
Abstract
The hand is the site of many different types of benign and malignant neoplasms. These lesions may originate in either soft tissues or bone. The most common benign tumours of the hand include enchondromas, ganglions, giant cell tumours of the tendon sheath, and epidermoid cysts while squamous cell carcinoma represents the most common hand malignancy. This chapter describes the clinical, radiological and histological findings, in addition to the treatment strategies of the most common tumours of the hand and wrist.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Hand Tumours
- Enchondroma
- Bizarre Parosteal Osteochondromatous Proliferation
- Giant Cell Tumour
- Acrometastases
- Epidermoid Cysts
- Nodular Fasciitis
- Mohs Micrographic Surgery
- Soft Tissue Sarcoma
- Squamous Cell Carcinoma
- Melanoma
- Synovial Sarcoma
- Epithelioid Sarcoma
Introduction
The hand is the site of many different types of benign and malignant neoplasms. These lesions may originate in either soft tissues or bone. The most common benign tumours of the hand include enchondromas, ganglions, giant cell tumours of the tendon sheath, and epidermoid cysts while squamous cell carcinoma represents the most common hand malignancy. This chapter describes the clinical, radiological and histological findings, in addition to the treatment strategies of the most common tumours of the hand and wrist.
Benign Bone Tumours of the Hand
Table 5.1.
Enchondroma
Enchondroma is a benign hyaline cartilage neoplasm of medullary bone. It is the most common primary bone tumour of the hand. The phalanges are affected in 80 % of hand cases, the metacarpals in 20 %, while enchondromas of the carpal bones are rare and account for less than 1 % of cases. The proximal phalanx is the most commonly involved tubular bone of the hand (40–50 %), while the thumb is the least affected digit. Patients present with painful swelling (40–51 %), pathological fracture (15–38 %), while 11–17 % present as an incidental finding on radiographs obtained for unrelated reasons [1–4].
Radiographs show a well-defined cystic, radiolucent intramedullary lesion containing thin internal trabeculations. Cortical thinning and medullary expansion are commonly seen, while chondroid calcifications i.e. rings-and-arcs, popcorn, flocculent, or stippled calcifications are seen less often than in enchondromas at other skeletal sites (Fig. 5.1). Magnetic resonance imaging shows multiple lobules of high signal intensity on T2-weighted and low signal intensity on T1-weighted images. Calcified foci appear as low signal intensity on both T1 and T2-weighted images. Macroscopically, enchondromas appear as lobules of blue-white hyaline cartilage with gritty yellow-white areas of calcification or ossification. Histologically, the tumour is hypocellular with abundant hyaline cartilage with chondrocytes that reside within lacunae. Enchondromas of the tubular bones of the hands and feet are generally more cellular and cytologically atypical than long bone tumours [5, 6].

Enchondroma of the distal phalanx. Posteroanterior and lateral radiographs show a lytic intramedullary lesion containing thin internal trabeculations and no visible chondroid calcifications
Simple curettage without augmentation is the treatment of choice of symptomatic enchondromas while small asymptomatic lesions could be observed [7, 8]. Schaller et al. [8], found no significant difference in the bone density or the functional results in those patients who had no bone grafting of their enchondromas as compared to those who had. Local recurrence rates of 1.4–27.2 % have been reported in the literature including late recurrences of up to 17 years postoperatively [2–4, 9]. Malignant transformation has been observed particularly in recurrent cases [2, 10].
Clinical Pearl
-
Simple curettage without augmentation is the preferred treatment modality of enchondromas. Asymptomatic lesions need no treatment.
Enchondromatosis
Ollier’s disease is a rare, non-familial, developmental disorder caused by failure of normal enchondral ossification and is characterized by multiple enchondromatosis with an asymmetric distribution and varying degrees of bone deformity. Maffucci’s syndrome is distinguished from Ollier’s disease by the presence of haemangiomas in the soft tissues and/or viscera. Metachondromatosis is another autosomal dominant incompletely penetrant disorder leading to multiple enchondromas and osteochondromas [6, 11].
The hand is the most common site of involvement followed by the foot, femur, humerus, and forearm bones. The disease is frequently confined to one side of the body. Clinical manifestations include localised pain, palpable bony masses, leg length discrepancy, pathological fractures, and bowing deformities. Changes in the symptoms, cortical destruction, and soft tissue extension are suspicious for malignant transformation of these syndromic enchondromas as opposed to solitary enchondromas. Malignant transformation occurs in 25–30 % of syndromic cases [6, 11, 12].
Bizarre Parosteal Osteochondromatous Proliferation of Bone
These are best described as surface-based osteo-cartilaginous lesions typically affecting the hands and feet. Histologically, the lesion consists of a cartilage cap and underlying bone. The cartilaginous component is highly cellular with irregular bony-cartilaginous interfaces, and enlarged, bizarre, and binucleate chondrocytes, mimicking chondrosarcoma [13–16]. Radiologically, Nora’s lesions are well-defined calcium containing masses adjacent to the cortical surface of the affected bones [17]. The lack of continuity with the underlying medullar cavity differentiates the lesion from an osteochondroma. It usually affects the proximal and middle phalanges and is most commonly seen in the third and fourth decades [18, 19]. Wide resection is the treatment of choice for bizarre parosteal osteochondromatous proliferation. These lesions have a high tendency for local recurrence after excision and recurrence rates between 51 and 67 % have been reported in the literature [13, 19, 20].
Osteoid Osteomas
Osteoid osteomas are benign bone-forming tumours that constitute 12 % of all benign bone neoplasms. These lesions usually become symptomatic in the second and third decades of life. Osteoid osteomas have limited growth potential and their size rarely exceed 1.5 cm in maximum diameter. Approximately 10 % of osteoid osteomas occur in the hands and feet, and these can be extremely difficult to diagnose as the typical pain pattern may be absent, and the radiographic and histologic features may vary from the classic osteoid osteomas which occur in the long bones. Most of the hand lesions occur in the proximal phalanges, followed by the distal and middle phalanges, carpal bones, and the metacarpals [21–24].
Pain and local tenderness are the most common complaints. Pain tends to be more severe at night and usually is relieved by salicylates. The pain is probably due to the high levels of Prostaglandin E2 and prostacyclin found in the tumour. Rarely, osteoid osteoma may be painless and these patients usually present with localized swelling of the affected digit. Slight erythematous changes that mimic tenosynovitis or infection can be observed in some patients [21–23].
The typical radiographic appearance is that of a small central radiolucent lesion, often referred to as the ‘nidus’, usually surrounded by an area of dense, fusiform, reactive osteosclerosis [21, 22] (Fig. 5.2.).

Osteoid Osteoma of the distal phalanx. Posteroanterior radiograph shows a small central radiolucent nidus in the base of the distal phalanx
Radiofrequency ablation has now replaced surgical excision of the nidus as the treatment of choice of osteoid osteoma. This technique should be used cautiously in the treatment of superficial lesions and those lesions in close proximity to important structures as thermal burns and soft-tissue reactions with pain have been reported [25, 26].
Clinical Pearl
-
Osteoid osteomas typically present with night pain that is relieved by simple analgesics or anti-inflammatories. CT scan is often needed to identify the nidus. Radiofrequency ablation should be used with caution as thermal burns of important structures can occur.
Osteoblastoma
Osteoblastoma is a rare, benign bone-forming neoplasm which produces woven bone spicules, which are bordered by prominent osteoblasts. It accounts for less than 1 % of primary bone tumours. Osteoblastomas are histologically identical to osteoid osteoma, however they are larger in size (≥2 cm in diameter), and usually do not respond to NSAIDs. Osteoblastomas of the hand bones are very rare. Osteoblastomas of the carpal scaphoid, hamate, metacarpals, and phalanges were reported in the literature [27–31].
Radiographs often demonstrate an expansile, lytic well circumscribed oval or spherical lesion surrounded by a periosteal shell of reactive bone. Focal areas of calcification may be observed in some cases indicating tumour bone mineralisation. Curettage and bone grafting is the preferred treatment modality of the primary cases. Recurrent lesions, and cases associated with severe bone destruction are best treated by en bloc resection and reconstruction [27, 29, 30].
Aneurysmal Bone Cysts
Aneurysmal bone cyst (ABC) is a benign osteolytic expansile cystic and haemorrhagic lesion of bone composed of blood filled spaces separated by connective tissue septa containing fibroblasts, osteoclast-type giant cells and reactive woven bone. Aneurysmal bone cysts of the hand account for only 3–5 % of all ABCs, mainly affecting the metacarpals and the phalanges [32–35].
Patients present with pain and swelling, which are usually present for an average period of 2–5 months. Roentgenograms show a lytic, eccentric, expansile lesion surrounded by a thin shell of subperiosteal reactive bone. MRI shows septated multilobulated lesion with characteristic fluid-fluid levels. Aneurysmal bone cyst is the only benign bone lesion which can extend across a growth plate into the epiphysis [32, 33, 36].
Although some authors reported satisfactory results with intralesional injections of calcitonin, methylprednisolone or Ethibloc (a sclerosing agent that has been used in the treatment of haemangiomas, lymphangiomas, aneurysmal bone cysts, and other hypervascular lesions), surgery remains the treatment of choice of aneurysmal bone cysts of the hand. Curettage, with or without bone grafting, is usually recommended. En-bloc excision of the primary lesion with structural bone grafting should be reserved for cases when full bone invasion of the phalanx or the metacarpal had occurred. Local recurrence was reported in 33–67 % of those cases treated with intralesional curettage while no recurrences were reported in those cases treated by surgical excision. Repeat curettage or resection and reconstruction may be performed for recurrent cases [33, 37–40].
Giant Cell Tumour of Bone
Giant cell tumour (GCT) of bone is defined as a benign, locally aggressive neoplasm which is composed of sheets of neoplastic ovoid mononuclear cells interspersed with uniformly distributed large, osteoclast-like giant cells [41]. Primary involvement of the bones of the hand and wrist is rare [42]. The metacarpals and phalanges are more commonly affected while less than 10 % of the reported cases involved the carpal bones [43–45]. The tumour occurs in a more central location, which differs from the eccentric location of giant cell tumour at other sites possibly due to the limited volume of bone [46]. Radiographs demonstrate a centrally located lytic lesion with internal trabeculation and a narrow zone of transition at the metaphyseal margin of the lesion [46] (Fig. 5.3). Giant cell tumours of the small bones do occur in younger patients and appear to have greater propensity for local recurrence than lesions in the long bones [42, 47, 48] (Fig. 5.4). Curettage was found to be an ineffective treatment method with recurrence rates of 79–87 % reported in the literature [43, 44]. Wittig et al. [49], reported no local recurrences after curettage, cryosurgery, and cementation of 3 cases of GCT of the tubular bones of the hand. However, most authors recommend en bloc resection and reconstruction or ray resection as primary treatment modalities of GCT of the hand with significantly lower rates of local recurrence when compared to intralesional procedures [42–44].

(a, b) Giant cell tumour of the first metacarpal in a 36-year-old patient. (a) Preoperative radiograph shows a centrally located lytic lesion with internal trabeculations and a narrow zone of transition. (b) Follow-up radiograph 12 months after treatment by extended curettage shows evidence of consolidation of the lesion

(a, b) Giant cell tumour of the proximal phalanx in a 41-year-old patient. (a) Preoperative posteroanterior radiograph demonstrates a typical giant cell tumour of the phalanx. (b) Posteroanterior radiograph six months following treatment by extended curettage of the lesion shows extensive local recurrence. This was treated by ray amputation
Clinical Pearl
-
En bloc resection and reconstruction or ray resection is the treatment of choice of GCT of the tubular bones of the hand as curettage is associated with high rates of local recurrence.
Primary Malignant Bone Tumours of the Hand
Bone sarcomas are rare and represent only 0.2 % of all neoplasms. While most patients with benign bone tumours present with an intermittent aching pain or occasionally a pathological fracture, malignant bone tumours typically present with non-mechanical or night pain. Radiological features can help in distinguishing benign from malignant neoplasms. These include tumour size, location, pattern of bone destruction, periosteal new bone formation, cortical disruption and soft tissue involvement. Once the diagnosis of a malignant neoplasm is suspected, the patient should be referred to the local sarcoma unit for treatment. Needle biopsy will confirm the nature of the lesion. MRI is the main imaging modality for local staging while chest CT and radionuclide bone scan will complete the systemic staging of the tumour [50] (Table 5.2).
Chondrosarcoma
Chondrosarcoma is the most common primary malignant bone tumour of the hand [51, 52]. One to eight percent of chondrosarcomas are located in the bones of the hand [53, 54]. Palmieri [55] found that 78 % of chondrosarcomas of the hand originated without a pre-existing lesion. Secondary chondrosarcomas usually develop in patients with multiple enchondromas. A chondrosarcoma arising on top of a solitary enchondroma is exceedingly rare [52, 55]. Patients are usually 60–80 years of age and present with painful swelling, often of long duration (up to 30 years) [56–58].
Chondrosarcoma of the hand almost always affect the epiphyseal region of the proximal phalanx or the metacarpal. Typical radiographic features include cortical destruction, scattered punctate densities of dystrophic matrix calcification, wide zone of transition, pathological fracture, and soft tissue extension [5, 55] (Fig. 5.5). Histologically, chondrosarcomas are composed of hyaline cartilage with hypercellularity and double nucleated cells. Bertoni et al. [56], stated that the histological findings of increased cellularity, binucleated cells, hyperchromasia and myxoid change may all be present in enchondromas of the small bones of the hands and feet. They found that the most significant histologic feature of chondrosarcoma in this location is permeation through the cortex into soft tissues and a permeative pattern in the cancellous bone [56].

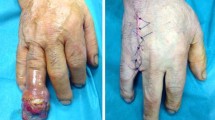
(a–e) Chondrosarcoma of the third metacarpal in an 80-year-old patient. (a) Posteroanterior preoperative radiograph demonstrates a pathological fracture through the lesion. (b) Sagittal T2-weighted MR image demonstrates the intraosseous and extraosseous extent of the lesion. (c–e) Pre-, intra-, and post-operative pictures of the hand showing that local control of the lesion was achieved by resection of the third ray
Chondrosarcomas of the hand have significantly lower metastatic potential when compared to chondrosarcomas of other locations. However, ray resection is still recommended to minimize the incidence of local recurrence [55, 57, 59].
Osteosarcoma
Osteosarcoma of the hand is exceedingly rare with less than 50 cases reported in the literature (0.18–0.39 % of all osteosarcomas). Twenty per cent of these reported cases represented secondary osteosarcomas after prior irradiation, Paget’s disease or as metastatic deposits from osteosarcomas elsewhere in the skeleton [58, 60, 61].
The site-specific distribution of these reported cases show that more than 70 % of the cases occur in either the metacarpal heads or the bases of the proximal phalanges. Osteosarcoma of the hand tends to occur at a relatively older age than in other skeletal sites, with more than 50 % of the reported cases occurring in patients 40 years of age or older [52, 60].
Pain and/or swelling are the most common presenting complaints with most cases showing an identifiable mass at the time of diagnosis. Radiographs show areas of sclerosis and periosteal reaction as well as areas of bone destruction (Fig. 5.6). The extent of extraosseous soft tissue extension generally corresponds to the size of the intramedullary tumour and the extent of cortical destruction [52, 58, 60, 61]. A complete tumour workup should include radiographs of the involved extremity, MRI of the involved region with gadolinium enhancement to evaluate the osseous and soft tissue extension of the lesion, a CT scan of the chest, and a bone scan to identify any skip lesions or possible metastatic disease.

Osteosarcoma of the proximal phalanx of the middle finger. Posteroanterior radiograph shows areas of sclerosis, periosteal reaction, and other areas of bone destruction
The diagnosis of osteosarcoma is based on the identification of tumour osteoid produced by atypical cells. Conventional osteosarcoma accounts for the majority of the cases while high grade surface osteosarcoma, periosteal osteosarcoma, and low grade parosteal osteosarcoma were rarely reported in the hand [52, 58]. Treatment of high-grade osteosarcoma should consist of neoadjuvant chemotherapy, surgical excision, followed by adjuvant chemotherapy. Ray amputation is the surgical procedure of choice for osteosarcoma of the phalanges while osteosarcoma of the metacarpals usually requires more extensive resections and reconstructions [52].
Ewing’s Sarcoma
Ewing’s sarcoma is the second most common primary malignant bone tumour of children and adolescents. The short tubular bones of the hands are rarely involved and these represent less than 1 % of all cases of Ewing’s sarcoma reported in the literature [58, 62].
Patients usually present with pain and swelling in the involved digit which may be associated with constitutional symptoms i.e. fever, leucocytosis, and malaise. Histologically, the tumour is composed of uniform round cells with small eosinophilic to clear cytoplasm, containing glycogen, and large round nuclei with fine chromatin and small nucleoli [58]. Radiographs demonstrate permeative lytic lesion with periosteal reaction and cortical destruction. Atypical radiographic findings have been reported in some cases including bony expansion, a cystic or honeycomb pattern, and lack of laminated or speculated periosteal reaction [62]. Treatment involves systemic chemotherapy in addition to local treatment in the form of radiotherapy, surgery, or a combination of both. Wide surgical excision with local reconstruction or ray amputation is the preferred local treatment modality with or without adjunctive postoperative radiotherapy. Although chemotherapy is the mainstay of treatment, post-operative radiation therapy should be considered as an essential part of the treatment programme, especially when wide margins cannot be achieved surgically or the response to chemotherapy, as noted in the resected specimen, is incomplete [63]. The overall survival rate and the event-free survival rate in patients with Ewing’s sarcoma and primitive neuroectodermal tumour (PNET) of the distal upper extremity was found by the cooperative Ewing’s sarcoma study group to be remarkably high as compared to other anatomical locations. This has been attributed to the average low tumour volume and the low incidence of metastatic disease at presentation [64].
Hand Metastasis
Metastatic lesions to the hand are rare and account for 0.1–0.2 % of all metastasis. The lung is the most common primary site (50 %), followed by the breast (15 %), and the kidney. The parenchyma of lung or liver tends to act as a filter in other malignancies. The absence of a filter may account for the relative frequency of lung cancer as a source in metastasis to the hand [65–68].
Acrometastases (metastases distal to the elbow and the knee) usually occur as rare, pre-terminal events and often are part of a widespread dissemination of metastases but it can be the first presenting sign of an occult carcinoma in about 15 % of the patients. The age of presentation is generally after the fifth decade, although a few patients as young as 18 months of age have been reported in the literature. The clinical findings commonly include pain, swelling, and erythema. Patients may occasionally present with pathological fracture. The terminal phalanges are the most frequent sites of metastasis, followed by the metacarpals and the proximal phalanges while the carpal bones are only rarely affected. The dominant hand is more commonly involved. This could be attributed to the fact that the dominant hand receives greater blood flow than the non-dominant one and may also be more prone to trauma [67–69].
Radiographs usually show a destructive permeative lesion without a marginal rim of sclerosis. Thyroid and renal carcinomas frequently produce lytic lesions, and prostate carcinoma is classically associated with blastic secondaries. Mixed lytic and blastic deposits are most often seen with malignancies of the lung and breast [67, 69].
Staging studies should be performed, since the extent of spread of primary tumour plays an important part in the prognosis of the patient. The aim of treatment of acrometastases is usually palliative. An exception is patients with solitary metastasis of renal cell carcinoma. These patients are appropriate candidates for wide surgical excision to improve survival [68–70].
Treatment modalities of acrometastases include radiotherapy, systemic chemotherapy, ray amputations, and curettage or limited marginal excision with adjuvant radiotherapy. Surgical excision of the lesion usually results in prompt relief of symptoms and early restoration of function. Ray resections to achieve wide surgical margins are often required. Patients with large unresectable lesions at presentation may be candidates for radiotherapy or intralesional surgery and adjuvant radiotherapy. Curettage of the lesion should be considered in patients with metastases in the carpal bones, or the thumb for whom amputation would result in an unacceptable degree of functional disability [67–69].
Tumours of the Soft Tissue
The large majority of soft tissue tumours are benign, with a very high cure rate after surgical excision. It is often very difficult to distinguish benign from malignant tumours of the soft tissue. Grimer et al. [71], recommended that any patient with a soft tissue mass that is increasing in size, has a size >5 cm or is deep to the deep fascia, whether or not it is painful, should be referred to a diagnostic centre with a suspected soft tissue sarcoma (STS). A core needle biopsy will confirm the nature of the lesion, however an excisional biopsy maybe the best option for superficial lesions <5 cm in diameter. MRI scan is the preferred imaging modality on investigating a soft tissue mass. Staging studies should be completed once the diagnosis of a STS is confirmed and these should include a high resolution CT scan of the chest to exclude pulmonary metastases prior to definitive treatment.
Clinical Pearl
-
Any patient with a soft tissue mass that is increasing in size, has a size >5 cm or is deep to the deep fascia, whether or not it is painful, should be referred to a diagnostic centre and managed by a specialist sarcoma multidisciplinary team for a suspected STS.
Benign Soft Tissue Tumours
Table 5.3.
Epidermoid Cysts
Epidermoid cysts or epidermal inclusion cysts are cysts lined by squamous epithelium and containing keratin [72–74]. These cysts can occur in subcutaneous, intratendinous, subungual, or intraosseous locations [73]. They are believed to be caused by traumatic implantation of epidermal fragments into the dermal tissue. They are therefore more commonly seen in adult males, especially manual workers. About 70–80 % of these cysts are located on the palm or the palmar aspect of the fingers [75].
Inclusion cysts are very common and may represent up to 16 % of all tumours of the hand [72, 73]. They are usually asymptomatic although symptoms of pain, tenderness, redness, and swelling are not infrequently seen [76]. Intraosseous epidermal inclusion cysts usually present as a well-defined unilocular lytic lesion with sclerotic margins. They are more commonly seen in the terminal phalanges [72–76].
Surgical excision is only indicated in symptomatic cases where the entire cyst, its intact lining, and any overlying scar tissue from any previous penetrating injury are marginally excised. Controversy exists as to whether the intraoperative spillage of the cyst contents increases the risk of local recurrence. Intraosseous epidermoid cysts should be treated with excision and curettage of the wall of the cavity [72, 73, 75]. Local recurrence rates of 11–17 % after surgical excision have been reported [72, 75].
Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF) is an uncommon benign proliferation of myofibroblasts with characteristic inclusion bodies in the cytoplasm of the neoplastic fibroblasts [77, 78]. Clinically, patients present in the first year of life with asymptomatic smooth nodular swellings, which may resemble a keloid, on the dorsal aspect of the fingers or toes. The tumour may undergo a spontaneous decrease in the number of inclusion bodies and becomes fibrotic with time [79, 80].
IDF does not have an aggressive nature and spontaneous involution is the rule. Wide local excision has been associated with a recurrence rate of up to 60 %. These lesions should therefore be observed without any aggressive treatment unless it causes marked dysfunction of the affected digit [77–80].
Clinical Pearl
-
IDF should not be excised as aggressive local recurrence could be seen in up to 60 % of the patients. Biopsy should be followed by watchful waiting as spontaneous involution is the rule.
Nodular Fasciitis
Nodular fasciitis is a rapidly growing benign, self-limiting, reactive lesion that can be mistaken for a malignant neoplasm [81, 82]. This lesion is commonly seen on the volar surface of the forearm but is rare in the hand. It is most commonly seen in young adults between 20 and 40 years of age. Nodular fasciitis of the hand seems to have a close association with trauma as compared to other locations. Macroscopically, it appears as a solitary round to oval well circumscribed nodule usually measuring less than 2 cm in diameter. Histologically, lesions show marked hypercellularity and high mitotic activity but lack nuclear hyperchromatism and pleomorphism. They are composed of spindle-shaped fibroblasts or myofibroblasts separated by intercellular myxoid material mixed with areas of prominent eosinophilic hyalinised stroma and incompletely formed storiform pattern. These lesions might grow much faster than Dupuytren nodules and therefore are often initially misdiagnosed as sarcomas. Diagnosis of nodular fasciitis requires histologic confirmation, and both diagnosis and treatment are accomplished by excisional biopsy. Nodular fasciitis has a very low recurrence rate of 1–2 % [81–83].
Fibroma of Tendon Sheath
They are more commonly seen in male patients in their fourth decade. They usually present as well-defined multilobated lesions, measuring less than 3 cm in diameter. Symptoms of nerve entrapment, finger triggering, or pain are not infrequently seen. Histologically, the lesion is composed of a collagenous stroma containing spindle fibroblasts in a moderate degree of cellularity with numerous slit-like vascular channels. Magnetic resonance imaging reveals a focal nodular mass adjacent to a tendon sheath with decreased signal on all pulse sequences and little or no enhancement. The aim of surgical excision is to relieve symptoms while preserving the function of the involved tendon. Recurrence has been reported in 11–24 % of cases [84–87].
Benign Adipocytic Tumours
Benign fatty tumours of the upper extremity can be classified based on the cell origin and the location of the fatty tumour. There are three histologically distinct types of fat cells: the immature fat cells (lipoblasts) which give rise to lipoblastomas, the mature brown fat cells which give rise to hibernomas, and the mature white fat cells (lipocytes) which give rise to lipomas. Lipomas only comprise 1–3 % of hand tumours. They usually present as a gradually enlarging, soft and resilient non-tender mass. Localised pain and symptoms of nerve compression are not infrequent complaints at presentation. Magnetic resonance imaging shows homogeneous and high signal intensity on T1-weighted images and an intermediate intensity on T2-weighted images. The intramuscular variety can be divided into two types: the infiltrative type and the well-circumscribed type. Local recurrence after excision is not uncommon in the infiltrative type. Malignant transformation has not been reported in lipomas of the hand [74, 88–90].
Malignant Soft Tissue Tumours
Soft Tissue Sarcomas
Table 5.4.
The management of soft-tissue sarcomas of the hand is controversial to both the hand surgeon and the oncologist. Wide surgical excision is often difficult to achieve in the hand because of the complex anatomy and the lack of readily expendable soft tissues [91]. Wide excision may also result in significant functional loss. Marginal excision, aiming to preserve the function can compromise the oncological outcome. Moreover, radiotherapy to the hand is poorly tolerated and could be associated with long term toxicity and poor functional outcome [92]. The importance of early diagnosis cannot therefore be overemphasized. Pradhan et al. [91], have shown that inadequate excision margins result in a 12 times greater risk of local recurrence, a three times greater risk of developing metastases and a five times greater risk of death than for those with clear margins. Bray et al. [92], and Jyothirmayi et al. [93], demonstrated that limb-salvage surgery, with adjuvant radiotherapy when necessary, is an effective alternative to amputation in the majority of patients with sarcoma of the forearm and hand. Post-operative radiotherapy to a dose of 55–60 Gy is well tolerated, with a low incidence of late toxicity [93, 94].
Local recurrence of soft tissue sarcomas is related to surgical margins, grade, and the use of postoperative radiotherapy. The overall 5-year survival rate for STS in the limbs is now being in the order of 65–75 % and is based on patient’s age, grade, depth, size, and histological diagnosis. Patients with intermediate/high grade tumours should be followed every 3–4 months in the first 2–3 years, then twice a year up to the fifth year, and once a year thereafter. It is recommended that patients with low grade tumours should be followed up every 4–6 months for 35 years, then annually thereafter [52, 71, 91, 95].
Approximately 5 % of soft tissue sarcomas occur in the hand region [94]. The most common histologic types are epithelioid sarcoma, clear cell sarcoma, synovial sarcoma, and malignant fibrous histiocytoma [91, 96, 97].
Clinical Pearl
-
En bloc excision with clear margins is the standard treatment for all patients with adult-type, localised STS. In some situations amputation may be the most appropriate surgical option to obtain local control and offer the best chance of cure. Postoperative radiotherapy should be considered in all cases of intermediate to high-grade STS despite the anticipated significant morbidity and poor functional outcome.
Epithelioid Sarcoma
Epithelioid sarcoma is one of the most common soft tissue sarcomas of the hand [52, 91, 96, 98–100]. It was first described by Enzinger in 1970 [101]. These tumours may represent up to 30 % of all malignant soft tissue tumours of the hand in the 16–25-year-old group [97]. Males are more commonly affected with a male-to-female ratio of 2:1 [52, 98]. The volar aspect of the fingers, hand, and wrist are most commonly involved. Epithelioid sarcoma usually presents as a painless, slowly growing mass or multiple palpable nodules [52, 96]. Ulceration of the epithelioid sarcoma is frequently seen, and thus can be initially misdiagnosed as an infection [52]. The macroscopic appearance of epithelioid sarcoma is that of multiple, ill defined, indurated, greyish-white nodules [52, 96, 102]. Histologically, epithelioid sarcoma has a characteristic nodular growth pattern and is composed of a mixed proliferation of eosinophilic epithelioid and spindle cells exhibiting slight nuclear atypia, vesicular nuclei, and small nucleoli [102]. Epithelioid sarcomas are highly malignant tumours that spread in an unpredictable fashion along the lymphatic system, tendons, nerve sheaths and fascial planes [52, 102]. The tumour also carries a high risk of local recurrence (35–70 %), regional and distant metastases (40 %) [97, 101, 102]. The most frequent sites of metastasis are the lungs, regional lymph nodes, scalp, bone, and brain [95, 102]. Aggressive surgical management with wide margins of excision is therefore indicated and amputation may be necessary in selected patients. Sentinel lymph node biopsy is recommended because of the frequent regional treatment failures. The roles of chemotherapy and radiation remain unclear [52, 96, 98, 100–102].
Synovial Sarcoma
Synovial sarcoma is a rare and aggressive soft tissue tumour that accounts for 5–10 % of all soft tissue sarcomas. They may represent 8–35 % of all soft tissue sarcomas of the hand and wrist, and are predominantly seen in males under the age of thirty [91, 97, 100, 103, 104].
Clinically, synovial sarcomas are similar to epithelioid sarcomas in that they can be initially misdiagnosed as benign lesions, owing to their usual presentation as painless, slowly-growing masses that have been present for months or even years. Focal or irregular calcifications may be visible on plain radiographs [52, 105].
Histologically, synovial sarcoma is either monophasic or biphasic. Biphasic synovial sarcoma has epithelial and spindle cell components while the spindle cell component occurs alone in the monophasic type [52, 105].
Synovial sarcomas are locally aggressive with unexplained natural history of late recurrence and distant metastasis (up to 30 years after diagnosis) [105, 106]. Complete surgical excision with negative margins is the treatment of choice. The difference between marginal and wide tumour resection appears to influence the outcome. Positive microscopic margins increases the risk of local recurrence associated with increased risk of metastatic spread and decreased disease free-survival. Radiotherapy is often required for local control of the disease but the role of adjuvant chemotherapy remains controversial [107, 108].
Clear Cell Sarcoma
Clear cell sarcoma is defined as a soft tissue sarcoma of young adults with melanocytic differentiation, typically involving tendons and aponeuroses [109]. These tumours usually present as a slowly growing mass that has been present for long duration of up to several years. Macroscopically, clear cell sarcomas have uniform fascicular growth pattern with lobular arrangement of cells delimited by delicate fibrous septa intimately bound to tendons or aponeuroses. Histologically, tumoral cells are round or spindle-shaped with abundant clear to slightly basophilic cytoplasm, with round vesicular nuclei and prominent nucleoli [109, 110]. Surgical excision is the preferred treatment modality. Tumour size, local recurrence and the presence of necrosis are statistically significant predictors of prognosis. Three-year, 5-year, and 20-year survival rates averaging 72, 62, and 10 % were reported in the literature [109–111].
Rhabdomyosarcoma
Rhabdomyosarcoma represents the largest category of soft tissue sarcomas in children and adolescents, accounting for more than 50 % of soft tissue sarcomas in this age group, with an annual incidence of 4–4.6 cases per million people under 15–20 years of age [112, 113]. The alveolar rhabdomyosarcoma is the type more commonly seen in the hand and more often occurring in adolescents and young adults (Fig. 5.7), while the embryonal subtype is typically seen in children less than 10 years of age [52, 112, 113].

(a–c) Alveolar Rhabdomyosarcoma of the hand in a 41-year-old male patient. a Posteroanterior radiograph, b Coronal fat-saturated T2-weighted MR image and c axial T1-weighted MR image demonstrate the extent of the lesion
Rhabdomyosarcoma is an aggressive tumour that tends to invade contiguous structures and becomes disseminated via the lymphatics and blood stream [112]. Histologically, embryonal rhabdomyosarcomas are composed of primitive mesenchymal cells in various stages of myogenesis, while the alveolar subtype exhibits round cell cytological features reminiscent of lymphomas but with primitive myoblastic differentiation [113, 114].
Patients are usually treated using a multimodality approach including surgery, chemotherapy, and radiotherapy. Radiotherapy should only be considered in patients at risk of local failure (e.g. marginal/incomplete resection, alveolar subtype) [112]. Due to the technical difficulties in achieving complete surgical resection without amputation in the hand region, there has been a recent report which recommends either definitive radiotherapy or surgical resection that maintains form and function as primary local therapy with no difference in the overall survival between the two groups [115].
Dermatofibrosarcoma Protuberans
Dermatofibrosarcoma protuberans (DFSP) is a rare fibroblastic slowly growing low-grade soft tissue sarcoma of the dermis layer of the skin. The lesion appears as a painless nodule that develops into a multinodular red-blue plaque. [52, 96] It is difficult to differentiate between DFSP, keloids, and hypertrophic scars on the basis of clinical presentation and therefore a biopsy is recommended. The locally infiltrative growth pattern features clinically inapparent extensions which often extend for long distances in a horizontal direction [116]. Consequently, the recurrence rate following excision of these tumours historically has been high, with reported recurrence rates of up to 60 % [117, 118]. Although adjuvant radiotherapy has been used in patients with positive margins, the preferred treatment modality is re-excision of the tumour aiming at a 3 cm margin. The use of Mohs micrographic surgery (MMS) has significantly decreased the recurrence rate of DFSP with a mean recurrence rate of 2.4 % reported in a literature review of 169 cases [116, 118–121].
Malignant Fibrous Histiocytoma
Malignant fibrous histiocytoma (MFH) is one of the three most prevalent histologic forms of soft tissue sarcomas in the hand [96]. MFH was found to represent 13 % of the distal upper extremity lesions in a large demographic study of malignant soft tissue tumours [97]. These are high grade malignant neoplasms that are best treated with wide local excision, combined with adjuvant radiotherapy. The role of chemotherapy remains controversial.
Malignant Skin Tumours
Table 5.5.
Squamous Cell Carcinoma
Squamous cell carcinoma (SCC) is the most common malignant skin tumour of the hand, representing 35–90 % of malignant hand tumours [122–125]. Multiple risk factors have been identified including ultraviolet radiation, immuno-compromised individuals, irradiated skin, human papillomavirus infection, chemical exposures, chronic wounds, previous burn injury, and certain genetic diseases such as xeroderma pigmentosum [126].
SCC of the hand appears to have higher rates of local recurrence, and metastasis, usually to regional lymph nodes. Schiavon et al. [124], reported a 22 % local recurrence rate, and a 28 % incidence of distant metastasis. Rayner [127] described the “danger zone” of the hand to include the dorsal skin of the proximal phalanges, interdigital clefts, and the first webspace. Tumours of this region of the hand are associated with an increased incidence of local recurrence and distant metastasis.
Many authors have recommended wide excisions with 1- to 2-cm margins. This radical treatment can lead to severe functional morbidity, undesirable cosmetic appearance with the potential need for amputation [126].
In order to minimize the functional morbidity caused by the wide surgical excision and to prevent the significant surgical injury to the surrounding tissues, other authors have favoured Mohs micrographic surgery (MMS) for treatment of cutaneous squamous and basal cell carcinomas [128]. MMS is a surgical technique that allows precise microscopic marginal control by using horizontal uninterrupted frozen sections. Joyner et al. [126], recommended marginal excision of squamous cell carcinoma of the hand with a 2- to 4-mm normal margin of tissue based on gross appearance by intraoperative evaluation with loupe magnification. Both techniques have been associated with 0 % incidence of local recurrence.
Keratoacanthoma
Keratoacanthoma is a relatively common low-grade malignant skin lesion of the head and neck. It seldom presents in the hand and may be difficult to differentiate from squamous cell carcinoma. Keratoacanthoma is characterized by rapid growth over a short period of time, followed by spontaneous resolution in most cases. Despite this spontaneous resolution, surgical excision is the treatment of choice and large or recurrent cases can be treated with Mohs micrographic surgery. Intralesional injections of methotrexate (MTX), bleomycin and steroids have been used with success in patients who are poor surgical candidates because of patient, tumour, or treatment-related factors [96, 129–133].
Subungual Keratoacanthoma is a rare benign neoplasm which most commonly occurs in middle-aged Caucasians [134]. It is a squamoproliferative neoplasm arising at the nail bed. Radiography consistently demonstrates a well-defined cup-shaped erosion of the underlying bone [134, 135]. Local surgical excision with curettage of the underlying bone is the preferred treatment modality of these rather aggressive lesions [134, 135].
Basal Cell Carcinoma
Basal cell carcinoma (BCC) is the most common overall cutaneous malignancy. Although extremely frequent elsewhere on the body, basal cell carcinomas are infrequent on the hands (<1 % of all basal cell carcinomas). Chronic sun exposure is the main etiologic factor but the rare occurrence in the hand cannot be explained [136, 137]. BCC is a slowly-growing tumour that patients tend to neglect for years. Males are more commonly affected and most patients would report a history of multiple skin malignancies. Metastatic spread is rare and local recurrence rate is influenced by the treatment method and the adequacy of the resection margins. Small lesions can be treated with curettage and electrodessication, laser therapy, or cryosurgery. Surgical excision with negative margins is the preferred treatment modality for BCC of the hand [96, 136, 137].
Melanoma
Melanoma is a highly malignant skin tumour that arises from melanocytes. Melanoma accounts for approximately 3 % of primary malignant hand tumours. Risk factors include ultraviolet radiation, fair skin, congenital nevi, multiple freckles, and positive family history of melanoma. These lesions have a variable clinical appearance and the diagnosis is often delayed resulting in more advanced stages of disease at presentation [138, 139]. Hove et al. [140], reported on 19 cases of melanoma of the hand with 21 % of these patients presenting with local spread of the disease (clinical stage II) and another 21 % presenting with regional lymph node metastases (clinical stage III).
Histologically, some features of the benign pigmented lesions of the palms may raise the suspicion of melanoma if considered alone. The presence of severe melanocytic atypia is possibly the most valuable feature in distinguishing between naevi and melanomas [141].
Most patients can be treated without radical amputations, even for melanomas of the digits. Roh et al. [139], found no significant difference in the survival rate between the patients treated by amputation versus wide local excision. Biopsy of all unexplained pigmented lesions on the hands can lead to early diagnosis thus improving the overall survival [140, 142, 143].
References
Gaulke R. The distribution of solitary enchondromata at the hand. J Hand Surg Br. 2002;27(5):444–5.
Löhrer L, Vordemvenne T, Neuber M, Krueger KU, Schult M, Ochman S. Enchondroma of the hand - treatment and long-term outcome. Z Orthop Unfall. 2010;148(6):709–15.
Machens HG, Brenner P, Wienbergen H, Pallua N, Mailänder P, Berger A. Enchondroma of the hand. Clinical evaluation study of diagnosis, surgery and functional outcome. Unfallchirurg. 1997;100(9):711–4.
Grünert J, Strobel M, Brug E. Enchondroma of the hand. Z Orthop Ihre Grenzgeb. 1995;133(2):180–6.
Kotnis NA, Davies AM, James SLJ. Hand and wrist. In: Davies AM, Sundaram M, James SLJ, editors. Imaging of bone tumors and tumor-like lesions: techniques and applications. Berlin: Springer; 2009. p. 621–36.
Lucas DR, Bridge JA. Chondromas: enchondroma, periosteal chondroma, and enchondromatosis. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 237–40.
Morii T, Mochizuki K, Tajima T, Satomi K. Treatment outcome of enchondroma by simple curettage without augmentation. J Orthop Sci. 2010;15(1):112–7.
Schaller P, Baer W. Operative treatment of enchondromas of the hand: is cancellous bone grafting necessary? Scand J Plast Reconstr Surg Hand Surg. 2009;43(5):279–85.
Gaulke R, Suppelna G, Hildebrand F, Brand J, Hüfner T, Krettek C. Recurrence of solitary enchondroma at the hand after operative treatment. Handchir Mikrochir Plast Chir. 2008;40(5):304–9.
O’Connor MI, Bancroft LW. Benign and malignant cartilage tumors of the hand. Hand Clin. 2004;20(3):317–23, vi.
D’Angelo L, Massimi L, Narducci A, Di Rocco C. Ollier disease. Childs Nerv Syst. 2009;25(6):647–53.
Bertucci V, Krafchik BR. What syndrome is this? Ollier disease+vascular lesions: maffucci syndrome. Pediatr Dermatol. 1995;12(1):55–8.
Nora FE, Dahlin DC, Beabout JW. Bizarre parosteal osteochondromatous proliferations of the hands and feet. Am J Surg Pathol. 1983;7(3):245–50.
Gursel E, Jarrahnejad P, Arneja JS, Malamet M, Akinfolarin J, Chang YJ. Nora’s lesion: case report and literature review of a bizarre parosteal osteochondromatous proliferation of a small finger. Can J Plast Surg. 2008;16(4):232–5.
Berber O, Dawson-Bowling S, Jalgaonkar A, Miles J, Pollock RC, Skinner JA, Aston WJ, Briggs TW. Bizarre parosteal osteochondromatous proliferation of bone: clinical management of a series of 22 cases. J Bone Joint Surg Br. 2011;93(8):1118–21.
Efstathopoulos NE, Papagelopoulos PJ, Lazarettos IT, Savvidou OD, Kaseta MA, Giannakou N, Papachristou GK. Bizarre parosteal osteochondromatous proliferation of the second metatarsal bone (Nora’s lesion). Orthopedics. 2005;28(2):168–70.
Torreggiani WC, Munk PL, Al-Ismail K, O’Connell JX, Nicolaou S, Lee MJ, Masri BA. MR imaging features of bizarre parosteal osteochondromatous proliferation of bone (Nora’s lesion). Eur J Radiol. 2001;40(3):224–31.
Michelsen H, Abramovici L, Steiner G, Posner MA. Bizarre parosteal osteochondromatous proliferation (Nora’s lesion) in the hand. J Hand Surg Am. 2004;29(3):520–5.
Gruber G, Giessauf C, Leithner A, Zacherl M, Clar H, Bodo K, Windhager R. Bizarre parosteal osteochondromatous proliferation (Nora lesion): a report of 3 cases and a review of the literature. Can J Surg. 2008;51(6):486–9.
Meneses MF, Unni KK, Swee RG. Bizarre parosteal osteochondromatous proliferation of bone (Nora’s lesion). Am J Surg Pathol. 1993;17(7):691–7.
Bilgin SS, Yildiz Y, Güçlü B, Sağlik Y. Osteoid osteoma of the hand: an evaluation of eight patients. Acta Orthop Traumatol Turc. 2004;38(3):206–11.
Burger IM, McCarthy EF. Phalangeal osteoid osteomas in the hand: a diagnostic problem. Clin Orthop Relat Res. 2004;427:198–203.
Plate AM, Lee SJ, Steiner G, Posner MA. Tumorlike lesions and benign tumors of the hand and wrist. J Am Acad Orthop Surg. 2003;11(2):129–41. Review.
Ostrowski ML, Spjut HJ. Lesions of the bones of the hands and feet. Am J Surg Pathol. 1997;21(6):676–90.
Harrod CC, Boykin RE, Jupiter JB. Pain and swelling after radiofrequency treatment of proximal phalanx osteoid osteoma: case report. J Hand Surg Am. 2010;35(6):990–4. Epub 2010 May 7.
Ramos L, Santos JA, Santos G, Guiral J. Radiofrequency ablation in osteoid osteoma of the finger. J Hand Surg Am. 2005;30(4):798–802.
Malcolm AJ, Schiller AL, Schneider-Sock R. Osteoblastoma. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 262–3.
Herrera Tenorio JG, Núnuez Fernández AI, Mendoza Quiroga JJ, Sesma Villalpando RA. Benign osteoblastoma in the proximal phalanx. A case report. Acta Ortop Mex. 2009;23(5):298–301.
Meade RA, Allende CA, Tsai TM. Osteoblastoma of the scaphoid: a case report. J Surg Orthop Adv. 2005;14(3):125–8.
Ragois P, Leclerc P, Hallonet D. Aggressive osteoblastoma of the carpal scaphoid bone. Rev Chir Orthop Reparatrice Appar Mot. 2000;86(1):94–7.
van Dijk M, Winters HA, Wuisman PI. Recurrent osteoblastoma of the hamate bone. A two-stage reconstruction with a free vascularized iliac crest flap. J Hand Surg Br. 1999;24(4):501–5.
Rosenberg AE, Nielsen GP, Fletcher JA. Aneurysmal bone cyst. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 338–9.
Başarir K, Saglik Y, Yildiz Y, Tezen E. Aneurysmal bone cyst of the hand: a report of four cases. Hand Surg. 2006;11(1–2):35–41.
Barbieri CH. Aneurysmal bone cyst of the hand. An unusual situation. J Hand Surg Br. 1984;9(1):89–92.
Frassica FJ, Amadio PC, Wold LE, Beabout JW. Aneurysmal bone cyst: clinicopathologic features and treatment of ten cases involving the hand. J Hand Surg Am. 1988;13(5):676–83.
Bogar WC. Aneurysmal bone cyst of the hand. J Manipulative Physiol Ther. 1993;16(3):182–4.
Dahlin DC. Giant-cell-bearing lesions of bone of the hands. Hand Clin. 1987;3(2):291–7.
Ropars M, Kaila R, Briggs T, Cannon S. Aneurysmal bone cysts of the metacarpals and phalanges of the hand. A 6 case series and literature review. Chir Main. 2007;26(4–5):214–7.
Adamsbaum C, Leclet H, Kalifa G. Intralesional ethibloc injections in bone cysts. Semin Musculoskelet Radiol. 1997;1(2):301–4.
Szendröi M, Antal I, Liszka G, Kónya A. Calcitonin therapy of aneurysmal bone cysts. J Cancer Res Clin Oncol. 1992;119(1):61–5.
Reid R, Banerjee SS, Sciot R. Giant cell tumour. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 310–2.
Ropars M, Kaila R, Cannon SR, Briggs TW. Primary giant cell tumours of the digital bones of the hand. J Hand Surg Eur Vol. 2007;32(2):160–4.
Athanasian EA, Wold LE, Amadio PC. Giant cell tumors of the bones of the hand. J Hand Surg Am. 1997;22(1):91–8.
Averill RM, Smith RJ, Campbell CJ. Giant-cell tumors of the bones of the hand. J Hand Surg Am. 1980;5(1):39–50.
Uppin SG, Sundaram C, Umamahesh M, Chandrashekar P, Rani YJ, Prasad VB. Lesions of the bones of the hands and feet: a study of 50 cases. Arch Pathol Lab Med. 2008;132(5):800–12.
James SL, Davies AM. Giant-cell tumours of bone of the hand and wrist: a review of imaging findings and differential diagnoses. Eur Radiol. 2005;15(9):1855–66.
Wold LE, Swee RG. Giant cell tumor of the small bones of the hands and feet. Semin Diagn Pathol. 1984;1(3):173–84.
Biscaglia R, Bacchini P, Bertoni F. Giant cell tumor of the bones of the hand and foot. Cancer. 2000;88(9):2022–32.
Wittig JC, Simpson BM, Bickels J, Kellar-Graney KL, Malawer MM. Giant cell tumor of the hand: superior results with curettage, cryosurgery, and cementation. J Hand Surg Am. 2001;26(3):546–55.
Grimer RJ, Hogendoorn PCW, Vanel D. Tumours of bone: introduction. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO classification of tumors of soft tissue and bone. Lyon: IARC Press; 2013. p. 244–7.
Tos P, Artiaco S, Linari A, Battiston B. Chondrosarcoma in the distal phalanx of index finger: clinical report and literature review. Chir Main. 2009;28(4):265–9.
Plate AM, Steiner G, Posner MA. Malignant tumors of the hand and wrist. J Am Acad Orthop Surg. 2006;14(12):680–92.
Nigrisoli M, Ferraro A, De Cristofaro R, Picci P. Chondrosarcoma of the hand and foot. Chir Organi Mov. 1990;75(4):315–23.
Mittermayer F, Dominkus M, Krepler P, Schwameis E, Sluga M, Toma C, Lang S, Grampp S, Kotz R. Chondrosarcoma of the hand: is a wide surgical resection necessary? Clin Orthop Relat Res. 2004;424:211–5.
Palmieri TJ. Chondrosarcoma of the hand. J Hand Surg Am. 1984;9(3):332–8.
Bertoni F, Bacchini P, Hogendoorn PCW. Chondrosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 247–51.
Gohla T, van Schoonhoven J, Prommersberger KJ, Lanz U. Chondrosarcomas of the hand. Handchir Mikrochir Plast Chir. 2004;36(5):328–32.
Baumhoer D, Jundt G. Tumours of the hand: a review on histology of bone malignancies. J Hand Surg Eur Vol. 2010;35(5):354–61. Epub 2010 Feb 24. Review.
Hatori M, Watanabe M, Kotake H, Kokubun S. Chondrosarcoma of the ring finger: a case report and review of the literature. Tohoku J Exp Med. 2006;208(3):275–81.
Fowble VA, Pae R, Vitale A, Bryk E, Vigorita VJ. Case reports: osteosarcoma of the hand: one case and a literature review. Clin Orthop Relat Res. 2005;440:255–61.
Okada K, Wold LE, Beabout JW, Shives TC. Osteosarcoma of the hand. A clinicopathologic study of 12 cases. Cancer. 1993;72(3):719–25.
Escobedo EM, Bjorkengren AG, Moore SG. Ewing’s sarcoma of the hand. AJR Am J Roentgenol. 1992;159(1):101–2.
Anakwenze OA, Parker WL, Wold LE, Amrami KK, Amadio PC. Ewing’s sarcoma of the hand. J Hand Surg Eur Vol. 2009;34(1):35–9. Epub 2008 Dec 17.
Daecke W, Ahrens S, Juergens H, Martini AK, Ewerbeck V, Kotz R, Winkelmann W, Bernd L. Ewing’s sarcoma and primitive neuroectodermal tumor of hand and forearm. Experience of the Cooperative Ewing’s Sarcoma Study Group. J Cancer Res Clin Oncol. 2005;131(4):219–25.
Kerin R. The hand in metastatic disease. J Hand Surg Am. 1987;12(1):77–83.
Kumar PP, Kovi J. Metastases to bones of the hands and feet. J Natl Med Assoc. 1978;70(11):837–40.
Vijayakumar S, Creditor M. Metastasis to the hand. J Natl Med Assoc. 1986;78(5):441–2.
Healey JH, Turnbull AD, Miedema B, Lane JM. Acrometastases. A study of twenty-nine patients with osseous involvement of the hands and feet. J Bone Joint Surg Am. 1986;68(5):743–6.
Spiteri V, Bibra A, Ashwood N, Cobb J. Managing acrometastases treatment strategy with a case illustration. Ann R Coll Surg Engl. 2008;90(7):W8–11.
Jung ST, Ghert MA, Harrelson JM, Scully SP. Treatment of osseous metastases in patients with renal cell carcinoma. Clin Orthop. 2003;409:223–31.
Grimer R, Judson I, Peake D, Seddon B. Guidelines for the management of soft tissue sarcomas. Sarcoma. 2010;2010:506182.
Lincoski CJ, Bush DC, Millon SJ. Epidermoid cysts in the hand. J Hand Surg Eur Vol. 2009;34(6):792–6.
Sobanko JF, Dagum AB, Davis IC, Kriegel DA. Soft tissue tumors of the hand. 1. Benign. Dermatol Surg. 2007;33(6):651–67.
Wong CH, Chow L, Yen CH, Ho PC, Yip R, Hung LK. Uncommon hand tumours. Hand Surg. 2001;6(1):67–80.
Lucas GL. Epidermoid inclusion cysts of the hand. J South Orthop Assoc. 1999;8(3):188–92.
Simon K, Leithner A, Bodo K, Windhager R. Intraosseous epidermoid cysts of the hand skeleton: a series of eight patients. J Hand Surg Eur Vol. 2011;36(5):376–8.
Spingardi O, Zoccolan A, Venturino E. Infantile digital fibromatosis: our experience and long-term results. Chir Main. 2011;30(1):62–5. Epub 2011 Jan 12.
Grenier N, Liang C, Capaldi L, Ney A, Lapidus C, Schappell D, Katarincic J, Robinson-Bostom L. A range of histologic findings in infantile digital fibromatosis. Pediatr Dermatol. 2008;25(1):72–5.
Hayashi T, Tsuda N, Chowdhury PR, Anami M, Kishikawa M, Iseki M, Kobayashi K. Infantile digital fibromatosis: a study of the development and regression of cytoplasmic inclusion bodies. Mod Pathol. 1995;8(5):548–52.
Kawaguchi M, Mitsuhashi Y, Hozumi Y, Kondo S. A case of infantile digital fibromatosis with spontaneous regression. J Dermatol. 1998;25(8):523–6.
Plaza JA, Mayerson J, Wakely Jr PE. Nodular fasciitis of the hand: a potential diagnostic pitfall in fine-needle aspiration cytopathology. Am J Clin Pathol. 2005;123(3):388–93.
Hara H, Fujita I, Fujimoto T, Hanioka K, Akisue T, Kurosaka M. Nodular fasciitis of the hand in a young athlete. A case report. Ups J Med Sci. 2010;115(4):291–6. Epub 2010 Sep 16.
Rankin G, Kuschner SH, Gellman H. Nodular fasciitis: a rapidly growing tumor of the hand. J Hand Surg Am. 1991;16(5):791–5.
Smith PS, Pieterse AS, McClure J. Fibroma of tendon sheath. J Clin Pathol. 1982;35(8):842–8.
Farshid G, Bridge JA. Fibroma of tendon sheath. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 205–7.
Chung EB, Enzinger FM. Fibroma of tendon sheath. Cancer. 1979;44(5):1945–54.
Fox MG, Kransdorf MJ, Bancroft LW, Peterson JJ, Flemming DJ. MR imaging of fibroma of the tendon sheath. AJR Am J Roentgenol. 2003;180(5):1449–53.
Al-Qattan MM, Al-Lazzam AM, Al Thunayan A, Al Namlah A, Mahmoud S, Hashem F, Tulbah A. Classification of benign fatty tumours of the upper limb. Hand Surg. 2005;10(1):43–59.
Lee YH, Jung JM, Baek GH, Chung MS. Intramuscular lipoma in thenar or hypothenar muscles. Hand Surg. 2004;9(1):49–54.
Fletcher CD, Martin-Bates E. Intramuscular and intermuscular lipoma: neglected diagnoses. Histopathology. 1988;12(3):275–87.
Pradhan A, Cheung YC, Grimer RJ, Peake D, Al-Muderis OA, Thomas JM, Smith M. Soft-tissue sarcomas of the hand: oncological outcome and prognostic factors. J Bone Joint Surg Br. 2008;90(2):209–14.
Bray PW, Bell RS, Bowen CV, Davis A, O’Sullivan B. Limb salvage surgery and adjuvant radiotherapy for soft tissue sarcomas of the forearm and hand. J Hand Surg Am. 1997;22(3):495–503.
Jyothirmayi R, Sittampalam Y, Harmer C. Soft tissue sarcoma of the hand or foot: conservative surgery and radiotherapy. Sarcoma. 1999;3(1):17–24.
Terek RM, Brien EW. Soft-tissue sarcomas of the hand and wrist. Hand Clin. 1995;11(2):287–305.
Casali PG, Jost L, Sleijfer S, Verweij J, Blay JY. Soft tissue sarcomas: ESMO clinical recommendations for diagnosis, treatment and follow-up. ESMO Guidelines Working Group. Ann Oncol. 2008;19 Suppl 2:ii89–93.
Sobanko JF, Dagum AB, Davis IC, Kriegel DA. Soft tissue tumors of the hand. 1. Malignant. Dermatol Surg. 2007;33(7):771–85.
Kransdorf MJ. Malignant soft-tissue tumors in a large referral population: distribution of diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995;164(1):129–34.
Herr MJ, Harmsen WS, Amadio PC, Scully SP. Epithelioid sarcoma of the hand. Clin Orthop Relat Res. 2005;431:193–200.
Bryan RS, Soule EH, Dobyns JH, Pritchard DJ, Linscheid RL. Primary epithelioid sarcoma of the hand and forearm. A review of thirteen cases. J Bone Joint Surg Am. 1974;56(3):458–65.
Brien EW, Terek RM, Geer RJ, Caldwell G, Brennan MF, Healey JH. Treatment of soft-tissue sarcomas of the hand. J Bone Joint Surg Am. 1995;77(4):564–71.
Enzinger FM. Epitheloid sarcoma. A sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26(5):1029–41.
Guillou L, Kaneko Y. Epithelioid sarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 205–7.
Buecker PJ, Villafuerte JE, Hornicek FJ, Gebhardt MC, Mankin HJ. Improved survival for sarcomas of the wrist and hand. J Hand Surg Am. 2006;31(3):452–5.
Dreyfuss UY, Boome RS, Kranold DH. Synovial sarcoma of the hand–a literature study. J Hand Surg Br. 1986;11(3):471–4.
Fisher C, de Bruijn DRH, Geurts van Kessel A. Synovial sarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 200–4.
Dick HM. Synovial sarcoma of the hand. Hand Clin. 1987;3(2):241–5.
Andrassy RJ, Okcu MF, Despa S, Raney RB. Synovial sarcoma in children: surgical lessons from a single institution and review of the literature. J Am Coll Surg. 2001;192(3):305–13.
Lewis JJ, Antonescu CR, Leung DH, Blumberg D, Healey JH, Woodruff JM, Brennan MF. Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J Clin Oncol. 2000;18(10):2087–94.
Sciot R, Speleman F. Clear cell sarcoma of soft tissue. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 211–2.
Marquès B, Terrier P, Voigt JJ, Mihura J, Coindre JM. Clear cell soft tissue sarcoma. Clinical, histopathological and prognostic study of 36 cases. Ann Pathol. 2000;20(4):298–303.
Lucas DR, Nascimento AG, Sim FH. Clear cell sarcoma of soft tissues. Mayo clinic experience with 35 cases. Am J Surg Pathol. 1992;16(12):1197–204.
Casanova M, Meazza C, Favini F, Fiore M, Morosi C, Ferrari A. Rhabdomyosarcoma of the extremities: a focus on tumors arising in the hand and foot. Pediatr Hematol Oncol. 2009;26(5):321–31.
Parham DM, Barr FG. Embryonal rhabdomyosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 146–9.
Parham DM, Barr FG. Alveolar rhabdomyosarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. WHO classification of tumors. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 150–2.
La TH, Wolden SL, Su Z, Linardic C, Randall RL, Hawkins DS, Donaldson SS. Local therapy for Rhabdomyosarcoma of the hands and feet: is amputation necessary? A report from the Children’s Oncology Group. Int J Radiat Oncol Biol Phys. 2011;80(1):206–12.
Breuninger H, Sebastian G, Garbe C. Dermatofibrosarcoma protuberans–an update. J Dtsch Dermatol Ges. 2004;2(8):661–7.
Smola MG, Soyer HP, Scharnagl E. Surgical treatment of dermatofibrosarcoma protuberans. A retrospective study of 20 cases with review of literature. Eur J Surg Oncol. 1991;17(5):447–53.
Mark RJ, Bailet JW, Tran LM, Poen J, Fu YS, Calcaterra TC. Dermatofibrosarcoma protuberans of the head and neck. A report of 16 cases. Arch Otolaryngol Head Neck Surg. 1993;119(8):891–6.
Nouri K, Lodha R, Jimenez G, Robins P. Mohs micrographic surgery for dermatofibrosarcoma protuberans: University of Miami and NYU experience. Dermatol Surg. 2002;28(11):1060–4; discussion 1064.
Haycox CL, Odland PB, Olbricht SM, Casey B. Dermatofibrosarcoma protuberans (DFSP): growth characteristics based on tumor modeling and a review of cases treated with Mohs micrographic surgery. Ann Plast Surg. 1997;38(3):246–51.
Ah-Weng A, Marsden JR, Sanders DS, Waters R. Dermatofibrosarcoma protuberans treated by micrographic surgery. Br J Cancer. 2002;87(12):1386–9. Review.
TerKonda SP, Perdikis G. Non-melanotic skin tumors of the upper extremity. Hand Clin. 2004;20(3):293–301, vi.
Chakrabarti I, Watson JD, Dorrance H. Skin tumors of the hand: a 10-year review. J Hand Surg. 1992;18-B:484–6.
Schiavon M, Mazzoleni F, Chiarelli A, Matano P. Squamous cell carcinoma of the hand: fifty-five case reports. J Hand Surg Am. 1988;13(3):401–4.
Fink JA, Akelman E. Nonmelanotic malignant skin tumors of the hand. Hand Clin. 1995;11(2):255–64.
Joyner KS, Wilson B, Wagner RF, Viegas SF. Marginal excision of squamous cell carcinomas of the hand. Orthopedics. 2008;31(1):79.
Rayner CR. The results of treatment of two hundred and seventy-three carcinomas of the hand. Hand. 1981;13:183–6.
Wagner RF, Cottel WI. Treatment of squamous and basal cell carcinomas of the hand with Mohs micrographic surgery: analysis of 53 consecutive patients. Skin Cancer. 1989;4:73–7.
Virnelli FR. Keratoacanthoma: excision or spontaneous resolution? J Dermatol Surg Oncol. 1982;8(3):162. 221.
Larson PO. Keratoacanthomas treated with Mohs’ micrographic surgery (chemosurgery). a review of forty-three cases. J Am Acad Dermatol. 1987;16(5 Pt 1):1040–4.
Patel NP, Cervino AL. Treatment of keratoacanthoma: is intralesional methotrexate an option? Can J Plastic Surg. 2011;19(2):e15–8.
Sayama S, Tagami H. Treatment of keratoacanthoma with intralesional bleomycin. Br J Dermatol. 1983;109(4):449–52.
Sanders S, Busam KJ, Halpern AC, Nehal KS. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28(10):954–8.
Oliwiecki S, Peachey RD, Bradfield JW, Ellis J, Lovell CR. Subungual keratoacanthoma–a report of four cases and review of the literature. Clin Exp Dermatol. 1994;19(3):230–5.
Choi JH, Shin DH, Shin DS, Cho KH. Subungual keratoacanthoma: ultrasound and magnetic resonance imaging findings. Skeletal Radiol. 2007;36(8):769–72. Epub 2007 Mar 23.
van Zuuren EJ, Bastiaens MT, Posma AN, Bouwes Bavinck JN. Basal cell carcinoma on the dorsum of the hand: report of 11 cases. J Eur Acad Dermatol Venereol. 2000;14(4):307–10.
Vandeweyer E, Herszkowicz A. Basal cell carcinoma of the dorsum of the hand. Acta Chir Belg. 2003;103(3):300–3.
Perdikis G, TerKonda SP. Pigmented skin lesions of the upper extremity. Hand Clin. 2004;20(3):283–91, vi.
Roh MR, Kim J, Chung KY. Treatment and outcomes of melanoma in acral location in Korean patients. Yonsei Med J. 2010;51(4):562–8.
Hove LM, Akslen LA. Clinicopathological characteristics of melanomas of the hand. J Hand Surg Br. 1999;24(4):460–4.
Fallowfield ME, Collina G, Cook MG. Melanocytic lesions of the palm and sole. Histopathology. 1994;24(5):463–7.
Warso M, Gray T, Gonzalez M. Melanoma of the hand. J Hand Surg Am. 1997;22(2):354–60.
Hughes LE, Horgan K, Taylor BA, Laidler P. Malignant melanoma of the hand and foot: diagnosis and management. Br J Surg. 1985;72(10):811–5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag London
About this chapter
Cite this chapter
Wafa, H.Y.M., Tillman, R.M. (2015). Tumours of the Hand. In: Trail, I., Fleming, A. (eds) Disorders of the Hand. Springer, London. https://doi.org/10.1007/978-1-4471-6563-7_5
Download citation
DOI: https://doi.org/10.1007/978-1-4471-6563-7_5
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-6562-0
Online ISBN: 978-1-4471-6563-7
eBook Packages: MedicineMedicine (R0)