
Abstract
Genetically modified mouse models have been instrumental in deciphering pathomechanisms in a large variety of human conditions. Similarly, transgenic and knockout mice have contributed to understanding neurodegenerative processes in Alzheimer’s disease (AD) and frontotemporal lobar degeneration (FTLD). While the first models for AD and FTLD, based on mutations in APP and tau, respectively, have been generated more than a decade ago, recent years have seen the identification of new genes involved in the disease. This led to the generation of a large number of new transgenic mouse models for FTLD. This chapter provides an overview of APP and tau-based mouse models of AD and FTLD and discusses in detail the more recent FTLD models expressing novel disease genes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Different Methods to Genetically Modify Mice
Transgenesis techniques to generate mouse models of disease rely on both gene transfer methods as well as methods to manipulate the early mouse embryo [1]. To date, the most commonly used technique involves microinjection of DNA constructs into the pronucleus of a developing zygote, leading to random integration of a transgene into the endogenous DNA [2]. This then produces a “transgenic animal” that has a foreign gene(s) stably incorporated into its genome through human intervention. This integrated recombinant double-stranded DNA is called a “transgene.”
Over the years, the development of more sophisticated models has allowed for better control of transgene expression, both temporally and spatially. This includes both inducible and conditional mouse models. Inducible mouse models enable the study of transgene expression in a strictly regulated manner, as they drive transgene expression exclusively upon induction, by either the presence or absence of a drug, in a dose-dependent manner. This allows researchers to overcome some of the problems associated with constitutive transgene expression, such as embryonic lethality. Conditional models involve the generation of mice with altered gene expression in a cell specific manner, through the expression of recombinase enzymes, which are under the control of a selected promoter, that can remove, invert, or translocate DNA segments.
Site-specific manipulation of the genome (gene targeting) allows for the disruption of a specific gene (knockout approach) or the insertion of a transgene in a defined locus (knock-in approach). Very recently, targeted transgenesis has been introduced, which relies on a core technology based on the use of engineered nucleases, such as zinc finger nucleases (ZfN) [3] or transcription activator-like effector nucleases (TALEN) [4]. This new technology enables investigators to manipulate virtually any gene in a diverse range of cell types and organisms with extreme precision (single base pair). Targeted transgenesis, used either for stable overexpression of a transgene or for disruption of endogenous genes, ultimately remains the most powerful tool to understand the mechanisms underlying physiological processes and their pathological counterparts.
Mouse Models of Alzheimer’s Disease
The past two decades have seen the generation of a large number of transgenic mouse models of AD, with a focus on amyloid-β (beta) (Aβ [beta])-forming models. These have assisted in a large number of studies investigating mechanisms underlying neuronal dysfunction and neurodegeneration in AD, as well as in developing and testing novel treatments. Aβ (beta)-forming transgenic mouse models have been extensively reviewed before [e.g., [5]]. Therefore, this part of the chapter will provide a rather general overview and highlight only some discoveries made using AD mouse models.
Amyloid-β (Beta) Precursor Protein (APP) Models
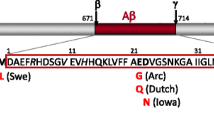
Intensive efforts have been made to develop transgenic mouse models that recapitulate the pathology and symptoms of AD over the past decades. While overexpression of human nonmutant APP did not result in plaque formation and memory deficits, it was the identification of pathogenic mutations in APP in familial cases of AD that paved the way for generating the first disease models [6]. Since then, expression of human mutant APP reproduced Aβ (beta) plaque pathology in a large number of transgenic mouse models [5]. In most models, expression of mutant APP results in the production of Aβ (beta) throughout the brain with plaque formation, affecting memory performance of mice in different test paradigms, such as the Morris water maze. APP transgenic models have also been the basis for showing a prion-like transfer of Aβ (beta) pathology between APP transgenic mice in a strain-dependent manner [7].
While initial studies did not report an overt neuronal loss, a limited number of subsequent studies of established lines reported a decrease in numbers of neurons in certain brain areas [8, 9]. However, the absence of pronounced neuronal loss remains a limitation of Aβ (beta)-forming APP transgenic mice.
To determine if loss of APP function contributes to the development of AD, APP knockout mice have been generated. However, their phenotypes are rather mild and possibly due to developmental anomalies [10]. Interestingly, early postnatal death of double knockout mice with deletion of APP and APLP2, the latter belonging to the same protein family, suggests a functional overlap between the family members during development [11]. APP-deficient mice have contributed to the understanding of the possible physiological functions of APP, some of which have implications for the disease [12–14].
In summary, APP transgenic mice have been instrumental in reproducing aspects of AD pathology in vivo and in deciphering mechanisms underlying disease. Over the past decades, APP transgenic mice have become a central in vivo tool in studying pathomechanisms and developing treatments for AD.
Combinatorial Models
In an attempt to accelerate Aβ (beta) pathology onset and progression and to more closely model the human pathology, mutant APP transgenic mice have been crossed with other mutation-harboring mice. For instance, mutations in the presenilin-encoding PSEN genes alter the activity of the γ (gamma)-secretase complex which presenilins are part of. Expression of mutant PSEN1 in mice crossed with Aβ (beta)-forming APP transgenic mice resulted in accelerated Aβ (beta) formation and earlier onset of behavioral deficits as well as neuronal loss [15, 16]. Interestingly, the effects of mutant PSEN was even more pronounced in the absence of the murine PSEN, achieved by a mutant human PSEN1 knock-in approach [17]. Conversely, reduced β (beta)-secretase activity in BACE-deficient mice reduced Aβ (beta) formation and ameliorated behavioral deficits when crossed on an Aβ (beta)-forming APP transgenic strain [18–20], while overexpression of BACE on an APP background increased pathology [21].
Carriers of the APOEε (epsilon)4 allele have a 20-fold increased risk of developing AD, making it the number one risk gene for developing sporadic late-onset AD [22]. In support of a role for ApoE in Aβ (beta) pathology, crossing APP transgenic mice on a ApoE-/- background reduced both Aβ (beta) levels and its deposition [23]. Conversely, expressing human ApoE4 in APP transgenic mice, by viral gene delivery, increased pathology [24].
Aβ (beta)-forming APP mice were used to provide the first in vivo evidence for the amyloid cascade hypothesis that places Aβ (beta) upstream of tau pathology and neurodegeneration in the sequence of pathogenic events. Accordingly, crossing of APP transgenic mice with human mutant tau-expressing mice resulted in increased neurofibrillary tangle (NFT) formation [25]. A similar result has been achieved by injecting synthetic aggregated Aβ (beta)1-42 into brains of P301L mutant tau transgenic pR5 mice [26].
The central role of tau in AD development, particularly in mediating neuronal deficits induced by Aβ (beta), has been shown when APP transgenic mice were crossed on a tau-deficient background [27]. This approach prevented premature mortality and behavioral deficits associated with Aβ (beta) formation, though the levels of Aβ (beta) and numbers of plaques were unchanged. In this context, we showed that tau mediates Aβ (beta)-induced excitotoxicity by controlling Fyn levels at the postsynapse and sensitizing NMDA receptors to become easily hyperexcited [28]. This work provides the first evidence for a non-axonal function of tau in the dendritic compartment of neurons [29], which has since been supported by other studies since [30, 31].
Taken together, combinatorial approaches using APP transgenic mice together with additional mutant strains have provided exciting new insight into the pathogenesis of AD. Although only a selected small number of studies have been presented here, it is reasonable to expect that combinatorial approaches using APP-based AD mouse models will continue to extend our understanding of AD.
Mouse Models of Frontotemporal Lobar Degeneration
Frontotemporal lobar degeneration (FTLD; also referred to as frontotemporal dementia [FTD]) umbrellas a large number of related neurodegenerative conditions with overlapping clinical symptoms. This is paralleled by an increasing number of proteins that have been found to be present in deposits in FTLD brains as well as the identification of more and more genes carrying pathogenic mutations, further distinguishing subforms of FTLD [32]. This chapter will discuss transgenic mouse models generated by expressing or deleting different genes, with an emphasis on more recent models. Tau models, some of which have been around for many years, will be addressed rather generally and by highlighting some of the recent findings in these mice.
Tau Models
While tau deposits in neurons together with the formation of extracellular Aβ (beta) plaques in AD patient brains, tau forms inclusions in the absence of overt Aβ (beta) pathology in FTLD. To model the tau pathology of AD and FTLD in mice, the first transgenic strain was generated to express the longest human isoform of tau without mutations in neurons [33]. These mice presented with accumulation of hyperphosphorylated forms of tau, resembling a pre-tangle state, but they failed to reproduce NFT formation. Interestingly, aged mice of this tau transgenic line developed motor deficits together with a Wallerian degeneration of axonal tracks in the spinal cord, indicating that pre-tangle hyperphosphorylated tau suffices to impair neuronal function and integrity without deposition.
It took close to five more years after the first tau model had been published, until transgenic expression of human tau carrying a pathogenic FTDP-17 mutation, P301L, achieved NFT formation in vivo [34]. These mice are characterized by severe motor and behavioral deficits, axonal degeneration, and early death, resembling aspects of the human disease. Since the generation of this first mutant tau-expressing mouse model, many additional lines have been generated that recapitulated different aspects of the human condition [5]. Interestingly, neuronal loss that characterizes the human disease has not been reproduced in the earlier mutant tau transgenic mice. But eventually, this has been achieved, when mice expressing distinct mutations (N279K [35] or P301S [36, 37]) using conventional neuronal promoters or particularly high levels of P301L mutant human tau using an inducible modified CMV promoter [38] showed pronounced neuronal loss. These lines are characterized by early-onset NFT formation. Neuronal loss has also been achieved in an elegant transgenic model expressing a mutant but truncated tau that is limited to the microtubule-binding repeats and characterized by rapid tau fibril formation and deposition [39]. This model used an inducible modified CMV promoter too and in combination with a complementary model that expresses the same truncated tau variant but with inclusion of two aggregation-preventing point mutations (I277P and I308P) forms an excellent in vivo tool to study tau fibril formation and test anti-aggregation drugs [39].
Since tau pathology in human FTLD is not limited to neurons, transgenic mouse models with non-neuronal mutant tau expression have been generated [40, 41]. Interestingly, both expression in astrocytes and in oligodendrocytes resulted in neuronal dysfunction and axonal degeneration. This is possibly due to impairment of glia in supporting neuronal function and integrity.
Mutant tau transgenic mice have become a highly valuable tool for studying pathomechanisms underlying tau pathology and neurodegeneration in FTLD but also in AD. Accordingly, transgenic mice are currently extensively used to investigate the prion-like disease progression hypothesis for tau, which includes release of distinct tau species from diseased neurons that are then taken up by healthy neurons to form a seed for disease propagation [42]. So far, it has been shown that tau pathology can be transferred from a mutant tau transgenic line with NFT formation to a transgenic strain that expresses nonmutant human tau and does not form NFTs unless inoculated with brain extracts from NFT-forming mice [43] or human patient brains with tau pathology [44] by stereotaxic injection. Furthermore, inducible mutant tau expression limited to a distinct brain area (entorhinal cortex) leads to NFT formation in connected brain areas (hippocampus) as mice age [45].
Mutant tau transgenic mice are also regularly used for preclinical drug development and testing. For instance, more recently, several groups have developed vaccination strategies targeting pathological tau, either by active or passive immunization [46–49]. Each of these studies used different mutant tau transgenic mouse lines to show efficacy and safety of this approach, providing the preclinical evidence needed to further this approach to clinical trials. Similarly, mutant tau transgenic mice have been used to determine the effects of compounds on different aspect of tau pathology [37, 50].
Taken together, it was the generation of mutant tau transgenic mice that provided in vivo evidence that pathogenic FTLD mutations accelerate tau aggregate formation and deposition and drive neuronal dysfunction and loss. Furthermore, mutant tau transgenic mice are important tools for studying pathomechanisms in vivo and to develop and test new therapeutic approaches. Finally, although the pathogenic mutations expressed in these lines originate from FTLD patients, tau transgenic mice are also valuable for studying tau-related aspects of AD, given the similarity of tau pathology in AD and FTLD.
TAR DNA-Binding Protein 43 (TDP-43) Models
In 2006, Neumann and colleagues identified in a groundbreaking publication TDP-43 as the major component of, until then, unidentified ubiquitin-positive deposits in FTLD [51]. Moreover, they showed that similar deposits in amyotrophic lateral sclerosis (ALS) (also referred to as Lou Gehrig’s disease or motor neuron disease [MND]) are also made up of TDP-43. TDP-43 is a nuclear protein with two RNA/DNA binding motifs. Consistent with these domains, TDP-43 is involved in RNA/DNA-related processes in cells, including RNA trafficking, RNA splicing, and promoter binding [52]. In disease, TDP-43 accumulates in the cytoplasm due to unknown reasons and undergoes secondary modifications, such as truncation, phosphorylation, and ubiquitination, eventually leading to the formation of aggregates [53].
Similar to tau transgenic mice, the identification of mutations in the TDP-43-encoding TARDBP gene has paved the way for the generation of a number of transgenic mouse models with mutant TDP-43 expression. Furthermore, non-disease mutants of TDP-43 with deletion of function domains have been expressed in mice.
The first TDP-43 mouse model published in 2009 expressed human TDP-43 carrying the A315T mutation under the murine prion protein promoter to generate the Prp-TDP43A315T mice [54]. These mice have an approximate threefold expression over endogenous TDP-43 with highest expression present in the brain and spinal cord. Ubiquitination of proteins in layer V neurons of the cortex concomitantly occurs with loss of nuclear staining of TDP-43 in selective neurons in these mice. Reactive gliosis is also present in this region of degenerating neurons.
This initial TDP-43 transgenic line [54] was followed by several new models generated over the past years [55–59]. Wils and colleagues expressed nonmutant human TDP-43 under the neuronal murine Thy1 promoter to generate the TDP-43WT lines TAR4 and TAR6 [55]. Hemizygous TAR4 and TAR6 have 2.8- and 1.9-fold, and homozygous TAR4/4 and TAR6/6 have 5.1- and 3.8-fold expression over endogenous TDP-43. These mice have nuclear and cytoplasmic inclusions in cortical layer V neurons that are ubiquinated and phosphorylated as well as a marked astrogliosis. The limited neuronal loss observed in these mice correlated with expression levels of TDP-43. In addition, homozygous TAR4 have an accumulation of cytoplasmic full length TDP-43 as well as the 25 kDa and 35 kDa C-terminal fragments. Phenotypically, these mice exhibit complex motor impairments, with hind limb clasping, reduced footstep length, reduced motor performance on the Rota-Rod, as well as reduced survival rate with disease onset and severity dependent on TDP-43 expression levels.
Xu and colleagues expressed nonmutant human TDP-43 under the murine prion protein promoter to generate the TDP-43PrP with a 1.9–2.5-fold expression over endogenous TDP-43 [56]. An increased human TDP-43 mRNA level was observed with a concomitant decrease in mouse TDP-43 mRNA levels. These mice produce ~25 kDa C-terminal TDP-43 fragments, which are urea insoluble, as well as phosphorylated and ubiquinated cytoplasmic inclusions, reactive gliosis, and argyrophilic degenerating neurites and neurons in the spinal cord. Interestingly, these mice also have abnormal clustering and degeneration of mitochondria in their spinal cord neurons. TDP-43PrP mice display lower body weights compared to wild-type littermates at 14 days, together with hindlimb clasping, body tremors, and a “swimming” gait at 21 days. Their survival is limited as they die between 1 and 2 months of age.
Swarup and colleagues generated three TDP-43 transgenic mice (nonmutant human TDP-43, TDP-43A315T and TDP-43G348C) from DNA subcloned from TARDBP bacterial artificial chromosomes containing the endogenous Δ4 kB promoter [59]. These mice present with an approximately threefold overexpression of transgenic TDP-43 over the endogenous protein. Significantly more ~25 kDa and 35 kDa C-terminal fragments were observed in TDP-43A315T and TDP-43G348C compared to nonmutant TDP-43 expressing mice. Ubiquitination of cytoplasmic TDP-43 was observed only in the mutant TDP-43 lines. Abnormal aggregates containing peripherin and neurofilament proteins were also present in TDP-43G348C mice. In addition, gliosis and neuroinflammation were observed in all lines. Furthermore, all lines presented with cognitive and motor deficits in the passive avoidance test, Barnes maze test, and Rota-Rod at 7–10 months with these impairments being most severe in the TDP-43G348C line. Interestingly, they revealed that there is significant increase of GFAP promoter activity or astrogliosis before the onset of behavioral impairments.
Igaz and colleagues generated transgenic mice with inducible overexpression of either nonmutant human TDP-43 (hTDP-43 WT) or human TDP-43 with mutated nuclear localization signal (hTDP-43-ΔNLS) [57]. Mutation of the NLS prevents TDP-43 from entering the nucleus, and, hence, it accumulates in the cytoplasm [60]. Neuronal expression was achieved by using a CaMK2α promoter to drive tet-off rTA and a tetracycline responsive promoter to drive hTDP43 expression. hTDP-43 WT mice had an 8- to 9-fold expression over endogenous TDP-43 and hTDP-43-ΔNLS mice 0.4- to 1.7-fold, respectively. Doxycycline treatment started at birth to suppress expression during postnatal brain development was removed at weaning (3 weeks of age) and mice were analyzed at various time points after doxycycline removal. Both models present with urea-insoluble TDP-43 with no concomitant presence of C-terminal fragments. In addition, ubiquitinated and phosphorylated TDP-43 aggregates were found to be present in hTDP-43-ΔNLS mice. Significant neuronal loss was observed in the dentate gyrus of both lines with the hTDP-43-ΔNLS mice having a more acute and severe dentate gyrus degeneration. The presence of axonal loss and gliosis of the corticospinal tract of hTDP-43-ΔNLS mice occur in a time-dependent manner relative to the developments of motor deficits.
Since the abnormal localization of TDP-43 in disease means that the protein is depleted from the nucleus, TDP-43 might not be able to execute its normal functions (=loss of function). To test this in vivo, Kraemer and colleagues employed a gene trap insertion strategy to generate mice lacking TDP-43 [61]. Heterozygous mice are viable in contrast to homozygous mice, which is embryonically lethal. Heterozygous (Tardbp+/−) mice have reduced grip strength with no reportable differences in pathology observed.
Progranulin (PGRN) Models
Mutations in the progranulin (PGRN) gene have been shown to cause tau-negative, ubiquitin-positive, and TDP-43-positive FTLD [62, 63]. The majority of these mutations are known to cause messenger RNA (mRNA) instability (resulting in degradation), while other mutations can cause loss of the entire mutant allele [63], cause prematurely truncated protein [63], or result in the generation of mutant PGRN protein that cannot be secreted efficiently [64] or appropriately cleaved [65]. Therefore, through a variety of mechanisms, these mutations all result in either reduced PGRN levels or loss of PGRN function. It is for this reason that PGRN knockout mice have been used to study this particular disorder.
A variety of PGRN knockout strains have been generated [66–70]. Except for one report [71], all of these knockout strains produce offspring with genotypes at an expected Mendelian ratio, suggesting that loss of PGRN does not impair embryonic development and/or survival. One common feature of all of these strains is that aged, homozygote mice all develop severe astrogliosis and microgliosis that increases with age (generally first detected around 12 months of age). Hence, neuroinflammation may play a role in the disease process. Interestingly, PGRN homozygote knockout mice react less efficiently and with more severe inflammation to bacterial listeria infections [67], and both PGRN-deficient microglia and macrophages are more cytotoxic to cultured neurons [67, 69]. In addition to this, hippocampal slices from PGRN homozygote knockout mice show greater neuronal sensitivity to glucose and oxygen starvation [67]. This suggests that FTLD-PGRN may arise from a combination of deregulated inflammation as well as increased neuronal vulnerability to certain stressors.
In all but one strain [68], homozygote PGRN knockout mice have been found to display significantly more ubiquitinated structures in various brain regions by as early as 7 months (ranging from 7 to 18 months), which increase with age. In support of a compromised ubiquitin-proteasome system, increased p62 and cathepsin D (markers of autophagy and lysosomes) were found in addition to increases in neuronal ubiquitin in PGRN knockout mice [70]. These pathological changes are common features of FTLD-TDP but are also associated with aging. Furthermore, in three of the PGRN knockout strains, levels of lipofuscin, a marker of cellular aging, were significantly increased (throughout the brain and also in the liver in one strain) by as early as 8 months. Hence, PGRN knockout mice may undergo accelerated aging, thereby potentially contributing to the disease process. Interestingly, levels of PGRN progressively increased in the brains of aging wild-type animals, suggesting a role for PGRN in aging [71]. However, no neuronal loss or markers of apoptosis have been observed in any of the knockout strains, though some lines have shorter life spans [70, 72].
Although PGRN mutations are associated with TDP-43 neuropathology in humans, it is not clear whether this is also the case in PGRN knockout mice. To date, only some pathologically phosphorylated TDP-43 has been identified in the brains of two strains [67, 70, 73]. It therefore remains unclear what role PGRN mutations play in the development of TDP-43 pathology, though it does not appear that loss of PGRN alone suffices to cause TDP-43 relocalization or aggregation.
The behavioral assessment of different PGRN knockout lines has produced variable results. This could be the result of variation in genetic background or differences in protocols and equipment used. PGRN knockout mice do not have any significant motor impairments (although reduced muscle strength has been reported by Ghoshal and colleagues); however, there have been multiple reports of reduced social engagement and aggression [68, 72, 73] and depression-like behavior and disinhibition [73], which mimics several major behavioral hallmarks of FTLD. In addition, aged PGRN knockout mice show reduced performance during Morris water maze testing [70, 72, 73] and novel object testing [68], suggesting late-onset learning and memory impairments. Although the mechanism by which PGRN deficiency causes these behavioral phenotypes is unclear, Petkau and colleagues (2012) utilized electrophysiological recordings to demonstrate that hippocampal slices from PGRN homozygote knockout mice display reduced postsynaptic responsiveness and occasional LTP dysfunction. Furthermore, CA1 pyramidal neurons showed reduced dendritic length and reduced spine density. Therefore, synaptic dysfunction may play a role in the disease process underlying FTLD-PGRN.
It should be noted that the majority of studies discussed above utilized homozygote PGRN knockout mice, despite the fact that PGRN mutations cause haploinsufficiency in humans. For this reason, it is important to highlight some results obtained from heterozygote PGRN knockout mice [74]. These mice express approximately 50 % less PGRN mRNA and protein (and were maintained on two different genetic backgrounds), but unlike homozygote PGRN knockout mice, they do not develop any significant astrogliosis, microgliosis, and lipofuscinosis or show any electrophysiological changes, nor do they have any motor impairments or memory and learning impairments. Despite this, these animals (regardless of the genetic background) still show social and emotional dysfunction.
In summary, PGRN knockout mice recapitulate a number of hallmark features of FTLD-TDP43, including neuroinflammation, ubiquitinated aggregates, and behavioral impairments. However, the exact role of TDP-43 in this disease and the effects of PGRN haploinsufficiency versus homozygous deficiency remain to be determined.
Valosin-Containing Protein (VCP) Models
Mutations in the valosin-containing protein (VCP) gene are known to cause the multisystem degenerative disorder inclusion body myopathy associated with Paget’s disease of the bone and frontotemporal dementia (IBMPFD) [75]. Although muscle weakness and myopathy are the most common clinical features of this disorder, approximately 30 % of patients also develop language and behavioral impairments typical of FTLD [76]. Furthermore, TDP-43- and ubiquitin-positive inclusions are found in both the brain and muscle of IBMPFD patients. Interestingly, some reports also link VCP mutations to amyotrophic lateral sclerosis [77, 78]. Over 20 mutations have been identified in VCP, all of which are thought to alter the 3D structure of VCP and thereby perturb the interactions between VCP and its various substrates [79]. Substitution of arginine 155 to histidine (R155H) is the mutation most commonly associated with IBMPFD. It is for this reason that the majority of mouse models utilize this particular mutation.
To develop an animal model of IBMPFD, a number of groups have generated transgenic mice that express mutant VCP [80–84]. Although these strains all express a similar mutant protein, there are a number of inherent differences among the strains. For example, because mouse VCP differs from the human protein by only one amino acid, some groups chose to express human mutant VCP in the mouse model, whereas other models express mutant mouse VCP; various promoters have been used to generate mice that overexpress the mutant protein exclusively in the muscle [81], the brain [80], or ubiquitous expression in all tissues [82]; while other groups generated knock-in mice that express mutant VCP at levels similar to that of the endogenous protein [83, 84].
Despite these inherent differences, however, all mutant VCP mouse strains have been reported to develop VCP-negative, TDP-43-positive, and ubiquitin-positive aggregates. These develop in regions where the mutant protein is expressed, i.e., the muscle, brain, and spinal cord. In heterozygote animals these aggregates appear at around 10–15 months in the muscle and the spinal cord, and at 14–20 months in the brain; and in homozygote mice [84] TDP-43 aggregates were observed as early as 15 days in the muscle, brain, and spinal cord. In some strains, cytoplasmic and nuclear clearance of TDP-43 was observed, as well as insoluble and high molecular weight TDP-43 species [80, 82, 85]. In one particular strain, TDP-43 aggregates were observed to co-localize with the stress granule marker TiA-1, and overall levels of TiA-1 were increased, suggesting an increased stress response, which could potentially alter mRNA transport and translation. Altered stress granule dynamics and/or altered mRNA metabolism may therefore play a role in the disease processes associated with TDP-43 proteinopathies. Despite the presence of TDP-43 aggregates, however, none of the strains show any sign of neurodegeneration in the brain [80, 82, 83], although loss of motor neurons in the spinal cord has been reported [85].
Other pathological features commonly observed in these mice include significant increases in levels of general protein ubiquitination [80, 81, 84, 85] and upregulation of markers of autophagy [83–85] in the muscle, brain, and spinal cord. Combined with the knowledge that VCP is known to play a role in regulating ubiquitin degradation of a number of proteins, this data suggest that dysfunctional protein degradation and accumulation of ubiquitinated proteins may play a role in the development of this disorder. In addition to this, high molecular weight species of TDP-43 were found to pull down with VCP, suggesting a direct interaction between VCP and high molecular weight TDP-43 isoforms in these mice [80]. One possible explanation for this interaction is that VCP may be trying to direct TDP-43 to the proteasome for degradation, and that disruptions to this interaction may cause TDP-43 to accumulate in the cytoplasm and eventually aggregate.
IBMPFD is most commonly characterized by myopathy. In accordance with this, in all the mutant VCP mice strains that express the transgene in muscle tissue, significant pathology is observed. This includes the following: vacuoles, disordered architecture, variation in muscle fiber size, and swollen mitochondria [81–85]. On average, these features were observed at around 6–15 months of age; however, in mice that were bred to homozygosity, muscle abnormalities were already observed after 15 days. Radiographic and biochemical bone deformities consistent with Paget’s disease are also commonly observed in IBMPFD. Similar characteristics have been reproduced in the mutant VCP mice, including loss of bone structure, decreased bone density, hypomineralization, and sclerotic lesions at around 13–16 months of age [82–84]. Therefore, these mice recapitulate the wide range of pathological features associated with IBMPFD within the muscle, brain, and bone.
In general, all mutant VCP mouse strains show signs of muscle weakness and reduced Rota-Rod performance, which is in accordance with the clinical presentation in human patients [81–84]. Although some reports show weight loss and reduced survival in certain strains [82, 85], particularly in the homozygote mice which only survive to 14–21 days [84], this has not been observed in all strains. Custer and colleagues (2010) reported increased anxiety in these mice in the elevated zero maze and reduced performance in the novel object test, while other strains did not show any memory deficits [82–84]. Rodriguez-Ortiz and colleagues (2013) used a neuron-specific promoter to overexpress mutant VCP specifically in the forebrain [80]. These mice showed no difference in swim speed and distance in the Morris water maze but showed significant impairment in the probe trial, as well as impairments in object recognition testing, indicating learning and memory impairments. Furthermore, in these studies, higher expressing mutant VCP mice were shown to have greater cognitive deficits than lower expressing mice, and both lines showed greater impairment with age, suggesting that neuronal, mutant VCP expression impairs cognition in an age- and dose-dependent manner in these mice.
In summary, mutant VCP mice develop muscle and brain pathology as well as bone abnormalities that closely match with that observed in human IBMPFD patients. In addition, the spinal cord pathology closely matched that observed in human ALS patients. This therefore raises the question whether inclusion body myopathy, Paget’s disease, ALS, and FTLD share a common underlying mechanism. Because these mice develop ubiquitin-positive, TDP-43 aggregates and show relocalization of TDP-43, they can be used not only to study IBMPFD but also mechanisms underlying the development of TDP-43 pathology in general, particularly the neuron-specific expressing mice.
Charged Multivesicular Body Protein 2B (CMBP2B) Models
Although rare, mutations in the charged multivesicular body protein 2B (CHMP2B) gene are associated with familial forms of FTLD that display ubiquitin- and p62-positive inclusions that are negative for tau, FUS, and TDP-43 [86]. All mutations identified have been shown to cause a loss of the C-terminus of CHMP2B; therefore, the disease pathogenesis could be caused by either loss of normal CHMP2B function or, more specifically, loss of the CHMP2B C-terminus. To investigate this in greater depth, Ghazi-Noori and colleagues (2012) generated both wild-type (CHMP2Bwt) and C-terminally truncated (CHMP2Btrunc) CHMP2B transgenic mice, as well as CHMP2B knockout mice [87]. Initially, both the CHMP2B transgenic and knockout mice showed normal survival curves; however, after 500 days the CHMP2Btrunc mice showed increased mortality. Interestingly, the CHMP2Btrunc mice were shown to develop p62- and ubiquitin-positive inclusions (but TDP-43- and FUS-negative) that were absent in the CHMP2Bwt and knockout mice, suggesting that the formation of these inclusions was dependent on the expression of mutant CHMP2B. Furthermore, these inclusions were absent in the knockout mice, this suggests that the pathology is not caused by a loss of function but rather a gain of toxic function. These inclusions were found in a number of brain regions and motor neurons in the spinal cord, as early as 6 months, and were found abundantly by 18 months of age. In addition to the formation of inclusions, the CHMP2Btrunc were also shown to develop astrogliosis and microgliosis, which was absent in the CHMP2Bwt and knockout mice. Interestingly, there were no signs of astrogliosis in the CHMP2Btrunc mice until 12 months of age and thus occurred only after the formation of inclusions, whereas reactive microglia were already present at 6 months of age and therefore coincided with the formation of inclusions. Another feature that was found to develop exclusively in the CHMP2Btrunc mice was axonal swellings. These swellings were apparent at 6 months and increased with age and were found to contain mitochondria as well as vesicles from the lysosomal and autophagy degradation pathways. This suggests that axonal dysfunction and impairment, and possibly even axonal transport, may play a role in the disease process underlying FTLD caused by CMHP2B mutations.
Fused in Sarcoma (FUS) Models
Mutations in the fused in sarcoma (FUS) gene have been identified not only in rare cases of FTLD [88] but also in a number of familial ALS cases [89, 90]. In contrast to the pathology in ALS, however, FUS-positive inclusions identified in cases of FTLD co-localize with the RNA-binding proteins TAF15 and EWS and are also ubiquitinated. The majority of FUS mutations cluster within the extreme C-terminus of the protein and interfere with the nuclear localization sequence residing in the C-terminus [91]. However, it has been demonstrated that overexpression of nonmutant FUS is sufficient to cause an aggressive phenotype and neuropathology in mice [92] as well as in rats [93].
Mitchell and colleagues (2013) generated both heterozygote (FUStg/+) and homozygote (FUStg/tg) mice overexpressing human nonmutant FUS in the brain, spinal cord, and testis [92]. Although the FUStg/tg mice expressed higher levels of transgenic human FUS, this was found to decrease endogenous levels of murine FUS. FUStg/tg mice were found to have a significantly shorter life span that only averaged 82 days, whereas FUStg/+ mice seem to have similar survival to that of nontransgenic littermates. Both the FUStg/tg and (to a lesser extent) FUStg/+ mice show significant increases in overall levels of nuclear FUS. In addition, the FUStg/tg mice also show significant increases in levels of cytoplasmic FUS. This matches with the histological finding of numerous perinuclear inclusions throughout the brain and spinal cord and cytoplasmic FUS within cortical neurons of end-stage FUStg/tg mice, whereas only perinuclear inclusions are found (to a lesser extent) in the brains of FUStg/+ mice. Because nuclear levels of FUS are higher in the FUStg/tg mice, this suggests that localization of FUS to the cytoplasm is dependent upon the levels of nuclear FUS. Despite the formation of the cytoplasmic FUS aggregates, no nuclear clearance of FUS was observed. In addition to FUS aggregates, significant increases in the levels of ubiquitin were observed in the FUStg/tg mice, and to a lesser extent in the FUStg/+ mice. However, there was no obvious co-localization between FUS and ubiquitin. Furthermore, these FUS aggregates do not co-localize with EWS and TAF15, as is observed in FTLD. The molecular composition of the FUS aggregates in these mice therefore more closely resembles that observed in ALS rather than in FTLD.
Despite the formation of FUS aggregates, FUS transgenic mice showed no signs of neuronal loss or gliosis in the brain. However, in the spinal cords of FUStg/tg mice (and to a lesser extent in FUStg/+ mice), a significant decrease was observed in the number of motor neurons, as well as astrogliosis and microgliosis. The FUStg/tg mice also showed significant muscle atrophy and reduced muscle force, while FUStg/+ mice showed significant disorganization of muscle fibers. This suggests that overexpression of nonmutant FUS is more toxic to motor neurons than cortical neurons, particularly when it aggregates in the cytoplasm.
FUStg/tg mice develop a severe early-onset motor phenotype, whereas FUStg/+ mice show no signs of motor dysfunction. By 4 weeks the FUStg/tg mice developed a tremor and stilted gait, and from 4 weeks onward they failed to gain weight and showed significant impairments on the Rota-Rod. By 8 weeks of age, the mice showed significant decreases in general locomotor activity, clenching, and hindlimb paralysis.
In summary, these mice recapitulate various pathological and behavioral features of both ALS and FTLD patients, making them a good model to study these disorders. Exactly how overexpression of FUS causes these features, and whether a similar process occurs in the presence of mutant FUS, and whether the same process occurs in both ALS and FTLD remain to be determined [94].
Conclusion
Genetically modified mouse models are central to in vivo studies in AD and FTLD. Such models have provided insight and in some aspects a detailed understanding of pathological processes. With the identification of new proteins that form intracellular inclusion and novel pathogenic mutations in genes in FTLD, the number of different mouse models has dramatically increased. However, keeping in mind that each of the models reproduces and addresses only certain aspect of the human condition, it is likely that we will see a lot more transgenic models of even long-known candidates such as APP and tau. In addition, many of the new models of FTLD await being used in combination with other genetically modified strains to address complex pathological processes in vivo.
References
Brinster RL, Cross PC. Effect of copper on the preimplantation mouse embryo. Nature. 1972;238(5364):398–9.
Ittner LM, Gotz J. Pronuclear injection for the production of transgenic mice. Nat Protoc. 2007;2:1206–15.
Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325(5939):433.
Sung YH, Baek IJ, Kim DH, Jeon J, Lee J, Lee K, et al. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol. 2013;31:23–4.
Gotz J, Ittner LM. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9:532–44.
Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373(6514):523–7.
Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313(5794):1781–4.
Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, et al. Neuron loss in APP transgenic mice. Nature. 1998;395(6704):755–6.
Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, et al. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS One. 2013;8(4):e59586.
Guo Q, Wang Z, Li H, Wiese M, Zheng H. APP physiological and pathophysiological functions: insights from animal models. Cell Res. 2012;22(1):78–89.
Wang P, Yang G, Mosier DR, Chang P, Zaidi T, Gong YD, et al. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. J Neurosci. 2005;25:1219–25.
Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;8:525–31.
Li ZW, Stark G, Gotz J, Rulicke T, Gschwind M, Huber G, et al. Generation of mice with a 200-kb amyloid precursor protein gene deletion by Cre recombinase-mediated site-specific recombination in embryonic stem cells. Proc Natl Acad Sci U S A. 1996;93:6158–62.
Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell. 2010;142:857–67.
Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4(1):97–100.
Schmitz C, Rutten BP, Pielen A, Schafer S, Wirths O, Tremp G, et al. Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. Am J Pathol. 2004;164:1495–502.
Wang R, Wang B, He W, Zheng H. Wild-type presenilin 1 protects against Alzheimer disease mutation-induced amyloid pathology. J Biol Chem. 2006;281:15330–6.
Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, et al. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron. 2004;41:27–33.
McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, et al. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP Transgenic Mice. J Biol Chem. 2007;282:26326–34.
Ma H, Lesne S, Kotilinek L, Steidl-Nichols JV, Sherman M, Younkin L, et al. Involvement of beta-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:8167–72.
Willem M, Dewachter I, Smyth N, Van Dooren T, Borghgraef P, Haass C, et al. beta-site amyloid precursor protein cleaving enzyme 1 increases amyloid deposition in brain parenchyma but reduces cerebrovascular amyloid angiopathy in aging BACE x APP[V717I] double-transgenic mice. Am J Pathol. 2004;165:1621–31.
Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115:1449–57.
Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–4.
Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, et al. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2005;102:1211–6.
Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–91.
Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–5.
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–4.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–97.
Ittner LM, Gotz J. Amyloid-beta and tau—a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011;12:67–72.
Mondragon-Rodriguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, et al. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J Biol Chem. 2012;287(38):32040–53.
Nakanishi N, Ryan SD, Zhang X, Khan A, Holland T, Cho EG, et al. Synaptic protein alpha1-takusan mitigates amyloid-beta-induced synaptic loss via interaction with tau and postsynaptic density-95 at postsynaptic sites. J Neurosci. 2013;33:14170–83.
Mackenzie IR, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, et al. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 2011;121:207–18.
Gotz J, Probst A, Spillantini MG, Schafer T, Jakes R, Burki K, et al. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. Embo J. 1995;14:1304–13.
Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–5.
Dawson HN, Cantillana V, Chen L, Vitek MP. The tau N279K exon 10 splicing mutation recapitulates frontotemporal dementia and parkinsonism linked to chromosome 17 tauopathy in a mouse model. J Neurosci. 2007;27:9155–68.
Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–51.
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51.
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81.
Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, et al. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–48.
Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM, et al. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci. 2005;6(25):3539–50.
Higuchi M, Zhang B, Forman MS, Yoshiyama Y, Trojanowski JQ, Lee VM. Axonal degeneration induced by targeted expression of mutant human tau in oligodendrocytes of transgenic mice that model glial tauopathies. J Neurosci. 2005;25:9434–43.
Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–90.
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13.
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–40.
Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302.
Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27(34):9115–29.
Bi M, Ittner A, Ke YD, Gotz J, Ittner LM. Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One. 2011;6(12):e26860. doi:10.1371/journal.pone.0026860. Epub 2011 Dec 8.
Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010;224:472–85.
Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, et al. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem. 2011;286:34457–67.
van Eersel J, Ke YD, Liu X, Delerue F, Kril JJ, Gotz J, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci U S A. 2010;107:13888–93.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3.
Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–78.
Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol. 2008;15:772–80.
Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106:18809–14.
Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107:3858–63.
Xu YF, Gendron TF, Zhang YJ, Lin WL, D’Alton S, Sheng H, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30:10851–9.
Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121:726–38.
Stallings NR, Puttaparthi K, Luther CM, Burns DK, Elliott JL. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis. 2010;40:404–14.
Swarup V, Phaneuf D, Bareil C, Robertson J, Rouleau GA, Kriz J, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134:2610–26.
Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283:13302–9.
Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VM, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119:409–19.
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–9.
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–4.
Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, et al. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283:1744–53.
Wang J, Van Damme P, Cruchaga C, Gitcho MA, Vidal JM, Seijo-Martinez M, et al. Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J Neurochem. 2010;112:1305–15.
Kayasuga Y, Chiba S, Suzuki M, Kikusui T, Matsuwaki T, Yamanouchi K, et al. Alteration of behavioural phenotype in mice by targeted disruption of the progranulin gene. Behav Brain Res. 2007;185:110–8.
Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–28.
Petkau TL, Neal SJ, Milnerwood A, Mew A, Hill AM, Orban P, et al. Synaptic dysfunction in progranulin-deficient mice. Neurobiol Dis. 2012;45:711–22.
Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, et al. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest. 2012;122:3955–9.
Wils H, Kleinberger G, Pereson S, Janssens J, Capell A, Van Dam D, et al. Cellular ageing, increased mortality and FTLD-TDP-associated neuropathology in progranulin knockout mice. J Pathol. 2012;228(1):67–76. PubMed PMID: 22733568.
Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol. 2010;177(1):311–24. PubMed PMID: 20522652. Pubmed Central PMCID: 2893674.
Ghoshal N, Dearborn JT, Wozniak DF, Cairns NJ. Core features of frontotemporal dementia recapitulated in progranulin knockout mice. Neurobiol Dis. 2012;45(1):395–408.
Yin F, Dumont M, Banerjee R, Ma Y, Li H, Lin MT, et al. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. FASEB J. 2010;24:4639–47.
Filiano AJ, Martens LH, Young AH, Warmus BA, Zhou P, Diaz-Ramirez G, et al. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci. 2013;33:5352–61.
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–81.
Kimonis VE, Mehta SG, Fulchiero EC, Thomasova D, Pasquali M, Boycott K, et al. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A. 2008;146A:745–57.
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–64.
Abramzon Y, Johnson JO, Scholz SW, Taylor JP, Brunetti M, Calvo A, et al. Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33:2231 e1–e6.
Tang WK, Li D, Li CC, Esser L, Dai R, Guo L, et al. A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants. EMBO J. 2010;29:2217–29.
Rodriguez-Ortiz CJ, Hoshino H, Cheng D, Liu-Yescevitz L, Blurton-Jones M, Wolozin B, et al. Neuronal-specific overexpression of a mutant valosin-containing protein associated with IBMPFD promotes aberrant ubiquitin and TDP-43 accumulation and cognitive dysfunction in transgenic mice. Am J Pathol. 2013;183:504–15.
Weihl CC, Miller SE, Hanson PI, Pestronk A. Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum Mol Genet. 2007;16:919–28.
Custer SK, Neumann M, Lu H, Wright AC, Taylor JP. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet. 2010;19:1741–55.
Badadani M, Nalbandian A, Watts GD, Vesa J, Kitazawa M, Su H, et al. VCP associated inclusion body myopathy and Paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS One. 2010;5(10):e13183.
Nalbandian A, Llewellyn KJ, Badadani M, Yin HZ, Nguyen C, Katheria V, et al. A progressive translational mouse model of human valosin-containing protein disease: the VCP(R155H/+) mouse. Muscle Nerve. 2013;47(2):260–70.
Yin HZ, Nalbandian A, Hsu CI, Li S, Llewellyn KJ, Mozaffar T, et al. Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. 2012;3:e374.
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–8.
Ghazi-Noori S, Froud KE, Mizielinska S, Powell C, Smidak M, Fernandez de Marco M, et al. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain. 2012;135(Pt 3):819–32.
Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74:366–71.
Kwiatkowski Jr TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–8.
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–11.
Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–57.
Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B, et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013;125:273–88.
Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D, et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7:e1002011.
Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus-Hernandez M, Ansorge O, et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134(Pt 9):2595–609.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag London
About this chapter
Cite this chapter
van Eersel, J., Delerue, F., Ittner, L.M., Ke, Y.D. (2014). Alzheimer’s Disease and Frontotemporal Lobar Degeneration: Mouse Models. In: Galimberti, D., Scarpini, E. (eds) Neurodegenerative Diseases. Springer, London. https://doi.org/10.1007/978-1-4471-6380-0_8
Download citation
DOI: https://doi.org/10.1007/978-1-4471-6380-0_8
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-6379-4
Online ISBN: 978-1-4471-6380-0
eBook Packages: MedicineMedicine (R0)