
Abstract
High-intensity afferent input, tissue injury and inflammation, and injury to the peripheral nerve will initiate pain states with characteristic psychophysical properties. As will be considered below, this information processing can be modified to change the content of the message generated by a given stimulus to enhance the pain state (e.g., produce hyperalgesia), normalize a hyperalgesic state, or produce a decrease in pain sensitivity (e.g., produce analgesia). Management of that pain state is addressed by the use of agents or interventions which though specific targets at the level of the sensory afferent, the spinal dorsal horn or at higher-order levels (supraspinal) modify the contents of the sensory message generated by that physical stimulus. The important advances in the development of pain therapeutics have reflected upon the role played by specific underlying mechanisms which regulate these events.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Background
High-intensity afferent input, tissue injury and inflammation, and injury to the peripheral nerve will initiate pain states with characteristic psychophysical properties. As will be considered below, this information processing can be modified to change the content of the message generated by a given stimulus to enhance the pain state (e.g., produce hyperalgesia), normalize a hyperalgesic state, or produce a decrease in pain sensitivity (e.g., produce analgesia). Management of that pain state is addressed by the use of agents or interventions which though specific targets at the level of the sensory afferent, the spinal dorsal horn or at higher-order levels (supraspinal) modify the contents of the sensory message generated by that physical stimulus. The important advances in the development of pain therapeutics have reflected upon the role played by specific underlying mechanisms which regulate these events. The aim of this overview chapter is to provide a context for the more detailed discussion of analgesics and their actions, which occur in accompanying chapters.
Overview of the Psychophysics of Nociception
Acute Stimulation
Transient thermal or mechanical stimulus of an intensity as to potentially yield injury evokes an escape response and an autonomic reaction (increased blood pressure and heart rate). The functional phenotype typically has four characteristics. (i) The response magnitude or pain report covaries with stimulus intensity. (ii) Removal of the stimulus immediately terminates the sensation and/or attendant behaviors. (iii) The sensation/ behavior is referred specifically to the site of stimulation, for example, it is somatotopically delimited, typically to the dermatome to which the stimulus is applied and the response, for example, the stimulated paw is the paw that is withdrawn. (iv) Often with an acute stimulus (such as a thermal probe), there are two perceived components to the aversive sensation: an immediate sharp stinging sensation followed shortly by a dull throbbing sensation (Fig. 1.1).

Schematic displays the defining psychophysical properties that characterize the pain report after an acute high-intensity stimulus (left) and that after a local tissue injury (right). As indicated, in the inset plotting pain score vs. stimulus intensity, with the acute stimulus, the pain report for a given displays a threshold above which there is a monotonic increase in the magnitude of the reported pain state. After a tissue injury, there is an ongoing pain, and a stimulus applied to the injury site reveals that there is a greater pain report for a given stimulus (e.g., a primary hyperalgesia). In addition, it is appreciated that an innocuous mechanical stimuli applied to an adjacent uninjured area yields an enhanced response referred to secondary tactile allodynia
Tissue Injury
The psychophysics of pain associated with a tissue injury and inflammation has several distinct psychophysical elements that distinguish it from the events initiated by an acute high-intensity stimulus (Fig. 1.1):
-
(i)
With local tissue injury (such as burn, abrasion, or incision or the generation of a focal inflammatory state as in the joint), an acute sensation is generated by the injuring stimulus which is followed by an ongoing dull throbbing aching sensation which typically referred to the injury site—skin, soft tissue, or joint—and which evolves as the local inflammatory state progresses.
-
(ii)
Application of a thermal or mechanical stimulus to the injury sited will initiate a pain state wherein the pain sensation is reported to be more intense than would be expected when that stimulus was applied to a non-injured site. That is to say, as shown in Fig. 1.1, the stimulus response curve is shifted up (e.g., an ongoing pain) and to the left. This lowered threshold of stimulus intensity required to elicit an aversive response to a stimuli applied to the injury site is referred to as primary hyperalgesia. Thus, modest flexion of an inflamed joint or moderate distention of the gastrointestinal (GI) track will lead to behavioral reports of pain.
-
(iii)
Local injury and a low-intensity stimuli applied to regions adjacent to the injury may also produce a pain condition, and this is referred to as 2° hyperalgesia or allodynia. Thus, light touch may be reported as being aversive and is referred to as tactile allodynia.
These examples of “sensitization” secondary to local injury and inflammation are observed in all organ systems. Common examples would be sunburn (skin inflammation) leads to extreme sensitivity to warm water, inflammation of the pleura leads to pain secondary to respiration, and eyelid closure is painful secondary to corneal abrasion [1, 2].
In the case of inflammation of the viscera, the ongoing pain sensations are typically referred to specific somatic dermatomes. Thus, cardiac ischemia is referred to the left arm and shoulder, while inflammation of the bowel is associated with ongoing pain and hypersensitivity to light touch applied to the various quadrants of the abdomen. Such “referred” pain states reflect the convergence of somatic and visceral pain systems [3].
Nerve Injury
As described by Silas Weir Mitchell in 1864, frank trauma leads to two identifying elements: ongoing dysesthetic pain typically referred to the dermatome innervated by the injured nerve and prominent increase in the sensitivity to light touch applied to these regions. Injury to the nerve may be initiated by a wide variety of physical (extruded intervertebral disc compression section), toxic (chemotherapy), viral (postherpetic neuralgia: HIV), and metabolic (diabetes). In most of these syndromes, these two elements are expressed to varying degrees [4].
Encoding of Acute Nociception
As outlined in Fig. 1.2, the systems underlying these effects of acute high-intensity stimulation may be considered in terms of the afferents, the dorsal horn, and projection components. Under normal conditions, activity in sensory afferents is largely absent. However, peripheral mechanical and thermal stimuli will evoke intensity-dependent increases in firing rates of lightly myelinated (A∂) or unmyelinated (C) afferents. Based on differential blockade, these two fiber types, differing markedly in conduction velocity, are thought to underlie the acute sharp pain and subsequent dull throbbing sensation, respectively.

This schematic provides an overview of the organization of events that initiate pain state after an acute high-intensity stimulus applied to the skin. (a) A physical stimulus activates channels such as the TRP channels on the terminal of small diameter afferents (light line). (b) There are two classes of afferents: large low-threshold afferents (Ab: dark line) and small high-threshold afferents (A∂/C: light line). As the stimulus intensity increases, there is a monotonic increase in the discharge rate of each class of afferents with the low-threshold afferents showing an increase at low intensities, whereas the high-intensity afferents show an increase at higher intensities. The low and high threshold afferents project respectively into the deep and superficial dorsal horn. (c) The afferent input leads to depolarization of these spinal afferent terminals (d) which release excitatory transmitters yielding an (e) intensity-dependent depolarization of the second-order neuron. (Shown here is an example of the response of a neuron receiving convergent input from high and low threshold afferents.) Populations of these neurons project into the contralateral ventrolateral pathways to project to higher centers. (f) Broadly speaking, there are two classes of outflow. There are those which project into the somatosensory (ventrobasal) thalamus which then sends projection to the somatosensory cortex. A second type of projections goes to more medial thalamic regions and sends projection into areas of the old limbic forebrain (e.g., anterior cingulate/inferior insula)
Transduction Channels and Afferent Terminal Activation
The transduction of the physical stimulus is mediated by specific channels which increase their conductance when certain stimulus properties are present. Channels vary in the range of temperatures which activate them, ranging from hot (such as the TRPV1) to cool to cold (TRPM1). The acute response properties of the afferent are thus defined by the collection of transducer channels that are expressed on its terminals. Activation of these channels increases inward sodium and calcium currents and progressive depolarization of the terminal (Fig. 1.2a) [5, 6].
Chemical Sensitivity of Temperature Channels
An important element regarding these channels is that while they are to varying degrees temperature sensitive, they also show sensitivity to specific chemicals. Thus, the TRPV1 responds to capsaicin, while the TRPM1 responds to menthol. Accordingly, when these agents are applied to the tongue, the sensation associated with their application corresponds with the sensation produced by the fibers which normally activate these fibers, hot and cool, respectively [7].
Action Potential Generation
Peripheral terminal depolarization leads to activation of the voltage-gated sodium channels which then leads to action potentials in the respective afferent. Subtypes of sodium channels (designated as NaV 1.1 through NaV 1.9 channels) have been identified.
These channels differ in terms of their activation properties as well as their pharmacology (e.g., tetrodotoxin sensitive or insensitive) and their distribution. Thus, some channels may be found principally on unmyelinated afferents (Nav 1.7) or distributed widely on all types of excitable membranes ranging from myocytes to brain neurons to a variety of afferents. Importantly, the frequency of afferent discharge is proportional to terminal depolarization which is proportional to stimulus intensity (Fig. 1.2b) [8, 9].
Encoding Properties of Primary Afferents
There are three important properties that define the encoding properties of any given class of primary afferents (Fig. 1.2b) [10]:
-
(i)
Under resting conditions, the primary afferent, whether A or C, shows little or no spontaneous activity.
-
(ii)
Primary afferents typically begin to respond to their respective stimulus modality (e.g., Aβ-tactile or C-thermal/mechanical/chemical) at some minimal intensity (e.g., threshold).
-
(iii)
Above threshold, the frequency of firing evoked in the afferent axon will be proportional to stimulus intensity over a range of intensities. “Low-threshold” afferents will typically discharge at intensities that are considered to be nonnoxious. “High-threshold” or nociceptive afferents will discharge at intensities that are considered to be aversive in character.
Afferent Synaptic Transmission
Afferent action potentials invade the spinal terminal and depolarize these terminals. Such activation opens voltage-sensitive calcium channels which activate a variety of synaptic proteins which mediate the mobilization of synaptic vesicles and thereby initiate transmitter release.
Calcium Channels and Afferent Transmitter Release
There are a variety of voltage-sensitive calcium channels that regulate terminal transmitter release (referred to as CaV channels). These channels are distinguished on the basis of their voltage sensitivity, their location, and the agents which block them. The best known of these channels are the N-type calcium channel blocked by the therapeutic agent ziconotide. Block of this channel will block the release of many afferent terminal transmitters [11].
Spinal Afferent Terminal Transmitters
Sensory afferent uniformly releases excitatory transmitters. In terms of transmitters (Table 1.1), virtually all afferents contain and release the excitatory amino acid glutamate. Small afferent releases not only glutamate but also one or more of several peptides, such as substance P or calcitonin gene-related peptide (CGRP) (Fig. 1.2c). These transmitters in turn act postsynaptically upon eponymous receptors present on several of populations of dorsal horn neurons (Fig. 1.2d) [12]:
-
(i)
Glutamate exerts the primary depolarizing effect through the activation of the AMPA receptor which leads to a short lasting increase in sodium conductance yielding a potent and short lasting depolarization of the membrane.
-
(ii)
Substance P acts upon a G protein-coupled receptor that leads to a long slow depolarization of the membrane. Importantly, such receptors lead not only to a depolarization of the membrane, but to an increase in intracellular calcium, the glutamate by activating voltage-sensitive calcium channel and the NK1 by mobilizing intracellular calcium stores.
Laminar Organization of Spinal Dorsal Horn
The spinal dorsal horn is organized transversely in laminae (Rexed laminae), ranging from the most superficial dorsal horn marginal layer (lamina I), the substantia gelatinosa (lamina II), and the deeper nucleus proprius (laminae III–VI) [13].
Primary Afferent Projections
There are several important principles reflecting the pattern of termination of afferent terminals in the dorsal horn (Fig. 1.3) [14]:

Schematic presenting the spinal cord in transverse section (top) and horizontal (bottom) and showing: (i) (left) the ramification of C fibers in the superficial dorsal horn (laminae I and II) and collateralization into the tract of Lissauer and (ii) (right) the ramification of A fibers in the dorsal horn (terminating in the deep dorsal horn) and collateralization rostrocaudally into the dorsal columns and at each segment into the dorsal horn. The densest terminations are within the segment of entry. There are less dense collateralizations into the dorsal horns at the more distal spinal segments. This density of collateralization corresponds to the potency of the excitatory drive into these distal segments. Thus, distal segments may receive input from a given segment, but the input is not sufficiently robust to initiate activation of the neurons in the distal segment under normal circumstances
-
(i)
Small afferents (A∂/C) terminate superficially in the lamina I (marginal layer) and lamina II. In contrast, the large afferents (Aβ) project deep into the dorsal horn and curve upwards to terminate just deep to lamina III.
-
(ii)
Observing the spinal cord from the dorsal surface, it is noted that the central processes of the afferents collateralize, sending processes rostrally and caudally up to several segments into the dorsal columns (large afferents) or in the tract of Lissauer (small C-fiber afferents). Periodically, these collaterals send sprays into the dorsal horn at distal segments. Thus, neurons up to several segments distal to a given root entry zone of any given segment will receive afferent input from a given root (e.g., the L5 root will make synaptic contact with dorsal horn cells as far rostral as spinal segment L1). Importantly, the primary excitation occurs at the level of entry where the synaptic connections are strongest. At more distal segments, the degree of excitation from the proximal root is progressively reduced.
Dorsal Horn Neurons
Based on the organization of afferent termination, one can appreciate that superficial lamina I marginal neurons are primarily activated by small, high-threshold afferent input; hence they are “nociceptive specific.” In contrast, the deeper lying cells have their cell bodies in lamina V and are hence called lamina V neurons but send their dendrites up into the superficial laminae. Interestingly, they receive input from Aβ (low-threshold) input on their ascending dendrites and C-fiber (high-threshold) input on their distal terminals (Fig. 1.4). Accordingly, these cells with their convergent input show activation at low intensities (mediated by the Aβ input) and increasing activation as the intensity rise (mediated by the C-fiber input). Accordingly, as shown in Fig. 1.2e, the cell shows increasing discharge rates over the range from very low to very high-threshold stimuli. Accordingly, these cells are referred to as wide-dynamic-range (WDR) neurons [15].

Schematic displays (left) two principal classes of second-order neurons. As indicated, small afferents tend to terminate superficially (laminae I and II), while large afferents tend to project deep into the dorsal horn and terminate below lamina II. Accordingly, cells lying in lamina I (marginal layer) receive largely high-threshold input. Cells lying deeper (lamina V) received input from large afferents on their proximal dendrites and can receive excitatory input directly or through excitatory interneurons on their distal dendrites. (right) Single-unit recording from spinal dorsal horn, showing firing pattern (impulses/s) of a (top) high-threshold (nociceptive-specific marginal) neuron and (bottom) a horn wide-dynamic-range neuron, (WDR) located primarily in lamina V in response to graded intensities of mechanical stimulation (brush, pressure, pinch, squeeze) applied to the receptive fields of each cell. Both cells project supraspinally. Note the relationship between firing patterns and the response properties of the afferents with which each cell makes contact
Dorsal Horn Projections
These lamina I and lamina V neurons then project via the ventrolateral tracts to higher centers and thence to cortical levels. Projections may occur ipsilaterally or contralaterally in the ventrolateral tracts. Ipsilaterally projecting axons typically project to terminate in the medial brainstem reticular nuclei. Cells receiving these projections then project to the thalamus. Contralateral axons project into several thalamic nuclei [13, 16].
Supraspinal Organization
The supraspinal projections can be broadly classified in two motifs (Fig. 1.2e) [13]:
-
(i)
Dorsal horn, ventrobasal thalamic complex-somatosensory cortex. This is the classic somatosensory pathway. In these cases, the nervous system undertakes to maintain a specific intensity-, spatial-, and modality-linked encoding of the somatic stimulus, as summarized in Fig. 1.2. This pathway possesses the characteristics that relate to the psychophysical report of pain sensation in humans and the vigor of the escape response in animals. In the absence of tissue injury, removal of the stimulus leads to a rapid abatement of the afferent input and disappearance of the pain sensation. At all levels, the intensity of the message is reflected by the specific populations of axons which are activated and by the frequency of depolarization: the more intense the stimulation, the more frequent is the firing of the afferent; the greater is the dorsal horn transmitter release, the greater is the evoked discharge and the higher is the frequency of firing in the ascending pathway.
-
(ii)
Dorsal horn-medial thalamus-limbic cortex. Here, there appears to be little precise anatomical mapping. Cells in this region project to regions such as the anterior cingulate cortex or inferior insula. The anterior cingulate is part of the older limbic cortex and is believed to be associated with emotional content.
The above subdivision reflects the orthogonal component of the pain experience, notably the “sensory-discriminative” components (“I hurt here on a scale of 1–10, 6”) and the “affective-motivational” component of the pain pathway (“I have cancer, I am mortal”) as proposed by Ronald Melzack and Kenneth Casey.
Encoding of Nociception After Tissue Injury
As reviewed above, with tissue injury, a distinct pattern of aversive sensations is observed. The psychophysical profile noted with injury or inflammation is composed of (i) an ongoing sensory experience that is described as dull throbbing aching ongoing pain, (ii) enhanced responsiveness to subsequent stimulation (e.g., hyperalgesia/tactile allodynia), and (iii) secondary pain referral (e.g., sensations which are aversive when applied to adjacent uninjured areas).
Peripheral Changes in Afferent Transmission Resulting from Tissue
As described in Fig. 1.1, in the event that a stimulus leads to a local injury, as in a tissue crush (trauma) or an incision, such stimuli may lead to the subsequent local elaboration of active products that directly activate the local terminals of afferents (that are otherwise silent) innervating the injury region and facilitating their discharge in response to otherwise submaximal stimuli. This then leads to an ongoing afferent barrage and enhanced response to any given stimulus (e.g., peripheral sensitization) (Fig. 1.5).

This schematic provides an overview of the organization of events that initiate pain state after a tissue injuring stimulus of the skin. (a) Local tissue injury leads to the initiation of an innate immune response that yields the release of a variety of active factors. The factors acting through eponymous receptors on the terminals of C fibers lead to an activation of the C fiber and a state of sensitization. Accordingly, such products initiate an ongoing activity and an enhanced response to an otherwise innocuous stimulus. (b) The injury thus leads to an ongoing activity in small afferent. (c) The ongoing activity activates dorsal horn neurons and initiates a state of facilitation (windup). (d) The ongoing afferent traffic and injury products lead to a change in the tropic functions of the dorsal root ganglion leading to changes in protein synthesis and the expression of various receptors and channels which serve to enhance afferent responsiveness. (e) In the dorsal horn, the ongoing afferent drive initiates additional changes related to the activation of microglia and astrocytes as well as the invasion of typically nonneuronal cells including neutrophils and lymphocytes in the extreme. The net effect is to enhance the outflow initiated by any given stimulus, for example, hyperalgesia and allodynia. (f) With facilitation, the wide dynamic range neurons (WDR) are activated in response to stimuli that would normally not activate these neurons.[AW1]
Origin of Ongoing Activity and Enhanced Terminal Responsiveness After Tissue Injury
The source of these active factors may be considered in terms of their source including the following (Fig. 1.5):
-
(i)
Damaged cells which yield increased extracellular contents (potassium).
-
(ii)
Products of plasma extravasation (clotting factors, cellular products such as platelets and erythrocytes which release products including amines (5HT), peptides (bradykinin), and various lipidic acids (prostaglandins)).
-
(iii)
Innate immune cascade wherein given the chemoattractants present in the injury site, there will be a migration of inflammatory cells including neutrophils and macrophages. These contribute products such as myeloperoxidases, cytokines (TNF/IL1β), nerve growth factors (NGF), and serine proteases (trypsin).
-
(iv)
Terminal of primary afferent C fibers activated by the local milieu will lead to a local release of sP and CGRP which respectively cause vasodilation (erythema) and capillary leakiness (e.g., tissue swelling).
Importantly, these products have several effects on terminal function that are dependent upon the presence of the eponymous receptors on those terminals (e.g., trypsin activates proteinase-activated receptors: PARs; TNF) and the concentrations of the ligand (Fig. 1.6) [17].

This schematic provides an overview of the organization of events that initiate pain state after an injury to soft tissue. In the face of tissue injury, a variety of active products are released from local tissues, inflammatory cells, and the blood. These products exert a direct effect upon the small afferent terminal, free nerve endings, through specific receptors on the terminal. These receptors are coupled through a variety of second messengers which can lead to a local depolarization because of increased sodium and calcium influx. This leads to the activation of voltage-sensitive sodium channels (NaV) that initiate the regenerative action potential. In addition, the kinases and the increased intracellular calcium can initiate phosphorylation (PK) of channels and receptors, leading to an enhanced responsiveness of these channels and receptors. The net effect is to initiate an ongoing activity after the injuring stimulus has been removed and an increase in the discharge arising from any given stimulus
-
(i)
Activate the sensory terminal, increase intracellular calcium, and initiate a conducted action potential.
-
(ii)
Activate terminal kinases which serve to phosphorylate many membrane channels (e.g., sodium channels) and receptors (TRPV receptors) to increase their excitability. These actions are generally considered to result in spontaneous “afferent discharges” and to an enhanced responsiveness of the terminal to subsequent stimuli manifested by a left shift in the stimulus response curve for the sensory afferent. Overall, these properties are consistent with an ongoing pain stimulus and the ability of a given stimulus applied to that afferent in innervating the injured tissue to show a greater response (Fig. 1.2a).
It should be noted that these events are ubiquitous. This scenario has been demonstrated in numerous body systems, for example, cornea of the eye (sensitivity to light touch after abrasion), joint (pain of modest movement after inflammation of the knee), tooth pulp (sensory experience of cardiac-induced pressure changes in the tooth after inflammation of the pulp), and migraine (activation of the meningeal afferents which, like those in the tooth pulp, are not activated by normal mechanical movement or vascular pulsation). Indeed, think of any disease pathology described by the suffix “-itis” (Fig. 1.6).
Spinal Changes in Afferent Transmission Resulting from Tissue Injury
As reviewed above, acute activation of small afferents by extreme stimuli results in a spinal activation of dorsal horn neurons, the magnitude of which is proportional to the frequency (and identity) of the afferent input (Fig. 1.2e). Factors increasing that input-output relationship will cause a given stimulus to appear more intense (e.g., hyperalgesia). Conversely, factors reducing that function will cause a more intense stimulus to be encoded as less intense (e.g., analgesia). In the preceding section, it was appreciated that inflammation causes an enhanced response at the peripheral level. It is appreciated that there is also an enhanced response mediated in the spinal dorsal horn.
Central (Spinal) Facilitation
Animal research has demonstrated that repetitive afferent activation causes dorsal horn wide-dynamic-range (WDR) neurons to show evident signs of facilitation, labeled “windup” by Lorne Mendell and Patrick Wall (Fig. 1.4). This facilitation is characterized by following properties [18]:
-
(i)
High-frequency repetitive stimulation of C (but not A) fibers results in a progressively facilitated discharge of the WDR neurons.
-
(ii)
The receptive field of the WDR neuron showing windup was significantly expanded acutely following the conditioning afferent stimulation, for example, stimulation of an adjacent dermatome which hitherto did not activate that cell, would now lead to activity in that neuron (Fig. 1.7).
Fig. 1.7 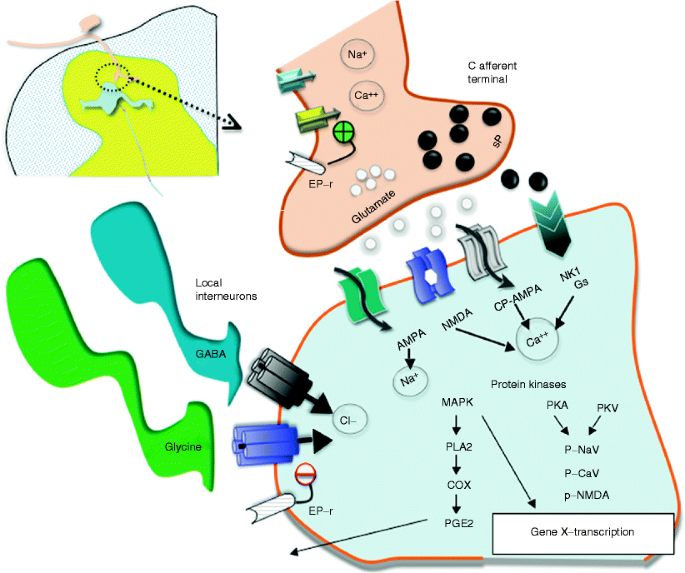
This schematic provides an overview of the organization of the events transpiring at the level of the first-order synapse. (i) As indicated, the presynaptic effects of depolarization lead to opening of voltage-sensitive calcium and sodium channels with increases in intracellular sodium and calcium and mobilization and release of transmitters (sP and glutamate). (ii) These act upon eponymous receptors (see text), leading to depolarization and increase in intracellular calcium. (iii) Activation of kinases which phosphorylate a variety of channels and receptors activates intracellular enzyme cascades such as for PLA2 and increasing gene transcription. (iv) Release of products such as prostanoids (PGE2) which can act upon the local membrane through their eponymous receptors (EP-r) where presynaptically they enhance the opening of voltage-sensitive calcium channels and postsynaptically reduce the activity of glycine receptors. (v). As indicated in addition, the first-order synapse is regulated by inhibitor interneurons such as those release GABA and glycine. These interneurons can be activated by afferent collaterals and by descending pathways to downregulate the excitability of this synapse

Enhanced Response of WDR Neuron
The enhanced responsiveness of the cell was shown by intracellular recording to reflect a progressive and sustained (after termination of the stimulation) excitability of the neuron of the cell, rendering the membrane increasingly susceptible to even weak afferent inputs.
The enlarged receptive field can be explained by the ability of subliminal input coming from afferent input arising from an adjacent non-injured receptive field which was otherwise insufficient to activate a normally excitable cell.
Pharmacology of Central Facilitation
The enhanced excitability of dorsal horn neurons secondary to repetitive small afferent input reflects a series of complex mechanistic motifs that have a diverse pharmacology which will be briefly reviewed below. These can be broadly considered in terms of those systems which are (i) postsynaptic to the primary afferent and (ii) mediated by local neuronal networks, extraspinal networks, and nonneuronal networks. Examples will be reviewed below (Fig. 1.8).

Schematic presents the spinal cord in horizontal section (see Fig. 1.3). Receptive field of dorsal horn neuron depends upon the origin of its segmental input and the input from other segments, which can activate it. Thus, neuron 1 receives strong input from RF1 and very weak (ineffective) input from RF2. After injury in receptive field (RF) 1, neuron 1 becomes “sensitized.” Collateral input from RF2 normally is unable to initiate sufficient excitatory activity to activate neuron 1, but after sensitization, RF2 input is sufficient. Now, the RF of neuron 1 is effectively RF1 + RF2. Thus, local injury can by a spinal mechanism leads acutely to increased receptive fields such that stimuli applied to a non-injured RF can contribute to the post-tissue injury sensation
Primary Afferents
Small afferents release peptides (e.g., sP/CGRP) and excitatory amino acids (glutamate) which evoke excitation in second-order neurons through their eponymous receptors (Table 1.2; Fig. 1.7) [19–22].
-
(i)
AMPA. Activation of the AMPA receptor leads to a short lasting but prominent increase in sodium conductance, yielding a robust, transient depolarization. Direct monosynaptic afferent-evoked excitation is largely mediated by the AMPA receptors, for example, AMPA receptor antagonists will block most acute excitatory input and produce an acute analgesia. A subtype of AMPA receptor is Ca permeable. For example, activation of these receptors leads to large increases in intracellular calcium.
-
(ii)
NMDA. NMDA is a glutamate-activated ionophore that passes sodium and calcium. At normal resting membrane potential, the NMDA receptor is blocked by a magnesium ion. In this condition, occupancy by glutamate will not activate the ionophore. If there is a modest depolarization of the membrane (as produced during repetitive stimulation secondary to the activation of AMPA (glutamate) and neurokinin 1 (NK1) (substance P) receptors), the Mg block is removed, permitting glutamate to now activate the NMDA receptor. When this happens, the NMDA channel permits the passage of Ca. Accordingly, block of the NMDA receptor has no effect upon acute activation but will prevent windup.
-
(iii)
NK1 and CGRP. For sP and CGRP, excitation is through G protein-coupled receptors, neurokinin 1 (NK1) and CGRP, the effects of which are cAMP dependent and couple through the activation of phospholipase C. Activation of these receptors leads to slow, relatively long-lasting membrane depolarization accompanied by an increase in intracellular calcium. Agents which block the NK1 or CGRP receptor will produce minor effects upon the behavior evoked by acute excitation but will reduce the onset of the facilitated state and behaviorally defined hyperalgesia.
-
(iv)
Growth factors. In addition to classic transmitters, growth factors such as brain-derived nerve growth factor (BDNF) is synthesized by small DRGs and released from spinal terminals, packaged in dense-cored vesicles, and transported within axons into terminals in the dorsal horn of the spinal cord. BDNF has potent sensitizing effect on spinal neurons mediated through TRK receptors.
As noted, with ongoing afferent drive, a progressive increase in excitation is noted. Aside from activation of the NMDA receptors, other components to this facilitatory process can be noted. These can be broadly considered in terms of those systems which are local to the neuronal networks in the dorsal horn, extraspinal networks, and nonneuronal networks. Several examples of each will be reviewed below.
Postsynaptic to the Primary Afferents
Repetitive activation of the primary afferent yields membrane depolarization and a significant increase in intracellular calcium. The increased intracellular calcium activates a series of intracellular cascades. Several examples are given below (Fig. 1.6):
-
(i)
Activation of kinases. Persistent afferent input leads to a marked increase in intracellular Ca++ which leads to activation of a wide variety of phosphorylating enzymes, including protein kinase A and C, calcium calmodulin-dependent protein kinases, as well as mitogen-activated kinases (MAPKs) including p38 MAP kinase and ERK. Each of these kinases leads to a variety of downstream events which serve to increase the excitability of the neuron [23, 24].
-
(ii)
Channel phosphorylation. The excitability of many channels is controlled by phosphorylation. Several examples may be cited. (1) PKA- and PKC-mediated phosphorylation of the NMDA ionophore leads to a facilitated removal of the Mg++ block and an increase in calcium current. (2) P38 leads MAPK activation to activation of phospholipase A2 (PLA2) which initiates the release of arachidonic acid and provides the substrate for cyclooxygenase (COX) to synthesize prostaglandins. In addition, this MAPK activates transcription factors such as NFKβ, which in turn activates synthesis of a variety of proteins, such as the inducible cyclooxygenase, COX2. Spinal P38 MAPK inhibitors thus reduce acutely initiated hyperalgesia and reduce the upregulation of COX2 otherwise produced by injury [23, 24].
-
(iii)
Lipid cascades. A variety of phospholipases, cyclooxygenases, and lipoxygenases are constitutively expressed in the dorsal horn in both neuronal and nonneuronal cells. Lipid products including prostaglandins and other eicosanoids are synthesized and released after small afferent input. They serve to enhance the opening of voltage-sensitive calcium channels, augmenting afferent transmitter release. In addition, prostaglandins act postsynaptically to reduce glycine-mediated inhibition on second-order dorsal horn neurons. Such reduction in glycine or GABA interneuron activity leads to an increase in dorsal horn excitability (to be discussed further below). Spinal delivery of PGE will increase, while PLA2 or COX inhibitors will reduce, injury-induced hyperalgesia [25, 26].
-
(iv)
Nitric oxide synthase (NOS). The neuronal and inducible forms of NOS are found in the spinal cord, and NO plays a facilitatory role, acting presynaptically through cGMP to enhance transmitter release. Spinal NOS inhibitors reduce post-tissue injury hyperalgesia [27].
Local Interneuronal Networks
The spinal dorsal horn has many local interneuronal circuits which are activated by primary afferent input:
-
(i)
These interneurons may contain and release glutamate to act upon AMPA and NMDA receptors and are intrinsically excitatory. This polyneuronal chain can enhance the excitatory drive from a given afferent.
-
(ii)
In addition, there are a wide variety of local interneurons which contain and release inhibitory amino acids such as GABA and glycine which act respectively on GABA A receptors and glycine receptors which are chloride ionophores that serve typically to downregulate the excitability of the membrane. These interneurons may project onto primary afferent terminals (presynaptic) and onto higher-order neurons (postsynaptic inhibition). The net excitatory outflow from the dorsal horn depends upon this local inhibitory regulation. Anything that increases that activity will diminish outflow, while events that inhibit the functionality of these inhibitory circuits will increase excitatory outflow.
As noted above, second-order deep dorsal horn neurons can receive excitatory input from large (Aβ) afferents. In spite of this afferent input onto dorsal horn neurons which are believed to play a role in nociceptive processing, this Aβ input will not typically evoke a pain state. However, after tissue injury such low-threshold mechanical stimuli may initiate a pain state (tactile allodynia). An element of this transition is believed to reflect a loss of local GABA or glycine inhibition. Thus, block of spinal GABA A and glycine receptors yields a markedly enhanced response of these WDR neurons to Aβ input and a behaviorally defined tactile allodynia. As noted above, repetitive small afferent input leads to a dorsal horn release of PGE2 which in turn reduces glycine-mediated opening of the glycine receptor and leads to a reduction in this local inhibition. The net effect is a corresponding increase in excitation evoked by low-threshold afferents.
Bulbospinal Systems
Serotonergic pathways (arising from the midline raphe nuclei of the medulla) project into the spinal dorsal horn. The effects of this bulbospinal projection are mediated by the presence of a variety of dorsal horn 5HT receptors. Some are inhibitory (5HT1a,b), and some are directly excitatory (5HT 2,3,7). The net effect is complexly defined by the nature of the neurons upon which the receptor is located. Inhibitory receptors on an excitatory neuron will lead to an inhibition of excitation. Conversely, inhibition of an inhibition will lead to an excitation.
The most prominent effect however appears to be a net increase in excitability mediated by 5HT3 bearing dorsal horn neurons. A particularly interesting circuit involves the observation that lamina I (marginal) neurons project into the medullary brainstem to activate these bulbospinal serotonin neurons to activate deep dorsal horn neurons through the 5HT3 receptor. This spino-bulbo-spinal feedback pathway is believed to play an important role in afferent-driven spinal facilitation (Fig. 1.9) [28].

Schematic shows bulbospinal 5HT arising from caudal raphe projects to the dorsal horn to synapse on 5HT3 cells and enhance excitability. This pathway may be activated by projections from lamina I neurons projecting to the raphe resulting in a spino-bulbo-spinal positive feedback loop
Nonneuronal Cells
Within the spinal parenchyma, there are a variety of nonneuronal cells. These include (i) astrocytes which arise from a multipotent neural stem cells, (ii) monocyte-derived cells (e.g., macrophages) which enter the nervous systems around parturition to become resident microglia, and (iii) circulating cells which enter the nervous systems during the course of peripheral injury and inflammation (neutrophils, lymphocytes, and macrophages). Classically, astrocytes were believed to play a role in trophic systems function. The microglia were considered to be activated by CNS injury, and the circulating cells were part of the response to catastrophic injury and infection.
Current thinking now emphasizes the enormous constitutive contributions of these cells to the excitability of local neuronal circuits. While there are no direct synaptic linkages, neuraxial astrocytes and microglia can be activated by several linkages [29–32]:
-
(i)
High-intensity afferent input leading to synaptic overflow of products such as glutamate, substance P, and BDNF.
-
(ii)
Networks of astrocytes which may communicate over a distance by the spread of excitation through local nonsynaptic contacts (“gap” junctions) and by ATP acting on purine receptors on the glia.
-
(iii)
Release of products from neurons. Microglia can be activated by release of chemokines (fractalkine) from the neuronal membrane. In addition to afferent input after tissue injury and inflammation, circulating cytokines (such as IL1β/TNF) can activate perivascular astrocytes/microglia.
-
(iv)
Circulating products such as cytokines and lipids can activate these perivascular nonneuronal cells.
-
(v)
Activation of spinal innate immune systems. It is appreciated that glial cells express a variety of toll-like receptors (TLRs). These TLRs are primitive recognition sites (first discovered in fruit flies) that can lead to glial activation. While these recognition sites have classically been considered relevant to recognizing membrane or molecular components of nonself entities such as viruses and bacteria, it is now appreciated that in the course of inflammation there are products that are released that can also activate these TLRs and their intracellular cascades. Activation of these receptors can initiate hyperalgesic states, while their blockade or knockout can minimize post-inflammatory hyperalgesia [33].
When activated, these glial cells can regulate synaptic excitability by (i) releasing excitatory products including ATP, free radicals, nitric oxide, lipid mediators, and cytokines and (ii) regulating extracellular parenchymal glutamate (by transporter-mediated uptake and release).
Preclinical work with spinal inhibitors of microglial activation such as minocycline (a second-generation tetracycline) and pentoxyfiline that have been reported to block indices of an acute or chronic glial activation and diminish hyperalgesic states has supported the role of nonneuronal cells in inflammation and injury-induced pain. These agents, while not clinically useful, suggest important directions in drug therapy development [34].
These events outlined above involving the nonneuronal cells are referred to broadly as “neuroinflammation.” The work emphasizes that astrocytes and microglia are constitutively active and contributing to acute changes in spinal network excitability and can contribute to the enhanced response of the dorsal horn after peripheral tissue injury and inflammation.
Evolution of a Chronic Pain State After Acute Injury
In the preceding sections, we have focused on the events which occur after tissue injury and inflammation. After such tissue injury and inflammation, for example, as after trauma and surgery, pain typically resolves with a time course that is typically consistent with the resolution of the inflammation, a consequence which parallels the healing process. In a variable but significant fraction of patients, a failure to resolve the pain state in spite of healing may be noted. The persistency may be the result of an occult inflammation (e.g., failure to heal) or perhaps injury to the nerve which leads to events that are evidently unable to heal (see below). Alternatively, there is increasing evidence that in the face of persistent inflammation (as say in arthritis) that there may be fundamental changes in the functionality of the afferent/DRG to yield a state of persistent sensitization. For example, in the face of a persistent (weeks) inflammation in animal models, an allodynic state is noted that continues after the resolution of the inflammation. Importantly, the knockout of the TLR4 receptors has no effects upon the inflammation but prevents the evolution of the persistent tactile allodynia. This is an important area of ongoing research [35, 36].
Summary
Tissue injury and inflammation initiate a behavioral phenotype characterized by ongoing pain and the appearance of states where mildly aversive or innocuous stimuli lead to an enhanced pain state at the site of injury (primary) and adjacent to the site of injury (secondary). The mechanisms underlying these behavioral states reflect release of “active factors” at the injury site initiating afferent traffic and sensitizing the afferent terminal, yielding an enhanced response to a given stimulus. The ongoing afferent activity leads to a complex series of events in the dorsal horn representing local changes in membrane excitability, activation of local facilitatory circuits, blocking local inhibitory circuits, activation of spino-bulbo-spinal links, and engaging a complex “neuroinflammatory” process involving spinal nonneuronal cells.
Encoding of Nociception After Nerve Injury
The mechanisms underlying the spontaneous pain and the miscoding of low-threshold tactile input are not completely understood. However, the organizing concept is that these events reflect (i) an increase in spontaneous activity the injured afferent and (ii) an exaggerated response of spinal neurons to low-threshold afferent input (Fig. 1.10).

This schematic provides an overview of the organization of events that initiate pain state after a peripheral nerve injury. (a) Nerve injury leads to retrograde chromatolysis and then sprouting to form local neuromas. (b) In addition to the changes in the terminals, there are trophic changes in the DRG leading to significant changes in the expression of a variety of channel and receptor proteins. (c) Over time, there is the appearance of ectopic activity in the injured axon. This activity arises from both the neuroma as well as the dorsal root ganglion. (d, e) In the dorsal horn, there are a series of reactive changes which lead to a reorganization of nociceptive processing. These changes include changes in the excitability of the second-order neurons, changes in the inhibitory control which normally regulates dorsal horn excitability, and then the activation of nonneuronal cells which contribute to the pro-excitatory nature of the nerve injury
Events Initiated by Nerve Injury
Injury leads to prominent changes at the site of nerve injury and in the DRG of the injured axon [8, 9, 37, 38]:
-
(i)
Injury site: After acute injury of the peripheral afferent axon, there is an initial dying back (retrograde chromatolysis) until the axon begins to sprout, sending growth cones forward. Such axonal growth cone often fails to contact with the original target, and these sprouts show proliferation. Collections of these proliferated sprouts form neuromas.
-
(ii)
DRG: Although the original injury is restricted to the peripheral nerve site, the distal injury has an enormous impact upon the dorsal root ganglion. Several events should be emphasized. (i) Markers of neuronal injury (such as ATF-3, an injury-evoked transcription factor) show a large-scale increase in expression in the DRG of the injured axons. (ii) There is an increased activation of the glial satellite cells (expressing GFAP) present. The DRG neurons are markedly enhanced. (iii) The DRG neurons show prominent increases in the expression of a variety of proteins such as those for sodium channels, calcium channels, and auxiliary calcium channel proteins (such as the alpha 2 delta subunit) and (iv) conversely decreases in the expression of other proteins such as those for certain potassium channels.
Origins of Spontaneous Pain State
As reviewed in the preceding sections, under normal conditions, the normal primary afferent axons show little of any spontaneous activity. After acute injury, the afferent axons display (i) an initial burst of afferent firing secondary to the injury, (ii) silence for intervals of hours to days, and (iii) development over time of spontaneous afferent traffic in both myelinated and unmyelinated axons.
Ongoing afferent input origin of ongoing pain. The ongoing afferent input is believed to provide the source of the afferent activity that leads to spontaneous ongoing sensation (Fig. 1.4). Evidence for this assertion that the ectopic afferent activity is in part responsible for the associated pain behavior is based on the observations that (i) parallel onset of pain and ectopic activity in neuroma and DRG, (ii) pain behavior blocked by application of TTX/local anesthetics to neuroma/DRGs, (iii) dorsal rhizotomy transiently reverse the pain behavior, and (iv) irritants applied to DRG initiate activity and importantly, evoke pain behavior [39].
Site of Origin of Spontaneous Afferent Traffic
Recording from the afferent axon has indicated that origin of the spontaneous activity in the injured afferent arises both from the neuroma and from the DRG of the injured axon (Fig. 1.4).
Mechanisms of Ongoing Activity
The generation of ongoing activity in the neuroma/DRG of the injured axon results from upregulation of excitable channels/receptors and appearance of excitatory substances in the DRG/neuroma.
Increased Sodium Channel Expression
Cloning shows that there are multiple populations of sodium channels, differing in their current activation properties and structure contributing to the action potential [8, 9].
Multiple sodium channels have been identified based on structure (NaV 1.1–NaV 1.9), whether they are tetrodotoxin sensitive (TTX), and their activation kinetics. Based on these designations, some subtypes are spatially limited in their distribution. Thus, NaV 1.8 and 1.9 are present in small primary afferents.
Importance of sodium channel subtypes in humans has been shown in identified loss- and gain-of-function mutations. The SCN9A gene encodes the voltage-gated sodium channel NaV 1.7, a protein highly expressed in pain-sensing dorsal root ganglion neurons and sympathetic ganglion neurons. Mutations in SCN9A cause three human pain disorders:
-
(i)
Loss of function: Loss-of-function mutations results in insensitivity to pain, no pain perception, and anosmia, but patients are otherwise normal.
-
(ii)
Gain of function: Activating mutations cause severe episodic pain in paroxysmal extreme pain disorders with episodic burning pain in mandibular, ocular, and rectal areas as well as flushing, and primary erythermalgia, a peripheral pain disorder in which blood vessels are episodically blocked then become hyperemic with associated with severe burning pain.
Peripheral nerve injury increases the expression of many sodium channels in the DRG, and these channels are transported to the distal terminals. Increased channel increases ionic conductance and appears to increase spontaneous activity in the sprouting axon terminal. Note that systemic (IV/IP) lidocaine at concentrations which do not block conducted action potentials will block the “ectopic” discharges originating in DRG and neuroma. These concentrations are notable in that they will correspondingly block hyperpathia in the nerve jury pain state otherwise observed in humans and in animal models.
Decreased K Channel Expression
Many classes of types of gated K + channels have been described. Opening of K + channels yields membrane hyperpolarization and a reduced excitability. In the face of nerve injury, a reduced expression of such channels has been described, and it is hypothesized that this may contribute to the increased ectopic afferent activity observed after nerve injury [40].
Inflammatory Products
The sprouted terminals of the injured afferent axon display transduction properties that were not possessed by the original axon, including mechanical (e.g., compression) and chemical sensitivity. Thus, neuromas display sensitivity humoral factors, such as prostanoids, catecholamines, and cytokines (TNF). DRGs also respond to these products.
These products are released from local sources such as satellite cells in the DRG and Schwann cells in the periphery. The DRG is of particular interest as it lies outside the blood-brain barrier, for example, it can be influenced by circulating factors. This evolving sensitivity is of particular importance given that following local nerve injury, there is the release of a variety of cytokines, particularly TNF, which can thus directly activate the nerve and neuroma.
Following nerve injury, there is an important sprouting of postganglionic sympathetic efferents that can lead to the local release of catecholamines. This scenario is consistent with the observation that following nerve injury, the postganglionic axons can initiate excitation in the injured axon (see below). These events are believed to contribute to the development of spontaneous afferent traffic after peripheral nerve injury.
Origins of Evoke Hyperpathia
The observation that low-threshold tactile stimulation yields a pain states has been the subject of considerable interest. The psychophysical properties of this state emphasize that the pain results from activation of low-threshold mechanoreceptors (Aβ afferents). This ability of light touch evoking this anomalous pain state is de facto evidence that the peripheral nerve injury has led to a reorganization of central processing, that is, it is not a simple case of a peripheral sensitization of otherwise high-threshold afferents. In addition to these behavioral changes, the neuropathic pain condition may display other contrasting anomalies, including on occasion an ameliorating effect of sympathectomy of the afflicted limb and an attenuated responsiveness to spinal analgesics such as opiates. Several underlying mechanisms have been proposed to account for this seemingly anomalous linkage.
Dorsal Root Ganglion Cell Cross Talk
Following nerve injury, evidence suggests that “cross talk” develops between afferents in the DRG and in the neuroma. Here, action potentials in one axon generate depolarizing currents in an adjacent quiescent axon. Thus, activity arising in one axon (a large afferent) would drive activity on a second axon (small C fiber) [41].
Afferent Sprouting
Under normal circumstances, large myelinated (Aβ) afferents project into the spinal Rexed lamina III and deeper (see above). Small afferents (C fibers) tend to project into spinal laminae II and I, a region consisting mostly of nocisponsive neurons. Following peripheral nerve injury, it has been argued that the central terminals of these myelinated afferents (A fibers) sprout into lamina II of the spinal cord. With this synaptic reorganization, stimulation of low-threshold mechanoreceptors (Aβ fibers) could produce excitation of these neurons and be perceived as painful. The degree to which this sprouting occurs is a point of current discussion, and while it appears to occur, it is less prominent than originally reported.
Loss of Intrinsic GABAergic/Glycinergic Inhibitory Control
As reviewed above, GABA/glycinergic interneurons display a potent regulation of large afferent-evoked WDR excitation. The relevance of this intrinsic inhibition to pain processing is evidenced by the observation that spinal delivery of GABA A receptor or glycine receptor antagonists yields a powerful behaviorally defined tactile allodynia [42, 43].
In general, while there are changes in dorsal horn after nerve injury, the predominant evidence does not support a loss of dorsal horn inhibitory amino acids circuitry. Recent observations now suggest an important alternative. After nerve injury, spinal neurons regress to a neonatal phenotype in which GABA A activation becomes excitatory. As noted, the GABA A and glycine channels are chloride ionophores, wherein their activation (increasing Cl permeability) normally leads to a mild hyperpolarization of the postsynaptic membrane as Cl moves inside the cell. After injury, there is a loss of the Cl exporter (so-called KCC2), and there is an accumulation of Cl inside the cell. Now, increasing conductance leads to an extracellular movement of the Cl. This loss of negative charge causes the cell to mildly hypopolarize. This accordingly would turn an inhibitory regulation circuit for larger afferent to a facilitatory circuit for large afferent drive of the WDR neuron [44, 45].
Nonneuronal Cells and Nerve Injury
Nerve section or compression leads to activation of spinal microglia and astrocytes in spinal segments receiving input from injured nerves with a time course that parallels the changes in pain states. While the origin of this activation is not clear, it will lead to an increased spinal expression of COX/NOS/glutamate transporters/proteinases. The effects of such changes in spinal cord afferent processing have been previously reviewed above [46].
Sympathetic Dependency
Following peripheral nerve injury, an increased innervation by postganglionic sympathetic terminals of the neuroma and of the DRG of the injured axons is reliably noted. In the DRG, these postganglionic fibers form baskets of terminals around the ganglion cells. Several properties of this hyperinnervation are noteworthy [47, 48]:
-
(i)
They invest ganglion cells of all sizes, but particularly large ganglion cells (so-called type A).
-
(ii)
Postganglionic innervation occurs largely in the ipsilateral DRG but also occurs to a lesser degree in the contralateral DRG.
-
(iii)
Activation of the preganglionic efferents (traveling in the ventral roots) will activate the sensory axon by an interaction at the site of injury or at the level of the DRG.
-
(iv)
Activation is blocked by intravenous phentolamine, emphasizing an adrenergic effect.
Generalization to Many Nerve Injury Pain States
After nerve injury, there evolves an increase in ongoing dysesthesia and an enhanced response to low-threshold mechanical stimuli (allodynia). These effects are believed to reflect an increase in ectopic activity that arises from the neuromas well as the injured axon. The origin of the ectopic activity is believed to reflect an increased expression of sodium channel, decreased expression of K channels in the neuroma, and DRG leading to enhanced excitability. The allodynia is considered to reflect an alteration in the activation produced by large low-threshold afferents (Aβ). This alteration may result from cross talk between axons and/or a loss of inhibitory regulation.
It should be noted that the above review generically considers the “injured” axon. These changes reviewed above have been observed in animal models following chemotherapy, varicella zoster, extruded intervertebral disks (compressing the nerve root), and osteosarcoma. Accordingly, these changes described in preclinical models are believed to have a great likelihood of being relevant to the human condition.
Conclusions
In the preceding sections, we have provided an overview of the various systems that underlie the three heuristic subdivisions of acute, post-tissue injury and post-nerve injury pain states. An important concept is that in many clinical conditions, it is virtually certain that the clinical state is not one or the other, but rather a combination. Table 1.3 presents a superficial analysis of the types of mechanisms which may be involved in, for example, cancer pain. It is compelling to consider that such a patient may experience a pain state that reflects all three conditions between the events that arise from the tumor itself, the chemotherapy and the surgery (Tables 1.3 and 1.4).
The likelihood of multiple mechanisms mediating a particular pain state has an important ramification when it comes to the appropriateness of any particular analgesic therapy. Table 1.3 presents a summary of the basic mechanisms of actions of several classes of analgesic agents. Though not specifically discussed in this chapter (see elsewhere in this text), it is appreciated that they act to alter nociceptive transmission in a variety of ways. Opiates have a potent effect upon spinal transmission initiated by small primary afferents, whereas an NSAID largely has an effect when there is a facilitated state initiated by local inflammation. As reviewed above, there is in addition a central role for NSAIDs because of the constitutive expression of COX in the spinal dorsal horn and the role of prostaglandins in enhancing presynaptic transmitter release and diminish the inhibitory efficacy of the glycine receptor. In the face of multiple pain mechanisms, it can be appreciated that to minimize any pain state may well require addressing multiple therapeutic targets. Hence, it is not surprising that the profile of analgesic management of complex states, such as cancer, often shows 3–4 analgesic agents being employed.
References
Dougherty PM. Central sensitization and cutaneous hyperalgesia. Semin Pain Med. 2003;1:121–31.
Johanek L, Shim B, Meyer RA. Chapter 4 Primary hyperalgesia and nociceptor sensitization. Handb Clin Neurol. 2006;81:35–47.
Mayer EA, Gebhart GF. Basic and clinical aspects of visceral hyperalgesia. Gastroenterology. 1994;107:271–93.
Baron R. Neuropathic pain: a clinical perspective. Handb Exp Pharmacol. 2009;194:3–30.
Stucky CL, Dubin AE, Jeske NA, Malin SA, McKemy DD, Story GM. Roles of transient receptor potential channels in pain. Brain Res Rev. 2009;60:2–23.
Binshtok AM. Mechanisms of nociceptive transduction and transmission: a machinery for pain sensation and tools for selective analgesia. Int Rev Neurobiol. 2011;97:143–77.
Cortright DN, Szallasi A. TRP channels and pain. Curr Pharm Des. 2009;15(15):1736–49.
Dib-Hajj SD, Black JA, Waxman SG. Voltage-gated sodium channels: therapeutic targets for pain. Pain Med. 2009;10(7):1260–9.
Cohen CJ. Targeting voltage-gated sodium channels for treating neuropathic and inflammatory pain. Curr Pharm Biotechnol. 2011;12:1715–9.
Raja SN, Meyer RA, Campbell JN. Peripheral mechanisms of somatic pain. Anesthesiology. 1988;68:571–90.
Yaksh TL. Calcium channels as therapeutic targets in neuropathic pain. J Pain. 2006;7(1 Suppl 1):S13–30.
Ruscheweyh R, Forsthuber L, Schoffnegger D, Sandkühler J. Modification of classical neurochemical markers in identified primary afferent neurons with abeta-, adelta-, and C-fibers after chronic constriction injury in mice. J Comp Neurol. 2007;502:325–36.
Willis Jr WD. The somatosensory system, with emphasis on structures important for pain. Brain Res Rev. 2007;55:297–313.
Todd AJ, Spike RC. The localization of classical transmitters and neuropeptides within neurons in laminae I-III of the mammalian spinal dorsal horn. Prog Neurobiol. 1993;41:609–45.
Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11:823–36.
Ralston 3rd HJ. Pain and the primate thalamus. Prog Brain Res. 2005;149:1–10.
Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–8.
Herrero JF, Laird JM, López-García JA. Wind-up of spinal cord neurones and pain sensation: much ado about something? Prog Neurobiol. 2000;61:169–203.
Dickenson AH, Chapman V, Green GM. The pharmacology of excitatory and inhibitory amino acid-mediated events in the transmission and modulation of pain in the spinal cord. Gen Pharmacol. 1997;28:633–8.
Bleakman D, Alt A, Nisenbaum ES. Glutamate receptors and pain. Semin Cell Dev Biol. 2006;17:592–604.
Luo C, Seeburg PH, Sprengel R, Kuner R. Activity-dependent potentiation of calcium signals in spinal sensory networks in inflammatory pain states. Pain. 2008;140:358–67.
Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926.
Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Zhang YQ. Protein kinases as potential targets for the treatment of pathological pain. Handb Exp Pharmacol. 2007;177:359–89.
Velázquez KT, Mohammad H, Sweitzer SM. Protein kinase C in pain: involvement of multiple isoforms. Pharmacol Res. 2007;55:578–89.
Svensson CI, Yaksh TL. The spinal phospholipase-cyclooxygenase-prostanoid cascade in nociceptive processing. Annu Rev Pharmacol Toxicol. 2002;42:553–83.
Zeilhofer HU. The glycinergic control of spinal pain processing. Cell Mol Life Sci. 2005;62:2027–35.
Tang Q, Svensson CI, Fitzsimmons B, Webb M, Yaksh TL, Hua XY. Inhibition of spinal constitutive NOS-2 by 1400 W attenuates tissue injury and inflammation-induced hyperalgesia and spinal p38 activation. Eur J Neurosci. 2007;25:2964–72.
Suzuki R, Rygh LJ, Dickenson AH. Bad news from the brain: descending 5-HT pathways that control spinal pain processing. Trends Pharmacol Sci. 2004;25:613–7.
Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36.
Ren K, Dubner R. Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr Opin Anaesthesiol. 2008;21:570–9.
Abbadie C, Bhangoo S, De Koninck Y, Malcangio M, Melik-Parsadaniantz S, White FA. Chemokines and pain mechanisms. Brain Res Rev. 2009;60:125–34.
Clark AK, Staniland AA, Malcangio M. Fractalkine/CX3CR1 signalling in chronic pain and inflammation. Curr Pharm Biotechnol. 2011;12:1707–14.
Grace PM, Rolan PE, Hutchinson MR. Peripheral immune contributions to the maintenance of central glial activation underlying neuropathic pain. Brain Behav Immun. 2011;25:1322–32.
Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83.
Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367:1618–25.
Xu Q, Yaksh TL. A brief comparison of the pathophysiology of inflammatory versus neuropathic pain. Curr Opin Anaesthesiol. 2011;24:400–7.
Tuchman M, Barrett JA, Donevan S, Hedberg TG, Taylor CP. Central sensitization and Ca(V) α2δ ligands in chronic pain syndromes: pathologic processes and pharmacologic effect. J Pain. 2010;12:1241–9.
Bráz JM, Ackerman L, Basbaum AI. Sciatic nerve transection triggers release and intercellular transfer of a genetically expressed macromolecular tracer in dorsal root ganglia. J Comp Neurol. 2011;519:2648–57.
Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37.
Takeda M, Tsuboi Y, Kitagawa J, Nakagawa K, Iwata K, Matsumoto S. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol Pain. 2011;7:5.
Devor M, Wall PD. Cross-excitation in dorsal root ganglia of nerve-injured and intact rats. J Neurophysiol. 1990;64(6):1733–46.
Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37:111–23.
Sivilotti L, Woolf CJ. The contribution of GABAA and glycine receptors to central sensitization: disinhibition and touch-evoked allodynia in the spinal cord. J Neurophysiol. 1994;72:169–79.
Polgár E, Hughes DI, Riddell JS, Maxwell DJ, Puskár Z, Todd AJ. Selective loss of spinal GABAergic or glycinergic neurons is not necessary for development of thermal hyperalgesia in the chronic constriction injury model of neuropathic pain. Pain. 2003;104:229–39.
Price TJ, Cervero F, de Koninck Y. Role of cation-chloride-cotransporters (CCC) in pain and hyperalgesia. Curr Top Med Chem. 2005;5(6):547–55.
Cao H, Zhang YQ. Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev. 2008;32(5):972–83.
McLachlan EM, Jänig W, Devor M, Michaelis M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal-root ganglia. Nature. 1993;363:543–6.
Drummond PD. Involvement of the sympathetic nervous system in complex regional pain syndrome. Int J Low Extrem Wounds. 2004;3:35–42.
Borsook D, Becerra L. CNS animal fMRI in pain and analgesia. Neurosci Biobehav Rev. 2011;35:1125–43.
D’Souza WN, Ng GY, Youngblood BD, Tsuji W, Lehto SG. A review of current animal models of osteoarthritis pain. Curr Pharm Biotechnol. 2011;12:1596–612.
Mogil JS. Animal models of pain: progress and challenges. Nat Rev Neurosci. 2009;10:283–94.
Waszkielewicz AM, Gunia A, Słoczyńska K, Marona H. Evaluation of anticonvulsants for possible use in neuropathic pain. Curr Med Chem. 2011;18:4344–58.
Xu J, Brennan TJ. The pathophysiology of acute pain: animal models. Curr Opin Anaesthesiol. 2011;24:508–14.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 American Academy of Pain Medicine
About this chapter
Cite this chapter
Yaksh, T.L., Wiese, A.J. (2013). A Survey of Systems Involved in Nociceptive Processing. In: Deer, T., et al. Comprehensive Treatment of Chronic Pain by Medical, Interventional, and Integrative Approaches. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-1560-2_1
Download citation
DOI: https://doi.org/10.1007/978-1-4614-1560-2_1
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-1559-6
Online ISBN: 978-1-4614-1560-2
eBook Packages: MedicineMedicine (R0)