
Abstract
L-3,4-dihydroxyphenylalanine (l-DOPA) treatment in Parkinson’s disease (PD) patients commonly leads to dyskinesia, a hyperkinetic movement disorder that remains an unsolved clinical problem. The unravelling of key pathophysiological mechanisms in PD and dyskinesia has led to updated models of the basal ganglia motor circuit, capturing nonlinear neuronal information processing in a dynamical network architecture. Our understanding into the functional organization of the basal ganglia motor system is further supported by recent computational models that focus on neuronal activations within distinct closed feedback loops. Together, these models of the basal ganglia circuitry compose a more comprehensive and detailed insight into the diverse neuronal dysfunctions in the pathophysiology of PD and LID.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Parkinson’s Disease and Levodopa-Induced Dyskinesia
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that affects approximately 1 % of the population over the age of 55 years, with highest prevalence in ages of 85 years and over [1]. PD is commonly characterized by a clinical syndrome of motor symptoms (bradykinesia, postural abnormalities, and resting tremor) [2] that occur due to an extensive loss of nigrostriatal neurons which release dopamine [3], a modulatory neurotransmitter of the basal ganglia motor circuit [4].
In the early 1960s, studies showed that dopamine replacement with its immediate metabolic precursor, l-3,4-dihydroxyphenylalanine (l-DOPA), dramatically alleviated PD motor symptoms [5, 6]. Following this, l-DOPA was introduced to PD patients [7] and has since been widely used for the treatment of PD. However, following long-term use of l-DOPA, the initial beneficial effects of treatment are compromised by unpredictable “on-off” fluctuations of therapeutic effects [8, 9], gradual “wearing off” of therapeutic efficacy [10, 11], and l-DOPA-induced dyskinesia (LID) [12]. The latter is a severe hyperkinetic motor complication that is commonly expressed as an idiosyncratic mixture of chorea (irregular flow of muscular movements in rapid and slow phases) and dystonia (slow twisted movements from abnormal muscular contractions) [13]. LID occurs in approximately 90 % of PD patients after 9 years of l-DOPA treatment [14], and once established, dyskinesia is elicited upon each administration of l-DOPA, or dopamine agonist [15]. Moreover, LID increases in severity with further l-DOPA treatment [16] and can become as debilitating as PD itself, causing a negative impact on quality of life [17].
Understanding the pathophysiology of LID is an important step in developing a suitable treatment that can resolve the clinical need of treating dyskinesia. In this review, we discuss the pathophysiology of PD and LID using the basal ganglia circuitry model of the motor circuit. We also describe a recent computational model that demonstrates subtle dysfunctions in neural processing within the basal ganglia following the loss of dopamine. In addition, we highlight recent experimental findings of molecular adaptations that occur in the nuclei outside of the basal ganglia, which may have important roles in the expression of LID.
Basal Ganglia
The basal ganglia are a group of subcortical nuclei that include the striatum (caudate nucleus and putamen), subthalamic nucleus (STN), substantia nigra (pars reticulata, SNr, and pars compacta, SNc), ventral tegmental area, and globus pallidus (internal, GPi, and external, GPe, segments) [18]. These interconnected nuclei are modulated by dopamine and together form a neural network that relays information from the cortex to the thalamus. These so-called corticobasal ganglia-thalamocortical loops functionally convey information for both motor and non-motor processes [4]. Several of these loops exist for motor, oculomotor, associative, limbic, and orbitofrontal functions. Moreover, each loop projects from largely segregated regions of the basal ganglia and thalamus to different cortical target areas of the cerebral hemisphere [19].
Classic “Input” and “Output” Stations of the Basal Ganglia
The striatum is a major input station of the basal ganglia receiving different afferent projections, which include dopaminergic fibers from the midbrain [20], serotonergic fibers from dorsal and medial raphe nucleus [21], noradrenergic fibers from the locus coeruleus [22], acetylcholinergic fibers from the pedunculopontine nucleus, and glutamatergic fibers from the thalamus, STN, and cortex [23, 24]. The glutamatergic fibers of the cortex project massively to the striatum in a somatotopically organized manner [25–27]. In the motor cortico-basal ganglia-thalamo-cortical loop of the primate brain, sensorimotor afferents of the primary motor and somatosensory cortices project to the posterolateral putamen [26, 28]. Here, the dorsal region is occupied by somatotopic representation of the leg, which is followed by the arm, while the facial representation lays most ventral [28]. The putamen projects via γ-aminobutyric acid (GABA)-ergic medium spiny neurons (MSNs) to the GPi/SNr [29, 30], which are the output nuclei of the basal ganglia. These nuclei send GABAergic efferent neurons to the motor nuclei of the thalamus (ventralis anterior and lateralis) and brain stem [19, 31–33]. In turn, the motor nuclei convey excitatory glutamatergic projections to motor-related cortical areas, completing the motor corticobasal ganglia-thalamocortical loop [34–37].
Striatal “Direct” and “Indirect” Pathways
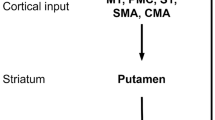
In the 1980s, a model of the basal ganglia circuitry was proposed based on the available anatomical, neurochemical, and electrophysiological data (see Fig. 7.1a) [19, 32]. This now “classic” model describes two main efferent projections from the striatum to the output of the basal ganglia, the so-called direct and indirect pathways. The direct pathway refers to the monosynaptic neuronal connection between the striatum and GPi/SNr. The neurons of this pathway primarily express dopamine D1 receptors and preproenkephalin-B (PPE-B), an opioid peptide that is subsequently cleaved to produce co-transmitters substance P, dynorphins, leucine-enkephalins, and α-neoendorphin [38]. The “indirect” pathway describes the polysynaptic neuronal connection of the striatum to GPi/SNr. Striatofugal neurons of this pathway project to the GPe and, in turn, the GPe sends GABAergic efferent fibers to the STN. From here, glutamatergic efferent fibers of the STN project to the GPi/SNr. The striatopallidal neurons of the indirect pathway primarily express dopamine D2 receptors and preproenkephalin-A (PPE-A), an opioid peptide that is subsequently cleaved to enkephalin [38]. Both the direct and indirect pathways are modulated by dopamine, which activates striatonigral neurons of the direct pathway and inhibits striatopallidal neurons of the indirect pathway (see Fig. 7.1a).


Schematic diagrams of the classic basal ganglia circuitry model illustrating linear, feed-forward information processing. Dopamine mediates opposing functional effects on the two major projection pathways of the striatum for (a) normal motor function. Loss of endogenous dopamine in (b) Parkinson’s disease (PD) causes abnormal neuronal activity leading to reduced excitatory feedback to the cortex. Repeated treatment with l-DOPA in PD induces (c) dyskinesia, causing increased activity in the cortex. Arrow size corresponds to activity of neuronal projections. l-DOPA L-3,4-dihydroxyphenylalanine, D 1 R dopamine D1 receptor, D 2 R dopamine D2 receptor, Enk enkephalin, Dyn prodynorphin, STR striatum, GPi internal segment of the globus pallidus, GPe external segment of the globus pallidus, STN subthalamic nucleus, SNr substantia nigra pars reticulata, SNc substantia nigra pars compacta, VA/VL ventralis anterior and lateralis nuclei
Based on segregated pathways, the classic functional model of the basal ganglia describes the processing of neural information in a feed-forward manner for achieving a behavioral outcome. Using the motor circuit as an example, it was suggested that the direct pathway facilitates the execution of desired motor sequences, while the indirect pathway mediates blocking of unwanted motor programs to “smooth” cortical-initiated motor sequences [4, 39–41]. Both the direct and indirect pathways lead to inhibition of the basal ganglia output nuclei for normal motor function. Accordingly, electrophysiological studies of saccadic eye and limb movements in awake monkeys have shown GPi/SNr neurons are tonically active (50–100 Hz) during rest and exhibit reduced activity during movement [42–45].
Basal Ganglia Circuitry in Parkinson’s Disease
The classic model of the basal ganglia circuitry has been used to describe the pathophysiology of PD (see Fig. 7.1b) [32, 33, 46]. From this model, PD motor symptoms occur as a result of an imbalance between direct and indirect pathways caused by extensive degeneration of nigrostriatal dopaminergic neurons. While striatonigral neurons of the direct pathway become underactive, striatopallidal neurons of the indirect pathway become overactive leading to inhibition of GPe and subsequent disinhibition of the glutamatergic efferent fibers of the STN [32]. Thus, with loss of dopamine, both pathways lead to increased activation of the GPi/SNr, thereby inhibiting the motor thalamic nuclei. The resulting effect is reduced activation of motor cortical areas, which is seen to occur in the primary sensory motor cortex [47] and supplementary motor area [48] in the parkinsonian state.
In the late 1980s, several groundbreaking studies were conducted that helped uncover key mechanisms in the pathophysiology of PD. In these experiments conducted by Mitchell et al., neuronal metabolic marker 2-deoxyglucose (2-DG) was used to reveal the activity states of the basal ganglia subnuclei in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned nonhuman primate (nhp) model of PD. It was found that the STN was hyperactivated, while the GPe, thalamic ventralis anterior, and lateralis nuclei were hyper-inhibited [49–51]. Accordingly, these major discoveries suggested that there was hyperactivation of the basal ganglia output structures in PD [51], which was later confirmed through measurements of electrophysiological activity [52–54] and mRNA expression of neuronal activity marker, cytochrome oxidase subunit I [55]. Additionally, the activation states of the striatofugal pathways in PD have been demonstrated through the expression of striatal PPE precursors, where reports have consistently shown reduced PPE-B and increased PPE-A mRNA expression in the striatum [56–58]. These molecular data demonstrate underactivation of striatonigral neurons and hyperactivation of striatopallidal neurons, which both favor the overactivation of the basal ganglia output nuclei in PD.
Following the original neuronal metabolic activity studies, behavioral experiments carried out in MPTP-lesioned nhps further characterized the pathophysiological changes in PD. Fundamentally, these studies revealed the causal role of the STN in production of parkinsonian motor symptoms [59, 60], which were dramatically abolished following surgical or neurochemical (muscimol or kainic acid) lesion of this structure. At the cellular level, subthalamotomy was also shown to reduce the overactivation of the basal ganglia output nuclei in PD [61, 62]. Collectively, these revolutionary findings led to a resurgence of neurosurgical procedures for the treatment of parkinsonism, which included the ablation of the GPi [63, 64] or STN [65], and deep brain stimulation (DBS) of the STN [66–68].
Basal Ganglia Circuitry in Levodopa-Induced Dyskinesia
Early suggestions put forward on the pathophysiology of LID essentially described the opposite functional state to that of PD (see Fig. 7.1c) [69]. Particular emphasis was originally placed on the indirect pathway in the pathogenesis of the dyskinetic state, where it was proposed that the underactivation of the striatopallidal neurons caused disinhibition of the GPe, leading to subsequent over-inhibition of the STN. In turn, disinhibition of thalamic motor nuclei resulted in excessive excitatory input to motor cortical areas, which is found to occur in PD patients expressing LID [70, 71]. Pioneering experiments, again conducted by Mitchell et al. [72] in MPTP-lesioned nhps, showed that at peak dose of dopamine agonist-induced dyskinesia, there was an increased uptake of 2-DG in the STN and GPi, demonstrating that these structures were hyper-inhibited. This study also showed that 2-DG was reduced in the motor thalamic nuclei, reflecting its hyperactivated state in dyskinesia [72].
The role of the direct pathway in the pathogenesis of LID was later emphasized by Bezard et al. [86]. In this key review, it was suggested that underactive/abnormal firing of the basal ganglia output nuclei in dyskinesia [52, 73–78] was primarily caused by overactivated striatonigral neurons of the direct pathway. Indeed, functional hyperactivation of the direct pathway in LID has been demonstrated at the cellular level from (1) dramatic elevations of striatal mRNA expression of PPE-B and prodynorphin [58, 79–82] and (2) supersensitization of striatal dopamine D1 receptors [83]. In addition, it has been reported that treatment with selective dopamine D1 receptor agonist, ABT-431, in PD patients elicits dyskinesia to a similar extent to that of l-DOPA [84], supporting the hypothesis of a hyperactivated direct pathway in the pathogenesis of dyskinesia. It should be noted that these data are inline with the mechanism suggested in the classic functional model, whereby an overactivated direct pathway mediates over-inhibition of the basal ganglia output, causing the underactivation of these nuclei (see Fig. 7.1c).
On the contrary, the proposed underactivation of the indirect pathway in the pathophysiology in LID has been, somewhat, inconsistent with several experimental findings, which has presented some limitations of the classic functional model (discussed in more detail in the section below). For example, the underactivation of the indirect pathway due an overactivated GPe is not consistently seen in dyskinetic MPTP-lesioned nhps [55]. In addition, striatopallidal neurons of the indirect pathway are not underactive, as demonstrated by the levels of striatal PPE-A mRNA, which are actually further upregulated, rather than downregulated, in dyskinesia compared to PD [79, 85]. It has since been suggested that increased striatal PPE-A mRNA in LID may occur due to reduced parkinsonism following l-DOPA treatment, rather than LID itself [86]. This is consistent with clinical findings that have shown dopamine D2 receptor agonists are effective antiparkinsonian agents with a reduced risk of inducing dyskinesia [87].
At this point, it is worth mentioning that the classic functional basal ganglia model has provided an excellent basis for describing the functional mechanisms involved in normal and disease states (see Fig. 7.1a–c). However, the classic model remains too simplistic, and its use is limited when describing the pathophysiological mechanisms in PD and LID. In the next section, we outline some of the main inconsistencies that have arisen between experimental data and the classic functional basal ganglia model.
Developments of the Basal Ganglia Circuitry Model
Progressing from the original descriptions of the classic functional model by Alexander and Crutcher [4], experimental reports have revealed a greater complexity in the neural organization and information processing within the basal ganglia. These data have led to the development of the functional model, which has engaged the highly dynamic nature of neural networking in the basal ganglia circuitry.
Organization and Structure
In the classic model, the separate direct and indirect pathway organization has been widely accepted, but the actual degree of segregation and opposing functional activity of the striatal neuronal projections remains unclear [88]. Firstly, striatofugal axons show consistent collateralization to both the GPe and GPi [89], suggesting an interconnected, rather than segregated, organization. Secondly, the response of the striatofugal pathways to dopamine cannot be simply viewed as an activating or deactivating effect caused exclusively via actions on dopamine D1 or D2 receptors, respectively, as (1) a high percentage of striatal MSNs expresses both subtypes of dopamine receptors [90–92] and (2) because dopamine D1 and D2 receptor responses are not consistently opposite [93, 94]. It is also worth mentioning that the modulatory effects of dopamine in the basal ganglia are not only restricted to the striatum. In fact, extensive dopaminergic SNc projections are found to innervate most, if not all, of the other basal ganglia subnuclei [95–100]. Thus, taking this into consideration, another level of complexity is added, as these dopaminergic innervations can bypass the feed-forward processing mediated by striatofugal neuronal activity [101–103].
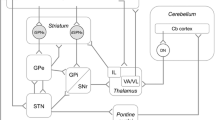
From the early 2000s, the mechanism of neural processing in the basal ganglia has been reevaluated [104, 105]. The concept of a linear feed-forward mechanism, solely based on altered firing rate of each basal ganglia subnucleus, has been found to be inconsistent with preclinical and clinical data, sparking the reorganization of the basal ganglia circuitry. In particular, the model now incorporates the numerous internal feedback loops [104, 106], which have been found to exist through reciprocal connections between the many of the basal ganglia subnuclei [107–110]. This reformed organization of basal ganglia circuitry has brought drastic changes to the arrangement of the classic indirect pathway, which include (1) the GPe as now occupying a central position and being viewed as a key structure for inhibitory modulation of the striatum, GPi and STN (see Fig. 7.2) [111–113], and (2) the STN being considered as another major input station of the basal ganglia, receiving afferent glutamatergic projections from the cortex [114, 115] and thalamus [116], while sending glutamatergic efferent projections to the GPe, GPi/SNr, ventral thalamic nuclei, and pedunculopontine nucleus [117, 118]. Importantly, the overall structural reorganization of the motor circuit now sees the functional dual disynaptic control of the GPe and GPi (see Fig. 7.2) [104, 113] via parallel cortical projections to the striatum and STN. The corticostriatal projection uses the striatum to mediate inhibitory control of the pallidal segments, while the cortico-STN projection, also known as the hyper-direct pathway [115], uses the STN to mediate fast excitatory input to these structures [119].

A schematic diagram showing the functional organization of the basal ganglia. This updated model proposed by Obeso et al. [113] illustrates the dual disynaptic control of the internal and external segments of the globus pallidus, originating from corticostriatal and cortico-subthalamic projections. The position of the external segment of the globus pallidus in this model has been emphasized to mediate important inhibitory control of the basal ganglia output nuclei, while modulating the activity of the striatum and subthalamic nucleus via reciprocal connections. DRs dopamine receptors, STR striatum, GPi internal segment of the globus pallidus, GPe external segment of the globus pallidus, STN subthalamic nucleus
Mechanism of Neural Information Processing
The mechanism of neural processing in the classic functional model, which is based only on the firing rate of each individual subnucleus in the basal ganglia, is unable to explain several major experimental findings in PD and LID. Most notably, simple underactivation of the basal ganglia output nuclei in dyskinesia [73, 76, 77] cannot fully explain the pathogenesis of LID [75]. This is because lesion of the GPi does not result in dyskinesia [120, 121]. In fact, pallidotomy of the internal segment effectively alleviates LID in MPTP-lesioned nhps [122] and PD patients [123, 124], which is opposite to the proposed outcome of the classic functional model. Other major inconsistencies of the classic model have been identified in PD, which include: (1) lesions of the motor thalamic nuclei do not exacerbate parkinsonism in patients [120] and (2) lesions of the GPe do not induce PD motor symptoms [54]. Collectively, these data have indicated that the processing of neural information for motor behavior in the basal ganglia is much more complex than originally thought [19], which cannot rely exclusively on firing rates. Instead, the firing rate model has been developed to incorporate the functional roles of neuronal firing patterns, such as synchronicity and oscillatory activations, in motor function, which are characteristically different in established PD and LID [75, 125–127].
Neuronal Firing Patterns in the Basal Ganglia
The study of neuronal firing patterns is commonly conducted through electrophysiological measurements of single/multiunit activity or local field potentials (LFPs). Single/multiunit recordings show the action potentials of one or multiple neurons, while LFPs typically reflect subthreshold synchronized afferent activations of a larger group of neurons [128]. These sets of neuronal data are analyzed for either synchronous or oscillatory patterns of activity, which are determined using a range of statistical tools on the time and/or frequency domains [129]. While the oscillatory activity can modulate neuronal synchronization in cortical and subcortical regions [130, 131], these patterns of neuronal activity are not mutually exclusive, i.e., synchronized firing can occur in the absence of periodic firing or vice versa [129]. The presence of synchronized activity between networks in distinct neuroanatomical regions is hypothesized to “bind” or “couple” neural ensembles, as part of a wider functional integration process [130–132]. Such synchronized firing is suggested to be a mechanism of neural processing in cortical and thalamic regions. On the contrary, the function of the basal ganglia, as an intermediate in corticobasal ganglia-thalamocortical loop, has been suggested to mediate so-called dimensional reduction, which describes the funnelling of redundant cortical neuronal inputs for efficient action planning [133]. This has been indicated from electrophysiological studies conducted in normal animals that have revealed the activity of neurons in the GPe [134], GPi [135], and STN [136] are generally asynchronous, with approximately 90 % or more of recorded neurons displaying uncorrelated firing patterns [53, 137, 138]. The oscillations in spike activity of MSNs in the striatum are typically weak [139, 140].
Abnormal Neuronal Firing Patterns in the Pathophysiology of PD and LID
Following original reports of abnormal neuronal firing patterns in experimental parkinsonism [53, 141], patterns in burst firing, synchronization, and oscillatory activity have been well studied in PD and LID. Initial experiments conducted single-unit measurements of neuronal activity in the subnuclei of the basal ganglia in MPTP-lesioned nhps and showed that the incidence of burst firing was increased in the GPe, GPi, and STN [53, 141, 142]. Additionally, neurons within these structures were found to demonstrate hyper-synchronized oscillatory activity that was characteristically <30 Hz (within the β (beta)-band) [53, 135, 143]. Similar findings have been reported in clinical studies, following measurements of LFPs via macroelectrodes during neurosurgery [125]. Although these data are not directly comparable to single-unit measurements, LFPs in the STN and GPi of PD patients in the off-state showed dominant low-frequency (<30 Hz) oscillations, with increased coherence in activity (at 6 and 20 Hz) between these structures. The origin of these low-frequency oscillations in PD has been suggested to arise from an abnormal network effect following extensive loss of dopamine in the basal ganglia, which may occur from imbalanced activity between the direct and hyper-direct pathways [105] or due to rebound firing of STN [144–146] caused by abnormal inhibitory input from the GPe [147–149]. The STN is likely to impose enhanced β (beta)-band oscillations on the GPi through its direct synaptic connection, which then reverberates through the motor corticobasal ganglia-thalamocortical loop, as indicated from coherent β (beta)-band oscillations between the basal ganglia subnuclei and motor cortical regions [150–153].
The functional relevance of enhanced β (beta)-band oscillations in the motor corticobasal ganglia-thalamocortical loop has been postulated to disrupt information processing, contributing to the expression of PD motor symptoms [154]. In-line with this suggestion, low-frequency (5–20 Hz) stimulations of the STN typically worsen akinesia in PD patients [155–157], while neuronal firing of GPi cells at 4–6 Hz in MPTP-lesioned nhps [53] and PD patients [158] has been correlated to the frequency of resting tremor. Moreover, studies have shown that treatment with dopaminergic agents in PD suppresses the low-frequency β (beta)-band oscillations in the basal ganglia [125, 159, 160] and motor cortical regions [151, 152], causing several marked changes in neuronal activity, such as the uncoupling of high-frequency oscillations (HFO) (>300 Hz) to low-frequency β (beta)-band oscillations [160] and a shift to a new prominent peak in activity at ~70 Hz (γ (gamma)-band) [125], as parkinsonism is alleviated. The presence of γ (gamma)-band oscillations (70–85 Hz) in the basal ganglia, particularly the STN, may be reflective of an improved motor state in PD as clinical studies have shown (1) HFS of the STN >70 Hz alleviates PD motor symptoms in patients [155] which also suppresses low-frequency β (beta)-band oscillations in the GPi [161] and (2) increased coherence between the STN and GPi in the γ (gamma)-band frequency following l-DOPA treatment in PD patients, which also augments with movement [151]. However, in PD patients that exhibit dyskinesia following treatment with dopaminergic medication, different patterns of neuronal activities are induced [127, 162]. While LFPs in the STN of these patients do show increased (17.8 %) logarithmic power of activity in the γ (gamma)-frequency range, there is a more striking increment (77.6 %) in activity at 4–10 Hz (θ (theta)/α (alpha)-band) that is specifically associated with dyskinesia [127, 163]. Clinical data have also revealed that the coherence between the STN and GPi at <10 Hz is increased in the dyskinetic state [162].
As discussed above, neuronal patterns of burst firing, synchronization, and oscillatory activity within the motor corticobasal ganglia-thalamocortical loop are associated with different motor states. It has been postulated that tonic levels of endogenous dopamine in the normal basal ganglia may mediate desynchronized neuronal firing for the processing of motor commands [126, 164]. However, in PD, when there is extensive loss of dopamine, these motor commands are not, or inefficiently, processed within the basal ganglia. Prominent changes, such as enhanced hyper-synchronization and oscillatory patterns of firing at β (beta)-band frequencies, may represent increased threshold levels of activity, acting as a “barrier” that ultimately impedes information processing for movement [126, 164]. On the contrary, hyper-synchronization of neuronal activity at θ (theta)/α (alpha)-band frequencies, as recorded in LID [127, 162, 163], may allow for release of involuntary motor sequences that become expressed as dyskinesia. Thus, neurosurgical procedures for symptomatic treatments of PD and LID can be viewed as a method of alleviating, or resetting, abnormal subcortical activations [165], allowing the resumption of neuronal processing for motor programming in the absence of an endogenous dopamine tone. In the next section, we discuss a recently developed computational model of the basal ganglia circuit for action selection, which describes the loss of motor function and emergence of low-frequency oscillations following striatal dopamine denervation.
Computational Models of the Basal Ganglia Motor Circuit
Although the precise functional consequences of abnormal neuronal firing patterns in the basal ganglia in parkinsonian and dyskinetic states remain unclear, recent advances have been made in our understanding of neural processing in the expression of the motor symptoms. Studies conducted by Boraud’s group identified key functional changes in the basal ganglia output nuclei that related to the onset of parkinsonism [166]. Their work demonstrated that hyper-synchronized β (beta)-band oscillations in the GPi occurred following the establishment of PD motor symptoms in MPTP-lesioned nhps [166]. In the same study, the authors identified that onset of experimental parkinsonism was closely related to a shift in the firing profile of GPi neurons, where there was an increased (~1.5-fold) proportion of excitable neurons and a decreased (~0.5-fold) number of functional inhibitory neurons [166]. Moreover, in an earlier study conducted by the same research group, GPi neurons in MPTP-lesioned nhps were found to have altered firing activities that were related to the spatiotemporal aspects of motor processing [167]. In these MPTP-treated nhps, it was shown that the number of GPi neurons responsive to manipulated limb movements was increased (~5-fold), demonstrating a loss of somatosensory selectivity [167]. Neurons within the GPi also displayed premature firing in relation to onset of muscular activity, suggesting dysfunctional neural processing for movement [167]. These key experimental findings indicated that changes in neural activity at the level of the GPi could be instrumental in disrupting motor processing in action selection, leading to the motor disabilities seen in PD.
In 2006, Boraud’s team put forward a dynamic computational model of the basal ganglia network that described the neural processing for action selection from closed feedback loops (see Fig. 7.3) [105]. Interestingly, in-line with experimental findings in MPTP-treated nhps [166], this computational model described how reduced dopamine levels caused loss of action selection that correlated with a shift in the proportion of activated neurons in the GPi, prior to the development of synchronized low-frequency oscillations within the basal ganglia [105]. This functional model of the basal ganglia uses two main feedback loops that are each arranged in a somatotopic manner [168–171]: (i) the hyper-direct pathway (cortex-STN-GPi-thalamus-cortex) [114] and (ii) the direct pathway (cortex-striatum-GPi-thalamus-cortex). These two pathways have opposing effects on cortical activity; the hyper-direct inhibits the thalamic nuclei which causes reduced cortical firing (global negative), while the direct pathway disinhibits the thalamic nuclei leading to increased cortical firing (global positive). Leblois et al. [105] described two somatotopic channels from one corticobasal ganglia-thalamocortical loop (each composed of two feedback loops) that act in parallel and compete to execute action selection (see Fig. 7.3) [170–172]. This is achieved when the activity of one cortical population overcomes the threshold activity of that in the other. Importantly, the projections from the STN to the GPi have pivotal roles in the execution of a motor program, as the STN mediates “cross-path” activity, modulating GPi neuronal activity in its own loop and also in the competing circuit [105]. This functional model also describes the effects of dopamine in the striatum, where it mediates potentiation of corticostriatal synapses in sensorimotor regions [173], strengthening direct pathway activity for biasing selection of the motor program in the corresponding circuit. When dopamine levels are normal, i.e., 100 %, the computational model demonstrates how action selection can occur as “symmetry breaking,” the transition of activity when one neuronal population becomes greater than the other, takes place. Such a situation arises when motor planning information, sent from sensorimotor cortical areas, produces strong positive feedback activity in the direct pathway and inhibitory feedback activity in the hyper-direct pathway, causing asymmetric activations of neuronal populations in the GPi. In turn, cortical activity in one population is enhanced, while the other is attenuated leading to the selection of an action [105].

A schematic diagram of basal ganglia neuronal connections in a model for action selection proposed by Leblois et al. [105]. In this circuit, two neuronal populations (black and gray), each composed of (i) cortex-STR-GPi-thalamus-cortex and (ii) cortex-STN-GPi-thalamus-cortex pathways compete to execute action selection. This is achieved when activity in of one cortical population overcomes threshold activity in the other cortical population. Importantly, projections from the STN cross over, modulating GPi neuronal activity in the competing loop. Dopamine in the striatum mediates potentiation of corticostriatal synapses, strengthening the activity of a specific striatal projection. This can lead to increased feedback to the cortex in the same neuronal population to cause action selection. DA dopamine, STR striatum, GPi internal segment of the globus pallidus, STN subthalamic nucleus
An extension to the computational model proposed by Leblois et al. has recently been described for the processing of neural information in the basal ganglia for two level decision-making (i.e., cognitive and motor) [174]. Based on electrophysiological data in nhps [175], the updated model demonstrated how task-related decisions made at the cognitive level can influence the motor level for action selection. The model architecture of the updated model is more sophisticated, describing two action selection modules, i.e., one cognition and one motor, which act in parallel. Each action selection module arises from distinct regions of the cortex, consisting of the direct and hyper-direct pathways, in a corticobasal ganglia-thalamocortical loop. For each loop, channels composed of separate ensembles in cortical areas are representative of decision choices that compete for action selection [174]. This computational model incorporates the idea of multiple corticobasal ganglia-thalamocortical loops for different aspects of neural processing [4], which interact in the striatum as afferent fibers converge in specific overlapping regions [174]. In this model, symmetry breaking for action selection can be initiated by internal noise prior to learning, which is followed by dopamine-mediated effects at corticostriatal synapses. Subsequently, synaptic gain in the direct pathway at the striatal level mediates positive feedback of that channel, while negative feedback of the hyper-direct pathway suppresses competing channels [174], in a center-surround inhibitory fashion [176]. Thus, activities of both circuits promote action selection of a specific channel.
These novel dynamic computational models of neural networks may prove to be important tools in the study of basal ganglia disorders. In the original computational model, Leblois et al. [105] demonstrated that striatal dopamine denervation leads to complete loss of action selection ability. Initial changes included a marked reduction in the ratio of inhibited GPi neurons by the direct pathway, which occurred following ~30 % dopamine loss [105]. As a result, feedback from the direct pathway was reduced, preventing the mechanism of symmetry breaking. Interestingly, after approximately 70–80 % striatal dopamine denervation, the inability of the direct pathway to counteract negative feedback of the hyper-direct pathway resulted in synchronized oscillatory neuronal activity (frequency of 10–12 Hz) [105]. As these predictions are in-line with experimental findings in PD [166], the model provides an excellent tool for studying the pathogenesis of disease states, with the advantage of incorporating more parallel loops and additional anatomical subnuclei [105, 174]. Although the pathophysiological changes in LID have yet to be modelled in these computational models, it would be particularly interesting to investigate whether synchronized θ (theta)/α (alpha)-band oscillations are produced in the basal ganglia in the dyskinetic state, as reported in patients [127, 162]. Speculatively, the dyskinesia could be modelled by incorporating pathophysiological hallmarks of LID, such as dysfunctional LTP at corticostriatal synapses [177, 178]. If this is possible, the current computational model could help elucidate the precise consequences of abnormal neuronal oscillatory activations in the basal ganglia subnuclei on action selection or identify the subtle changes in firing activities that lead to the expression of dyskinesia. In addition, we suggest that future basal ganglia models should be extended for describing the pathophysiology of LID. This is because recent studies have demonstrated molecular and functional adaptations associated with the expression of dyskinesia also occur in anatomical regions beyond the subnuclei of the basal ganglia. In a recent study conducted by Halje et al. [179], it was shown that dyskinetic motor symptoms in the unilateral 6-hydroxydopamine (6-OHDA)-lesioned rat model of LID were alleviated as abnormal motor cortical oscillations (80 Hz) were attenuated, following application of a dopamine D1 receptor antagonist to specific cortical regions. These data, as well as our recent findings [180], highlight the need to look beyond the basal ganglia subnuclei for functional changes that can directly impact motor function.
Additional Nuclei in the Pathophysiology of LID
Molecular changes in the pathophysiology of LID have been well studied in the basal ganglia subnuclei [181], but little remains known of the adaptations that occur in other structures. A previous report identified the bed nucleus of the stria terminalis (BST) was hyperactivated in dyskinetic MPTP-lesioned nhps [182], suggesting a potential role of this structure in the pathophysiology of LID. Using the unilateral 6-OHDA-lesioned rat model of LID, we recently investigated the molecular adaptations in the whole brain by quantifying the expression of four immediate early genes (IEGs) (ΔFosB, ARC, FRA2, Zif268/EGR1) [180]. We found that dyskinesia severity in l-DOPA-treated unilateral 6-OHDA-lesioned rats correlated to the overexpression of these specific IEGs in the following structures: oval (oBST), juxta (jBST), and medial (mBST) bed nucleus of the stria terminalis, lateral habenula (lHb), pontine nuclei (Pn), and cuneiform nucleus (CnF). Such molecular adaptations in these nuclei could stem from irregular activities of afferent fibers. For example, serotonergic afferents [183] to the oBST and jBST may facilitate unregulated fluctuations of dopamine release in LID [184–187], which is likely to cause an abnormal functional state of these nuclei [188]. The molecular adaptations in the lHb nuclei, a structure that projects to different monoaminergic regions including the serotonergic dorsal and medial raphe, could be involved in the aberrant release of dopamine from serotonergic (5-HT) terminals in LID, contributing to the pathogenesis of dyskinesia [189–192]. Further studies are currently being conducted to fully elucidate the functional roles of these additional nuclei in the pathogenesis of dyskinesia. It is important to note that recent findings from our group and others [179] highlight the need to evaluate regions outside of the basal ganglia for fully uncovering the pathophysiological mechanisms in LID.
Conclusions
The functional basal ganglia circuitry model for describing the pathophysiology of PD and LID has developed quite considerably over the past few decades. Critical evaluation of functional mechanisms has proved an important step in progressing from the original descriptions of basic box-arrow circuitry and feed-forward information processing [4] to more updated basal ganglia models, which have captured complex neural network connections [104] and the dynamic nonlinear neuronal processing in disease states [105]. While our understanding of the pathophysiological mechanisms of PD and LID motor symptoms remains incomplete, the road to uncovering subtle dysfunctional neuronal processes will undoubtedly be guided by accurately modelling the latest experimental findings. Recent technological advancements that allow for the simultaneous measurements of single-unit neuronal activity, whole body kinematics, and muscular activities in freely moving nhps [193] are likely to be at the forefront of relating specific motor abnormalities that occur in PD and LID to abnormal neural processing in the basal ganglia and other anatomical regions. By striving to understand the complex mechanisms involved, we hope to make solid progress in the development of novel clinical treatments for PD and LID, to ultimately improve the quality of life of patients suffering from these movement disorders.
References
de Rijk MC, Tzourio C, Breteler MM, Dartigues JF, Amaducci L, Lopez-Pousa S, et al. Prevalence of parkinsonism and Parkinson’s disease in Europe: the EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1997;62(1):10–5.
Marsden CD. Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1994;57(6):672–81.
Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin Wochenschr. 1960;38:1236–9.
Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci. 1990;13(7):266–71.
Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180(4596):1200.
Friedhoff AJ, Hekimian L, Alpert M, Tobach E. Dihydroxyphenylalanine in extrapyramidal disease. JAMA. 1963;184:285–6.
Birkmayer W, Hornykiewicz O. The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wien Klin Wochenschr. 1961;73:787–8.
Fahn S. “On-off” phenomenon with levodopa therapy in Parkinsonism. Clinical and pharmacologic correlations and the effect of intramuscular pyridoxine. Neurology. 1974;24(5):431–41.
Duvoisin RC. Variations in the “on-off” phenomenon. Adv Neurol. 1974;5:339–40.
Shoulson I, Glaubiger GA, Chase TN. On-off response. Clinical and biochemical correlations during oral and intravenous levodopa administration in parkinsonian patients. Neurology. 1975;25(12):1144–8.
Marsden CD, Parkes JD. “On-off” effects in patients with Parkinson’s disease on chronic levodopa therapy. Lancet. 1976;1(7954):292–6.
Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism–chronic treatment with L-dopa. N Engl J Med. 1969;280(7):337–45.
Fahn S. The spectrum of levodopa-induced dyskinesias. Ann Neurol. 2000;47(4 Suppl 1):S2–9; discussion S-11.
Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16(3):448–58.
Rascol O. Medical treatment of levodopa-induced dyskinesias. Ann Neurol. 2000;47(4 Suppl 1):S179–88.
Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351(24):2498–508.
Chapuis S, Ouchchane L, Metz O, Gerbaud L, Durif F. Impact of the motor complications of Parkinson’s disease on the quality of life. Mov Disord. 2005;20(2):224–30.
Crossman AR, Neary D. Neuroanatomy: an illustrated colour text. Edinburgh: Churchill Livingstone; 2000.
Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–81.
Lavoie B, Smith Y, Parent A. Dopaminergic innervation of the basal ganglia in the squirrel monkey as revealed by tyrosine hydroxylase immunohistochemistry. J Comp Neurol. 1989;289(1):36–52.
Lavoie B, Parent A. Immunohistochemical study of the serotoninergic innervation of the basal ganglia in the squirrel monkey. J Comp Neurol. 1990;299(1):1–16.
Delfs JM, Zhu Y, Druhan JP, Aston-Jones GS. Origin of noradrenergic afferents to the shell subregion of the nucleus accumbens: anterograde and retrograde tract-tracing studies in the rat. Brain Res. 1998;806(2):127–40.
Groenewegen HJ, Galis-de Graaf Y, Smeets WJ. Integration and segregation of limbic cortico-striatal loops at the thalamic level: an experimental tracing study in rats. J Chem Neuroanat. 1999;16(3):167–85.
Castle M, Aymerich MS, Sanchez-Escobar C, Gonzalo N, Obeso JA, Lanciego JL. Thalamic innervation of the direct and indirect basal ganglia pathways in the rat: Ipsi- and contralateral projections. J Comp Neurol. 2005;483(2):143–53.
Kunzle H. Bilateral projections from precentral motor cortex to the putamen and other parts of the basal ganglia. An autoradiographic study in Macaca fascicularis. Brain Res. 1975;88(2):195–209.
Kunzle H. Projections from the primary somatosensory cortex to basal ganglia and thalamus in the monkey. Exp Brain Res. 1977;30(4):481–92.
McGeorge AJ, Faull RL. The organization of the projection from the cerebral cortex to the striatum in the rat. Neuroscience. 1989;29(3):503–37.
Romanelli P, Esposito V, Schaal DW, Heit G. Somatotopy in the basal ganglia: experimental and clinical evidence for segregated sensorimotor channels. Brain Res Brain Res Rev. 2005;48(1):112–28.
Somogyi P, Smith AD. Projection of neostriatal spiny neurons to the substantia nigra. Application of a combined Golgi-staining and horseradish peroxidase transport procedure at both light and electron microscopic levels. Brain Res. 1979;178(1):3–15.
Somogyi P, Bolam JP, Totterdell S, Smith AD. Monosynaptic input from the nucleus accumbens–ventral striatum region to retrogradely labelled nigrostriatal neurones. Brain Res. 1981;217(2):245–63.
Carpenter MB, Nakano K, Kim R. Nigrothalamic projections in the monkey demonstrated by autoradiographic technics. J Comp Neurol. 1976;165(4):401–15.
Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12(10):366–75.
DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13(7):281–5.
Clavier RM, Atmadja S, Fibiger HC. Nigrothalamic projections in the rat as demonstrated by orthograde and retrograde tracing echniques. Brain Res Bull. 1976;1(4):379–84.
Herkenham M. The afferent and efferent connections of the ventromedial thalamic nucleus in the rat. J Comp Neurol. 1979;183(3):487–517.
Deniau JM, Chevalier G. The lamellar organization of the rat substantia nigra pars reticulata: distribution of projection neurons. Neuroscience. 1992;46(2):361–77.
Deniau JM, Mailly P, Maurice N, Charpier S. The pars reticulata of the substantia nigra: a window to basal ganglia output. Prog Brain Res. 2007;160:151–72.
Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320.
Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci. 1990;13(7):277–80.
Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 1990;13(7):244–54.
Agid Y. Parkinson’s disease: pathophysiology. Lancet. 1991;337(8753):1321–4.
Hikosaka O, Wurtz RH. Visual and oculomotor functions of monkey substantia nigra pars reticulata. I. Relation of visual and auditory responses to saccades. J Neurophysiol. 1983;49(5):1230–53.
Hikosaka O, Wurtz RH. Visual and oculomotor functions of monkey substantia nigra pars reticulata. II. Visual responses related to fixation of gaze. J Neurophysiol. 1983;49(5):1254–67.
Hikosaka O, Wurtz RH. Visual and oculomotor functions of monkey substantia nigra pars reticulata. III. Memory-contingent visual and saccade responses. J Neurophysiol. 1983;49(5):1268–84.
Turner RS, Anderson ME. Context-dependent modulation of movement-related discharge in the primate globus pallidus. J Neurosci. 2005;25(11):2965–76.
Penney Jr JB, Young AB. Speculations on the functional anatomy of basal ganglia disorders. Annu Rev Neurosci. 1983;6:73–94.
Rascol O, Sabatini U, Chollet F, Celsis P, Montastruc JL, Marc-Vergnes JP, et al. Supplementary and primary sensory motor area activity in Parkinson’s disease. Regional cerebral blood flow changes during finger movements and effects of apomorphine. Arch Neurol. 1992;49(2):144–8.
Bezard E, Crossman AR, Gross CE, Brotchie JM. Structures outside the basal ganglia may compensate for dopamine loss in the presymptomatic stages of Parkinson’s disease. FASEB J. 2001;15(6):1092–4.
Crossman AR, Mitchell IJ, Sambrook MA. Regional brain uptake of 2-deoxyglucose in N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the macaque monkey. Neuropharmacology. 1985;24(6):587–91.
Mitchell IJ, Cross AJ, Sambrook MA, Crossman AR. N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in the monkey: neurochemical pathology and regional brain metabolism. J Neural Transm Suppl. 1986;20:41–6.
Mitchell IJ, Clarke CE, Boyce S, Robertson RG, Peggs D, Sambrook MA, et al. Neural mechanisms underlying parkinsonian symptoms based upon regional uptake of 2-deoxyglucose in monkeys exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience. 1989;32(1):213–26.
Filion M, Tremblay L, Bedard PJ. Effects of dopamine agonists on the spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991;547(1):152–61.
Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72(2):507–20.
Soares J, Kliem MA, Betarbet R, Greenamyre JT, Yamamoto B, Wichmann T. Role of external pallidal segment in primate parkinsonism: comparison of the effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism and lesions of the external pallidal segment. J Neurosci. 2004;24(29):6417–26.
Vila M, Levy R, Herrero MT, Ruberg M, Faucheux B, Obeso JA, et al. Consequences of nigrostriatal denervation on the functioning of the basal ganglia in human and nonhuman primates: an in situ hybridization study of cytochrome oxidase subunit I mRNA. J Neurosci. 1997;17(2):765–73.
Gerfen CR, McGinty JF, Young 3rd WS. Dopamine differentially regulates dynorphin, substance P, and enkephalin expression in striatal neurons: in situ hybridization histochemical analysis. J Neurosci. 1991;11(4):1016–31.
Henry B, Crossman AR, Brotchie JM. Characterization of enhanced behavioral responses to L-DOPA following repeated administration in the 6-hydroxydopamine-lesioned rat model of Parkinson’s disease. Exp Neurol. 1998;151(2):334–42.
Ravenscroft P, Chalon S, Brotchie JM, Crossman AR. Ropinirole versus L-DOPA effects on striatal opioid peptide precursors in a rodent model of Parkinson’s disease: implications for dyskinesia. Exp Neurol. 2004;185(1):36–46.
Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249(4975):1436–8.
Aziz TZ, Peggs D, Sambrook MA, Crossman AR. Lesion of the subthalamic nucleus for the alleviation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the primate. Mov Disord. 1991;6(4):288–92.
Wichmann T, Bergman H, DeLong MR. The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induced by subthalamic inactivation in the MPTP model of parkinsonism. J Neurophysiol. 1994;72(2):521–30.
Guridi J, Herrero MT, Luquin MR, Guillen J, Ruberg M, Laguna J, et al. Subthalamotomy in parkinsonian monkeys. Behavioural and biochemical analysis. Brain. 1996;119(Pt 5):1717–27.
Laitinen LV, Bergenheim AT, Hariz MI. Leksell’s posteroventral pallidotomy in the treatment of Parkinson’s disease. J Neurosurg. 1992;76(1):53–61.
Baron MS, Vitek JL, Bakay RA, Green J, Kaneoke Y, Hashimoto T, et al. Treatment of advanced Parkinson’s disease by posterior GPi pallidotomy: 1-year results of a pilot study. Ann Neurol. 1996;40(3):355–66.
Gill SS, Heywood P. Bilateral dorsolateral subthalamotomy for advanced Parkinson’s disease. Lancet. 1997;350(9086):1224.
Benabid AL, Pollak P, Gross C, Hoffmann D, Benazzouz A, Gao DM, et al. Acute and long-term effects of subthalamic nucleus stimulation in Parkinson’s disease. Stereotact Funct Neurosurg. 1994;62(1–4):76–84.
Limousin P, Pollak P, Benazzouz A, Hoffmann D, Broussolle E, Perret JE, et al. Bilateral subthalamic nucleus stimulation for severe Parkinson’s disease. Mov Disord. 1995;10(5):672–4.
Limousin P, Krack P, Pollak P, Benazzouz A, Ardouin C, Hoffmann D, et al. Electrical stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N Engl J Med. 1998;339(16):1105–11.
Crossman AR. A hypothesis on the pathophysiological mechanisms that underlie levodopa- or dopamine agonist-induced dyskinesia in Parkinson’s disease: implications for future strategies in treatment. Mov Disord. 1990;5(2):100–8.
Brooks DJ, Piccini P, Turjanski N, Samuel M. Neuroimaging of dyskinesia. Ann Neurol. 2000;47(4 Suppl 1):S154–8; discussion S8–9.
Rascol O, Sabatini U, Brefel C, Fabre N, Rai S, Senard JM, et al. Cortical motor overactivation in parkinsonian patients with L-dopa-induced peak-dose dyskinesia. Brain. 1998;121(Pt 3):527–33.
Mitchell IJ, Boyce S, Sambrook MA, Crossman AR. A 2-deoxyglucose study of the effects of dopamine agonists on the parkinsonian primate brain. Implications for the neural mechanisms that mediate dopamine agonist-induced dyskinesia. Brain. 1992;115(Pt 3):809–24.
Papa SM, Desimone R, Fiorani M, Oldfield EH. Internal globus pallidus discharge is nearly suppressed during levodopa-induced dyskinesias. Ann Neurol. 1999;46(5):732–8.
Boraud T, Bezard E, Guehl D, Bioulac B, Gross C. Effects of L-DOPA on neuronal activity of the globus pallidus externalis (GPe) and globus pallidus internalis (GPi) in the MPTP-treated monkey. Brain Res. 1998;787(1):157–60.
Boraud T, Bezard E, Bioulac B, Gross CE. Dopamine agonist-induced dyskinesias are correlated to both firing pattern and frequency alterations of pallidal neurones in the MPTP-treated monkey. Brain. 2001;124(Pt 3):546–57.
Merello M, Balej J, Delfino M, Cammarota A, Betti O, Leiguarda R. Apomorphine induces changes in GPi spontaneous outflow in patients with Parkinson’s disease. Mov Disord. 1999;14(1):45–9.
Lozano AM, Lang AE, Levy R, Hutchison W, Dostrovsky J. Neuronal recordings in Parkinson’s disease patients with dyskinesias induced by apomorphine. Ann Neurol. 2000;47(4 Suppl 1):S141–6.
Stefani A, Stanzione P, Bassi A, Mazzone P, Vangelista T, Bernardi G. Effects of increasing doses of apomorphine during stereotaxic neurosurgery in Parkinson’s disease: clinical score and internal globus pallidus activity. Short communication. J Neural Transm. 1997;104(8–9):895–904.
Cenci MA, Lee CS, Bjorklund A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur J Neurosci. 1998;10(8):2694–706.
Henry B, Crossman AR, Brotchie JM. Effect of repeated L-DOPA, bromocriptine, or lisuride administration on preproenkephalin-A and preproenkephalin-B mRNA levels in the striatum of the 6-hydroxydopamine-lesioned rat. Exp Neurol. 1999;155(2):204–20.
Lundblad M, Picconi B, Lindgren H, Cenci MA. A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2004;16(1):110–23.
Aubert I, Guigoni C, Li Q, Dovero S, Bioulac BH, Gross CE, et al. Enhanced preproenkephalin-B-derived opioid transmission in striatum and subthalamic nucleus converges upon globus pallidus internalis in L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol Psychiatry. 2007;61(7):836–44.
Aubert I, Guigoni C, Hakansson K, Li Q, Dovero S, Barthe N, et al. Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann Neurol. 2005;57(1):17–26.
Rascol O, Nutt JG, Blin O, Goetz CG, Trugman JM, Soubrouillard C, et al. Induction by dopamine D1 receptor agonist ABT-431 of dyskinesia similar to levodopa in patients with Parkinson disease. Arch Neurol. 2001;58(2):249–54.
Calon F, Birdi S, Rajput AH, Hornykiewicz O, Bedard PJ, Di Paolo T. Increase of preproenkephalin mRNA levels in the putamen of Parkinson disease patients with levodopa-induced dyskinesias. J Neuropathol Exp Neurol. 2002;61(2):186–96.
Bezard E, Brotchie JM, Gross CE. Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci. 2001;2(8):577–88.
Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE, et al. Development of dyskinesias in a 5-year trial of ropinirole and L-dopa. Mov Disord. 2006;21(11):1844–50.
Nadjar A, Brotchie JM, Guigoni C, Li Q, Zhou SB, Wang GJ, et al. Phenotype of striatofugal medium spiny neurons in parkinsonian and dyskinetic nonhuman primates: a call for a reappraisal of the functional organization of the basal ganglia. J Neurosci. 2006;26(34):8653–61.
Levesque M, Parent A. The striatofugal fiber system in primates: a reevaluation of its organization based on single-axon tracing studies. Proc Natl Acad Sci U S A. 2005;102(33):11888–93.
Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16(20):6579–91.
Yung KK, Smith AD, Levey AI, Bolam JP. Synaptic connections between spiny neurons of the direct and indirect pathways in the neostriatum of the rat: evidence from dopamine receptor and neuropeptide immunostaining. Eur J Neurosci. 1996;8(5):861–9.
Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, et al. Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci. 2000;3(3):226–30.
Kerr JN, Wickens JR. Dopamine D-1/D-5 receptor activation is required for long-term potentiation in the rat neostriatum in vitro. J Neurophysiol. 2001;85(1):117–24.
Nicola SM, Hopf FW, Hjelmstad GO. Contrast enhancement: a physiological effect of striatal dopamine? Cell Tissue Res. 2004;318(1):93–106.
Joel D, Weiner I. The connections of the dopaminergic system with the striatum in rats and primates: an analysis with respect to the functional and compartmental organization of the striatum. Neuroscience. 2000;96(3):451–74.
Prensa L, Cossette M, Parent A. Dopaminergic innervation of human basal ganglia. J Chem Neuroanat. 2000;20(3–4):207–13.
Smith Y, Kieval JZ. Anatomy of the dopamine system in the basal ganglia. Trends Neurosci. 2000;23(10 Suppl):S28–33.
Sanchez-Gonzalez MA, Garcia-Cabezas MA, Rico B, Cavada C. The primate thalamus is a key target for brain dopamine. J Neurosci. 2005;25(26):6076–83.
Smith Y, Villalba R. Striatal and extrastriatal dopamine in the basal ganglia: an overview of its anatomical organization in normal and Parkinsonian brains. Mov Disord. 2008;23 Suppl 3:S534–47.
Rommelfanger KS, Wichmann T. Extrastriatal dopaminergic circuits of the Basal Ganglia. Front Neuroanat. 2010;4:139.
Kreiss DS, Anderson LA, Walters JR. Apomorphine and dopamine D(1) receptor agonists increase the firing rates of subthalamic nucleus neurons. Neuroscience. 1996;72(3):863–76.
Francois C, Savy C, Jan C, Tande D, Hirsch EC, Yelnik J. Dopaminergic innervation of the subthalamic nucleus in the normal state, in MPTP-treated monkeys, and in Parkinson’s disease patients. J Comp Neurol. 2000;425(1):121–9.
Jan C, Francois C, Tande D, Yelnik J, Tremblay L, Agid Y, et al. Dopaminergic innervation of the pallidum in the normal state, in MPTP-treated monkeys and in parkinsonian patients. Eur J Neurosci. 2000;12(12):4525–35.
Obeso JA, Rodriguez-Oroz MC, Rodriguez M, Lanciego JL, Artieda J, Gonzalo N, et al. Pathophysiology of the basal ganglia in Parkinson’s disease. Trends Neurosci. 2000;23(10 Suppl):S8–19.
Leblois A, Boraud T, Meissner W, Bergman H, Hansel D. Competition between feedback loops underlies normal and pathological dynamics in the basal ganglia. J Neurosci. 2006;26(13):3567–83.
McHaffie JG, Stanford TR, Stein BE, Coizet V, Redgrave P. Subcortical loops through the basal ganglia. Trends Neurosci. 2005;28(8):401–7.
Haber SN, Fudge JL, McFarland NR. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 2000;20(6):2369–82.
Mallet N, Micklem BR, Henny P, Brown MT, Williams C, Bolam JP, et al. Dichotomous organization of the external globus pallidus. Neuron. 2012;74(6):1075–86.
Sato F, Lavallee P, Levesque M, Parent A. Single-axon tracing study of neurons of the external segment of the globus pallidus in primate. J Comp Neurol. 2000;417(1):17–31.
Shink E, Bevan MD, Bolam JP, Smith Y. The subthalamic nucleus and the external pallidum: two tightly interconnected structures that control the output of the basal ganglia in the monkey. Neuroscience. 1996;73(2):335–57.
Obeso JA, Rodriguez-Oroz MC, Javier Blesa F, Guridi J. The globus pallidus pars externa and Parkinson’s disease. Ready for prime time? Exp Neurol. 2006;202(1):1–7.
Kita H. Globus pallidus external segment. Prog Brain Res. 2007;160:111–33.
Obeso JA, Rodriguez-Oroz MC, Benitez-Temino B, Blesa FJ, Guridi J, Marin C, et al. Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov Disord. 2008;23 Suppl 3:S548–59.
Nambu A, Tokuno H, Hamada I, Kita H, Imanishi M, Akazawa T, et al. Excitatory cortical inputs to pallidal neurons via the subthalamic nucleus in the monkey. J Neurophysiol. 2000;84(1):289–300.
Nambu A, Tokuno H, Takada M. Functional significance of the cortico-subthalamo-pallidal ‘hyperdirect’ pathway. Neurosci Res. 2002;43(2):111–7.
Lanciego JL, Gonzalo N, Castle M, Sanchez-Escobar C, Aymerich MS, Obeso JA. Thalamic innervation of striatal and subthalamic neurons projecting to the rat entopeduncular nucleus. Eur J Neurosci. 2004;19(5):1267–77.
Mena-Segovia J, Bolam JP, Magill PJ. Pedunculopontine nucleus and basal ganglia: distant relatives or part of the same family? Trends Neurosci. 2004;27(10):585–8.
Rico AJ, Barroso-Chinea P, Conte-Perales L, Roda E, Gomez-Bautista V, Gendive M, et al. A direct projection from the subthalamic nucleus to the ventral thalamus in monkeys. Neurobiol Dis. 2010;39(3):381–92.
Kita H, Tachibana Y, Nambu A, Chiken S. Balance of monosynaptic excitatory and disynaptic inhibitory responses of the globus pallidus induced after stimulation of the subthalamic nucleus in the monkey. J Neurosci. 2005;25(38):8611–9.
Marsden CD, Obeso JA. The functions of the basal ganglia and the paradox of stereotaxic surgery in Parkinson’s disease. Brain. 1994;117(Pt 4):877–97.
Obeso JA, Rodriguez MC, DeLong MR. Basal ganglia pathophysiology. A critical review. Adv Neurol. 1997;74:3–18.
Iravani MM, Costa S, Al-Bargouthy G, Jackson MJ, Zeng BY, Kuoppamaki M, et al. Unilateral pallidotomy in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated common marmosets exhibiting levodopa-induced dyskinesia. Eur J Neurosci. 2005;22(6):1305–18.
Lang AE. Surgery for levodopa-induced dyskinesias. Ann Neurol. 2000;47(4 Suppl 1):S193–9; discussion S9–202.
Parkin SG, Gregory RP, Scott R, Bain P, Silburn P, Hall B, et al. Unilateral and bilateral pallidotomy for idiopathic Parkinson’s disease: a case series of 115 patients. Mov Disord. 2002;17(4):682–92.
Brown P, Oliviero A, Mazzone P, Insola A, Tonali P, Di Lazzaro V. Dopamine dependency of oscillations between subthalamic nucleus and pallidum in Parkinson’s disease. J Neurosci. 2001;21(3):1033–8.
Brown P. Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson’s disease. Mov Disord. 2003;18(4):357–63.
Alonso-Frech F, Zamarbide I, Alegre M, Rodriguez-Oroz MC, Guridi J, Manrique M, et al. Slow oscillatory activity and levodopa-induced dyskinesias in Parkinson’s disease. Brain. 2006;129(Pt 7):1748–57.
Magill PJ, Sharott A, Bevan MD, Brown P, Bolam JP. Synchronous unit activity and local field potentials evoked in the subthalamic nucleus by cortical stimulation. J Neurophysiol. 2004;92(2):700–14.
Boraud T, Bezard E, Bioulac B, Gross CE. From single extracellular unit recording in experimental and human Parkinsonism to the development of a functional concept of the role played by the basal ganglia in motor control. Prog Neurobiol. 2002;66(4):265–83.
Engel AK, Singer W. Temporal binding and the neural correlates of sensory awareness. Trends Cogn Sci. 2001;5(1):16–25.
Engel AK, Fries P, Singer W. Dynamic predictions: oscillations and synchrony in top-down processing. Nat Rev Neurosci. 2001;2(10):704–16.
Mackay WA. Synchronized neuronal oscillations and their role in motor processes. Trends Cogn Sci. 1997;1(5):176–83.
Bar-Gad I, Morris G, Bergman H. Information processing, dimensionality reduction and reinforcement learning in the basal ganglia. Prog Neurobiol. 2003;71(6):439–73.
Raz A, Vaadia E, Bergman H. Firing patterns and correlations of spontaneous discharge of pallidal neurons in the normal and the tremulous 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine vervet model of parkinsonism. J Neurosci. 2000;20(22):8559–71.
Nini A, Feingold A, Slovin H, Bergman H. Neurons in the globus pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of parkinsonism. J Neurophysiol. 1995;74(4):1800–5.
Wichmann T, Bergman H, DeLong MR. The primate subthalamic nucleus. I. Functional properties in intact animals. J Neurophysiol. 1994;72(2):494–506.
Bar-Gad I, Heimer G, Ritov Y, Bergman H. Functional correlations between neighboring neurons in the primate globus pallidus are weak or nonexistent. J Neurosci. 2003;23(10):4012–6.
Bergman H, Feingold A, Nini A, Raz A, Slovin H, Abeles M, et al. Physiological aspects of information processing in the basal ganglia of normal and parkinsonian primates. Trends Neurosci. 1998;21(1):32–8.
Berke JD, Okatan M, Skurski J, Eichenbaum HB. Oscillatory entrainment of striatal neurons in freely moving rats. Neuron. 2004;43(6):883–96.
Dejean C, Gross CE, Bioulac B, Boraud T. Synchronous high-voltage spindles in the cortex-basal ganglia network of awake and unrestrained rats. Eur J Neurosci. 2007;25(3):772–84.
Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991;547(1):142–51.
Wichmann T, Soares J. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J Neurophysiol. 2006;95(4):2120–33.
Heimer G, Rivlin-Etzion M, Bar-Gad I, Goldberg JA, Haber SN, Bergman H. Dopamine replacement therapy does not restore the full spectrum of normal pallidal activity in the 1-methyl-4-phenyl-1,2,3,6-tetra-hydropyridine primate model of Parkinsonism. J Neurosci. 2006;26(31):8101–14.
Bevan MD, Wilson CJ, Bolam JP, Magill PJ. Equilibrium potential of GABA(A) current and implications for rebound burst firing in rat subthalamic neurons in vitro. J Neurophysiol. 2000;83(5):3169–72.
Hallworth NE, Bevan MD. Globus pallidus neurons dynamically regulate the activity pattern of subthalamic nucleus neurons through the frequency-dependent activation of postsynaptic GABAA and GABAB receptors. J Neurosci. 2005;25(27):6304–15.
Bevan MD, Hallworth NE, Baufreton J. GABAergic control of the subthalamic nucleus. Prog Brain Res. 2007;160:173–88.
Cruz AV, Mallet N, Magill PJ, Brown P, Averbeck BB. Effects of dopamine depletion on information flow between the subthalamic nucleus and external globus pallidus. J Neurophysiol. 2011;106(4):2012–23.
Tachibana Y, Iwamuro H, Kita H, Takada M, Nambu A. Subthalamo-pallidal interactions underlying parkinsonian neuronal oscillations in the primate basal ganglia. Eur J Neurosci. 2011;34(9):1470–84.
Schwab BC, Heida T, Zhao Y, Marani E, van Gils SA, van Wezel RJ. Synchrony in Parkinson’s disease: importance of intrinsic properties of the external globus pallidus. Front Syst Neurosci. 2013;7:60.
Marsden JF, Limousin-Dowsey P, Ashby P, Pollak P, Brown P. Subthalamic nucleus, sensorimotor cortex and muscle interrelationships in Parkinson’s disease. Brain. 2001;124(Pt 2):378–88.
Cassidy M, Mazzone P, Oliviero A, Insola A, Tonali P, Di Lazzaro V, et al. Movement-related changes in synchronization in the human basal ganglia. Brain. 2002;125(Pt 6):1235–46.
Williams D, Tijssen M, Van Bruggen G, Bosch A, Insola A, Di Lazzaro V, et al. Dopamine-dependent changes in the functional connectivity between basal ganglia and cerebral cortex in humans. Brain. 2002;125(Pt 7):1558–69.
Shimamoto SA, Ryapolova-Webb ES, Ostrem JL, Galifianakis NB, Miller KJ, Starr PA. Subthalamic nucleus neurons are synchronized to primary motor cortex local field potentials in Parkinson’s disease. J Neurosci. 2013;33(17):7220–33.
Gatev P, Darbin O, Wichmann T. Oscillations in the basal ganglia under normal conditions and in movement disorders. Mov Disord. 2006;21(10):1566–77.
Moro E, Esselink RJ, Xie J, Hommel M, Benabid AL, Pollak P. The impact on Parkinson’s disease of electrical parameter settings in STN stimulation. Neurology. 2002;59(5):706–13.
Timmermann L, Wojtecki L, Gross J, Lehrke R, Voges J, Maarouf M, et al. Ten-Hertz stimulation of subthalamic nucleus deteriorates motor symptoms in Parkinson’s disease. Mov Disord. 2004;19(11):1328–33.
Fogelson N, Kuhn AA, Silberstein P, Limousin PD, Hariz M, Trottenberg T, et al. Frequency dependent effects of subthalamic nucleus stimulation in Parkinson’s disease. Neurosci Lett. 2005;382(1–2):5–9.
Hutchison WD, Lozano AM, Tasker RR, Lang AE, Dostrovsky JO. Identification and characterization of neurons with tremor-frequency activity in human globus pallidus. Exp Brain Res. 1997;113(3):557–63.
Kuhn AA, Tsui A, Aziz T, Ray N, Brucke C, Kupsch A, et al. Pathological synchronisation in the subthalamic nucleus of patients with Parkinson’s disease relates to both bradykinesia and rigidity. Exp Neurol. 2009;215(2):380–7.
Lopez-Azcarate J, Tainta M, Rodriguez-Oroz MC, Valencia M, Gonzalez R, Guridi J, et al. Coupling between beta and high-frequency activity in the human subthalamic nucleus may be a pathophysiological mechanism in Parkinson’s disease. J Neurosci. 2010;30(19):6667–77.
Brown P, Mazzone P, Oliviero A, Altibrandi MG, Pilato F, Tonali PA, et al. Effects of stimulation of the subthalamic area on oscillatory pallidal activity in Parkinson’s disease. Exp Neurol. 2004;188(2):480–90.
Foffani G, Ardolino G, Meda B, Egidi M, Rampini P, Caputo E, et al. Altered subthalamo-pallidal synchronisation in parkinsonian dyskinesias. J Neurol Neurosurg Psychiatry. 2005;76(3):426–8.
Meissner W, Ravenscroft P, Reese R, Harnack D, Morgenstern R, Kupsch A, et al. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6-OHDA-lesioned rat in the presence of excessive extracellular striatal dopamine. Neurobiol Dis. 2006;22(3):586–98.
Brown P. Abnormal oscillatory synchronisation in the motor system leads to impaired movement. Curr Opin Neurobiol. 2007;17(6):656–64.
Brown P, Eusebio A. Paradoxes of functional neurosurgery: clues from basal ganglia recordings. Mov Disord. 2008;23(1):12–20; quiz 158.
Leblois A, Meissner W, Bioulac B, Gross CE, Hansel D, Boraud T. Late emergence of synchronized oscillatory activity in the pallidum during progressive Parkinsonism. Eur J Neurosci. 2007;26(6):1701–13.
Leblois A, Meissner W, Bezard E, Bioulac B, Gross CE, Boraud T. Temporal and spatial alterations in GPi neuronal encoding might contribute to slow down movement in Parkinsonian monkeys. Eur J Neurosci. 2006;24(4):1201–8.
Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res Brain Res Rev. 1995;20(1):91–127.
Kayahara T, Nakano K. Pallido-thalamo-motor cortical connections: an electron microscopic study in the macaque monkey. Brain Res. 1996;706(2):337–42.
Hoover JE, Strick PL. The organization of cerebellar and basal ganglia outputs to primary motor cortex as revealed by retrograde transneuronal transport of herpes simplex virus type 1. J Neurosci. 1999;19(4):1446–63.
Kelly RM, Strick PL. Macro-architecture of basal ganglia loops with the cerebral cortex: use of rabies virus to reveal multisynaptic circuits. Prog Brain Res. 2004;143:449–59.
Deniau JM, Menetrey A, Charpier S. The lamellar organization of the rat substantia nigra pars reticulata: segregated patterns of striatal afferents and relationship to the topography of corticostriatal projections. Neuroscience. 1996;73(3):761–81.
Calabresi P, Centonze D, Bernardi G. Electrophysiology of dopamine in normal and denervated striatal neurons. Trends Neurosci. 2000;23(10 Suppl):S57–63.
Guthrie M, Leblois A, Garenne A, Boraud T. Interaction between cognitive and motor cortico-basal ganglia loops during decision making: a computational study. J Neurophysiol. 2013;109(12):3025–40.
Pasquereau B, Nadjar A, Arkadir D, Bezard E, Goillandeau M, Bioulac B, et al. Shaping of motor responses by incentive values through the basal ganglia. J Neurosci. 2007;27(5):1176–83.
Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol. 1996;50(4):381–425.
Picconi B, Centonze D, Hakansson K, Bernardi G, Greengard P, Fisone G, et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6(5):501–6.
Belujon P, Lodge DJ, Grace AA. Aberrant striatal plasticity is specifically associated with dyskinesia following levodopa treatment. Mov Disord. 2010;25(11):1568–76.
Halje P, Tamte M, Richter U, Mohammed M, Cenci MA, Petersson P. Levodopa-induced dyskinesia is strongly associated with resonant cortical oscillations. J Neurosci. 2012;32(47):16541–51.
Bastide MF, Dovero S, Charron G, Porras G, Gross CE, Fernagut PO, et al. Immediate-early gene expression in structures outside the basal ganglia is associated to l-DOPA-induced dyskinesia. Neurobiol Dis. 2013;62:179–92.
Cenci MA, Konradi C. Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog Brain Res. 2010;183:209–33.
Guigoni C, Li Q, Aubert I, Dovero S, Bioulac BH, Bloch B, et al. Involvement of sensorimotor, limbic, and associative basal ganglia domains in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J Neurosci. 2005;25(8):2102–7.
Phelix CF, Liposits Z, Paull WK. Monoamine innervation of bed nucleus of stria terminalis: an electron microscopic investigation. Brain Res Bull. 1992;28(6):949–65.
Cenci MA, Lundblad M. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J Neurochem. 2006;99(2):381–92.
Carta M, Carlsson T, Munoz A, Kirik D, Bjorklund A. Involvement of the serotonin system in L-dopa-induced dyskinesias. Parkinsonism Relat Disord. 2008;14 Suppl 2:S154–8.
Navailles S, Bioulac B, Gross C, De Deurwaerdere P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson’s disease. Neurobiol Dis. 2010;38(1):136–43.
Carta M, Bezard E. Contribution of pre-synaptic mechanisms to L-DOPA-induced dyskinesia. Neuroscience. 2011;198:245–51.
Krawczyk M, Georges F, Sharma R, Mason X, Berthet A, Bezard E, et al. Double-dissociation of the catecholaminergic modulation of synaptic transmission in the oval bed nucleus of the stria terminalis. J Neurophysiol. 2011;105(1):145–53.
Carta M, Carlsson T, Kirik D, Bjorklund A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain. 2007;130(Pt 7):1819–33.
Carta M, Carlsson T, Munoz A, Kirik D, Bjorklund A. Serotonin-dopamine interaction in the induction and maintenance of L-DOPA-induced dyskinesias. Prog Brain Res. 2008;172:465–78.
Rylander D, Parent M, O’Sullivan SS, Dovero S, Lees AJ, Bezard E, et al. Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann Neurol. 2010;68(5):619–28.
Navailles S, Bioulac B, Gross C, De Deurwaerdere P. Chronic L-DOPA therapy alters central serotonergic function and L-DOPA-induced dopamine release in a region-dependent manner in a rat model of Parkinson’s disease. Neurobiol Dis. 2011;41(2):585–90.
Vollenweider I, Lang Y, Borton D, Ko D, Li Q, Courtine G, et al. Translational analysis platform for neuromotor disease research and therapeutic validation: application to Parkinson’s disease. In: Society of Neuroscience conference. San Diego; 2013. Poster 241.18/O1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag London
About this chapter
Cite this chapter
Ko, W.K.D., Bastide, M., Bezard, E. (2014). Basal Ganglia Circuitry Models of Levodopa-Induced Dyskinesia. In: Fox, S., Brotchie, J. (eds) Levodopa-Induced Dyskinesia in Parkinson's Disease. Springer, London. https://doi.org/10.1007/978-1-4471-6503-3_7
Download citation
DOI: https://doi.org/10.1007/978-1-4471-6503-3_7
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-6502-6
Online ISBN: 978-1-4471-6503-3
eBook Packages: MedicineMedicine (R0)