation, produces potent behavioral analgesia. Further, both opiate drugs and electrical stimulation produce tolerance with repeated treatments, show cross-tolerance to each other’s effects, and demonstrate analgesic summation following combined sub-analgesic treatments. In electrophysiological studies, both procedures inhibit nociceptive, but not non-nociceptive, cells in the dorsal horn.
The Endogenous Opioid System of Peptides and Receptors Maps Well onto Pain-Modulating Circuits
The PAG-RVM system employs endogenous opioids at multiple levels. Opioid peptides are abundant in both the PAG and RVM. Mu, delta, and, to a lesser degree, kappa opioid receptors are densely distributed in the PAG and RVM. These major opioid receptor families are further biochemically and pharmacologically subdivided into subtypes (mu: μ1, μ2; delta: δ1, δ2; kappa: κ1, κ2, κ3), and this finer characterization was found to be important in further distinguishing receptor mediation of supraspinal opioid analgesia, particularly by the laboratories of Gavril Pasternak, Frank Porreca, and Richard Bodnar. First, whereas selective agonists for μ, d1, d2, and κ opioid receptors produce analgesia following ventricular administration, only morphine and deltorphin produce analgesia when applied directly to the vlPAG or RVM. Second, whereas ventricular pretreatment with the general opioid antagonist naltrexone blocked analgesia elicited by ventricular μ, δ, and κ agonists, antagonists selective for specific receptor subtypes block only the analgesic response elicited by the corresponding agonist. Third, the genes for the μ (MOR-1), δ (DOR-1), and κ (KOR-1) receptors were identified and cloned in the 1990s, with the MOR-1 gene encoding four exons and the DOR-1 and KOR-1 genes containing three coding exons. The immunohistochemical mapping of gene products in the vlPAG and RVM corresponded well with autoradiographic receptor localization. A gene knockdown technique using antisense oligodeoxynucleotides (AS ODN) against each exon of each opioid receptor gene allowed correlations between gene activity and in vivo pharmacology and provided converging evidence for opioid receptor subtype analgesic selectivity. Thus, ventricular AS ODN probes directed against DOR-1 blocked DPDPE and deltorphin analgesia but failed to affect μ- and κ-agonist-mediated analgesia. Correspondingly, AS ODN probes directed against KOR-1 blocked U50488H analgesia but failed to affect μ- and δ-agonist-mediated analgesia. Interestingly, AS ODN probes directed against exons 1 and 4 of MOR-1 selectively blocked morphine analgesia, whereas probes directed against exons 2 and 3 of MOR-1 selectively blocked M6G and heroin analgesia (Table 89.1).
Interactions Occur Among Opioid-Sensitive Sites
Interactions between the opioid-sensitive substrates in the PAG and RVM have also been defined. Two approaches have been used in vivo. The first approach determined whether opioid analgesia elicited from one supraspinal site (e.g., PAG) is blocked by receptor antagonist pretreatment into a second site (e.g., RVM). The second tested whether sub-analgesic doses of opioid agonists in pairs of sites produce analgesic synergy following combined treatment.
Opioid analgesia elicited from the vlPAG is dependent upon the integrity of serotonergic, opioid, excitatory amino acid, and cholinergic receptor function in the RVM. Reversible and irreversible inactivation of the RVM blocked opioid analgesia elicited from the vlPAG, and the previously described neurochemical projection systems were involved in analgesic responses elicited from vlPAG-RVM pathways as well as intrinsic RVM mechanisms. The serotonergic (5HT) pathway from the vlPAG to the RVM was implicated given that RVM pretreatment with 5HT1, 5HT2, or 5HT3 antagonists blocked vlPAG morphine analgesia. The enkephalinergic projection from the vlPAG to RVM and RVM populations of μ- and δ-opioid receptors were implicated given that RVM pretreatment with general μ- and δ-opioid antagonists blocked vlPAG morphine analgesia. In addition to opiates, l-glutamate or NMDA elicits analgesia when applied to the vlPAG or RVM, and RVM pretreatment with competitive or noncompetitive NMDA but not kainate/AMPA receptor antagonists blocked vlPAG morphine analgesia. Moreover, cholinergic agonists also elicit analgesia when applied to the vlPAG and RVM, and RVM pretreatment with muscarinic or nicotinic cholinergic receptor antagonists also blocked vlPAG morphine analgesia.
μ-Opioid analgesic responses elicited individually from the vlPAG and RVM form a coordinated synergistic system. Opiate administration into the vlPAG or RVM produces dose-dependent analgesic responses. Sub-analgesic doses of morphine or the μ-selective agonist, DAMGO, in the vlPAG or RVM alone produced marked analgesia when simultaneously delivered to both sites and shifted the analgesic dose-response curve in each site three- to tenfold to the left. Moreover, μ- and δ2-opioid receptors in the vlPAG and RVM display between-site synergy such that a sub-analgesic dose of morphine delivered to one site and a sub-analgesic dose of deltorphin delivered to the second site produce marked analgesia and leftward shifts in each dose-response curve. These data speak to the complex heterogeneous neurochemical interrelationships occurring in the PAG-RVM axis in mediating pain-inhibitory responses. The next section provides possible behavioral contexts in which such a system might be activated.
Environmental Analgesia: Severe Stress and Other Biologically Significant Events Engage Pain-Modulating Systems
Pain is a high-priority sensory system that serves as a biological warning device. It commands attention, triggers automatic defensive motor responses, and motivates organized behaviors to escape or avoid injury. However, pain cannot be the highest behavioral priority at all times. The pain-inhibiting influence of the PAG-RVM system allows other biologically significant behaviors to proceed without being preempted by pain. Thus, rodents exhibit hypoalgesia in the presence of a predator or aggressive conspecific, and similar processes presumably explain Henry Beecher’s seminal observations of pain suppression in wartime casualties mentioned in section “Central Pain-Modulatory Systems.” Other biologically significant situations in which descending inhibition contributes to analgesia or hypoalgesia include mating, pregnancy and parturition, feeding, and micturition. The function of pain-modulating systems therefore is to regulate transmission of the pain signal in accord with other behavioral and physiological demands. Because the earliest demonstrations of analgesia related to a particular behavioral context involved severe acute stress, the term “stress-induced analgesia” was widely adopted. However, recognition of the more general role of pain-modulating systems as allowing the organism to achieve other biologically significant goals suggested the broader term “environmental analgesia,” which is now often used.
In the laboratory, acute exposure to severe stressful stimuli, such as inescapable foot shock, forced cold-water swims, immobilization, or glucoprivation, has often been used to suppress nociception. Analgesia in these paradigms appears to be the result of the stressful consequences of the stimuli, and not the physical characteristics of the different stimuli per se. This is demonstrated by the observations that the analgesic response adapts with repeated exposure to the stressor, whereas other responses (e.g., hypothermia following acute and chronic cold-water swims, hyperphagia following acute and chronic glucoprivation) fail to show adaptation. Similarly, Klaus Miczek and others found that defeat-induced analgesia in male mice is elicited in the “resident-intruder paradigm” wherein an “intruder” mouse is introduced into the home cage of the “resident” mouse. In subsequent aggressive encounters, the “resident” almost always wins. Importantly, despite the fact that both “residents” and “intruders” sustain wounds, only the “defeated intruder” displays analgesia, again indicating that stress is the critical trigger for analgesia.
The role of the PAG-RVM system in environmental analgesia is probably best studied in the “conditioned fear” paradigm in which animals exhibit hypoalgesia in a context previously associated with an aversive, fear-inducing event. Typically, this is tested in a distinctive chamber in which the animal previously received shock to the paws. Systematic studies by Michael Fanselow, Fred Helmstetter, and colleagues showed that hypoalgesia during conditioned fear is blocked by inactivation of the PAG or RVM and by local blockade of μ-opioid receptors, indicating that endogenous opioid peptides are engaged as part of this circuit. Importantly, the PAG-RVM system is engaged during fear by inputs from the amygdala. This structure has long recognized as important in emotional regulation, providing a clear demonstration of a top-down mechanism of pain modulation. A later section will discuss the role of the amygdala in top-down recruitment of the PAG-RVM system.
The Neurochemistry of Stress (Environmental)-Induced Analgesia Involves Opioid and Nonopioid Systems as well as Neurohormonal Mechanisms
Some forms of environmental analgesia are mediated by endogenous opioid release, whereas others are nonopioid mediated. Plasma and cerebrospinal levels of opioid peptides are elevated following acute exposure to many stressors that produce behavioral antinociception. In the cases of analgesia induced by glucoprivation, immobilization stress, and social conflict, as well as by vaginal probing, and late-stage pregnancy and parturition, analgesia can be attenuated or blocked by an opioid antagonist, demonstrating a causal role for opioid signaling in the behavior. John Liebeskind and colleagues showed that an opioid role in mediating foot shock-induced analgesia appeared to be related to a loss of controllability and the parameters of the foot shock. Prolonged, intermittent foot shocks, but not brief, continuous foot shocks, elicited analgesia that was naloxone reversible and cross-tolerant with morphine. Likewise, intermittent but not continuous cold-water swims produced analgesia that was naloxone reversible and cross-tolerant with morphine. Richard Bodnar and colleagues suggested that nonopioid analgesia can sometimes be potentiated by administration of an opioid receptor antagonist, suggesting that selective activation of one system relative to another is controlled through a process similar to Steven Kuffler’s classic model of collateral inhibition in the retina.
A common operational definition of stress involves engagement of the hypothalamo-pituitary-adrenal (HPA) axis. Some, but not all, forms of environmental analgesia, opioid and nonopioid, also require the HPA axis. Thus, analgesia elicited by prolonged, intermittent foot shock is blocked by hypophysectomy or lesions placed in the medial-basal hypothalamus, as well as adrenalectomy, dexamethasone pretreatment, and adrenal demedullation. Other opioid-mediated forms of stress-induced analgesia show a starkly different pattern, with glucoprivation-induced analgesia, for example, enhanced by medial-basal hypothalamic damage or hypophysectomy. Yet other forms of opioid-dependent analgesia, such as social defeat, are apparently entirely independent of the HPA axis. Further, some but not all forms of nonopioid environmental analgesia are dependent upon the HPA axis. Based on these patterns, Linda Watkins and David Mayer proposed that four different types of stress- and environmentally induced analgesia occur: opioid neurohormonal, nonopioid neurohormonal, opioid neural, and nonopioid neural.
Sex Differences Have Been Observed in the Central Control of Pain Inhibition in Animals
Sex differences in mu-opioid analgesia have been consistently observed in animal models with male rodents displaying greater analgesia than female rodents. This sex difference is not attributable to pharmacokinetic factors but appears to relate to the potency of opioid (μ, δ, and κ) agonists. The observations of opioid analgesic sex differences vary as a function of genotype. Thus, male Sprague-Dawley and Long-Evans rats, but not Wistar-Kyoto rats, are more sensitive to morphine analgesia than females, and some inbred mouse strains (AKR, C57BL/6, SWR) display greater effects in males as well. Ultralow doses of naltrexone respectively enhance and decrease morphine analgesia in female and male rats, and NMDA antagonism enhances morphine analgesia in male but not female rodents.
Gonadal Hormones Interact with Pain-Modulatory Systems to Mediate Analgesic Sex Differences
Sex differences in systemic and central opioid analgesia are mediated by the phase of the estrus cycle in female rodents, with the greatest degree of analgesia in the metestrus and proestrus phases and the least in the estrus phase. Removal of the reproductive organs modulates these sex differences; whereas adult gonadectomy interferes with the activational effects of gonadal hormones, neonatal gonadectomy interferes with organizational effects of gonadal hormones. Adult gonadectomy typically produced minimal alterations in systemic and central morphine analgesia in male and female animals.
By contrast, the laboratories of Theodore Cicero and Richard Bodnar demonstrated that neonatal gonadectomy produced more potent effects upon the sex differences in morphine analgesia. Thus, neonatal castration of male rat pups 1 day after birth significantly reduced subsequent adult systemic and central (vlPAG) morphine analgesia to a level similar to that of neonatal vehicle-treated females tested during the estrus phase. In contrast, neonatal treatment with testosterone propionate in female rat pups 1 day after birth significantly enhanced subsequent adult systemic and central (vlPAG) morphine analgesia to a level similar to that of sham-operated males. Activational-organizational gonadal hormone interactions were observed such that adult ovariectomy enhanced vlPAG morphine analgesia in female rats receiving neonatal testosterone treatment. Finally, intact female rats with excitatory ibotenic acid lesions placed in each of two hypothalamic (ventromedial and medial preoptic nuclei) sites involved in the central accumulation of estrogens displayed profound enhancements in the magnitude and potency of systemic morphine analgesia. In contrast, male rats or adult ovariectomized female rats with identical ibotenic acid lesions failed to display any changes in the magnitude or potency of systemic morphine analgesia.
Sex differences and sexual dimorphism are observed for analgesia induced by spinal administration of opiates such that μ-receptor activation elicits analgesia in males, but a combination of μ- and κ-opiate receptor activation is necessary to produce analgesia in females. Anatomical and physiological substrates corroborate the behavioral observations of sex differences in spinal circuits mediating analgesia and nociception. Thus, Alan Gintzler found that hormone-simulated pregnancy in female rats increases the number of spinal cells co-expressing dynorphin peptide and δ-opioid receptors. Although μ-like immunoreactivity in the lumbosacral dorsal horn was similar in males and females, a greater proportion of cytoplasmic κ-receptor labeling within axon terminals was denser in estrus and proestrus females relative to males.
Sex differences in rodents in the PAG-RVM axis mediating supraspinal opioid analgesia are controlled by organizational gonadal hormone effects. Hypothalamic enkephalinergic neurons, sensitive to changes in sex hormone levels, express enkephalin gene products in females to a greater degree than males and project to estrogen-binding PAG neurons. Anne Murphy found that male rats possess significantly greater numbers of androgen receptor-immunoreactive neurons in the PAG that project to the RVM than females.
Like systemic treatment, male rodents display greater μ-mediated analgesia following vlPAG or RVM administration than female rodents on thermal, mechanical, and inflammatory measures. Further, Anne Murphy produced corresponding anatomical data showing that although females displayed significantly more retrogradely labeled PAG-RVM output neurons than males, inflammatory pain activated more PAG-RVM cells in male than in female rats. In turn, morphine suppressed the ability of Inflammation to activate the PAG in male, but not, female rats. Further, morphine preferentially activated the PAG-RVM pathway 胺in the male rat. Finally, males displayed greater μ-opioid receptor expression in the PAG than females, and selective lesions of μ-opioid receptor-expressing PAG neurons blocked systemic morphine analgesia in males only, providing a potential central mechanism of action for morphine analgesic sex differences. Thus, the connections between hypothalamic estrogen-sensitive nuclei and the classic brain stem (vlPAG and RVM) nuclei involved in pain inhibition may reveal the anatomical underpinnings of the sex differences in opiate analgesia.
Sex Differences Are Also Observed for Opioid Tolerance and Dependence
Repeated opioid exposure reduces analgesic potency due to tolerance and, like opioid analgesia, is subject to sex differences. Ben Kest and Jeffrey Mogil found that male rats develop morphine tolerance more quickly than females, yet recover analgesic potency more rapidly after discontinuation of chronic morphine treatment. The slower onset of morphine tolerance in females is dependent upon the phase of the estrus cycle such that quicker morphine tolerance is observed in males and proestrus females but not in ovariectomized and intact females tested during metestrus, diestrus, or estrus. Activational and organizational mechanisms of gonadal hormones mediate opiate tolerance sex differences with adult gonadectomy abolishing this effect and neonatal gonadectomy yielding similar morphine tolerance in both sexes. Spinal sites of action have been implicated because intrathecal morphine administration produces quicker morphine tolerance in females.
Sex differences are also observed in opioid physical dependence. Physical dependence, an unwanted side effect of acute or chronic opioid exposure manifested by characteristic behavioral and physiological withdrawal signs, is observed more acutely in men relative to women. However, a different pattern emerges in animal studies. A common animal model of opioid dependence is naloxone-precipitated withdrawal in which the general opioid antagonist is acutely administered to an opioid-dependent animal. Male rodents display greater weight loss and behavioral withdrawal signs (wet-dog shakes, jumping) than females accompanied by a fourfold leftward shift in the ED50 for naloxone in males. This greater sensitivity in males has been attributed to organizational gonadal effects given its presence in prepubertal male mice as well as increased μ-opioid receptor density or decreased striatal or cortical dopamine and DOPAC (a dopamine metabolite) levels during withdrawal. Further, signs of spontaneous withdrawal from chronic morphine occur more quickly and last longer in male rodents.
Sex Differences in Analgesia Are Less Well Characterized in Humans
In contrast to the relatively straightforward observations of analgesic sex differences in animals, such differences are less clear in human studies and control of human clinical pain syndromes. Very often, the measures are indirect such as evaluating opioid consumption, defined by μ-opioid patient-controlled analgesia (PCA) for postoperative pain, which was higher in men than in women in 12 studies, but showed no sex differences in eight other studies. In many of these studies, sex comparisons were not the primary focus of investigation and included such confounds as underlying disease, age, opioid plasma concentrations, and the fact that opioid consumption involves other factors, including baseline pain sensitivity, onset efficacy, addiction fear, and nausea-vomiting. The μ-receptor opioids, alfentanil and morphine-6-glucuronide, induced similar magnitudes of analgesia in the two sexes, yet women experienced more pain and required more morphine than men to achieve a similar degree of analgesia. In contrast, Jon Levine found that opioid analgesics acting at the κ-opioid receptor (e.g., nalbuphine, butorphanol, pentazocine) but not morphine elicit greater and more prolonged pain relief in women specifically after dental surgery but not in other human experimental pain models. Interestingly, Jeffrey Mogil found that κ-agonist-mediated analgesia is sex-dependently modulated by MC1R, the gene that encodes melanocortin-1 receptors. In humans, variants of the MC1R gene are associated with red hair, fair skin, freckles and high chance of melanoma, and redheaded women, but not men with two or more variant alleles of the MC1R gene (and all with red hair) displayed significantly greater pentazocine analgesia, potentially mediated by melanocortin-1 receptor activation by a-MSH and dynorphin. Further specific human studies are needed to examine the possibility that human analgesic sex differences occur and are of clinical relevance.
The PAG-RVM System Facilitates, as well as Inhibits, Pain
The PAG-RVM system was initially viewed as an “analgesia system,” activated by electrical stimulation, intense stress, or opioid analgesic drugs to inhibit pain transmission. It is now recognized to be considerably more complex, with the capability of enhancing pain under certain conditions. For example, direct infusion of the neuropeptides neurotensin and cholecystokinin (CCK) in the RVM facilitates nociception. Similarly, although the effect of electrical stimulation in either the PAG or RVM is typically antinociceptive, stimulation with low currents at some sites in the RVM facilitates nociception. This indicates that the PAG-RVM system has both pain-inhibiting and pain-facilitating outputs, and suggests that the effect of the antinociceptive, inhibitory output is more potent, masking that of the pronociceptive, facilitating output when both are activated by electrical stimulation.
The pain-facilitating output from the PAG-RVM system can be activated as part of a positive feedback loop, such that “pain facilitates pain.” For example, an inflammatory stimulus localized to the lateral surface of the proximal hind limb leads to increased sensitivity to heat applied to the plantar surface of the paw of the same limb. This secondary hyperalgesia is reversed by NMDA receptor blockade in the RVM. The descending positive feedback mechanism is presumably adaptive and, like sensitization of pain-transmission pathways in the peripheral nerve and spinal cord, promotes recuperative behaviors and prevents use of injured body parts. However, the problem with any positive feedback loop is loss of control if mechanisms for terminating its action are inadequate. Indeed, there is evidence from a number of models of persistent pain states that the RVM maintains hypersensitivity. Thus, blocking the RVM reverses established hypersensitivity in acute and chronic inflammatory models, as well as following nerve injury, and in the dural inflammation model of migraine headache. BDNF (brain-derived neurotrophic factor), NMDA receptors, and the neuropeptide CCK are all implicated in RVM-mediated mechanisms of pain facilitation triggered by noxious stimulation as part of a positive feedback loop.
Interestingly, a facilitating output from the RVM contributes to “opioid-induced hyperalgesia.” This refers to a paradoxical increase in pain sensitivity triggered by either acute or chronic administration of opioid analgesic drugs, including morphine. Opioid-induced hyperalgesia has been documented in animals, human experimental subjects, and clinical populations. A number of mechanisms have been evoked, but Frank Porreca, Todd Vanderah, and colleagues have shown in animal studies that a facilitating output from the RVM contributes to behavioral hypersensitivity observed for relatively prolonged periods (days) after opioid exposure. This facilitating output must be driven by endogenous CCK release because local application of a CCK receptor antagonist in the RVM reverses the behavioral hyperalgesia. Because the RVM is also a critical site for opioid analgesia, the fact that this region mediates opioid-induced hyperalgesia highlights the dynamic balance between pain inhibition and pain facilitation exerted by the PAG-RVM system.
Perhaps not surprisingly, sex differences are observed in opioid-induced hyperalgesia, just as in previously described opioid analgesia. Female rodents display greater hyperalgesia than males following an acute morphine experience, a difference enhanced when the animals are given naltrexone, a nonselective opioid antagonist. This difference is due to activational effects of gonadal hormones because it is blocked by ovariectomy and reinstated by estrogen. In males and ovariectomized females, opioid-induced hyperalgesia involves NMDA receptor-mediated processes. In sum, whereas the analgesic effects of opioids are more pronounced in male rodents, the hyperalgesic sequelae of opioid administration are more pronounced in females. Finally, the pain-facilitating output from the PAG-RVM system is also triggered by “top-down” mechanisms, such as activation of the hypothalamus during sickness or mild stress. It is likely that the PAG, like the RVM, has some role in facilitating pain, although this region has received much less attention than the RVM. However, inhibition of cyclooxygenase, the rate-limiting enzyme in synthesis of prostaglandin, in the PAG has acute antinociceptive effects, which implies that endogenous prostanoids in the PAG are acting to facilitate pain.
Two RVM Cell Populations, “ON-Cells” and “OFF-Cells,” Are the Basis for Bidirectional Pain Control from this Region
Our understanding of the neurophysiological basis for pain modulation is most advanced for the RVM. Early electrophysiological analyses of the region revealed heterogeneous responses to noxious stimulation, with excitation, inhibition, or no response to noxious stimulation in deeply anesthetized animals. The focus of most experiments was on excitation, based at least in part on the idea of counterirritation, that is, that “pain inhibits pain.” The OFF-cell/ON-cell/NEUTRAL-cell classification, developed by Howard Fields, Mary Heinricher, and their colleagues beginning in the 1980s, was thus a significant advance in that it provided a defined framework relating RVM neuronal firing to the pain-modulating function of the region. ON, OFF-, and NEUTRAL-cells can be identified in lightly anesthetized and unanesthetized animals and are defined by changes in activity that occur beginning just before the animal responds behaviorally to a noxious stimulus. The stimulus modality (thermal, mechanical, or chemical) and the site of stimulation are not important. OFF-cells are identified by a pause in firing beginning just before an animal behaviorally withdraws from a noxious stimulus. ON-cells are defined by a burst of activity, again beginning just before the animal behaviorally withdraws from a noxious stimulus. The remaining cells in the RVM do not respond during nociceptive withdrawals or to noxious stimuli, and they are referred to as NEUTRAL-cells.
The behavior-related changes in OFF- and ON-cell activity pointed to some role for these two classes in pain modulation. Because nociceptive reflexes are inhibited when RVM neurons are activated nonselectively using electrical stimulation, the pause in OFF-cell firing just before an animal responds to a noxious stimulus suggested that OFF-cells inhibit pain and that they must stop firing in order for the pain-related information to be transmitted. By contrast, ON-cells are active just when the animal responds to the noxious stimulus, which indicated that they are not likely to inhibit pain. Subsequent work from the Heinricher laboratory using pharmacological tools to manipulate ON- and OFF-cell populations confirmed that OFF-cells suppress nociception and that ON-cells facilitate nociception. OFF-cells are the only RVM neurons activated by local infusions of μ-opioid agonists and by low doses of the GABAA-receptor antagonist, bicuculline, both of which produce behavioral antinociception. On the other hand, ON-cells, but not OFF-cells, are activated by CCK and low doses of the neuropeptide neurotensin. Selective activation of RVM ON-cells by local microinjection of these neuropeptides produces behavioral hyperalgesia. RVM OFF-cells and ON-cells thus provide a basis for bidirectional pain modulation from this region.
The organization of the RVM suggests that the neurons of each physiological class function as a whole, exerting global control over nociception, without somatotopic subdivisions. Individual RVM neurons have large, often total body, receptive fields, and project diffusely to multiple segments of the spinal cord. In awake unrestrained rats, ON- and OFF-cells respond briskly to light touch and to sudden sound, as well as to noxious inputs, and activity can be related to arousal and other behavioral states. These findings suggest that any behaviorally significant stimulus, noxious or innocuous, can modulate nociceptive processing through the PAG-RVM system.
NEUTRAL-Cells and Serotonin
The role of the RVM NEUTRAL-cells, if any, in pain modulation remains unclear. NEUTRAL-cells clearly represent an RVM population distinct from the ON- and OFF-cell classes. NEUTRAL-cells have a distinct pharmacology and are unresponsive to a range of neuromodulators that alter the firing of ON- and/or OFF-cells. Although an early report from Kenji Miki working with Ron Dubner and Ke Ren suggested that some NEUTRAL-cells develop ON-cell and OFF-cell-like properties over time, this concept has not to date garnered further support. One possible role for NEUTRAL-cells would be to mediate other aspects of RVM function (such as thermogenesis or cardiovascular control). The problem with this suggestion has been that early work, primarily from Peggy Mason and colleagues, indicated that a subset of NEUTRAL-cells were serotonergic. The significance of this is that spinal serotonin release is important in both descending facilitation and inhibition from the RVM. This suggested that at least this serotonergic subset of NEUTRAL cells has a role in pain modulation. However, Jean-Francois Bernard used juxtacellular recording techniques to sample from a large number of RVM neurons and found that serotonin is not restricted only to NEUTRAL-cells. Indeed, a substantial population of serotonergic RVM neurons has ON- and OFF-cell properties. Serotonin therefore has the potential to modulate spinal nociceptive transmission as a transmitter of the ON- and OFF-cell populations. Serotonergic NEUTRAL-cells could therefore mediate aspects of spinal serotonin function unrelated to pain.
Recruitment of OFF-Cells Suppresses Pain
Opioid Analgesic Drugs Disinhibit RVM OFF-Cells to Produce Analgesia
Activation of RVM OFF-cells is necessary and sufficient for the antinociceptive actions of opioids. OFF-cells are activated when morphine or a μ-opioid agonist is given systemically, in the PAG or locally in the RVM (Fig. 89.2). Preventing opioid activation of OFF-cells also blocks the analgesic action of these drugs. How do opioids activate OFF-cells? Although μ-opioids act postsynaptically to hyperpolarize neurons by opening potassium channels, they also act presynaptically to inhibit neurotransmitter release. The activation of OFF-cells is thus disinhibition: μ-opioids suppress GABA release responsible for the OFF-cell behavior-related pause through a presynaptic mechanism (Fig. 89.3). μ-opioids are known to suppress GABA release in the RVM, and the withdrawal-related OFF-cell pause is known to be mediated by GABA. This pause is blocked by morphine or μ-receptor agonists such as DAMGO, thus disinhibiting the OFF-cells (Fig. 89.2). The opioid-sensitive GABA input that produces the OFF-cell pause is not a local interneuron, but its source has not been identified.

OFF-cells are activated and ON-cell firing suppressed by local infusion of the μ-opioid receptor agonist DAMGO. OFF-cell (top trace) and ON-cell (lower trace) show reflex-related changes in activity and irregular spontaneous activity characteristic of these neurons. OFF-cell firing increased dramatically, and the ON-cell was almost silenced following DAMGO microinjection in the RVM. Both effects were reversed by systemically administered naloxone. Tail-flick trials, 5 min apart, are indicated by triangles below each trace, with filled symbols indicating that a reflex occurred, open triangles that there was no flick during heat application (heat was terminated at 10 s if there was no reflex response). RVM DAMGO microinjection inhibited the tail flick, and this antinociception was reversed by naloxone (Adapted with permission from Heinricher et al. (1994) Neuroscience 63:279–288)

μ-Opioid agonists have both direct and indirect actions in the RVM. ON-cells are inhibited, directly. OFF-cells are disinhibited. The OFF-cell reflex-related pause is mediated by a GABAergic input from outside of the RVM. This GABA input is inhibited by μ-receptor agonists, allowing the OFF-cells to become continuously active
μ-Opioids also suppress the firing of ON-cells (Fig. 89.2). Although removal of facilitation is not by itself sufficient to produce potent behavioral hyperalgesia under basal conditions, it likely contributes to the analgesic actions of these drugs.
It is interesting to contrast the actions of κ-agonists in the RVM with those of μ-receptor agonists. At the membrane level, in vitro studies show that κ-receptor agents hyperpolarize some RVM neurons that are presumed to be OFF-cells, but also reduce glutamatergic excitation by a presynaptic action. Functionally, κ-receptor agonists applied in the RVM are reported to produce hypoalgesia or to have no effect, but also to reverse μ-opioid analgesia. These apparently conflicting results can be explained by distinct effects on ON-cells and OFF-cells. If κ-agonists block glutamatergic excitation of ON-cells, one would expect a hypoalgesic or anti-hyperalgesic effect. If κ-agonists also inhibit OFF-cells, this would oppose activation of these neurons by μ-receptor agonists, preventing μ-mediated analgesia. The effects of κ-agonists in the RVM would thus be state dependent, attenuating or reversing hyperalgesia mediated by activation of ON-cells, but also blocking analgesia mediated by activation of OFF-cells. The actual observed effect would depend on whether ON-cells or OFF-cells were more active in the behavioral context under study.
Physiological Recruitment of RVM OFF-Cells Inhibit Pain
As already discussed, there is strong evidence that the PAG-RVM system contributes to environmentally induced changes in pain. Indeed, many forms of environmental analgesia are attenuated or blocked by lesions placed in the RVM. A specific role for OFF-cells is indicated by several observations. First, administration of a stressful foot-shock blocks the pause in firing that characterizes these neurons. Second, fear-related processes organized in the amygdala cause OFF-cells to become continuously active. The responses of these neurons to severe stress or fear thus mirror their response to opioid analgesic drugs. OFF-cells also appear to contribute to graded modulation of nociception during other biologically significant behaviors. For example, nociceptive threshold is elevated while an animal is urinating or feeding. Both behaviors may be associated with an increase in OFF-cell excitability. Interestingly, hypoalgesia during feeding is related to the degree of hunger, and when the food offered is highly palatable, the associated hypoalgesia is reversed by administration of an opioid receptor antagonist.
Recruitment of RVM ON-Cells Facilitate Pain
As described previously, pain facilitation from the RVM can be engaged by both bottom-up and top-down mechanisms. At the level of RVM neurons, such mechanisms recruit the ON-cell population. Thus, ON-cells are activated by any noxious stimulus sufficient to elicit a withdrawal response. As a result, behavioral responses to any subsequent stimuli are enhanced as long as the ON-cell population remains active (which can be for many minutes, even under anesthesia). ON-cells also exhibit prolonged activation during acute inflammation and contribute to secondary hyperalgesia. Selective pharmacological blockade of ON-cells under these conditions attenuates or blocks behavioral hyperalgesia. ON-cells are also recruited in top-down models of hyperalgesia, including sickness and mild stress, mediated by the medial preoptic area and dorsomedial nucleus of the hypothalamus, respectively.
ON-cells also contribute to chronic pain states. These neurons show enhanced sensitivity in chronic inflammation and nerve-injury models. Moreover, blocking CCK receptors in the RVM reverses hyperalgesia in these models. Because ON-cells are the only RVM neurons that are excited by the neuropeptide CCK, it is likely that the anti-hyperalgesic action of CCK receptor antagonists is mediated by ON-cells. The extent to which altered ON-cell responsiveness in chronic pain states reflects enhanced inputs from the spinal cord, inputs from other brain regions that are themselves altered in the chronic pain state, or plasticity of the RVM circuitry itself remains to be determined.
Top-Down Mechanisms Engage the PAG-RVM System
Much of the discussion thus far has emphasized how the PAG-RVM system responds to afferent noxious stimulation and local actions of analgesic drugs. However, recruitment of this system by higher structures is equally important and has the potential to explain much of the influence of cognitive and emotional factors on pain, particularly for human pain syndromes. Critical structures include the amygdala, anterior cingulate, and several nuclei of the hypothalamus.
The amygdala plays a central role in stress-induced analgesia. The amygdala has a dense reciprocal connection with the vlPAG, and the contribution of the PAG-RVM system to the analgesia observed following severe stress or fear has already been mentioned. The underlying neural circuitry has been studied intensively using the conditioned fear paradigm as a laboratory model. These studies demonstrate that μ-opioid-dependent fear-related processes organized in the amygdala play a critical role in hypoalgesia associated with conditioned fear. This conditioned hypoalgesia involves suppression of nociceptive processing at the dorsal horn, and it is blocked by lesions of the PAG or RVM. The output pathway uses endogenous opioids, since conditioned hypoalgesia is blocked by infusions of nonselective as well as selective μ- and δ2-receptor opioid antagonists in the vlPAG. Conversely, conditioned hypoalgesia is mimicked by direct infusion of morphine in the amygdala (basolateral nucleus), which interacts synergistically with opioid administration in the PAG and activates OFF-cells in the RVM.
In contrast with the “stress-induced analgesia” evoked by intense stress and fear, mild stress and anxiety can give rise to “stress-induced hyperalgesia,” consistent with the common clinical observation that everyday stress exacerbates chronic pain. The intensity of the stressor and the individual’s level of arousal are believed to interact to determine whether a particular stressor has an analgesic or hyperalgesic effect. The amygdala appears to be as important in stress-induced hyperalgesia as it is in stress-induced analgesia. The relevant output pathways mediating hyperalgesia are not as well defined as those for amygdala-mediated analgesia however. Nevertheless, the central nucleus of the amygdala likely contributes to hyperalgesia associated with chronic stress and anxiety. Thus, blocking the important stress-related peptide CRF (corticotropin releasing factor) in the central nucleus of the amygdala attenuates measures of pain intensity and aversiveness following nerve injury, and direct administration of the stress hormone corticosterone into the central nucleus produces hyperalgesia. Conversely, blocking certain metabotropic glutamate receptors in this region has an anxiolytic and anti-hyperalgesic effect that may be mediated through the RVM.
In addition to the amygdala, mild or chronic stress also recruits the dorsomedial nucleus of the hypothalamus, which projects to both the PAG and RVM. Blocking the dorsomedial hypothalamus interferes with autonomic responses to mild stress, and stimulation of this region produces not only tachycardia and hyperthermia but also hyperalgesia. That hyperalgesia has been shown to be mediated by RVM ON-cells.
The anterior cingulate cortex is another area that is strongly connected with the PAG-RVM system. Lesions placed in the cingulate cortex and associated fiber tracts have been used for decades as a treatment for intractable pain in patients. Such lesions reduce the suffering aspect of pain, with subtle, if any, effects on the sensory-discriminative component. Similar effects are seen in laboratory studies in animal models, with a reduction in pain-related affective behaviors accompanied by relatively little effect on detection thresholds. One interpretation of these findings would be that the emotional component of the pain experience is represented in anterior cingulate cortex. However, Min Zhuo and his colleagues have shown that the facilitation of pain behaviors produced by electrical stimulation in the anterior cingulate is eliminated by inactivation of the RVM. This suggests that cingulate lesions exert their effects at least in part by removing a drive to the pronociceptive output from the PAG-RVM system. Interpretation of this literature is complicated however, since some authors report a reduction in pain-related aversion following stimulation in the anterior cingulate. One possibility is that the stimulation is producing a functional lesion of the anterior cingulate by disrupting physiological firing patterns.
The medial preoptic area (MPO) is another rostral structure that can engage the PAG-RVM system to produce hyperalgesia. The MPO has long been known to mediate the “sickness response,” which includes fever, increased sleep, and aphagia as part of the adaptive response to infection. The essential elements of the sickness response are organized by prostaglandin E2 (PGE2), which is formed in the MPO following immune challenge. Direct administration of PGE2 in the MPO has now been shown to elicit hyperalgesia, as well as the classically recognized components of the sickness response. This hyperalgesia requires activation of RVM ON-cells. It has been suggested that activation of pain-facilitating systems as part of the sickness response explains the aches and myalgias associated with illness and infection, and contributes to recovery by promoting rest and recuperative behaviors.
Negative Feedback, or “Pain Inhibits Pain,” Is a Central Premise of “Diffuse Noxious Inhibitory Controls” (DNIC)
As described above, the PAG-RVM system acts as a positive feedback loop, with activation of RVM ON-cells by noxious stimulation, which enhances responses to subsequent inputs. In a sense, “pain facilitates pain” by triggering the facilitating influence from the PAG-RVM system, and as previously reviewed, this positive feedback system can be adaptive by enhancing vigilance in conditions where further injury is likely, and by promoting guarding and other recuperative behaviors. However, a different situation arises when noxious stimulation impinges simultaneously on two distinct regions of the body. In this case, “pain inhibits pain,” is a phenomenon that has long been recognized, and even employed therapeutically, in counterirritation techniques.
This inhibition of pain evoked by one stimulus when a second stimulus is applied to a part of the body innervated by a different spinal segment (thus an “extrasegmental” stimulus) has been referred to as “diffuse noxious inhibitory controls” (DNICs) by Daniel LeBars, Luis Villanueva, and colleagues. The term “DNIC” comes from the observation in animal studies that activation of a wide-dynamic range neuron in the dorsal horn by a noxious stimulus delivered to its excitatory receptive field (the “test” stimulus) can be attenuated by noxious stimulation almost anywhere else on the body (the “conditioning” stimulus). Innocuous conditioning stimuli do not have the same effect. The remote inhibition of wide-dynamic range dorsal horn neurons is mediated not by the PAG-RVM system but by a region in the caudal lateral medulla, the subnucleus reticularis dorsalis (SRD). The SRD receives direct spinoreticular input and, in turn, projects, back to the deep dorsal horn (Fig. 89.4) as well as to supraspinal sites such as ventromedial thalamus. Neurons in SRD respond to noxious stimulation delivered almost anywhere on the body and thus have the physiological properties needed to exert an inhibitory effect triggered by any noxious input. However, it is not yet known whether the counterirritation phenomenon seen in humans is explained by DNIC or by some other descending control mechanism. An international consortium of pain researchers including David Yarnitsky, Lars Arendt-Nielsen, and others has therefore recommended that counterirritation demonstrated in psychophysical studies in humans be referred to as “conditioned pain modulation” (CPM), rather than DNIC.

The SRD (subnucleus reticularis dorsalis) as a negative feedback loop. The SRD receives excitatory input from nociceptive dorsal horn neurons at all levels of the spinal cord and projects back to all levels, inhibiting activity of wide-dynamic range neurons. Ascending connections of the SRD are not shown
The physiological function of CPM is still an area of intense investigation. LeBars and colleagues suggest that remote inhibition of dorsal horn neurons serves as a contrast mechanism, enhancing the signal from one noxious stimulus at the expense of another. This inhibitory mechanism may also be important in coordinating motor responses to simultaneous noxious stimuli. Interestingly, recent studies in humans have used the efficacy of the CPM response as an index of the activity of central pain-modulating systems in different individuals. Relatively poor CPM is seen in a range of idiopathic pain syndromes and predicts the development of chronic postsurgical pain and opioid-induced hyperalgesia. Again, however, it must be emphasized that whether the CPM observed in humans reflects the “diffuse noxious inhibitory control” of wide-dynamic range neurons seen in animal studies or whether additional circuits are involved, including the PAG-RVM system and intracortical connections, remain to be determined.
Emerging Evidence for Action of Pain-Modulating Systems in Humans Includes Anatomical, Deep-Brain Stimulation, Imaging, and Psychophysical Approaches
Within a decade of the demonstration of stimulation-produced analgesia in animals, neurosurgeons documented analgesic effects of stimulating homologous areas in humans for treatment of intractable pain. The significance of this is twofold. First, it confirmed that the inhibition of nociceptive behaviors in animal studies was in fact suppression of pain and not merely confounding motor inhibition. Second, it demonstrated that the PAG-RVM circuit delineated in animals also functioned in a similar fashion in humans. Subsequent studies showed that endogenous opioid peptides are an important part of this circuit in humans, as already observed in animals and that exogenous opioids target the PAG-RVM system and its higher inputs, including the anterior cingulate cortex, to produce analgesia. Functional imaging studies indicate that the PAG-RVM system is activated in facilitated pain states, as well as during pharmacological and endogenous analgesia.
The clearest demonstration of recruitment of pain-modulating systems in humans is probably in the context of placebo analgesia. Placebo analgesia is analgesia produced by an inert procedure that the subject (or patient) has been induced to believe will be analgesic. In pharmacological studies, the placebo treatment would typically be a pill without the active ingredient. The mechanism underlying placebo analgesia is conditioned expectation of pain relief. A link between expectation and the PAG-RVM system is provided in imaging studies by Falk Eippert and colleagues. These authors found that the anterior cingulate cortex was functionally coupled to the PAG during placebo and that this coupling was opioid dependent. They also saw a decrease in pain-related activation at the spinal cord during placebo, confirming that the placebo effect involves not just cortico-cortical interactions but descending inhibition.
Interestingly, activation of the PAG is also seen in human subjects who are anticipating delivery of a painfully hot stimulus, and the PAG signal predicts the amount of pain experienced as a result of the stimulus. This highlights the complexity of interpreting functional imaging data: does activation in this case represent recruitment of a pain-facilitating output from the PAG or triggering of an attempted compensatory inhibition? Strong interpretation of these imaging data requires single-cell data from animal studies. However, CCK, implicated in RVM ON-cell activation in persistent pain states, has also been shown to be important for hypersensitivity in humans who are led to expect greater pain. This suggests that the activation seen in the functional imaging studies represents recruitment of a pain-facilitating output.
The alteration of pain experience by expectation is just one example of how cognitive and emotional factors can modulate pain sensation by engaging the PAG-RVM pain-modulating system. There is now increasing evidence that this system contributes to the effects of distraction, stress, and emotion on pain experience in humans. Experimental approaches that are firmly grounded in the animal literature and that combine functional imaging methods with sophisticated psychophysical testing are likely to provide increasing insights into the human pain experience in the coming years.
Outlook
In sum, the brain is no longer viewed as a passive receiver of a pain message relayed from damaged tissue. Transmission of signals related to pain is regulated by brain modulatory systems that can inhibit or facilitate the pain experience. These modulatory systems provide a mechanism through which prior experience, other behavioral priorities, contingencies in the physical or social environment, or stimulus history (e.g., prior injury leading to inflammation) can influence pain. Recognition of the role of centrifugal control of pain forces us to think more broadly about pain pathophysiology and suggests potential pharmacological and behavioral approaches to therapy for acute and chronic pain. The coming years should see increasing understanding of how the PAG-RVM system is altered in chronic pain states and how it is engaged by top-down mechanisms.
Abbreviations
- AS ODN:
-
Antisense oligodeoxynucleotide
- CCK:
-
Cholecystokinin
- CPM:
-
Conditioned pain modulation
- CRF:
-
Corticotropin releasing factor
- DAMGO:
-
[D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin
- DNIC:
-
Diffuse noxious inhibitory controls
- DOPAC:
-
3,4-Dihydroxyphenylacetic acid, a dopamine metabolite
- DPDPE:
-
[D-Pen2,D-Pen5]Enkephalin
- β-FNA:
-
β-funaltrexamine
- HPA:
-
Hypothalamo-pituitary axis
- 5HT:
-
5-hydroxytryptamine (serotonin)
- M6G:
-
Morphine-6-glucuronide
- MPO:
-
Medial preoptic area
- αMSH:
-
Alpha-melanocyte stimulating hormone
- norBNI:
-
Nor-binaltorphimine
- PAG:
-
Periaqueductal gray
- PGE2 :
-
Prostaglandin E2
- RVM:
-
Rostral ventromedial medulla
- SRD:
-
Subnucleus reticularis dorsalis
- vlPAG:
-
Ventrolateral periaqueductal gray
Further Reading
Bingel U, Tracey I (2008) Imaging CNS modulation of pain in humans. Physiology (Bethesda) 23:371–380
Bodnar RJ, Kest B (2010) Sex differences in opioid analgesia, hyperalgesia, tolerance and withdrawal: central mechanisms of action and roles of gonadal hormones. Horm Behav 58:72–81
Fields HL, Basbaum AI (1978) Brainstem control of spinal pain-transmission neurons. Annu Rev Physiol 40:217–248, Classic paper that helped define the field
Heinricher MM, Fields HL (2012) Central nervous system mechanisms of pain modulation. In: McMahon S, Koltzenburg M (eds) Wall and Melzack's Textbook of Pain, 6th edn. Elsevier, London
Heinricher MM, Tavares I, Leith JL, Lumb BM (2009) Descending control of nociception: specificity, recruitment and plasticity. Brain Res Rev 60:214–225
Le Bars D (2002) The whole body receptive field of dorsal horn multireceptive neurones. Brain Res Rev 40:29–44
Lovick TA (1997) The medullary raphe nuclei: a system for integration and gain control in autonomic and somatomotor responsiveness? Exp Physiol 82:31–41
Melzack R, Wall PD (1965) Pain mechanisms: a new theory. Science 150:971–979, Classic paper that helped define the field
Yaksh TL, Rudy TA (1978) Narcotic analgestics: CNS sites and mechanisms of action as revealed by intracerebral injection techniques. Pain 4:299–359, Classic paper that helped define the field
Yarnitsky D (2010) Conditioned pain modulation (the diffuse noxious inhibitory control-like effect): its relevance for acute and chronic pain states. Curr Opin Anaesthesiol 23:611–615
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this entry
Cite this entry
Bodnar, R., Heinricher, M.M. (2013). Central Mechanisms of Pain Suppression. In: Pfaff, D.W. (eds) Neuroscience in the 21st Century. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-1997-6_102
Download citation
DOI: https://doi.org/10.1007/978-1-4614-1997-6_102
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-1996-9
Online ISBN: 978-1-4614-1997-6
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences