
Abstract
Despite decades of research on Fe (or Co)-based electrocatalysts for the oxygen reduction reaction (ORR) in acidic medium, such as that in PEM fuel cells, the role of the metal is still one that raises a great deal of controversy. Consequently, the nature of the catalytic site in these non-noble metal ORR catalysts is still a topic of debate. One camp within the scientific community believes that the metal is an integral and electrochemically active part of the catalytic site, while the other believes that the metal is merely a chemical catalyst for the formation of special oxygen-reducing N-doped carbon structures. After presenting the case for the importance of non-noble catalysts at the cathode of PEM fuel cells, we introduce the three models of active sites that were advocated during the 1980s by van Veen, Yeager, and Wiesener and discuss how they have evolved, especially that of Yeager. Wiesener’s model is analyzed in detail through the work of several research groups that have been staunch supporters. The oxygen reduction mechanism on Fe-based and N-doped carbon catalytic sites is also reviewed. It is concluded that all the active sites proposed by van Veen, Yeager, and Wiesener in the 1980s, while different, are in fact simultaneously present in Fe (or Co)-based catalysts active for ORR in acidic medium, except that their activity and relative population in these catalysts are different, depending on the choice of the metal precursor, nitrogen precursor, structural properties of the carbon support, and the synthesis procedure.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
10.1 Introduction
It is a well-known fact that fossil fuels like coal, oil, and gas are the energy sources that account for about 90 % of all the energy consumed by the developed world. With a global consumption of 85 million barrels per day (1 barrel = 160 L), oil is the energy source of choice for transportation [1]. This sector represents more than one third of today’s total energy consumption per capita [2]. Despite a projected increase of oil consumption in the next few decades, known oil reserves are not overly abundant and may represent only about 40 years’ worth of energy at the present consumption rate. The projected life of natural gas reserves is of about 60 years, while it is about 250 years for coal [3]. In the not-so-distant future, we will therefore likely need an alternative energy source to fossil fuels. The latter may be more valuable and important as a source of carbon in the petrochemical, chemical, and pharmaceutical industries for future generations.
Among the possible sustainable replacements for fossil fuels are two key prospects: methanol and hydrogen. A methanol-based society, like the one advocated by G. Olah, would use the half-cycle of the oxidation of methanol to CO2 to produce energy and the other half-cycle to return CO2 back to methanol in a sustainable way [4]. Similarly, a hydrogen-based society would oxidize H2 to H2O to produce energy and reduce H2O back to H2 in a sustainable way. The advantage of using H2 as a fuel is that, unlike methanol, it is easily oxidizable electrochemically by O2 (Air), even at the low temperatures (around 80 °C) in polymer H2/O2 (Air) electrolyte membrane (PEM) fuel cells. The latter are very efficient electrical power generators that are especially well adapted for transportation applications, but also for smaller portable applications. Using (compressed) pure H2 as a fuel, PEM fuel cells would only produce H2O as a reaction product. Then, in an electrolyzer, water can be efficiently split back into H2 and O2 using stationary renewable electrical energy sources like solar, wind, and hydroelectricity.
In order to generate acceptable power levels, however, low-temperature PEM fuel cells need catalyzed electrodes. Thus far, the electrocatalysts used in PEM fuel cells have been exclusively Pt based. However, Pt is an industrial metal, mainly mined in South Africa (with about 75 % of the world production) and Russia (about 15 % of the world production). Today, 50 ± 10 % of Pt production of the 200,000 kg/year is used as chemical catalyst in catalytic converters for internal combustion engine vehicles. A mere glance at Fig. 10.1 immediately emphasizes the link between the cost of Pt, that of crude oil, and the health of the economy. In other words, one cannot hope for a strong economy and for Pt price to stay low at the same time.

Relationship between the costs of Pt, crude oil, and the health of the economy since the year 2000
Since Pt is a scarce and expensive metal, in 2002, the US Department of Energy (DOE) set targets for the maximum quantity of Pt to be used in H2/O2 (Air) PEM fuel cells. Initially, the 2015 target was set to 0.2 g Pt/kWe (with kWe for the rated electric power) for total anode and cathode Pt content combined. However, due to the considerable rise in Pt price, a new target—0.125 g Pt/kWe—was recently set for 2017 [5]. At around $1,700–1,800 per Pt Troy ounce (31.1 g), 0.125 g Pt/kWe would represent a Pt raw material cost of around $7 Pt/kWe. Meanwhile, the DOE also sets the cost target for the entire membrane electrode assembly to $9/kWe! There is, therefore, a strong case for replacing Pt with a lower cost non-noble metal-based electrocatalyst (or a metal-free electrocatalyst).
In the acid medium of PEM fuel cells (around pH 1), the oxidation of H2 on Pt is a much easier electrochemical reaction than the oxygen reduction reaction (ORR) on Pt. The latter reaction is notoriously slow. Consequently, about 5–10 times more Pt is needed at the cathode of PEM fuel cells than at their anode [6]. Therefore, a much greater cost reduction can be achieved by replacing Pt with a lower cost non-noble metal-based catalyst at the cathode of PEM fuel cells.
It is not possible to simply replace Pt with a non-noble metal as these metals (in their metallic state) will quickly corrode at the cathode of a PEM fuel cell. Oxides also have a strong tendency to dissolve in such an acidic environment. Some oxides, however, are acid resistant, but despite important advances recently reported in performing ORR in acid medium on these catalysts, they are still insufficiently active to replace Pt in fuel cells [7]. A detailed review on this topic can be found in Chap. 13. Promising contenders for the substitution of Pt-based catalysts for the cathode are Fe (or Co)-based electrocatalysts since they were recently reported to exhibit relatively high activity, stability, and high power in the potential range useful for transport and portable applications of PEM fuel cells [8–10].
The DOE has also set targets for non-platinum group metal (non-PGM) electrocatalysts. These targets are, of course, different from the DOE targets set for Pt-based catalysts because the nature of their catalytic sites is different from those of Pt nanoparticles or Pt layers on a carbon or inorganic support. Pt-based electrocatalysts are evaluated for their mass and specific activities at 900 mV iR-free in PEM fuel cells under operating conditions defined in ref. [6]. The 2015 target set by the DOE for non-PGM-based electrocatalysts for the cathode of H2/O2 (Air) PEM fuel cells was first set at a volumetric activity of 300 A/cm3 measured at 800 mViR-free and has remained so for the 2017 target [5]. The fuel cell operating conditions are the same as for Pt-based electrocatalysts. Today (2012), the highest volumetric activity reported was 230 A/cm3, achieved with a Fe-based electrocatalyst [10]. This is already quite close to and within grasp of the 2017 DOE target value of 300 A/cm3 for non-PGM catalysts. Note that it should be better now, that non-PGM catalysts have non-negligible volumetric activities at 900 mViR-free, to set their DOE target at that potential rather than at 800 mViR-free in order to avoid extended extrapolations to obtain their kinetic activity free of mass transport limitations.
It may seem unusual to measure and compare the catalytic activity in terms of the volume of the electrode (A/cm3), but contrary to Pt-based catalysts, which have a much higher raw material cost and are generally used with high Pt loadings on a conductive support in electrodes having a thickness lower than 10 μm, the lower-cost non-noble metal electrocatalysts may be used in much thicker cathodes (up to about 100 μm thick) before being subject to significant mass transport limitations. In an electrode using a non-noble metal electrocatalyst for ORR, the kinetically controlled current per unit of volume is given by Eq. (10.1):
where Current (E), in A/cm3, is a function of the applied potential E; SD is the number of catalytic sites/cm3; TOF (E), in electron per site and per second, is the average turnover frequency of the catalytic site(s) and is also a function of E; and e− is the charge of the electron in Coulombs. To increase the current per unit of volume at a specific potential, one needs to increase either (1) SD, (2) TOF (E), or (3) both SD and TOF (E). The turnover frequency for ORR of a catalytic site is an intrinsic property of this site. It is probably difficult to change it, let alone to increase it. It is much easier to increase the number of catalytic sites by modifying the synthesis of the catalyst. In order to do so, it is useful to have at least a basic understanding about the nature of the various catalytic site(s) at work in the catalyst.
Despite decades of research, the nature of the catalytic sites in non-noble metal ORR catalysts is still a topic of healthy debate and is the subject of this chapter. There are indeed two very different schools of thought regarding the nature of these catalytic sites: the metal in the non-noble metal electrocatalyst is either an active participant in the ORR or it is not. In the remainder of this chapter, a brief history of the various structures of the catalytic sites proposed in the early days will be presented. This will be followed by the contribution of our group and others to elucidate what we believe is the nature of the catalytic site(s) in these catalysts and why we (and others) believe that the metal is at the heart of the catalytic sites for ORR in these most active catalysts. This will be followed by a presentation of the arguments of the other schools of thought, which advocate that the metal is not electrocatalytically active, but instead only serves as a chemical catalyst for producing special carbon structures, which are the actual active site for ORR. The chapter continues with a discussion about these differing views regarding the nature of the catalytic site(s) and the mechanism for ORR on these catalytic sites to end up with concluding remarks.
10.2 The First Three Models of Catalytic Sites for ORR
Me–N4 macrocyclic complexes (with N4-macrocycles being, for instance, tetraazaannulene [TAA], tetramethoxyphenyl-porphyrin [TMPP], phthalocyanine [Pc]), and Me = Co or Fe were the first to be used as precursors of the catalytic sites. In particular, unsupported Co–N4 chelates (CoTAA, CoTMPP, and CoPc) or Co–N4 chelates adsorbed on a carbon support were used in the 1970s as ORR electrocatalysts [11]. However, both unsupported and carbon-supported Co–N4 chelate electrocatalysts underwent a rapid decline in the activity. An important discovery in these early years was that catalyst stability as well as activity toward ORR could be improved by subjecting Co–N4 chelate/carbon samples to thermal treatment in an inert gas like N2 or Ar [12].
The question then was the following: what happens to the Me–N4 catalytic site of the chelate after the heat treatment? This topic was strongly debated in the early days and is still not completely understood today. Essentially, three models of the catalytic site for ORR were presented, one each by van Veen, Yeager, and Wiesener, and their respective collaborators.
-
For van Veen, the thermal treatment at temperatures at which catalytic activity is maximum (500–600 °C) does not lead to the complete destruction of the macrocycles, but rather to a ligand modification which keeps the Me–N4 moiety intact (see Fig. 10.2) [13, 14].
Fig. 10.2 
Reaction steps of porphyrin with the carbon support during heat treatment (according to Fig. 9 in ref. [13]; reproduced with permission of the American Chemical Society)
-
Yeager and coworkers, who were mainly working at higher pyrolysis temperatures (800–850 °C), disagreed. They proposed that the decomposition of Me–N4 macrocycles starts at about 400–500 °C. At 800 °C, most of the metal becomes a mixture of oxides and metal, the latter spontaneously oxidizing when exposed to air at room temperature. Upon contact with an electrolyte solution, oxide species undergo dissolution. Metallic ions subsequently adsorb or coordinate to thermally formed sites on the carbon surface, most likely involving one or more nitrogen atoms bound to the carbon surface, but of a type different from the macrocycle-derived Me–N4 centers proposed by van Veen and coworkers. The resulting structure, C–N x –Me, was believed to be the true catalytic site for oxygen reduction [15–17].
-
A third model was proposed by Wiesener. In his view, the Co or Fe ions of the adsorbed N4 chelates promoted the decomposition of the chelate upon thermal heat treatment followed by the formation, at high temperature, of CNx, a special form of carbon, bearing nitrogen atoms that would be the true catalyst [18, 19]. In this scenario, the metal is believed to be only an intermediate and has no role in the electroreduction of oxygen.

In 1989, Yeager and coworkers reported the preparation and characterization of a novel catalyst active for oxygen reduction in acid medium that was made from the heat treatment at 800 °C of polyacrylonitrile (PAN) and a CoII or FeII salt dissolved in dimethylformamide and impregnated on a carbon support [20]. This novel preparation procedure allowed for the generalization of the ingredients necessary to obtain an ORR electrocatalyst in acid medium: (1) a Co or Fe precursor, (2) a nitrogen precursor, and (3) a carbon support for the adsorption or impregnation of the metal and nitrogen precursors. The resulting material had to be pyrolyzed at relatively high temperature, which, according to Yeager, was necessary to incorporate nitrogen functionalities onto the carbon support and to reduce the metal ions. The previous procedure using Me–N4 chelates was only a coincidental case in which the nitrogen and metal precursors were present in the same molecule, the N4-metal chelate. The possibility to use two separate precursors, one for the metal and the other for nitrogen, offered much more flexibility in the preparation of the catalysts and led to the use of much cheaper molecular precursors than the Me–N4 chelates. According to Yeager [20], the active site resulting from the novel procedure was the same as that previously proposed for the catalysts made from Me–N4 chelates. Once again, the catalytic site was C–N x –Me and was self-assembled when in contact with the electrolyte which dissolved the metal particles generated during pyrolysis. Here, however, as PAN was used, the nitrile nitrogen of PAN was converted to pyridinic nitrogen during the heat treatment and were believed to provide binding sites for the transition metal ions.
In the years following 1989, many groups including ours prepared catalysts for ORR using various Me–N4 chelates as well as new nitrogen and metal precursors. The overall work reported during these years is reviewed in ref. [21]. One work in particular stands out. It is a work by van Veen and collaborators [22], who used several pyrrole derivatives, as nitrogen precursors, which were loaded onto Vulcan and Norit BRX with Co acetate, as the metal precursor. The Co loading was 1.5 wt% for Vulcan and 4.5 wt% Co for Norit. The heat-treatment temperature was 700 °C in inert atmosphere. The best catalyst was obtained for a Co:N ratio of 1:10 with 2,5-dimethylpyrrole adsorbed on Vulcan. This catalyst was found to be equivalent to that obtained with CoTMPP loaded at 7 wt% chelate (0.6 wt% Co) on Vulcan and also heat treated at 700 °C. After EXAFS analysis of their catalysts, van Veen and coworkers concluded that a Co–N4 complex identical to that obtained with CoTMPP was, once again, the active catalytic site in their two catalysts. They also realized that the nitrogen precursor (2,5-dimethylpyrrole) had a structure that closely resembled one of the four metal-ligating groups in the proposed catalytic site (Fig. 10.2).
It is clear that, as far as the catalytic site is concerned, both Yeager and van Veen maintained their early positions. They did share a common point, however, which was that a metallic Fe or Co ion was at the heart of their proposed respective catalytic sites: Me–N4 bound to carbon for van Veen and C–N x –Me for Yeager. While van Veen and Yeager’s models for ORR catalytic sites were popular in the 1990s, the third model, proposed by Wiesener, had very few supporters during this same period. The most important work, invoking a metal-less catalytic site based on nitrogen, oxygen, and carbon atoms and on various oxidation states of the nitrogen atom, was that of Savy and coworkers [23]. Their catalyst was obtained by adsorbing CoTAA on Norit SX Ultra at a loading of 2.3 wt% Co and then heat treated up to 900 °C in inert atmosphere. Because no more Co was detectable by XPS after longevity testing in 0.5 M H2SO4 solution, while the catalyst was still active, they concluded that the catalytic site had to be metal-less. A sequence of reactions for the reduction of oxygen on a metal-less catalytic site is given in their paper. This is, however, a rare case where Savy invoked a metal-less catalytic site, because in most of his other work, he mainly agreed with the model proposed by van Veen. Wiesener’s CNx model of the catalytic site for ORR would, however, gain some interest after 2000 and rally many vocal supporters, as will be discussed later.
Using high non-noble metal loadings on carbon supports (following the example of Pt/C catalysts highly loaded in Pt) was characteristic of non-noble metal catalyst research in the 1990s, including for work done by our group (see ref. [21] for details). However, doing so resulted in high levels of inactive material being formed in the non-noble metal catalysts. The latter predominated the characterization of these catalysts and prevented any conclusions from being drawn about the identity or nature of the catalytic site(s). By the end of the 1990s, our group radically modified the procedure for synthesizing non-noble catalysts for ORR by using very low loadings (either as Fe or Co acetate or as Co- or ClFeTMPP) and replacing CH3–CN, used as nitrogen precursor, with NH3 gas during pyrolysis (in a mixture with H2 and Ar). We also used a synthetic carbon precursor, pyrolyzed perylene tetracarboxylic dianhydride (PTCDA) (see insert in Fig. 10.3), to create our own metal-free carbon supports. PTCDA (a dye) loses its carboxylic moiety upon heat treatment above 520 °C, a temperature at which PTCDA begins to polymerize into carbon fibers [25]. The reasons for using a carbon precursor like PTCDA were twofold. The first was that it was possible to fully purify the as-delivered PTCDA of most of its metallic impurities [26]. The second reason was that pyrolyzed PTCDA gave a carbon support with little background signal in ToF-SIMS, a surface analytical technique that we used to obtain important information about the catalytic site.

The catalytic activity for O2 reduction (in O2-saturated H2SO4 solution at pH 1) vs. the Fe content for catalysts prepared by adsorbing either iron acetate or ClFeTMPP on PTCDA and pyrolyzing the material at 900 °C in H2:Ar:NH3 (1:1:2) (according to Fig. 2 in ref. [24]; reproduced with permission of the American Chemical Society). Note: 5,000 ppm = 0.5 wt%
Figure 10.3 shows that, when iron acetate is used as the Fe precursor, the catalytic activity increases very quickly with Fe loading between 0 and 2,500 ppm and then levels off at loadings of 5,000 ppm (0.5 wt%) Fe and higher. Larger Fe loadings (up to about 2 wt%) could be reached before a leveling off of the catalytic activity when the Fe precursor was chloro-iron tetramethoxy phenyl porphyrin (ClFeTMPP). However, the maximum activity reached with ClFeTMPP and iron acetate as iron precursors was the same [24]. All catalysts in Fig. 10.3 were prepared via the impregnation of the Fe precursor on PTCDA. The resulting material was then pyrolyzed at 900 °C in H2:Ar:NH3. The difference between iron acetate and porphyrin precursors was interpreted in terms of a better interaction of the PTCDA with the Fe-porphyrin than with iron acetate, leading to a better dispersion of the iron ions on the carbon precursor. Given that similar activity results were obtained from catalysts made with iron acetate and the iron porphyrin, we mostly concentrated our efforts on the iron acetate as iron precursor in the preparation of these catalysts. Unless otherwise specified, all the results from our group that are described throughout the remainder of this chapter were obtained with iron acetate as the iron precursor.
The XPS spectra of the catalysts prepared with iron acetate were quite instructive [27]. Narrow-scan spectra of Fe 2p3/2 and 2p1/2 revealed that Fe was always found in the ionic form for all Fe loadings, but that a reduced form of Fe (either Fe0 or an iron carbide) was also detected above 8,100 ppm (0.81 wt%) Fe, in agreement with the leveling off of the catalytic activity observed in Fig. 10.3. Narrow-scan spectra of N1s of the same catalysts revealed that only the low-binding energy peak mainly assigned to pyridinic nitrogen [28, 29] (see Fig. 10.4 for the meaning of pyridinic nitrogen) was shifting toward higher binding energies for higher Fe loadings. A saturation of this effect at 0.3 eV appeared above 4,660 ppm Fe, again in agreement with the leveling off of the catalytic activity observed in Fig. 10.3. This shift has been interpreted in terms of a preferential interaction of Fe ion with pyridinic nitrogen atoms in the catalytic site. These nitrogen atoms are located at the edge of a graphene layer and contribute one electron to the π orbitals of the layer. Pyridinic nitrogen atoms also have an orbital in the plane of the graphene layer containing two nonbonding electrons, which are available to coordinate a metal ion such as iron.

Proposed moiety of the catalytic site referred to as FeN2/C consisting of two pyridinic N atoms in a 1,10-phenanthroline-type structure coordinating a Fe2+ ion [30]. The complete structure of this site was still undetermined in 2002; also shown are the various types of nitrogen atoms in a typical graphene plane
ToF-SIMS experiments performed on similar catalysts also prepared with iron acetate revealed (1) that the catalytic site contained at least two pyridinic nitrogen atoms and (2) that these nitrogen atoms were in a 1,10-phenanthroline-type structure like the one reproduced in Fig. 10.4 and proposed by our group in 2002 as part of the catalytic site in these catalysts [30]. This conclusion was reached because one ion—FeN2C4 +—came to the fore out of all ions detected by ToF-SIMS when the catalysts were prepared at various temperatures. The change in the relative intensity (abundance) of the latter is compared in Fig. 10.5 [24] with the evolution of the catalytic activity for the same Fe-based catalysts vs. the pyrolysis temperature. The coincidence of the two patterns indicated that FeN2C4 + is the ToF-SIMS signature of the main catalytic site found in such catalysts. We refer to this site as FeN2/C. The full coordination of the catalytic site proposed in Fig. 10.4 was unknown at that time. More about the coordination will be revealed later.

Comparison of the catalyst activity for ORR in O2 saturated H2SO4 solution at pH 1 (top panel) and the relative intensity of FeN2C4 + (bottom panel) vs. pyrolysis temperature for catalysts obtained by adsorbing iron acetate (0.2 wt% Fe) on pre-pyrolyzed PTCDA followed by heat treatment in inert atmosphere at various temperatures, ranging from 400 to 1,000 °C. Pre-pyrolyzed PTCDA is obtained by heat-treating PTCDA at 900 °C in H2:Ar:NH3 (1:1:2) (according to Fig. 8 in ref. [24]; reproduced with permission of the American Chemical Society)
Besides FeN2C4 +, other ions of the type FeN x C y + were also detected in the ToF-SIMS spectra of the catalysts prepared with iron acetate as Fe precursor (see Fig. 10.6). They were of much lower relative abundance than FeN2C4 + and could be divided into four families, each one having a different number of nitrogen atoms (from 1 to 4) bound to various numbers of carbon atoms. For instance, another ion—FeN4C8 +—was of relatively high abundance (up to 8 % at 500 °C, compared with the abundance of about 80 % for FeN2C4 + at 800 °C). It was assigned to a Fe ion coordinated to four nitrogen atoms, each nitrogen atom being itself bound to two carbon atoms (see Fig. 10.6). This ion is typical of the fragmentation of ClFeTMPP+ and is characteristic of the Fe–N4 moiety of the porphyrins [30]. It was, however, found in catalysts prepared with iron acetate! As FeN4C8 + is also reminiscent of the structure of the catalytic site proposed by van Veen and collaborators, which is illustrated in Fig. 10.2, the typical catalytic site referred to as FeN4/C.

Relative intensity for all FeN x C y + ions of the FeN2C y + family (open circles), FeN1C y + family (dark squares), FeN3C y + family (dark triangles), and FeN4C y + family (dark circles) as a function of the pyrolysis temperature for catalysts analyzed by ToF-SIMS. The catalysts were obtained by adsorbing iron acetate (0.2 wt% Fe) on pre-pyrolyzed PTCDA and heat-treating in inert atmosphere at various temperatures ranging from 400 to 1,000 °C. Pre-pyrolyzed PTCDA was obtained by heat-treating PTCDA at 900 °C in H2:Ar:NH3 (1:1:2) (according to Fig. 4 in ref. [30]: reproduced with permission of the American Chemical Society). FeN2C y +, which is mainly composed of FeN2C4 +, is the signature for the proposed FeN2/C structure for the major catalytic site in these catalysts. FeN4C y +, which is mainly composed of FeN4C8 +, and FeN1C y + and FeN3C y +, which are fragments of ionic products of FeN4C y +, are all the signatures of the proposed FeN4/C structure for the minor catalytic site in these catalysts
In a refined analysis of the ToF-SIMS spectra of catalysts prepared with iron acetate as the Fe precursor, it was assumed that all the ions having the same number of N atoms, originated from the same catalytic site(s). Figure 10.6 shows the relative intensity of each family of ions vs. the pyrolysis temperature. From Fig. 10.6, we observe two different behaviors. On the one hand, the family of ions of the type FeN2C y + increases in relative abundance with the pyrolysis temperature, then goes through a maximum at 800–900 °C, and decreases at 1,000 °C. On the other hand, the relative intensity of the three other families of ions behaves similarly, but with a maximum abundance at a low pyrolysis temperature. From this behavior, one may conclude that the ions belonging to the N1, N3, and N4 families have the same origin: FeN4/C (N1 and N3 ions would all originate from the fragmentation of N4 containing ions). On the other hand, the origin of the ions of the N2 family is FeN2/C, whose structure is presented in Fig. 10.4. Both FeN 4 /C and FeN 2 /C were therefore found simultaneously, but in various proportions based on the pyrolysis temperature, in catalysts obtained with iron acetate as the Fe precursor.
The simultaneous presence of the same two FeN2/C and FeN4/C catalytic sites was also observed when ToF-SIMS experiments were performed on catalysts made with an Fe-porphyrin (ClFeTMPP) with the same Fe loading as that of iron acetate (0.2 wt% Fe, nominal) and prepared under the same experimental conditions as those used for the catalysts made with iron acetate. In this case, however, the relative intensity of ions from the FeN4/C sites was always greater than that of the ions released by the FeN2/C sites. Four families of N-containing ions were again found, with the N2 family behaving differently from the N1, N3, and N4 families of ions. Two types of catalytic sites emerged once again from this analysis. As a general conclusion from these experiments, it appears that irrespective of the iron or nitrogen precursor used to obtain a catalyst for ORR, the resulting catalyst material always contains two types of catalytic sites referred to as Fe–N 2 /C and Fe–N 4 /C, the relative proportions of which depend on the pyrolysis temperature and the particular iron and nitrogen precursors used.
FeN4/C corresponds to the catalytic site proposed by van Veen. One may wonder, however, if FeN2/C corresponds to the catalytic site (C–N x –Me) proposed by Yeager. The answer to this question is no! Remember that, according to Yeager, C–N x –Me is not obtained during pyrolysis, but after contact of the pyrolyzed material with the acid electrolyte in which iron oxides and iron metal produced during pyrolysis are dissolved and re-coordinated to C–N x functionalities of the carbon to produce C–N x –Me sites for ORR. A recent experiment confirmed, however, that C–N x –Fe is only the precursor of the FeN2/C catalytic site [31]. In this experiment, when a commercial N234 carbon black was first pyrolyzed in NH3 at 950 °C, its N content increased from 0 to 2.2 at.%. When this N-enriched carbon was then impregnated with 0.2 wt% Fe (as iron acetate), a very poor catalyst for ORR was obtained. This is illustrated by the half-shaded star in Fig. 10.7. A second pyrolysis step in pure NH3 of this Fe-impregnated material, performed again at 950 °C, resulted in a sharp increase in its catalytic activity even after a second pyrolysis lasting only 1 min (black squares in Fig. 10.7). If the second pyrolysis was instead performed in Ar at the same temperature (black circles in Fig. 10.7), it took at least 40 min to reach the same activity as that reached with a pyrolysis in NH3. These experiments revealed the importance of the second heat-treatment step (and the nature of the gas used during the second pyrolysis) for producing catalytic sites under these conditions. In other words, the C–N x –Me structure proposed as a catalytic site by Yeager and collaborators was only the precursor of the actual catalytic site as C–N x –Me needed to be thermally activated at high temperature to become the actual catalytic site referred to as FeN2/C.

Catalytic activity in O2 saturated H2SO4 solution at pH 1 vs. pyrolysis time for N234 pristine carbon black that was first etched in NH3 at 950 °C and then impregnated with 0.2 wt% Fe (as iron acetate) (half-shaded star). A second pyrolysis of this material performed at 950 °C in pure NH3 (black squares) or in Ar (black circles) resulted in a sharp increase in the catalytic activity (according to Fig. 5 in ref. [31]; reproduced with permission of the American Chemical Society)
10.3 Influence of the Carbon Support
In the previous section, we learned that two catalytic sites, referred to as FeN4/C and FeN2/C, were simultaneously produced when pyrolyzing a carbon support on which 0.2 wt% (nominal) Fe was loaded as iron acetate. We also learned that FeN4/C is the catalytic site proposed by van Veen, while the C–N x –Me structure proposed by Yeager is the precursor of FeN2/C catalytic sites. The nitrogen atoms chelating the iron ion in FeN4/C are of pyrrolic character, while they are of pyridinic character in FeN2/C. FeN4/C is also a fully coordinated catalytic site, while FeN2/C is only the moiety of a more complex catalytic site for ORR. From these conclusions, it became clear that N-bearing functionalities on the carbon support are crucial because they are needed for ligating the iron ion of the catalytic site to the carbon support. Consequently, it seemed logical that increasing the number of N-bearing functionalities (either pyrrolic for FeN4/C or pyridinic for FeN2/C) on the carbon support would lead to more catalytic sites on this support. It was therefore of paramount importance to determine if N-bearing functionalities were the only factor or if other characteristics of carbon blacks were also important to maximize the ORR catalytic activity obtained with these carbon supports.
Starting in 2000 or so, few studies focused on the role of the carbon support and its influence on the activity of the electrocatalysts. In 2002, Bron and his colleagues compared the activity of catalysts made by the adsorption of Fe-phenanthroline on three different supports: Vulcan (254 m2/g), Printex (900 m2/g), and Black Pearls (1,500 m2/g) [32]. The Fe loading was 1.14 wt% for the three carbon supports, and all catalysts were obtained after pyrolysis at 900 °C in Ar. They found a difference of about one order of magnitude in the current densities measured at the same potentials for these catalysts on different carbon supports, with the catalyst on Black Pearls having the highest activity for ORR. This difference was attributed to the larger specific surface area of Black Pearls compared with that of Vulcan, which allowed for a larger density of catalytic sites. Our group also performed several studies on catalysts made with various carbon supports. In the first study [33] published in 2003, we used (1) six commercial carbons (Printex XE-2, Norit SX Ultra, Ketjenblack EC-600JD, acetylene black, Vulcan XC-72R, and Black Pearls 2000), (2) three developmental carbons (HS 300 from Lonza, RC1, and RC2 from Sid Richardson Carbon Corporation; RC1 was enriched in nitrogen, while RC2 was not), (3) the same nine previous supports pre-pyrolyzed at 900 °C in an atmosphere containing NH3 to increase their N content, and (4) a synthetic carbon made by pyrolyzing perylene tetracarboxylic dianhydride at 900 °C in an atmosphere containing NH3. The same Fe precursor, iron acetate, with a nominal loading of 0.2 wt% Fe, was impregnated on all these carbon supports, and the resulting materials were heat treated at 900 °C in an atmosphere containing NH3. The variation of activity measured in RDE or in fuel cell for these catalysts, all performed under the same experimental conditions, except for the carbon support, was significant. On the one hand, no correlation between the specific surface area and catalytic activity was found. On the other hand, there was a definite correlation between the catalytic activity and their surface nitrogen concentration measured by XPS.
In order to understand why different carbon supports had varying abilities in gaining N-bearing functionalities during pyrolysis in NH3, further studies were conducted on various commercially available furnace blacks as carbon supports. These materials were made of aggregates of sphere-like particles having a diameter varying between 23 and 375 nm with their ASTM grade. References to the properties of furnace blacks (which are used primarily in the rubber/tire industry) may be found in a book by Kinoshita [34] or by Donnet et al. [35].
The main reaction which occurs when a carbon support is pyrolyzed in NH3 at temperatures between 850 and 1,400 °C is the gasification of carbon by ammonia gas as given by Eq. (10.2) [36–38]:
In order to better understand the consequences of carbon gasification by reaction (10.2), this reaction was modeled for furnace carbon black particles [39, 40]. The premises of the model were that the particles of a pristine furnace black are composed of two solid phases: (1) an ordered phase containing graphitic crystallites and (2) a disordered phase resulting from the fast and out-of-equilibrium production of carbon black from oil feedstock. The assumed initial microstructure of a particle is represented in Fig. 10.8a where the gray area represents disordered carbon and the black rectangles graphitic crystallites. Before reacting with ammonia, the furnace black spherical particles have very little or no micro- or mesopores and their total surface area is about that of the combined exterior surface area of the carbon spheres. The model computed, as a function of the reaction time with NH3, how much mass of the graphitic and disordered carbon phase was left at various depths inside the particle. The model parameters were estimated from experimental data of the chosen specific carbon black as (1) the evolution, during heat treatment with NH3, of its specific surface area (available by BET measurements) and weight loss percentage; (2) the average initial diameter of the pristine carbon black particles (available by HRTEM or BET of pristine carbon black); and (3) the initial in-plane size of the graphitic crystallites of the pristine carbon black (available by Raman spectroscopy or Rietveld refinement of the XRD diffractogram).

(a) Schematic representation of the initial nonporous pristine carbon particle with its structure partitioned into m shells. Disordered phases (gray zone) and graphitic crystallites (black rectangles, not drawn to scale) (according to Fig. 1A in ref. [39]; reproduced with permission of the American Chemical Society). (b) Raman spectrum of the pristine carbon black N120 showing the deconvolution into five bands: P, D, Am, G, and D2 (according to Fig. 2A in ref. [41]; reproduced with permission of Elsevier). (c) N content, determined by XPS at the surface of the catalysts made with various pristine carbon blacks of Nxxx grade, vs. the width at half maximum W D of the Raman D peak. (d) Maximum catalytic activity for each series of catalysts based on carbon blacks of Nxxx grade vs. the N content determined by XPS at the surface of the catalysts
One of the important findings of the modeled gasification was that the disordered carbon in the particular carbon black studied reacts (or gasifies) about ten times faster with NH3 than the graphitic crystallites in the same carbon black. This leads to the creation of pores in the pristine nonporous spherical carbon black particles via the fast progressive gasification of disordered carbon by reaction with NH3, while the graphitic crystallites in the same particle are etched much more slowly. Furthermore, the reaction of NH3 with the disordered carbon content of these supports leaves nitrogen-bearing functionalities at the surface of the resulting carbon.
An estimate of the disordered carbon content in a pristine carbon may be obtained from its Raman spectrum. An example of the Raman spectrum of pristine carbon black of grade N120 is given in Fig. 10.8b [41]. This spectrum has been deconvoluted into five components [42]. The component of interest for disordered carbon content is peak D (the “defect band”). In polycrystalline carbonaceous materials consisting of a large number of small graphitic crystallites, like in pristine carbon black, it has been suggested that carbon atoms at the edge of a graphene layer are considered as the most probable origin of the D band [43]. Graphene layers in formation in a disordered carbon phase are therefore strong contributors to the D peak. It was found that W D, the full width at half maximum of the D band, measured experimentally in Raman, is a very good parameter to estimate the relative amount of disordered carbon phase in a carbon black [40]. Figure 10.8c shows the linear correlation found between the maximum nitrogen content in the catalysts vs. W D for the various pristine carbon blacks used to obtain these catalysts. The larger is W D (or the relative amount of disorder carbon phase in a specific carbon black), the larger is the nitrogen content measured at the surface of the catalyst prepared with this specific carbon black. Here, each carbon black was first loaded with 0.2 wt% of Fe as iron acetate then pyrolyzed in NH3 at 950 °C until an optimal weight loss corresponding to a maximum activity, as shown in Fig. 10.9a (right Y axis), was reached for each carbon black [41]. This optimum weight loss is reached when the thickness of the porous layer etched by NH3 in the pristine spherical carbon particle of Fig. 10.8a is maximum [39]. Further etching will lead to a decrease of the spherical particle diameter and a decrease of the catalytic activity for ORR. Figure 10.8d shows the correlation between the maximum catalytic activity obtained with specific pristine carbon blacks and the nitrogen content measured at the surface of these catalysts. It indicates that the catalytic activity first increases rapidly when the nitrogen content is small, but the effect saturates when the nitrogen content at the surface of the catalyst reaches about 2 at.%.

(a-right axis) ORR activity in O2 saturated H2SO4 solution at pH 1 of catalysts made in pure NH3 at 950 °C (filled circles) vs. the percentage weight loss by the catalyst during the heat treatment. All catalysts were made by impregnation of 0.2 wt% Fe as iron acetate (according to Fig. S5B in the supplementary information of [44]; reproduced with permission of The American Chemical Society). (a-left axis) Surface area of three types of pores and total specific area vs. weight loss percentage of carbon during the heat treatment in NH3 at 950 °C. Total surface area (open squares), surface area of micropores (width ≤ 2 nm; open circles), mesopores (up triangles), macropores (down triangles) (according to Fig. 5 in ref. [44]; reproduced with permission of The American Chemical Society). (b) Catalytic activity for O2 reduction in O2-saturated H2SO4 solution at pH 1 against microporous surface area of the catalysts of a, right axis (according to Fig. 6 in ref. [44]; reproduced with permission of The American Chemical society). The insert in b is a schematic representation of a slit-shaped micropore between two graphitic crystallites having La and Lc dimensions
Besides the reaction of NH3 with the disordered content of a carbon black leaving nitrogen-bearing functionalities at the surface of the etched carbon, there is another important consequence of the reaction of NH3 with carbon. As disordered carbon is mainly gasified, leaving behind a more crystalline structure of the carbon black, the occurrence of porosities in the carbon black follows the reaction front. Therefore, the total surface area of the carbon black greatly increases with the loss of carbon mass resulting from reaction (10.2). This is illustrated in Fig. 10.9a (left Y axis). The new porosities may be categorized as micropores (≤2 nm), mesopores (between 2 and 50 nm), and macropores (≥50 nm). Figure 10.9a (left Y axis) also shows the change in the surface areas assigned to the micropores, mesopores, and macropores versus the weight loss percentage. By comparing the trend of the catalytic activity in Fig. 10.9a (right Y axes) with the trends for the surface area of the various pore sizes, we can see that there is a direct correlation between the catalytic activity and the micropore surface area of the catalyst. This is illustrated more clearly in Fig. 10.9b. From Fig. 10.9b, and other results obtained with catalysts made with a wide range of commercial pristine carbons [41], we concluded that the most active catalytic sites that were previously referred to as FeN 2 /C are hosted in the micropores of the carbon support. These micropores were etched in the pristine nonporous carbon blacks by their reaction with NH3. For the catalyst presented in Fig. 10.9a, we also found that, at maximum catalytic activity, the nitrogen content of the catalyst was around 4 at.%, a value within the saturation region of Fig. 10.8d. In other words, the N content in this case was not a limiting factor to the catalytic activity. The latter was only limited by the availability of micropores for hosting the catalytic sites. A schematic structure of a slit-shaped micropore usually found in carbon blacks [45] is illustrated in the insert of Fig. 10.9b. This interstice between two graphitic crystallites was previously filled with disordered carbon. These graphitic crystallites consist of a few graphene layers characterized by La, the in-plane dimension of the graphene layers (between 2 and 3.5 nm), and a Lc, the height of the graphene stack (between 1 and 1.8 nm) for carbon black particles with diameters between 23 and 375 nm, respectively [41].
Following the discovery of the two important factors (the N and micropore contents) that govern the activity of catalysts made by the impregnation of a carbon black with 0.2 wt% Fe as iron acetate and its pyrolysis in NH3 at high temperature, in 2008, we proposed a structure that would replace FeN2/C, the incomplete catalytic site structure depicted in Fig. 10.4 that was previously introduced as a possible part of the most active type of catalytic site.
This new structure was referred to as FeN2 + 2/C and is depicted in Fig. 10.10. The label FeN2 + 2/C was chosen to distinguish this catalytic site from the van Veen type of site—FeN4/C (Fig. 10.2)—that was shown to be simultaneously present with FeN2/C (now FeN2 + 2/C) in Fe/N/C catalysts [30]. Here, however, the four nitrogen atoms chelating the iron ion in FeN2 + 2/C are of pyridinic character, while they are of pyrrolic character in the van Veen type FeN4/C site. The depicted structure of FeN2 + 2/C is based on several experimental findings, namely, (1) the previously found importance of pyridinic (and even phenanthrolic) nitrogen atoms in FeN2/C [27, 30], (2) the fact that these FeN2 + 2/C catalytic sites are hosted in micropores [44] which are slit shaped in carbon black [45] and that the width of the slit-shaped micropore hosting the catalytic site are between 1 and 2 nm wide [46], and (3) most EXAFS and Mössbauer experiments performed so far on Fe/N/C (and Co/N/C catalysts) always concluded that the metal ion of the catalytic sites was surrounded by four nitrogen atoms [22, 32, 47–50].

10.4 The Revival of the Wiesener Model
10.4.1 Stevenson and His Collaborators
In 2004, Maldonado and Stevenson published a paper about the electrocatalytic behavior of carbon nanofibers (CNFs) toward oxygen reduction obtained via the pyrolysis of ironII phthalocyanine [51]. They suggested that the disorder in the graphitic fibers, i.e., the presence of exposed edge phase defects, and the nitrogen functionalities were important factors influencing the adsorption of reactive intermediates generated during the reduction of oxygen. The enhancement of the ORR electrocatalysis on these materials was attributed to all of these factors. Their carbon nanofibers were obtained at 1,000 °C in a reducing atmosphere (Ar–H2) on a Ni mesh. The growth of the CNFs was catalyzed by a diffusion-controlled mechanism where iron particles that were left over from the pyrolysis of the phthalocyanine served as nucleation sites for the carbon fibers. Iron nanoparticles still present with the CNF were indeed observed by TEM and were predominantly encapsulated within graphitic envelopes. TEM also revealed that the nanofibers had a bamboo-like structure resulting from the substitution of N atoms into the graphene structure of the nanofibers. The possibility of N substitution in the CNFs was attributed to the formation of pentagonal-like defects, which were most likely responsible for the compartmentalized morphology of the carbon nanostructure. About 1 at.% of nitrogen atoms was detected by XPS as pyridinic, pyrrolic, and quaternary nitrogen at the surface of these catalysts, while the amount of Fe at the surface of CNFs was measured at about 0.1 at.% by the same analysis technique. From Raman spectroscopy of the N-doped CNFs, it was also deduced that significant edge plane sites were present in the catalyst.
All electrochemical measurements in the latter work were performed at pH 7, showing that oxygen reduction on N-containing carbon nanofibers was shifted by about 0.25 V toward more positive potentials compared with a glassy carbon electrode immersed in the same 1 mM O2-saturated solution of 1 M KNO3. The number of electrons transferred during ORR was determined to be 2. Maldonado and Stevenson concluded that it appeared highly unlikely that O2 reduction at their N-doped CNFs proceeded via a Fe-redox mediation-type mechanism typical for most intact iron macrocycle supported on a carbon electrode. They believed that the increased catalytic activity and more positive O2 reduction potential observed for their N-doped CNF electrode was predominantly the result of enhanced adsorption at nitrogen functional groups that accelerated ORR by a 2e process and improved the decomposition of peroxide. They also speculated that Fe predominantly acted to promote and stabilize nitrogen incorporation into the graphene sheets of CNFs rather than of being at the heart of the catalytic sites for oxygen reduction.
This paper was followed by another in 2005 [52] where the N-doped CNFs were prepared by a chemical vapor deposition at 800 °C using ferrocene and pyridine this time. Undoped CNFs were also prepared similarly with ferrocene and xylene. The residual bulk iron percentages for N-doped CNFs were obtained by TGA. It was 9 ± 1 wt% Fe for N-doped CNFs and 7 ± 1 wt% Fe for the undoped fibers. XPS surface analysis of the same fibers gave 1.1 and 1.2 at.% Fe, respectively, which corresponds to about 4.5 wt% Fe. The electrochemical properties of the two types of fibers were measured at pH 7 (1 M KNO3) and at pH 14 (1 M KOH), see Fig. 10.11a, b, respectively. From these figures, one may conclude that N-doped CNFs are better catalysts for ORR than the undoped ones. However, the potential difference between the two nanostructures decreases as the pH increases! Again, in this paper, they concluded that the activity of N-doped carbon nanofibers toward oxygen reduction and hydroperoxide decomposition is a direct result of nitrogen doping and is not related to the residual iron content. They added that they saw no evidence for the presence of iron-chelated surface sites (like FeN2/C or FeN4/C), but if these sites were indeed present on the surface of the N-doped CNFs, they should be easily poisoned by the presence of CO due to the strong irreversible binding of CO to the active iron site. Furthermore, nitrogen incorporation produced activated carbons with increased stability toward the fiber oxidation.

(a) Voltammetric responses for the oxygen reduction reaction of a non-doped carbon nanofiber electrode (dashed line) and a N-doped carbon nanofiber electrode (solid line) immersed in an O2 saturated 1 M KNO 3 solution (according to Fig. 6 in ref. [52]; reproduced with permission of the American Chemical Society). (b) Same as in (a), except the electrolyte used is an O2-saturated 1 M KOH solution (according to Fig. 10.7 in ref. [53]; reproduced with permission of Elsevier)
In an accompanying paper [53], the same N-doped carbon nanofibers were grown with ferrocene and pyridine, but this time, NH3 was added in addition to pyridine in order to increase their N content from 4 at.%, when no NH3 was added, up to 12 at.% with addition of NH3. At the same time, the residual Fe content increased from 9 wt% to about 20 wt%! Typically, most of the iron particles were encapsulated with layers of graphene. While both the amount of pyridinic and pyrrolic/graphitic nitrogen atoms increased with the Fe content, the pyridinic N peak showed a twofold greater increase in abundance compared to the pyrrolic/graphitic peaks. Raman spectra also indicated that the ratio of the D/G bands of CNFs also increased with an increase of the total N content in the fiber, suggesting an increase of the edge plane density with the N content as well. The prevalence of pyridinic functionalities in N-doped CNFs confers an alkaline character to these fibers with a pHpzc (pKa) of 9.3 for 4 at.% nitrogen in N-doped carbon nanofibers. These nitrogen atoms, located on the edge of graphitic planes, exhibit an extra lone pair of electrons which increase the electron density and electron-donating character of graphitic edge planes. It is reasonable to expect that these edge planes, which are commonly known to be reactive sites, will show an increased reactivity for ORR due to the electron density generated from the incorporation of nitrogen in pyridinic-like coordination. The reducing capability of these materials was indeed verified by iodimetric titration.
In subsequent years, Stevenson’s group continued the investigation of N-doped CNFs and carbon nanotubes (CNT) grown from ferrocene and pyridine [54, 55]. From these experiments, they concluded that O2 reduction at neutral pH was not catalyzed by surface-bound FeII/III species. In their last paper [56], they used the electroreducing properties of N-doped CNTs to reduce O2 at near neutral pH (pH 6.4) and generate hydroperoxide anions (HO2 −) that may further react with toxic CN− anions and oxidize them into OCN− nontoxic anionic species.
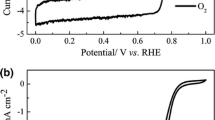
It is worth noting, after a thorough review of the publications by Stevenson and his collaborators, that they have always used N-doped carbon nanostructures as ORR electrocatalysts in neutral or in alkaline solutions, where the oxygen reduction reaction is facilitated and achievable even with carbon alone. The fundamental kinetic advantage in the alkaline medium arises primarily from the improved stabilization of the peroxide intermediates on the catalyst surface [57]. It is our opinion that it is not in studying non-noble electrocatalysts in alkaline or even in neutral media that it is possible to resolve the long-debated question about the controversial role of Fe (or Co) in the active site for ORR. The specific role of Fe (if any) can only be confirmed (or infirmed) from studies conducted in relatively strong acid medium, in which carbon is largely inactive for ORR [58]. This point is illustrated in Fig. 10.11 which demonstrates the decreased influence of nitrogen doping on the ORR activity for carbon nanofibers when measured at pH 14 vs. pH 7. On the contrary, when comparing the ORR activity of one metal-free, two Fe-based catalysts, and a Pt/C catalyst in pH 13 and pH 1, as illustrated in Fig. 10.12A (pH 13) and Fig. 10.12B (pH 1), the catalytic activities behave differently. In these figures, the curves labeled “d” are for an N-doped Black Pearls catalyst containing 0 wt% Fe (or a Fe content below the detection limit of 50 ppm for Fe in Neutron Activation Analysis), while curves “b” and “a” are for two Fe-based catalysts obtained with a nominal Fe loading of 0.2 wt% [41, 44] and 1 wt% [8], respectively. Curve “c” is for a 46 wt% Pt/C catalyst [60]. The blue arrows in Fig. 10.12A, B is an indication of the onset potential (V onset) of the catalysts, which will be defined in the next section. It is clear from Fig. 10.12A (at pH 13) that V onset is 0.85 V vs. RHE for curve “d,” which represents the metal-free N-doped Black Pearls, is practically the same as the V onset of 0.87 V vs. RHE for curve “b,” which represents the least active Fe/N/C catalyst in the same figure. On the other hand, in Fig. 10.12B (at pH 1), V onset is 0.63 V vs. RHE for curve “d” (metal-free N-doped Black Pearls), while it is 0.83 V vs. RHE for the least active Fe/N/C catalyst. Here we have a difference of 0.20 V between the two V onset values. With a Tafel slope of about 0.10 V per decade at pH 1(at 0.8 V vs. RHE) for the metal-free N-doped black Pearls [61], a factor of about 100.20/0.10 (or 100) is therefore expected at the same potential, between the mass activity (in A/g catalyst) of metal-free N-doped Black Pearls and that of the least active Fe/N/C catalyst in this acid medium!

(A) Disk currents at 1,600 rpm and 10 mV/s from RRDE measurements in O2-saturated KOH solution at pH 13 for the ball-milled Fe/N/C catalyst (curve a), the Fe/N/C catalyst made by impregnation (curve b), 46 wt% Pt/C (curve c), and heat-treated Black Pearls (curve d). Fe/N/C and Black Pearls were all heat treated in pure NH3 at 950 °C during the synthesis of the catalysts. Black arrows indicate the scan direction. Blue arrows pointing toward the axis of potentials give the V onset values (vs. RHE) for each catalyst (see also Fig. 10.13 for V onset definition). V onset are 0.85 V for curve d, 0.87 V for curve b, 0.93 V for curve a, and 0.97 V for curve c (according to Fig. 3b in ref. [60]; reproduced with permission of Elsevier). (B) Same as (A) except that RRDE measurements are performed in O2 saturated HClO4 solution at pH 1. The values of V onset vs. RHE for each catalyst are given by the blue arrows pointing to the axis of potentials. They are 0.63 V for curve d, 0.83 V for curve b, 0.90 V for curve a, and 1.03 V for curve c (according to Fig. 1b in ref. [60]; reproduced with permission of Elsevier)
10.4.2 Ozkan and Her Collaborators
The view that the role of Fe is merely a chemical catalyst for the formation of ORR-active CNx structures, first proposed by Wiesener and collaborators in 1986 [18], and again by Stevenson and collaborators in 2004, from their experiments on carbon nanostructures in neutral and alkaline media, was also held by Ozkan and collaborators in 2006 [62], but this time for ORR experiments in the acid medium (0.5 M H2SO4). Their first publication [62] has since been followed by many others who have arrived at the same conclusion: that the role of Fe is limited to catalyzing the growth of ORR-active CNx structures. To properly assess and compare reported ORR activity for catalysts measured in so many different conditions in many publications, we will require a tool or convention. This can be done by defining a consistent way to read the V onset for ORR of all these catalysts, as most researchers working in acid medium report at least the initial part of the ORR cyclic voltammogram (CV) starting at OCV and measured with a rotating disk electrode.
Figure 10.13 shows how these V onset values will be read on the reported CVs. Here straight lines (arrows) are drawn tangent to the inflection point (when possible) of the CV curves and extrapolated to zero disk current. For the purpose of this chapter, V onset shall therefore be defined as the potential at the arrow point. It will be expressed vs. RHE in order to compare experiments performed by different researchers in O2-saturated solutions of different acid concentrations [from 1 M to 0.005 M H2SO4 (or HClO4)]. V onset is not a perfect indicator of catalytic activity, but it is the only one which allows for a fair and meaningful comparison of the reported cyclic voltammograms of electrodes loaded with various catalyst loadings, as well as being recorded at various rotations and scan rates.

(a) Disk currents at 200 rpm and 10 mV/s from RRDE measurements in O2 saturated H2SO4 solution at pH 1 for as-received Ketjen Black, Vulcan, and Black Pearls. V onset values are obtained from a line tangent to the inflection point of the CV curve (when possible) and extrapolated to zero disk current. It is the potential at the tip of the arrow. V onset (vs. RHE) is 0.16 V for Ketjen Black, 0.23 V for Vulcan, and 0.34 V for Black Pearls (according to Figs. 1A, 2A, and 3A in ref. [59]; reproduced with permission of Elsevier). (b) Same as (a) except that all carbon blacks were heat treated at 900 °C in NH3:H2:Ar (2:1:2; in vol.). V onset (vs. RHE) is 0.50 V for Vulcan_NH3, 0.54 V for Ketjen Black_NH3, and 0.63 V for Black Pearls_NH3 (according to Figs. 1B, 2B, and 3B in ref. [59]; reproduced with permission of Elsevier)
Figure 10.13 was obtained by combining the CV curves measured at pH 1 that were published in 2004 by our group [59] for the ORR on three as-received carbon supports (Vulcan_as rec; Ketjen Black_as-rec; Black Pearls_as-rec, open symbols in Fig. 10.13a) and the same carbon supports after a heat treatment at 900 °C in Ar:H2:NH3 (Vulcan_NH3; Ketjen Black NH3; Black Pearls_NH3, gray symbols in Fig. 10.13b). The latter are all metal-free and N-doped carbons (Vulcan_NH3: 1.1 at.% N; Ketjen Black NH3: 0.8 at.% N; Black Pearls_NH3: 0.6 at.% N) [33]. The conversion from the potential vs. SCE reference electrode (0.24 V vs NHE) to vs RHE was done by adding 0.30 V at pH 1. So, for instance, V onset = −0.07 V vs. SCE for Vulcan_as-rec became 0.23 V vs. RHE and V onset = 0.20 V vs. SCE of Vulcan_NH3 became 0.50 V vs. RHE. All V onset values vs. RHE of Fig. 10.13a, b are given in their respective captions. V onset values were extrapolated similarly for both Fig. 10.12A, B. They are also given in their respective captions. Figure 10.13b shows that our group was already well aware of the beneficial role of N doping of metal-free carbon supports in 2004, but we were most interested in the much higher activities that could be achieved in the acid medium (pH 1) by adding a Fe (or Co) precursor to the synthesis of Fe-based catalysts for ORR as illustrated in Fig. 10.12B. Contrary to Wiesener and followers, we attributed this enhancement of activity to the presence of Fe-containing catalytic sites. Let us now go back to the catalysts obtained by Ozkan and collaborators.
The catalysts synthesized by Ozkan and her collaborators [62–77] were carbon nanostructures like multiwalled carbon nanotubes or carbon nanofibers with a herringbone structure, stacked platelets, or stacked cups. Onion-type carbon nanostructures were also obtained. They were always prepared by impregnating a metal acetate (usually iron or nickel acetate) onto an oxide support (alumina, silica, magnesia) or on Vulcan. The nominal metal loading on the support was always 2 wt%. This catalyst precursor was then pyrolyzed at 900 °C in acetonitrile (CH3–CN) vapors. Then the oxide support was removed by its dissolution in KOH, HF, or HCl, depending on the nature of the support. Finally, the resulting material was washed in HCl to remove the exposed metal and to obtain the CNx catalyst. The latter were labeled: CNx-Fe/Vulcan or CNx-Fe/Al2O3, or CNx-Ni/MgO to identify each type of catalyst based on of their synthesis procedure. Metal-free CNx catalysts were also prepared. In that case, the catalyst precursor contained no metal. All other steps of the above-described procedure remained the same. All these catalysts had a large distribution of N-doped nanostructures. For instance, CNx-Fe/Al2O3 was composed of 57 % stacked cups, 3 % fibers, and 40 % multiwalled carbon nanotubes, while CNx-Ni/Al2O3 was composed of 6 % stacked cups, 17 % fibers, 17 % multiwalled carbon nanotubes, and 60 % broken multiwalled carbon nanotubes [65, 71].
The carbon nanostructures were characterized using several techniques: BET (for the determination of their surface area), thermogravimetry and differential scanning calorimetry (to determine their stability toward oxidation), X-ray diffraction (to detect metallic and carbon phases), X-ray photoelectron spectroscopy (to obtain their surface nitrogen content), and transmission electronic microscopy (to identify the carbon nanostructures and observe the presence of remaining metal particles). All catalysts were also evaluated for their activity toward ORR in 0.5 M H2SO4 solutions at 1,000 rpm. Inks prepared by mixing the catalysts with Nafion were then deposited on the surface of a glassy carbon electrode for RDE characterization. Typical electrochemical results for ORR are shown in Fig. 10.14a, b. The three curves in Fig. 10.14a [71] are those of the oxygen reduction reaction on as-received Vulcan (V onset = 0.14 V vs. RHE), CNx-Ni/Al2O3 (V onset = 0.56 V vs. RHE), and CNx-Fe/Al2O3 (V onset = 0.72 V vs. RHE). The four curves in Fig. 10.14b [75] are those of the oxygen reduction reaction on as-received Vulcan (V onset = 0.16 V vs. RHE), metal-free CNx/MgO (V onset = 0.53 V vs. RHE), CNx-Fe/Vulcan (V onset = 0.60 V vs. RHE), and CNx-Fe/MgO (V onset = 0.63 V vs. RHE).

(a) Disk currents at 1,000 rpm from cyclic voltammetry experiments in O2-saturated 0.5 M H2SO4 solution for as-received Vulcan (V onset = 0.14 V vs. RHE), HF-washed CNx grown on Ni/Al2O3 (V onset = 0.56 V vs. RHE), and HF-washed CNx grown on Fe/Al2O3 (V onset = 0.72 V vs. RHE) (according to Fig. 1 in ref. [71]; reproduced with the permission of Springer). (b) Disk currents at 1,000 rpm and 10 mV/s from cyclic voltammetry experiments in O2 saturated 0.5 M H2SO4 solution for as-received Vulcan (V onset = 0.16 V vs. RHE), HCl-washed metal-free CNx grown on MgO (V onset = 0.53 V vs. RHE), CNx grown on Fe/Vulcan (V onset = 0.60 V vs. RHE), and HCl-washed CNx grown on Fe/MgO (V onset = 0.63 V vs. RHE) (according to Fig. 2b of [75]; reproduced with permission of Springer)
In all their reports, Ozkan and collaborators found that N-doped carbon nanostructures made with Ni were always less active than those made with Fe. Their explanation of this behavior was that, depending on the metal and the support used, carbon nanostructures were found to vary significantly developing into different nano-geometries. Depending on the obtained geometry, the exposed graphitic crystal planes and the nitrogen functional groups on the surface also varied significantly. A correlation was found between the edge plane exposure of the graphitic crystallites, the pyridinic nitrogen content of the catalysts, and the ORR activity. As CNx structures grown on Fe-containing supports were characterized by high amounts of edge plane exposure and high pyridinic nitrogen content, these CNx structures tended to be the most active ones for ORR. Their mean V onset value (when all results for Fe-containing catalysts published by Ozkan and collaborators are averaged) is 0.67 V vs. RHE. This is followed by CNx grown on metal-free supports, which are characterized by a mean value of their V onset of 0.61 V vs. RHE. Next are the CNx structures grown on Ni-containing supports with a mean V onset value of 0.52 V vs. RHE. While the mean value of V onset for all catalysts grown on Fe-containing support is 0.67 V vs. RHE, individual reports vary from a low V onset = 0.55 V [76] to a high V onset = 0.77 V vs. RHE [65]. There is less variation in the V onset (but also fewer samples) for the catalysts obtained without metal (from 0.53 [75] to 0.67 vs. RHE [65]) for a mean value of 0.61 V vs. RHE. The same is also true for the catalysts grown on Ni supports (from 0.46 [65] to 0.56 vs. RHE [71]) for a mean value of 0.52 V vs. RHE. The lowest, average, and highest V onset values from the paper by Ozkan and collaborators are illustrated in Fig. 10.15. The same figure also shows V onset values obtained from the publications of other groups.

Frame A—lowest, average (symbol), and highest V onset values (vs. RHE) reported by various groups for the oxygen reduction reaction in acid medium. The N-doped catalysts were either metal-free (open circles), grown on Ni/support (black triangle), Fe/support (black circles), or Co/support (black circles). Frame B—summary of the lowest, average (symbol), and highest V onset values (vs. RHE) reported for various groups for the oxygen reduction reaction in acid medium. Here, the open square represents the overall average of the open symbols in Frame A, while the black square represents the overall average of the black circles in Frame A. The gray diamond is the average value reported by various groups for all as-received carbon black supports
It should be noted that in Fig. 10.15, the metal-free CNx nanostructures grown by Ozkan and collaborators by pyrolyzing CH3–CN at 900 °C on Al2O3 and those obtained by Dodelet and collaborators by pyrolyzing carbon blacks in NH3 at 900 °C have practically the same V onset, despite the large difference in their total nitrogen content (5.2 at.% N for metal-free CNx/Al2O3 and between 0.6 and 1.1 at.% N [33] for all the metal-free catalysts of Fig. 10.13b). As a matter of fact, the N-doped Black Pearls in Figs. 10.13b and 10.12B are also both characterized by the same V onset: 0.63 V vs. RHE, despite the large difference in their total N contents. Indeed, the N-doped Black Pearls in Fig. 10.13b has a total N content of 0.6 at.%, while it is about 4 at.% for the N-doped Black Pearls in Fig. 10.12B [44]. The difference arises from the procedure used for their preparation: a pyrolysis step at 900 °C in a mixture of Ar:H2:NH3 for the former, while it was a pyrolysis step at 900 °C in pure NH3 for the latter. Furthermore, the percentage of their total N content that was pyridinic was always around 30 %, a value often obtained by Ozkan and collaborators. No improvement of the catalytic activity for ORR was therefore observed by our group when the total N content (or its pyridinic fraction) was increased from 0.6 to 4 at.% N (or from 0.2 to 1.3 at.% pyridinic N) for N-doped Black Pearls.
While the improvement of the catalytic activity observed by Ozkan and collaborators when their CNx nanostructures were grown on Fe-impregnated supports was interpreted in terms of an increased formation of graphitic edges stabilized by pyridinic nitrogen atoms (and perhaps also by graphitic nitrogen atoms), an alternative explanation to their ORR activity improvement may be the contribution of a few catalytic sites involving a Fe ion active for the ORR. For Ozkan and collaborators, the numerous metal particles observed by TEM in their materials only served to catalyze the formation of this special carbon and its stabilization by N doping, with N contents up to 9.3 at.% [70]. They used a similar argument for the lower catalytic activity of CNx nanostructures grown on Ni-impregnated supports. In this case, according to them, much less catalytic edges were formed and were also stabilized by a lower number of N atoms (down to 4.1 at.%) [71]. Our alternative explanation for this behavior would be that, even if sites involving Ni ions were obtained, all materials made with Ni as metal precursor have always been found to be inactive toward ORR [26] and these catalysts should therefore show an activity similar to that of metal-free CNx.
Besides doping their carbon nanostructures with nitrogen, Ozkan and collaborators also tried to increase their nitrogen doping of the graphitic edges in their catalysts with additional phosphorous atoms [72, 74] or with additional sulfur atoms [70]. CNx-Fe/MgO catalysts doped with a P/Fe = 1 ratio were prepared by pyrolysis at 900 °C in CH3–CN vapors. Both catalysts did not show much difference in their V onset (0.65 V without P and 0.67 V vs. RHE with P/Fe = 1). Here, P was introduced by adding triphenylphosphine to acetonitrile vapors in the reactor. The same conclusion was reached when S was added as thiophene to acetonitrile. In this case, V onset increased from 0.65 V vs. RHE for S = 0–0.68 V vs. RHE for 8.9 % thiophene in acetonitrile. Attempts to poison any Fe/N/C sites still present in purified CNx-Fe/MgO using a subsequent heat-treatment step at 350 °C in a mixture of 1,050 ppm H2S in H2 were unsuccessful [77]. As Fe/N/C sites were expected by Ozkan and her collaborators to be poisoned in these conditions, they concluded that no residual iron that might be catalytic for ORR was accessible for ORR in their material. This poisoning presumption of Fe/N/C by H2S has not, however, been verified with known structures, like unpyrolyzed iron phthalocyanine or porphyrins that contain a catalytic iron ion indeed able to reduce oxygen in acid medium. A similar poisoning presumption of Fe/N/C was made for CO to demonstrate that there was no iron active for ORR in these catalytic sites [52], until it was shown that CO, in fact, does not poison Fe sites in iron phthalocyanine [78]. Instead of H2S “poisoning” of their CNx catalysts, Ozkan and collaborators actually reported an improvement of their ORR catalytic after heat-treating their catalyst in the gas mixture containing H2S and H2! The V onset of the latter increased from 0.64 V vs. RHE before H2S heat treatment to 0.72 V vs. RHE after heat-treating CNx-Fe/MgO with H2S in H2. A similar improvement was also found by heat-treating CNx-Fe/MgO in 5.7 % H2 in N2. They suggested that the catalyst improvement was the result of its heat treatment in a reducing atmosphere. This may be so, but still does not explain why heat-treating a catalyst in a reducing atmosphere improves its activity for ORR. Further studies are necessary to rationalize this interesting observation.
10.4.3 Popov and His Collaborators
The view that the role of Fe (or Co) is limited to merely acting as chemical catalyst for the formation of active CNx structures for ORR in the acid medium is also shared by Popov and his collaborators. After a first publication in 2007 [79] about carbon-based metal-free catalysts that offered no details about their synthesis procedure, they published, in 2008, an interesting paper about the modification of carbon by Te as catalysts for ORR [80]. This catalyst was obtained by mixing Ketjenblack with telluric acid (H2TeO4) and then reducing the oxidized tellurium to Te with hydrazine. The resulting material was finally pyrolyzed in Ar to obtain Te/C. They showed by XRD that no Te particles existed above 600 °C and, by XPS, that above 600 °C Te formed C–Te species with catalytic activity toward ORR. The best activity was obtained after a pyrolysis at 1,000 °C. The V onset of this metal-free catalyst was 0.67 V vs. RHE. It was measured by RRDE at 900 rpm in 0.5 M H2SO4. The same catalyst produced only 0.6 % H2O2 at 0.6 V vs. RHE.
After this publication on Te/C, Popov and collaborators focused on producing N-doped carbons for ORR in the acid medium. In a 2008 publication [81], they reported the synthesis of several metal-free catalysts using the following procedure: Ketjenblack was first treated in HCl to remove metallic impurities before being oxidized in HNO3. The oxidized carbon black was then used as a support for several resins obtained by the polymerization of formaldehyde with (1) melamine, (2) urea, (3) thiourea, or (4) selenourea. The oxidized carbon covered with each of the polymerized resins was then pyrolyzed in N2 at various temperatures to obtain catalysts for ORR. The best catalyst was obtained with selenourea pyrolyzed at 800 °C (SeUF/C-800). Its V onset in 0.5 M H2SO4 (900 rpm) was 0.65–0.70 V vs. RHE, while a V onset of 0.69 V vs. RHE was obtained for the catalyst made with urea (UF/C-800; only composed of C and N). V onset was 0.22 V vs. RHE for unoxidized Ketjenblack. UF/C-800 showed only carbon diffraction peaks in XRD. The total N content of that catalyst was determined by XPS to be 2.2 wt% (~8.8 at.% N) with pyridinic, pyrrolic, and quaternary (graphitic) types of nitrogen atoms. In another publication [82], more details about the same catalyst were revealed. Here, the V onset of UF/C-800 was 0.67 V vs. RHE, with a %H2O2 of 3 % released at 0.5 V vs. RHE. The catalyst had a surface area of 321 m2/g. The Fe and Co contents of UF/C-800 were 0.4 and 0.3 ppm, respectively. As there was no Fe or Co in that catalyst, the origin of the measured activity was definitively attributed to pyridinic and quaternary nitrogen atoms substituting for C in the CNx structure. The strong Lewis basicity of carbon doped with pyridinic and quaternary nitrogen atoms facilitated the reductive adsorption reaction of O 2 without the irreversible formation of oxygen functionalities.
The next publication [83] represents a turning point in the publications of Popov’s group because this time they added Co and/or Fe to their synthesis procedure in order to improve the activity of CNx catalysts, while at the same time denying any direct metal participation toward ORR for these catalysts. In this paper, carbon metal-free catalysts were first prepared according to the procedure described previously [82], except for the pyrolysis temperature which was now 900 °C instead of 800 °C. V onset values of 0.28 V vs. RHE were obtained for Ketjenblack, 0.36 V vs. RHE for oxidized Ketjenblack, and 0.74 V vs. RHE for UF/C-900. A Ketjenblack treated with NH3 (preparation procedure not reported) with a V onset of 0.53 V vs. RHE was also synthesized. A peroxide yield of 3 % measured at 0.5 V vs. RHE was reported for UF/C-900. Carbon composite catalysts were also prepared. In the latter case, UF/C-900 was used as a carbon support on which a mixture of Co nitrate and Fe sulfate complexed with ethylene diamine was impregnated. The resulting material was pyrolyzed at 900 °C in Ar. Then it was washed with H2SO4 to remove excess metal. The best catalysts obtained with an equal amount of Co and Fe (labeled Co–Fe on metal-free/C-900) was characterized in 0.5 M H2SO4. Its V onset was 0.79 V vs. RHE, while the metal-free/C-900 catalyst had a V onset of 0.70 V vs. RHE. Co-Fe metal on metal-free/C-900 and metal-free/C-900 catalysts yielded 0.9 and 1.2 % H2O2 at 0.5 V vs. RHE, respectively.
These catalysts were characterized by the following techniques: (1) EXAFS on unleached Co metal-free/C-900 (no Co–N bonds were detected at temperatures ≥800 °C); (2) TEM and XRD on leached Co–Fe on metal-free/C-900 (Co, Fe, and Co x Fe y phases were detected as well as Fe3C, but the metal-containing phases were covered with graphitic layers); (3) ICP-MS and XPS on leached Co-Fe on metal-free/C-900 (9.6 wt% Co and 1.4 wt% Fe were measured by ICP-MS in the bulk of the catalyst, but no metal was detected by XPS at its surface); and (4) the N content of the leached Co–Fe on metal-free/C-900 was measured by XPS and found to be 1.9 wt% N (~7.6 at.% N). Only pyridinic and quaternary N1s peaks appeared in the XPS spectrum. From these observations, they concluded that:
-
1.
Despite of their presence at several wt% in the leached Co–Fe on metal-free/C, neither Co nor Fe metal was involved in the active catalytic sites for ORR
-
2.
The only catalytic species were pyridinic and quaternary nitrogen atoms. A similar publication [84] reached the same conclusions but also mentioned that, in fuel cell, catalysts predominated by pyridinic-type nitrogen atoms showed higher activity, but lower stability than catalysts predominated by quaternary nitrogen atoms, which showed lower activity but higher stability
The next publication [85] which is the most detailed one, offers all the necessary information to discuss Popov and collaborators’ position concerning the origin of the catalytic activity of these catalysts prepared with relatively high Co and/or Fe contents. In ref. [85], only Fe was used and the following catalysts were synthesized and characterized electrochemically for ORR in 0.5 M H2SO4 (see Fig. 10.16a), as well as by XPS for their N content:

(a) Disk currents at 900 rpm and 5 mV/s from cyclic voltammetry experiments in O2-saturated 0.5 M H2SO4 solution for: (1) a metal-free CNx grown on SiO2 at 900 °C (V onset = 0.47 V vs. RHE); (2) 1.2 Fe-CNx-Ar-1000 obtained by the impregnation of 1.2 wt% Fe as ironII acetate on CNx and then pyrolyzed at 1,000 °C in Ar (V onset = 0.80 V vs. RHE); (3) CNx-Ar-1000 obtained by the pyrolysis of CNx at 1,000 °C in Ar (V onset = 0.54 V vs. RHE); (4) 1.2 Fe-CNx-Ar-1000L is sample (2) leached in H2SO4 (V onset = 0.74 V vs. RHE); and (5) 1.2 Fe-CNx-Ar-1000LH is sample (4) reheat treated at 1,000 °C in Ar (V onset = 0.80 V vs. RHE) (according to Fig. 6 in ref. [85]; reproduced with the permission of Elsevier). (b) Mass activity, for the reduction of oxygen in an O2-saturated H2SO4 solution at pH 1 on Fe/N/C and Co/N/C catalysts, vs. the metal content of the catalysts. Fe/N/C and Co/N/C were obtained by impregnation of either ironII or cobaltII acetates onto a nonporous carbon black followed by the pyrolysis of these materials at 950 °C in pure NH3 (according to Fig. 3 in ref. [86]; reproduced with the permission of Elsevier)
-
1.
Silica was first impregnated with acrylonitrile (AN) and then polymerized as PAN and pyrolyzed in Ar at 900 °C. The resulting material was washed in NaOH to remove SiO2 leaving CNx-900 as metal-free catalyst. The V onset of CNx-900 was 0.47 vs. RHE with a total N content of 6.88 at.%, broken down as 1.85 at.% pyridinic, 2.06 at.% graphitic, 1.69 at.% pyrrolic, and 1.28 at.% N+–O−.
-
2.
CNx was also impregnated with 1.2 wt% Fe as ironII acetate and then pyrolyzed at 1,000 °C. The V onset of 1.2 wt% Fe-CNx-1000 was 0.80 V vs. RHE with a total N content of 3.51 at.%, broken down as 0.96 at.% pyridinic, 1.49 at.% graphitic, and 1.06 at.% N+–O−.
-
3.
A sample similar to (2) but with 0 wt% Fe was also prepared for comparison. The V onset of 0 wt% Fe-CNx-1000 was 0.54 V vs. RHE with a total N content of 3.64 at.%, broken down as 0.78 at.% pyridinic, 1.52 at.% quadratic, and 1.34 at.% N+–O−.
-
4.
Sample (2) was then leached (L) to remove excess metal. The V onset of 1.2 wt% Fe-CNx-1000L was 0.74 V vs. RHE, while its total N content was 3.44 at.%, broken down as 0.84 pyridinic, 1.97 quadratic, and 0.63 N+–O−.
-
5.
Sample (4) was then reheat treated (H) at 1,000 °C. The V onset of 1.2 wt%Fe-CNx-1000LH was 0.80 V vs. RHE with a total N content of 3.47 at.%, broken down as 0.68 at.% pyridinic, 2.26 quadratic, and 0.53 N+–O−.
Figure 10.16a shows the ORR behavior of these five samples. Here again, based on the data presented previously [83, 84], Popov and collaborators concluded that “it seems reasonable to suggest that pyridinic N and quadratic N may be the active sites for ORR.” On this basis, one would therefore expect that a sample with the highest content of pyridinic and quadratic atoms would also be the sample with the highest catalytic activity. This is not what is observed in Fig. 10.16 a! Indeed, with its highest content of 1.85 at.% of pyridinic and 2.06 at.% of quadratic nitrogen atoms for a total content of 3.91 at.% N active for ORR, CNx-900 is the sample with the lowest activity. Furthermore, its apparent number of electron transfer during ORR was only 2, while a sample like 1.2 wt% Fe-CNx-1000, which was the best performing catalyst in Fig. 10.16a, was characterized by a lower content of 0.96 at.% of pyridinic and 1.49 at.% of quadratic nitrogen atoms for a total content of 2.45 at.% N active for ORR. Its apparent number of electron transfer during ORR was 4! Even if we compare samples 3 and 2, prepared at the same pyrolysis temperature of 1,000 °C, but with 0 and 1.2 wt% Fe, respectively, sample 3 was characterized by a content of 0.78 at.% of pyridinic and 1.52 at.% of quadratic nitrogen atoms for a total N content of 2.30 at.% active for ORR, while sample 2 was characterized by a content of 0.96 at.% of pyridinic and 1.49 at.% of quadratic nitrogen atoms for a total content of 2.45 N active for ORR. These two values of total ORR-active N content only differ by 0.15 at.%, while their V onset differs by 0.80−0.54 = 0.26 V as seen in Fig. 10.16a! Furthermore, sample 3 with 0 at.% Fe reduces O2 with an apparent number of transferred electrons close to 2, while it is 4 for sample 2!
According to Popov and collaborators, “encapsulated Fe species in carbon substrate may exert some electron effect on N-modified active sites and facilitate ORR, but they are not active sites!” An alternative explanation would be that some Fe–Nx species are also generated upon pyrolysis of 1.2 wt% Fe-CNx-1000 and those Fe–Nx species are greatly increasing the activity toward ORR for these catalysts. Indeed, we show in Fig. 10.16b [86] that even small Fe loadings were enough to obtain a definite effect on the ORR activity of catalysts made by impregnation of ironII acetate onto a nonporous carbon black, followed by pyrolysis at 950 °C. The catalytic mass activity increases linearly with the Fe content from 0.005 to 0.10 wt%, where it reaches about 50 % of its maximum activity. The ORR activity of the catalysts presented in Fig. 10.16b are very similar to that presented in Fig. 10.12B, curve “b,” which is characterized by a V onset of 0.83 V vs. RHE, a value close to that attributed to 1.2 wt% Fe-CNx-1000 (0.80 V vs. RHE) in Fig. 10.16a. Because the actual content of 0.1 wt% Fe (measured by NAA) in Fig. 10.16b is only 8 % of the nominal Fe loading of 1.2 wt% Fe-CNx-1000 (Fig. 10.16a), any physical characterization of 1.2 wt% Fe-CNx-1000 will be predominated by the high content of Fe not involved in Fe–Nx bonds. It is also important to remember that 0.1 wt% Fe is only 0.025 at.% Fe, a level that is not easily detectable by most XPS spectrometers. An average value of V onset for metal-free catalysts and metal-containing catalysts from all the publications of Popov and collaborators is shown in Fig. 10.15, where it is compared with average V onset values obtained by other groups.
From the following publications [84–87] and from the recent minireview [88] published in 2011, we also learn that the catalysts rich in pyridinic nitrogen displayed higher activity but lower stability. This was attributed to the protonation of the basic pyridinic N. According to Popov and collaborators, the protonated pyridinium species (NH +) is no longer active because the interaction of NH + with O 2 would be prevented in such a case. The protonation of all the pyridinic nitrogen atoms was followed by recording the N1s XPS spectrum before and after stability experiments of the catalysts in 0.5 M H2SO4. Before protonation, the XPS spectrum displayed three components: pyridinic, quadratic, and N+–O−, while after stability experiments, only one peak was visible. This lone peak was attributed to the simultaneous presence of NH+ and quadratic N, which are both characterized by similar binding energies. Quadratic nitrogen atoms are not affected by their contact with an acid solution. It is for this reason that their catalytic behavior is quite stable [87–89].
10.4.4 The Japanese Consortium
A Japanese consortium following Wiesener’s view on CNx catalytic sites is also worthy of discussion. It started with a 2006 publication by Ozaki and collaborators who reported on the synthesis of metal-free catalysts for ORR in acid medium [90]. The catalysts were prepared by mixing a furan resin with various phthalocyanines (H2Pc, Li2Pc, MgPc) as nitrogen precursors and then pyrolyzing the resulting material at 1,000 °C in N2. The respective N/C loadings were 0.04, 0.05, and 0.04 for H2Pc, Li2Pc, and MgPc with a V onset in 1 M H2SO4 of 0.23, 0.33, and 0.23 V vs. RHE, respectively. All catalysts were amorphous carbons and the enhancement of ORR activity vs. that of the pyrolyzed furan resin alone (V onset= 0.15 V vs. RHE) was attributed to the introduction of edge-site nitrogen atoms on the surface of these amorphous carbons. In a second publication [91], the catalysts were prepared by pyrolyzing at 1,000 °C a mixture of polyfurfuryl alcohol and melamine. Two catalysts with different N dopings were obtained by varying the melamine content in the mixture. Their respective N/C loading was 0.011 and 0.032, and their V onset in 0.5 M H2SO4 were 0.26 V vs. RHE for the lowest N loading and 0.30 V vs. RHE for the highest one. The catalytic activity was attributed to the presence of N atoms at the edge of graphene planes obtained after pyrolyzing the precursors of carbon and nitrogen. Later on, in collaboration with other authors, they compared the catalysts synthesized with melamine in the previous publication with catalysts obtained by other synthesis procedures, in particular that of a nitrogen-doped carbon black reacted with NH3 for which they reported a N doping of 1.3 at.% and a V onset of 0.33 V vs. RHE [92]. A series of N-containing rod polymers—polyimides (PI), polyamides (PA), and polyazoles (AZ)—were also prepared as nitrogen and carbon precursors. Catalysts were obtained from these materials after their carbonization at 900 °C in N2 [93]. Their ORR properties were evaluated in 0.5 M H2SO4 and V onset = 0.48 V vs. RHE was measured for carbonized PI, while it was 0.52 and 0.62 V vs. RHE for carbonized PA and AZ, respectively. Relatively high N contents were measured by XPS in these catalysts (4.83 and 5.31 at.% N for catalysts obtained with PA and AZ). The XPS spectra were deconvoluted in several components and the catalytic activity was attributed to the presence of pyridinic and graphitic types of nitrogen atoms in the carbonized materials. For all of the studied materials, the graphitic nitrogen content was always higher than the pyridinic nitrogen one.
In parallel with these syntheses of metal-free catalysts, the Japanese consortium also prepared catalysts using either cobalt- or iron-containing precursors in their syntheses but denied the existence of metal-containing ORR catalytic sites in these catalysts. For instance, in 2009 [94], a Co complex (Co-[poly(4-vinylpyridine)]) was first loaded on Ketjenblack and pyrolyzed in N2 at 1,000 °C. The resulting material was acid washed to remove excess Co species. The same procedure was repeated with Ketjenblack alone for comparison. The V onset of the carbonized Co complex on Ketjenblack was measured in 0.5 M H2SO4 at 0.70 V vs. RHE, a potential much higher than for Ketjenblack alone (V onset = 0.26 V vs. RHE). As no Co was detected by XPS in the catalyst made with the Co complex, the drastic improvement in ORR activity for that catalyst vs. the ORR activity of Ketjenblack alone was explained in terms of nitrogen doping of the catalyst made with the Co complex. Similar catalysts were also prepared, but this time without carbon support, using either cobalt phthalocyanine (CoPc) or iron phthalocyanine (FePc) mixed with N-containing polymers or with phenolic resins that were carbonized at various temperatures, and then eventually washed with acids to remove the metal in excess. In a first publication [92], the mixture of CoPc and N-containing polymer were pyrolyzed at 900 °C. Again, the XPS of that catalyst did not show any Co 2p signal after acid washing and its V onset in 0.5 M H2SO4 was 0.60 V vs. RHE. Despite its relatively low N content (0.8 at.%), its catalytic activity was mostly attributed to the presence in this material of graphitic nitrogen atoms. It is important to note that 0.1 at.% metal is usually considered to be the detection limit for Co (or Fe) by XPS and that the detection of Co (or Fe) contents below this limit is not trivial and requires at least a very large number of scans to ascertain the presence (or absence) of the metal. It will be seen later in this section that even cobalt contents below 0.1 at.% Co (or ~0.4 wt% Co in these carbon-based catalysts) may indeed be responsible for an important ORR activity that can be attributed to Co–Nx active sites.
In 2010 [95], Ozaki and collaborators mixed a furan resin with iron, cobalt, or nickel acetylacetonates and carbonized the mixture in N2 at 600 to 1,000 °C. The resulting material was ball milled and then acid washed to remove excess metal. The best catalyst was obtained with the Co complex carbonized at 800 °C. Its V onset in 0.5 M H2SO4 was 0.62 V vs. RHE. The catalyst had a specific surface area of 211 m2/g and its N/C ratio was 0.035. Its ORR activity was explained by the formation of carbon nanoshells, but also by N doping of the catalyst carbonaceous material.
In two following publications [96, 97], a catalyst similar to that previously described in ref. [92] and obtained by the carbonization at 1,000 °C of a mixture of CoPc and phenolic resin followed by acid washing was studied in greater detail. Its nominal Co content was 3 wt%. The V onset of the latter, measured in 0.5 M H2SO4, was 0.68 V vs. RHE. Its N content was 0.37 at.%. The latter was determined by hard X-ray photoemission spectroscopy (HXPES), a surface analytic technique probing the first 10 nm of an analyzed material. Washing the catalyst with HCl did not change its V onset or its nitrogen content, which remained the same at 0.68 V vs. RHE and 0.37 at.% N, respectively, but somewhat decreased the Co detected by HXPES from 0.05 at.% in the as-synthesized catalyst to 0.03 at.% after acid washing. Also, the surface area of the as-synthesized catalyst, which was 287 m2/g, decreased to 242 m2/g after HCl washing. Most of the cobalt in these two catalysts formed clusters surrounded with a carbon shell structure made of an average number of eight graphene layers producing Co-containing nanostructures. XANES and EXAFS spectra at the Co edge indicated that, in both catalysts, Co was mainly in the metallic state with only a small amount of oxidized cobalt. The detection of a small amount of oxidized cobalt by XANES and EXAFS spectroscopies, both able to probe several micrometers, and the detection of the same oxidized cobalt by HXPES, characterized by a depth probed of about 10 nm, inferred that this oxidized cobalt was at the surface of the catalyst or accessible to HXPES in cobalt nanostructures encapsulated with only one or two graphitic layers. The Co 2p HXPES spectra of the as-synthesized and acid-washed catalysts are presented in Fig. 10.17. According to the authors, it is likely that such a low oxidized cobalt content (0.05 at.% detected by HXPES in the as-synthesized catalyst and 0.03 at.% after acid washing) cannot be responsible for the ORR activity of these catalysts. The latter was mainly attributed to carbon atoms that are the first neighbors of a quadratic nitrogen atom at the zigzag edge of graphene planes. These first carbon neighbors at favorable –C–N–C– sites had a density of 0.29 at.% in both catalysts. A theoretical study [98] of these sites concluded that their number may be increased by controlling the degree of hydrogenation of edge carbons by introducing some H2 gas in the reactor during the carbonization process. According to the authors, the presence of CoPc during the synthesis of these N-doped carbons will only catalyze the formation of the favored –C–N–C– sites, but neither metallic nor oxidized Co played any role in the ORR properties of these materials because their concentration, as detected by HXPES, was extremely low.

Co 2p HXPES spectra of as-grown (open circles) and acid-washed (filled circles) CoPc-PhRs1000 catalysts. As-grown CoPc-PhRs1000 was obtained by the carbonization at 1,000 °C of a mixture of CoPc and phenolic resin. The intensities are normalized to the total photon flux to enable comparison of Co contents between the as-grown and the acid-washed samples. Dashed lines are guides. As reference, Co 2p HXPES spectra of Co metal (black squares) and CoPc (open squares) are also shown. The arrow denotes oxidized Co components on the surface of the Co metal (according to Fig. 4 in ref. [97]; reproduced with the permission of Elsevier)
This conclusion seems to rule out the eventual presence of Co–Nx ORR-active sites in these catalysts. However, it can be seen in Fig. 10.16b [86] that oxidized Co contents in the 0.03–0.05 at.% range (and even below) may indeed exhibit a clear ORR activity in Co/N/C catalysts. When this Co content, expressed in at.%, is translated into Co wt%, it becomes 0.12–0.20 wt% Co. In Fig. 10.16b, it is shown that a maximum ORR mass activity of 0.40 A/g was measured at 0.8 V vs. RHE for Co/N/C catalysts containing 1 wt% Co (measured by neutron activation analysis). At 0.20 wt% Co, the mass activity was 0.16 A/g and it was 0.12 A/g at 0.12 wt% Co. This is 40 and 30 % of the maximum measured mass activity, respectively, and is not at all negligible. As a matter of fact, Co–Nx sites are detected in the HXPES spectrum of the as-synthesized catalyst presented in Fig. 10.17. Indeed, there is an obvious peak at 781–782 eV (see the vertical dashed line) that is at the same binding energy as that of oxidized cobalt in CoPc (bottom curve in Fig. 10.17). This peak is less obvious in the acid-washed catalyst, but is still there because, otherwise, a dip would be observed in the spectrum at the same binding energy as that shown by the curve for Co metal in Fig. 10.17. In conclusion, even if a fraction of all the Co atoms detected in Fig. 10.17 in these catalysts were in oxidized form, metal-containing Co–Nx sites, which are active for ORR, may actually contribute to part of their ORR activity!
The same remark also holds true for the following two publications. The same authors also used FePc (instead of CoPc) to improve the ORR activity of their catalysts [99, 100]. Here, Fe was loaded at 3 wt% as FePc with the phenolic resin and pyrolyzed at various temperatures, the best one being 600 °C. This material had a large number of metal-containing particles detected by TEM and a graphitic nanoshell morphology similar to that detected with CoPc. The metal-containing particles were mainly Fe3C (cementite) as concluded from XRD measurements. There was less than 0.5 at.% Fe in the catalyst. A comparison was made with materials produced with H2Pc instead of FePc. A large difference was observed in their V onset, measured in 0.5 M H2SO4: 0.70 V vs. RHE for the catalyst made with FePc, but only 0.46 V vs. RHE for the material made with H2Pc. Specific surface area and N contents were also measured. They were 400–500 m2/g and 3.7 wt% N for the catalyst made with FePc, while 500–600 m2/g and 2 wt% N were measured for the catalyst made with H2Pc. Pyridine and quaternary nitrogen atoms were the major nitrogen species in both catalysts. In the same publication [99], the authors also describe a model explaining the differences between both catalysts during carbonization: the decomposition of the phenolic resin in the mixture begins at 200–450 °C, while that of H2Pc and FePc starts around 400–600 °C. H2Pc and FePc decompose in phthalonitrile, but less phthalonitrile is lost, and more nitrogen atoms remain in the resulting carbonized material when FePc is present. The presence of iron enhances the growth of nanoshell structures and the N doping of the resulting graphitic material. It was assumed that quaternary nitrogen atoms increase the occurrence of zigzag edge graphene and edge exposure, therefore improving the ORR activity. It is also worth noting that the same catalyst and several catalysts, derived from the pyrolysis at 600 °C in N2 of the mixture of FePc with phenolic resins and then further activated in NH3 at 800 and 1,000 °C, resulted in catalysts that showed very good performance and/or stability when tested in fuel cells [100].
Finally, the same Japanese consortium also reported the synthesis and characterization of carbon nitride made without iron, as catalysts for ORR in acid medium (0.5 M H2SO4). In a seminal work [101], the synthesis of C3N4 was performed at 220 °C from the reaction of sodium azide with cyanuric chloride \( (3\mathrm{ Na}{{\mathrm{ N}}_3}+{{\mathrm{ C}}_3}{{\mathrm{ N}}_3}\mathrm{ C}{{\mathrm{ l}}_3}\to \mathrm{ g}\text{--}{{\mathrm{ C}}_3}{{\mathrm{ N}}_4}+\mathrm{ NaCl}+4{{\mathrm{ N}}_2}) \), where g-C3N4 is the planar phase of the nitride made up of condensed tri-s-triazine (melem) subunits. This material had a nitrogen content of 23 at.%. It was not pyrolyzed at higher temperature, but mixed with Vulcan to increase the electrical conductivity of the mixture. Its V onset was 0.30 V vs. RHE, while the V onset of Vulcan alone was 0.13 V vs. RHE. In a second and more detailed publication [102], the same synthesis procedure was used, but this time, carbon black was added in the reaction chamber as a high surface area catalyst support. Furthermore, the resulting g-C3N4 was pyrolyzed at 1,000 °C. The resulting material had a nitrogen content of 1.8 at.%. Its XPS spectrum was deconvoluted into 55 % pyridinic, 40 % graphitic, and 5 % amine nitrogen atoms.
V onset measured in 0.5 M H2SO4 was 0.76 V vs. RHE, as shown in Fig. 10.18a, with an apparent number of electrons of n = 3.55 transferred during ORR. The authors suggested that quaternary nitrogen atoms are responsible for the oxygen reduction in this carbon nitride catalyst via a 4e − process.

Disk currents at 1,500 rpm and 0.1 V/s from cyclic voltammetry experiments in O2-saturated 0.5 M H2SO4 solution for the following: (a) Carbon nitride (g-C3N4) supported on carbon black, synthesized in a stainless steel high-pressure reactor and pyrolyzed at various temperatures. The best catalyst is obtained at 1,000 °C with a V onset = 0.76 V vs. RHE. (b) Fe-free unsupported carbon nitride (g-C3N4) synthesized in a Teflon melting pot and pyrolyzed at 1,000 °C. V onset = 0.65 V vs. RHE (according to Fig. 6 in ref. [102] for (a) and Fig. 8 in the same reference for (b); reproduced with permission of The Electrochemical Society)
Curiously, some iron contaminated this otherwise “metal-free” g-C3N4 catalyst. This contamination was traced to the stainless steel high-pressure reactor used for the synthesis of the material. In order to produce g-C3N4 free of iron contamination, the synthesis was again performed, but this time without carbon black support and in a Teflon melting pot, using the same experimental conditions as the ones described previously. The V onset measured for this true iron-free catalyst (and according to the procedure described in Fig. 10.13) is 0.65 V vs. RHE (see Fig. 10.18b), and the apparent number of electrons transferred during ORR was now 3.4. This again illustrates the obvious influence that even small amounts of iron impurity may have on the V onset of these catalysts active for ORR in acid medium.
The mean values of metal-free catalysts prepared by the Japanese consortium with and without Co- or Fe-containing precursors are reported in Fig. 10.15.
10.4.5 Other Authors
Besides the work of Ozkan and Popov’s groups and the work of the Japanese consortium, other authors also defended the same view as Wiesener: that of a CNx catalytic site for ORR. Their results will now be summarized. It should be stressed that among these authors, some have made a particular effort to avoid any metal trace in the synthesis of their catalysts in order to determine the true ORR activity of the nitrogen-doped carbon materials. Their results will be presented first, followed by the results of authors using either Fe- or Co-containing molecules in the synthesis of their N-doped carbon catalysts.
10.4.5.1 ORR Catalysts Made Without Any Fe or Co
In 2009 [103] and 2010 [104], Takasu and his collaborators used carbonized silk to perform ORR in acid medium (0.5 M H2SO4). The catalysts were derived from a Bombix mori silk fibroin containing 18 different amino acids. The silk was first carbonized at 500 °C in N2 and then ball milled and heat treated in N2 at various temperatures between 500 and 1,500 °C. The resulting materials were finally steam activated at 850 °C. The best results were obtained for a heat treatment of the carbonized silk at 900 °C in N2. This catalyst had a surface area of 588 m2/g and an N content of 6 at.% with 29 % of pyridinic and 59 % of graphitic nitrogen atoms. Its V onset was 0.69 V vs. RHE [103] or 0.73 V vs. RHE [104]. By comparison, the V onset for Vulcan was 0.20 V vs. RHE. The catalyst was also characterized, for potentials below 0.5 V vs. RHE, of an apparent number of electron transfer of n = 3.5 which could be pushed up to n = 3.9 by adding ZrO2 in the catalytic layer. Its ORR activity was mostly attributed to quadratic N doping. This was the only type of nitrogen atom left in the N1s XPS spectrum of the catalyst obtained after a heat treatment at 1,200 °C, while some pyridinic nitrogen atoms were also detected in the catalyst heat treated at 900 °C. A single MEA fuel cell was performed with the best catalyst. It yielded a maximum of 150 mW cm2 at 80 °C in H2/O2 (0.2 MPa at both electrodes).
In 2012, the group of Rao and Ishikawa produced vertically aligned carbon nanotubes (with and without N doping) without metal impurities by coating various polymers with different nitrogen contents in the pores of an alumina membrane [105]. This was followed by the carbonization at 900 °C of the material and by a KOH treatment to remove alumina. The nitrogen-free polymer was poly(phenylacetylene). In 0.5 M H2SO4, it was characterized by a V onset of 0.23 V vs. RHE. The four other polymers, from poly(4-vinylpyridine) to poly(p-pyridazine-3,6-diyl) had an increasing N content that yielded V onset values of 0.36 V vs. RHE (N = 4.3 at.%), 0.47 V vs. RHE (N = 5.6 at.%), 0.56 V vs. RHE (N = 8.4 at.%) and 0.60 V vs. RHE (N = 10.7 at.%). The increase of ORR activity was closely correlated with the total content of pyridinic and quadratic nitrogen in the catalysts (N pyr + N quad = 3.9, 5.3, 8.0, 10 at.%, respectively).
In 2010, Liming Dai and his collaborators also synthesized, on a Si substrate and from SiO2 nanoparticles catalyst, absolutely metal-free carbon nanotubes by chemical vapor deposition at 900 °C from a mixture of CH4 and H2 [106]. The tubes were doped by adding NH3 in the gas mixture. The nanotubes were then transferred onto a glassy carbon electrode in order to test their ORR activity in 0.5 M H2SO4. A V onset of 0.53 V vs. RHE was measured for the tubes containing 3.6 at.% N. In this case, there was no graphitic nitrogen atom in the total nitrogen content, as only pyridinic (83 %) and pyrrolic (17 %) nitrogen atoms were detected by XPS in that material. The same year [107], metal-free N-doped carbons were also produced by impregnation of a mesoporous silica template (SBA-15) with pyrrole, which was then polymerized. The resulting material was heat treated at 800 °C. After removal of the silica template by KOH, the ORR activity of the metal-free catalyst was measured in 0.5 M H2SO4. Its V onset was 0.35 V vs. RHE. Its total surface area was 544 m2/g and a N/C ratio of 0.12 was measured, with 36 % pyridinic and 25 % graphitic nitrogen atoms.
The following results obtained by Sheng Dai and his collaborators deserve a special mention as these authors obtained the highest activity reported so far with truly N-doped metal-free carbon materials [108]. These catalysts were obtained by heat-treating at high temperature (950–1,050 °C) and with NH3 a 2D hexagonal-ordered mesoporous carbon having a total surface area of 658 m2/g before its reaction with NH3. When the latter material was measured in 0.05 M H2SO4 for its ORR activity, a V onset of 0.34 V vs. RHE was obtained. The V onset jumped to 0.72, 0.76, and 0.77 V vs. RHE after a heat treatment in NH3 at 950, 1,000, and 1,050 °C, respectively. This is illustrated in Fig. 10.19.

Disk currents at 1,600 rpm and 10 mV/s from cyclic voltammetry experiments in O2-saturated 0.05 M H2SO4 solution of an N-doped metal-free carbon catalyst heat treated in NH3 between 950 and 1,050 °C. The best catalyst is obtained at 1,050 °C. It has a V onset of 0.77 V vs. RHE (according to Fig. 2 in ref. [108]; reproduced with permission of The American Chemical Society)
The heat treatment greatly increased the total surface area of the three N-doped catalysts which reached 1,681, 2,121, and 1,923 m2/g, respectively, with a total N content of 6.0, 3.6, and 4.6 at.%, respectively. Their Fe content was also measured (by inductively coupled plasma spectrometry) and found to be below 100 ppm. The only particularity of these catalysts is their very high specific surface area which for the best catalyst (the one obtained at 1,050 °C) was nearly 2,000 m 2 /g. On the other hand, their total nitrogen content is not particularly high at 4.6 at.% N for the best catalyst with 46.9 and 10.0 % of pyridinic and graphitic nitrogen atoms, respectively. It is possible here that both the nitrogen content and the particular textured properties of the very high specific surface area of these materials are acting together to drastically improve the ORR activity of these materials. A comparative study of various carbon electrodes has previously shown that carbon substrates with a large number of edge plane sites have higher activity toward ORR [58]. In 0.1 M H2SO4, there is indeed a difference of 0.34 V for ORR on basal plane pyrolytic graphite and edge plane pyrolytic graphite substrates. When the V onset values of all N-doped metal-free carbon catalyst of this section are considered, an average V onset of 0.62 V vs. RHE is obtained with a minimum and maximum V onset of 0.35 and 0.78 V vs. RHE. This is plotted as OTHER in Fig. 10.15. This average V onset value of 0.62 V vs. RHE is however significantly boosted by the high V onset from the last series of catalysts reported by Sheng Dai and collaborators.
10.4.5.2 ORR Catalysts Made with Fe or Co
Similarly to Ozkan, Popov, and the Japanese consortium, other authors also obtained their ORR catalysts using Fe- or Co-containing precursors to improve their catalytic activity without attributing at least part of this activity to Fe–Nx or Co–Nx catalytic sites. In 2009 [109], Muhler and collaborators grew N-containing carbon nanotubes from acetonitrile over Co metal used as catalyst on a silica support. Two growth temperatures were used: 550 and 750 °C. The N-containing carbon nanotubes (N-CNT) were refluxed in KOH to remove the silica support and HCl to dissolve the metal in excess. The Co surface content, measured by XPS after sample purification, was 0.4 at.% for N-CNT550 and 0.2 at.% for N-CNT750. Some of this cobalt was fully covered by a carbon layer. Oxidized cobalt was also seen in the XPS spectrum of N-CNT550 and N-CNT750. The nitrogen content, also measured by XPS, was 6.3 at.% in N-CNT550 and was broken down as 43 % pyridinic and 28 % graphitic nitrogen atoms, while a nitrogen content of 7.5 at.% broken down as 20 % pyridinic and 46 % graphitic nitrogen atoms was found for N-CNT750. The ORR activity of these catalysts was measured in 0.5 M H2SO4 at 900 rpm. N-CNT550 was found to be more active, with a V onset of 0.70 V vs. RHE, than N-CNT750 with a V onset of 0.55 V vs. RHE. The higher activity of N-CNT550 was explained in terms of a higher edge plane exposure and a higher pyridinic content for the catalyst grown at lower temperature. The apparent number of electrons transferred during ORR was also n = 3.92 for N-CNT550, but only n = 2.48 for N-CNT750.
In 2010, Liming Dai and collaborators [110] grew on a silica/Si support, vertically aligned N-doped carbon nanotubes (N-CNT) at 850 °C and from a mixture of Ar, H2, and NH3 in the presence of iron phthalocyanine (FePc). The N-CNTs were removed from the SiO2/Si support and measured on a glassy carbon electrode. The V onset determined at pH 3 for this catalyst was 0.86 V vs. RHE. Its total N content was 4 at.%. This experiment was anterior to the one reported by this group and already commented in Sect. 10.4.5.1 [106]. In the latter experiment, similar N-doped carbon nanotubes were grown without FePc. The N content of that catalyst was 3.6 at.% and its V onset was only 0.53 V vs. RHE. The comparison between the two types of vertically grown N-doped carbon nanotubes clearly illustrates the important role played by FePc in the sharp rise in ORR activity when Fe-containing compounds are present in the reactor during the growth of the N-doped nanotubes!
In 2011, Woo and collaborators prepared ORR catalysts from the pyrolysis at 700, 800, and 900 °C of a mixture of iron oxide supported on Vulcan and dicyandiamide (C2H4N4; a dimer of cyanamide) [111]. TEM of the catalysts revealed that at 700 °C metal particles were encapsulated with a carbon layer, while they were mostly in carbon tubes at 900 °C. The total N content was 2.2, 3.5, and 6.6 at.% for the catalysts heat treated at 700, 800, and 900 °C, respectively. This N content was broken down as 54 % pyridinic and 0 % graphitic nitrogen atoms at 700 °C, while it was 61.4 % pyridinic and 10.7 % graphitic at 900 °C. The Fe content was also measured in these catalysts and also for Fe2O3/C, the catalyst iron precursor. It was 1.7, 1.4, and 0.5 at.% after heat treatments at 700, 800, and 900 °C, while it was 1.8 at.% Fe for FeO3/C. The V onset, measured in 1 M HClO4 at 2,000 rpm, was 0.78, 0.73 and 0.78 V vs. RHE for the three catalysts made at 700, 800, and 900 °C, respectively, but Fe2O3/C had a negligible catalytic activity for ORR. Since the Fe content in Fe2O3/C and in the catalyst heat treated at 700 °C were about the same (1.8 vs. 1.7 at.% Fe), but the activity increased dramatically after the heat treatment of Fe2O3/C mixed with dicyandiamide, it was concluded that (1) Fe-containing catalytic sites were not involved in this catalyst and (2) that the catalytic activity was the result of N doping (especially by pyridinic nitrogen atoms) of the carbon material.
In 2011, Sun and collaborators also prepared N-doped carbon nanotubes by the CVD at 950 °C of a mixture of Ar and CH4 on a ferrocene and melamine catalyst [112]. The N content in these N-CNT materials was varied from 0 to 7.7 at.%, while the Fe content was always between 0.3 ± 0.1 at.% in the same materials. The ORR activity of the N-CNT doped with 7.7 at.% was measured in 0.5 M H2SO4 at 900 rpm. Its V onset was 0.51 V vs. RHE.
The same year, Kim and collaborators [113] published results about catalysts made on three carbon supports: Ketjenblack (an amorphous carbon), platelet carbon nanofibers (P-CNF with many edges of graphene planes appearing at the surface of the support), and carbon nanotubes (CNT without edges along the surface of the tube). Urea and formaldehyde were first polymerized on these three carbon supports. This was followed by the impregnation on the polymerized material of a complex made by the reaction of cobalt nitrate with ethylene diamine. Finally, the three materials were pyrolyzed at 800 °C and then washed in acid to remove excess cobalt. The total N content increased from 0.7 wt% for the catalyst made on the CNT support, to 1.5 wt% for the catalyst made on Ketjenblack, and finally to 3.8 wt% for the catalyst obtained on the platelet carbon nanofibers. Their surface areas were 450, 906, and 45 m2/g, respectively. The V onset of the catalysts followed their N content with 0.53, 0.66, and 0.74 V vs. RHE. The ORR activity was attributed to the nitrogen content, the type of nitrogen (mostly pyridinic), and to the number of edges available on the carbon support. There was no mention of a possible role of Co–Nx sites in these catalysts, and the remaining Co content was not determined. These catalysts were also measured in fuel cell at 75 °C, with 2 atm. H2/2 atm. O2. The best catalyst provided a power density of 0.320 W/cm2 at 0.35 V. This is a power density typical of many catalysts containing Co or Fe precursors.
In a following publication [114], the same group polymerized pyrrole around carbon nanofibers and then impregnated this material with the complex made from cobalt nitrate and ethylene diamine before pyrolyzing the resulting material at 800 °C in Ar to obtain a catalyst for ORR. The final material was washed in acid to remove the metal in excess. The total N content of the catalyst was 4.5 wt% broken down as 49 % pyridinic, 22 % pyrrolic, and 30 % quadratic nitrogen atoms. The Co particles in the catalyst were protected from dissolution by a carbon layer formed during the heat treatment. The Co content of the leached catalyst was 2.7 wt%. Only fuel cell measurements at 75 °C, 2 atm. H2/2 atm. O2 were made with this catalyst, which yielded a maximum power density of 0.34 W/cm2 at 0.3 V, similar again to what was already obtained with the previous catalyst. Despite the possible contribution of Co–Nx sites to the activity, these authors attribute the ORR activity of their catalyst to pyridinic and quadratic nitrogen atoms, even suggesting a possible synergetic effect on increasing the N content and therefore the activity of the catalyst to the simultaneous presence of polypyrrole and ethylenediamine.
The last publication examined in this section will again give us the opportunity to illustrate that minute amounts of metal content (either Fe or Co) in N-doped carbons are capable of having a significant effect on their ORR activity in acid medium. In this publication by Zhang and collaborators [115], a nitrogen-doped carbon xerogel was made from a mixture of resorcinol formaldehyde and cobalt nitrate, where cobalt nitrate was considered an N-doping catalyst. The material was pyrolyzed at 800 °C in NH3, which was considered as the nitrogen source. The resulting material was leached in H2SO4 to remove all soluble species and yielded the catalyst labeled N-CX. When the activity of N-CX was measured for ORR in 0.5 M H2SO4 at 1,600 rpm, a V onset of 0.77 V vs. RHE was obtained. This is shown in Fig. 10.20. In these conditions, the catalyst reduced O2 with an apparent number n = 3.5–3.7 of electrons. In fuel cell at 80 °C, 0.2 MPa H2/0.2 MPa O2, it yielded a maximum power density of 0.36 W/cm2 at 0.35 V, a value similar to the two previous catalysts measured in fuel cell in this section. Curiously, the total N content is not reported, but a XPS spectrum at the N1s edge is shown and deconvoluted into pyridinic, quaternary, and oxidized nitrogen atoms with the quaternary peak predominating the spectrum. The ORR activity of this catalyst is substantially attributed to quaternary nitrogen atoms, which are doped in the carbon aerogel. The same quaternary nitrogen atoms also play an important role in the stability of the catalyst. This is also shown in Fig. 10.20 for N-CX after accelerated aging test (AAT) based on a continuous cycling of the potential for 1,000 cycles at room temperature from 0 to 1.2 V vs. RHE. The Co content in this catalyst is quite low and was measured at 0.07 wt%. This metal content is considered by the authors to be too low to have any effect on the catalyst activity. However, as it is shown in Fig. 10.16b, a Co content of 0.07 wt% may be responsible for 20 % of the maximum mass activity obtained with a catalyst made by the impregnation of 1 wt% of cobalt acetate on nonporous carbon black, followed by the pyrolysis of this material in NH3 at 950 °C. A value of 20 % of the maximum mass activity is obviously not negligible and Co–Nx catalytic sites may be contributing to the ORR activity of this catalyst. The average value of V onset for all the catalysts produced in the presence of Fe- or Co-containing molecules in this section is plotted as OTHERS in Fig. 10.15.

Disk currents at 1,600 rpm and 5 mV/s from cyclic voltammetry experiments in O2-saturated 0.5 M H2SO4 solution of an acid leached nitrogen-doped xerogel (N-CX) made from a mixture of resorcinol formaldehyde and cobalt nitrate. The material was pyrolyzed at 800 °C in NH3. The disk current for the same catalyst is also shown after accelerating aging tests (AAT). V onset for N-CX before AAT is 0.77 V vs. RHE. It is 0.75 V vs. RHE after AAT (according to Fig. 7a in ref. [115]; reproduced with permission of The Royal Society of Chemistry)
10.5 Discussion
A summary of the ORR activity, in terms of V onset in strong acid medium, is presented graphically in Fig. 10.15 for all catalysts prepared by the various research groups and discussed in this chapter. Figure 10.13 demonstrates how these V onset values were extrapolated from the various cyclic voltammograms reported in the literature. Figure 10.15 Frame A shows a cluster of the average V onset values (open circles) attributed to the catalysts based on N-doped carbon produced by various research groups. These catalysts were produced without any strategic metals, like Fe or Co, either in the precursor molecules used in their synthesis, or as chemical catalysts to generate their carbon nanostructures. Also shown in Frame A is a cluster of the average V onset values attributed to the catalysts produced by various research groups with strategic metals (Fe or Co), either in the precursor molecules used in their synthesis (dark circles) or as chemical catalysts to generate their carbon nanostructures (dark circles for Fe or Co and dark triangle for Ni). Each symbol includes a vertical line whose limits represent the highest and lowest individual V onset values reported for each group.
In Fig. 10.15a, the cluster of the average V onset values represented by the open circles is discernibly lower than the cluster of the average V onset values represented by dark circles. We may further simplify the presentation of the activity results for all these catalysts by calculating overall average values for each of these clusters. The overall average V onset value for all N-doped carbon catalysts prepared without strategic metal (Fe or Co) represented by open circles in Frame A is presented as an open square in Fig. 10.15b and has a value of 0.56 V vs. RHE. The overall average V onset value for the N-doped carbon catalysts prepared with strategic metal (Fe or Co) is presented as a dark square in Fig. 10.15b and has a value of 0.74 V vs. RHE. Each overall average V onset in Frame B includes a vertical line whose limits represent the highest and lowest individual V onset values reported for each cluster, respectively. In addition, Fig. 10.15b also includes the overall average, minimum, and maximum V onset values for ORR of all carbon blacks (Vulcan, Ketjenblack, Black Pearls, etc.) having RDE data in the publications reviewed in this chapter. The latter is presented as a gray diamond with a value of 0.20 V vs. RHE.
From Fig. 10.15b, we see a large potential difference (0.36 V) between the overall average onset potential of oxygen reduction on carbon (gray diamond) and on N-doped carbon obtained without any strategic metal (Fe or Co). According to the authors cited in this chapter, doping carbon with two types of nitrogen atoms (pyridinic and quadratic [also referred to as graphitic]) is an important factor for increasing the ORR catalytic activity. The presence of graphene plane edges in the catalyst is another important factor because carbon atoms at graphene plane edges are believed to have a better interaction with O2 than carbon atoms located in graphene basal planes. Since pyridinic-type nitrogen atoms are located on the edge of graphitic planes (see Fig. 10.4), it is difficult to attribute with certainty the observed positive effect on the catalytic activity for N-doped carbons to either the nitrogen atoms or to the graphene edges. The only case encountered in this chapter where graphene edges were the dominant factor in increasing the ORR activity was that of the catalyst presented in Fig. 10.19, for which a specific surface area of about 2,000 m2/g was measured. This very high surface area likely implied that O2 had access to a large number of graphene edges, while the total N content of the same catalyst was relatively low (4.6 at.%) compared to other N-doped catalysts discussed in this chapter. Furthermore, the iron content of the latter catalyst was only 100 ppm. Consequently, this catalyst is among the best examples of an N-doped carbon obtained without any strategic metal. The likely origin of its high ORR activity (V onset = 0.77 V vs. RHE) is therefore related to its large number of graphene edges available for O2 interaction and its reduction.
From the observation of Figs. 10.15a, b, one may also conclude that there is practically no difference between the average V onset value of 0.52 V vs. RHE for the reduction of O2 on N-doped carbon catalysts produced with Ni (dark triangle in Fig. 10.15a) and that of the overall average V onset value of 0.56 V vs. RHE of N-doped catalysts prepared without strategic metal. Since Ni is a strategic metal capable of chemically catalyzing the growth of carbon nanostructures, as shown by Ozkan and collaborators [65, 66, 68, 71], these authors have explained that the carbon nanostructures produced with Ni were mainly devoid of graphene plane edges. Therefore, these N-doped carbon materials were poorly interacting with O2. Our interpretation of the same results was based on the possible occurrence of Ni–Nx sites that were previously found to have some, but very low, ORR activity [26, 116]. According to our alternative explanation, these N-doped carbon nanostructures would therefore have an ORR activity similar to that of N-doped carbon catalysts made without any strategic metal like Fe or Co.
A third conclusion that may be deduced from Fig. 10.15b is that there is a difference of 0.18 V between the overall average V onset for the N-doped carbons catalysts prepared with Fe or Co in their synthesis (dark square) and the N-doped carbons obtained without either Fe or Co (open square). The latter may not seem large, but in fact means that a N-doped carbon made with a strategic metal will be able to generate (at about 0.8 V vs. RHE) 100.18/0.10 = 63 times more current density per unit mass compared to a N-doped catalyst made without a strategic metal. This calculation is based on a Tafel slope of 100 mV/decade (0.10 V/decade) at the same potential [61]. An experimental ratio of ORR activity at 0.8 V vs. RHE of about 60 was found between the mass activity (in A/g of catalyst) measured by RDE in H2SO4 at pH 1 for a Fe/N/C catalyst containing 0.27 wt% Fe (similar to that shown in Fig. 10.12B curve “b”) and a N-containing (but Fe-free) carbon prepared by pyrolysis in NH3 (see Fig. S7 in ref. [117]).
The reason for this factor of about 60 in ORR activity between the two types of N-doped carbon catalysts, i.e., N-doped carbons made with strategic metals like Fe or Co and those made without any strategic metal, is what divides the scientific community. One camp believes the presence of Fe–Nx or Co–Nx catalytic sites explains the drastic improvement while the other defends the hypothesis that a larger number of N atoms (pyridinic or graphitic) or a larger number of graphene edges catalyzed by Fe or Co is what explains the difference.
We have shown several examples in this chapter where residual strategic metals contents ≤0.2 wt% (Fe or Co) in N-doped catalysts, considered negligible by some researchers, can in fact result in non-negligible catalytic activity for ORR in acid medium, given that this activity increases quickly with the Fe or Co content in N-doped catalysts (see Figs. 10.3 and 10.16b). We also believe that the existence of Fe–Nx sites in Fe/N/C catalysts have now been proven with little or no doubt by ToF-SIMS, Mössbauer spectroscopy, STEM imaging and EELS analysis, and by some carefully performed poisoning experiments. These four types of proofs are now discussed.
10.5.1 ToF-SIMS
In Figs. 10.3 and 10.16b, the increase in catalytic activity with metal content in the Fe/N/C catalysts was attributed to the presence of Fe–Nx (or Co–Nx) catalytic sites capable of interacting with oxygen and reducing it to water. This assertion was based on the detection by ToF-SIMS in these Fe/N/C catalysts of several FeN x C y + ions (see Fig. 10.6) ejected from the top molecular surface layer of the probed catalyst. A strong correlation was found (see Fig. 10.5) between the relative intensity of the FeN2C4 + type ions detected by ToF-SIMS and the ORR catalytic activity for Fe/N/C catalysts prepared at various heat-treatment temperatures. This finding led us to propose the involvement of a FeN2/C catalytic site (see Fig. 10.4) as the most active site in these Fe/N/C catalysts, while also acknowledging that this proposed moiety was only part of the complete molecular site.
10.5.2 Mössbauer Spectroscopy
The second proof of the presence of Fe–Nx catalytic sites and their involvement in ORR in Fe/N/C catalysts comes from recent experiments performed by Mössbauer spectroscopy on these catalysts. The latter were obtained by adsorbing increasing amounts of iron acetate on a nonporous carbon black and heat-treating these materials in NH3 at 950 °C [117]. The change in the catalytic activity with the iron loading is presented in Fig. 10.21a. Figure 10.22 shows that several Fe-containing species were detected by Mössbauer spectroscopy in each of these catalysts. Three broad doublets, D1, D2, and D3, as well as two narrow doublets (D4 and D5) were found in all catalysts. Based on their Mössbauer parameters, D1, D2, and D3 have been assigned to three different FeN4-like species whose possible structures are illustrated in Fig. 10.23. D4 and D5 have both been attributed to iron nitride nanoparticles. The latter are also seen by high-resolution TEM. A last Fe species characterized by a broad singlet in the Mössbauer spectrum only appears when the actual Fe content in the catalyst reaches at least 0.27 wt% (see Fig. 10.22). This singlet has been assigned to incomplete FeN4-like sites that are irreversibly lost when the Fe/N/C catalyst becomes in contact with an acidic solution of H2SO4 at pH 1.

ORR activity and the relative composition of each Fe species vs. the bulk iron content. Also shown are the Energy Schemes of D1, D2, and D3, as well as the filling of the molecular orbitals for the ironII ion in the structure proposed for each site according to its proposed spin state: low in D1, intermediate in D2, and high in D3 [118] (according to Fig. 4 in ref. [117] for the left frame and to Fig. 2 in ref. [117] for the right frame; reproduced with permission of The Royal Society of Chemistry)

Deconvoluted Mössbauer spectra of the Fe/N/C catalysts. The iron content of the original catalyst is specified in each frame (according to Fig. 1 in ref. [117]; reproduced with permission of The Royal Society of Chemistry)

Top and side views of the proposed structures of the FeN4/C catalytic site in heat-treated, macrocycle-based catalysts assigned to Mössbauer doublet D1, the FeN2+2-like micropore-hosted site found in the catalyst prepared with iron acetate and heat treated in ammonia assigned to doublet D2, and the N-FeN2+2-like composite site, where N-FeN2+2 is assigned to doublet D3. In all side views, graphene planes are drawn as lines. For D2, the distance between the two nitrogen atoms belonging to the graphene planes above and below the FeN2+2-plane is similar to that in crystalline iron phthalocyanine, thus similarly influencing the lattice contribution to the quadrupole splitting. For D3, the ironII ion in N-FeN2+2 is coordinated by five pyridinic nitrogen atoms, one of them belonging to a plane located vertically below the N4-plane. This axial nitrogen coordination moves the ironII ion out of the N4-plane towards the fifth coordinating nitrogen atom. D3 may exists as N-FeN2+2/C or as a composite site N-FeN2+2 … Nprot/C, where Nprot is a protonable pyridinic nitrogen atom appearing at the edge of the upper plane in the side view of D3 [128]. Nprot is too far from N-FeN2+2 to have an influence on the Mössbauer spectrum of D3, but it is close enough to drastically improve the turnover frequency of the proposed composite site N-FeN2+2 … Nprot/C. According to Fig. 2 in ref. [117]; reproduced with permission of The Royal Society of Chemistry
Among the four different Fe species left after acid washing, only D1 and D3 have been found to be active for the ORR, the most active one being D3 as shown by the similarity between the ORR activity of the catalysts illustrated in Fig. 10.21a, d. Here, Fig. 10.21d represents the changes in the amount of iron ions (in wt%) assigned to D3 species vs. the total iron content in the Fe/N/C in the original catalyst (before acid washing). Although D2 in Fig. 10.21c shows the same trend as D3, it is not ORR active because the 3dz2 orbital of its ironII ion (the orbital interacting with O2 [119]) is already filled. This is illustrated in D2’s Energy Scheme in Fig. 10.21. On the other hand, the 3dz2 orbitals of the ironII ion in D3, with a population of only one electron, and that of the ironII ion in D1 with no electron, may both interact with O2 and therefore result in active sites for the reduction of oxygen. The similarity between Fig. 10.21c for D2 and Fig. 10.21d for D3 indicates that the two FeN4-like species are structurally related as shown in Fig. 10.23. Indeed, both D2 and D3 represent FeN4-like sites hosted in carbon micropores. The difference between D2 and D3 is that, according to the Mössbauer parameters of D3, a fifth coordinating nitrogen was postulated below the FeN4 plane pulling the Fe ion toward it. This modifies the sequence of the ironII ion energy levels [118] as shown in Fig. 10.21 and changes the spin of the FeN4-like site assigned to D3. The ironII in the site proposed for D3 is in a high spin state (S = 2), while it is in medium spin state (S = 1) for D2 and in low spin state for D1. These spin states affect the Mössbauer parameters of the FeN4-like doublets assigned to these sites as it is detailed in the literature [120–127].
From Fig. 10.23, the catalytic site illustrated in Fig. 10.2 has a D1 Mössbauer signature, while both D2 and D3 signatures represent sites whose incomplete structure was initially proposed as FeN2/C (Fig. 10.4), and later completed as FeN2 + 2/C (Fig. 10.10) and is now present either as an ORR inactive site, having the D2 signature, or as an ORR-active site (N–FeN2 + 2/C) having the D3 signature. Moreover, we have shown [128] that if there is a basic (pyridinic) nitrogen in the vicinity of an N–FeN2 + 2/C active site which is protonable, it may possibly act as a relay for quickly transferring protons to the iron ion of the N–FeN2 + 2/C catalytic site during ORR. The synergy between N–FeN2 + 2/C and the neighboring protonated pyridinic nitrogen drastically increases the turnover frequency of what is now the composite site: N–FeN2 + 2⋯NH+/C. A value of 11.4 electrons per site per second has been measured for the turnover frequency of N–FeN2 + 2⋯NH+/C [117]. This is only 2.2 times lower than the turnover frequency measured for Pt [6]. The high turnover frequency of N–FeN2 + 2⋯NH+/C is drastically reduced when the protonated NH+ in the composite site is neutralized by an anion. This was found to be the main mechanism affecting the ORR activity of the composite N–FeN2 + 2⋯NH+/C site in H2SO4 solution. The same phenomenon also occurs in fuel cell, where the neutralization of N–FeN2 + 2⋯NH+/C involves the sulfonate group (SO3 −) of the proton-conducting ionomer. This neutralization has been identified as the main problem leading to an apparent instability of these catalysts in fuel cells. “Apparent” is used to emphasize the fact that, in this neutralization reaction, the structure of the active site does not change, but the site is simply deactivated. Here, it should be noted that our interpretation of the apparent instability of these catalysts in fuel cells is at odds with that proposed by Popov and collaborators [88] as in their case, it is claimed that the protonation of the pyridinic nitrogen atoms of their N-doped catalysts leads to a decay of activity, while in our case, it is when N–FeN2 + 2⋯NH+/C is in its protonated form that the catalytic site is the most active.
Thus, to summarize the results obtained by Mössbauer spectroscopy, it is clear from the similarity between the changes in ORR activity of these Fe/N/C catalysts illustrated in Fig. 10.21a and the changes in the amount of N–FeN2 + 2⋯NH+/C highly active sites illustrated in Fig. 10.21d in the same catalysts that Fe–Nx catalytic sites are indeed active toward ORR in these Fe/N/C catalysts.
10.5.3 STEM Imaging and EELS Analysis
In a recent publication [129], Hongjie Dai and his collaborators reported the preparation of few-walled carbon nanotubes, with a high percentage of double-walled nanotubes and a small percentage of triple-walled nanotubes, grown from MgO-supported iron seeds by chemical vapor deposition. The outer walls of these few-walled carbon nanotubes were then partially opened by controlled oxidation, producing disordered graphene layers still partially attached to what was left of the carbon nanotube. When this material was subjected to a pyrolysis step in NH3 at 900 °C, N functionalities were generated at the defect sites in the tube and in the disordered graphene layers, and this, in conjunction with “iron impurities” left over from the nanotube growing process, produced Fe–Nx-type centers that were active for the oxygen reduction reaction in basic and acid solution. This catalyst was labeled NT-G (or NT-NG in the supplementary information).
Scanning transmission electron microscopy (STEM) imaging and simultaneous elemental analysis by electron energy loss spectroscopy (EELS) supplemented by annular dark field (ADF) imaging were performed on these catalysts. They indicated that Fe and N species were important to the high ORR electrocatalytic activity observed for NT-G and that this catalyst falls within the category of metal-nitrogen catalysts since its activity for ORR decreased when the “impurity” Fe content was lowered from 1 wt% to 0.1 wt%! Fig. 10.24 presents an example of bright field STEM images and their corresponding elemental analysis for a catalyst showing individual Fe and N atoms in close proximity as they should be in Fe–Nx catalytic sites. Note that each square in the elemental mapping represents a surface area of about 2 Å × 2 Å.

Distribution of Fe and N atoms in the NT-G ORR catalyst. (a) Bright field STEM and (b) corresponding ADF images of NT-G. The area indicated by a white square in (b) is further characterized by (c) ADF intensity mapping, (d) N EELS mapping, (e) Fe EELS mapping, and (f) overlaid Fe and N EELS maps. The ADF and EELS maps were recorded simultaneously (according to Fig. S8 in ref. [129]; reproduced with permission of Nature)
10.5.4 Poisoning Experiments
The most conclusive evidence to demonstrate the presence of Fe–Nx active sites for ORR in Fe/N/C catalysts would be to show the possibility of poisoning these sites with a strong ligand to the iron ion of Fe–Nx in contact with an O2 saturated electrolytic solution. In such a case, the poisoning ligand would prevent access of the site to O2 and hence suppress its subsequent reduction. CO would seem to be a good poison candidate since it is well known to poison hemoglobin in neutral pH. However, it has been shown that CO does not poison Fe-based catalysts in acid medium [78] (or even in neutral or basic media [unpublished results]). CO does not poison the oxygen reduction reaction on unpyrolyzed iron phthalocyanine (FePc) adsorbed on carbon in which the FeN4 moiety is known to have ORR activity [119].
Recent experiments by Gewirth and collaborators [130] have shown that the oxygen reduction activity of carbon-supported FePc or pyrolyzed (800 °C) carbon-supported FePc remain practically unaffected in pH 6 phosphate buffer containing NaF, KSCN, or ethane thiol (C2H5 SH). Only KCN is able to strongly decrease the oxygen reduction activity of the same catalysts in O2 saturated 0.1 M NaOH solution. Potassium cyanide is known to form very strong complexes with iron and poison the ORR at iron centers [131–133]. These results are consistent with those originally obtained by Yeager and collaborators on an unpyrolyzed FePc derivative showing a poisoning effect with cyanide in basic medium [131]. Rinsing cyanide-poisoned FePc and pyrolyzed FePc electrodes with water resulted in almost complete recovery of the ORR activity in fresh electrolyte without cyanide. This indicates that cyanide ions compete with O2 for access to the iron ion in the active site, but do not permanently bind to this site or alter it. Therefore, these results demonstrate that the active sites in these materials are Fe-centered both before and after pyrolysis. Similar poisoning experiments cannot, of course, be performed in an acidic medium, but if Fe–Nx active sites exist in basic medium, there is a strong presumption that they should also exist in acid medium.
10.6 Mechanism of O2 Reduction
10.6.1 O2 Reduction on Fe–Nx Active Sites
Given the fact that iron exists in several oxidation states and is the main transition metal found in many oxidoreductases (enzymes responsible for electron exchange in biological reactions) [134], including in the Fe-porphyrin-containing catalytic site of cytochrome c oxidase, the enzyme responsible for the oxygen reduction in the mammalian respiratory electron transfer chain embedded in the mitochondrial membrane [135], it is relatively easy to understand why iron in Fe/N/C catalysts should also be at the heart of the active sites that reduce O2 to H2O or O2 to H2O2 in acidic medium. A plausible mechanism for the electrochemical reduction of O2 on Fe/N/C catalysts may be similar to the one proposed by Boulatov for the catalyzed reduction of O2 on porphyrins [135]. This mechanism is illustrated in Fig. 10.25. In this figure, the solid lines on either side of the iron ion represent the porphyrin plane (that we will also refer to as L4 in this chapter, as it was also used in the theoretical work by Anderson and Sidik on the reduction of oxygen on L4Fe catalysts [136]; see below) with its four N atoms coordinating the iron ion. It is also proposed, in our case, that the fifth coordinating atom represented by X under the iron ion in L4Fe in Fig. 10.25 is a basic pyridinic nitrogen atom, as illustrated for D3 in Fig. 10.23.

A plausible mechanism for electrochemical reduction of O2 catalyzed by Fe-porphyrins (according to Fig. 12 in ref. [135]; reproduced with the permission of Springer Science + Business Media)
The oxygen reduction reaction starts when the iron ion is in the ferrous state (the L4FeII state indicated by a solid circle in Fig. 10.25). According to the calculations of Anderson and Sidik for oxygen reduction on iron-coordinated N4-chelates [136], L4FeII is most probably hydrated as L4FeII–OH2. The same calculations show that O2, in acid solution, is capable of displacing H2O from L4FeII–OH2 to form the ferric hydroperoxo species according to Eq. (10.3):
L4FeIII–O–OH, which is indicated by a dashed circle in Fig. 10.25, is a branching point, where one option (left arrow) is that the reduction reaction may release H2O and form an oxoferryl cation radical, then an oxoferryl species, then a ferric species, and finally back to the original ferrous species, ready to repeat the cycle. The following equations are involved in this long turnover cycle:
On the other hand (right arrow), L4FeIII–O–OH in Fig. 10.25 may release H2O2 and returning to the starting point, according to
Following the left arrow option therefore leads to the reduction of O2 to H2O with the transfer of 4e− and 4H+. It requires, however, going through the highly oxidized state of FeIV in the catalytic site. On the other hand, following the right arrow leads to the reduction of O2 to H2O2 with the transfer of 2e− and 2H+. For van Veen and his collaborators [137], the ability for the transition metal center to stabilize L4FeIV=O gives these catalysts the possibility, at high applied potential, to reduce oxygen to water by a direct 4e− reaction. According to them, it may explain why, for most Co–N4 chelates, no direct water formation was observed, other than the apparent formation caused by H2O2 decomposition. Anderson and Sidik [136] did not agree with the obliged passage of the Fe-centered site through L4FeIV=O, but instead suggested the replacement of Eqs. (10.3)–(10.6) with Eqs. (10.9)–(10.12):
Here, Eq. (10.9) is the same as Eq. (10.3), ending with the ferric hydroperoxo species as indicated by the dashed circle in Fig. 10.25. Equations (10.10–10.12) describe a reduction reaction path which does not appear on Fig. 10.25, where L4FeII (OH–OH) represents H2O2 bound to the L4FeII catalytic site. According to our experimental results on the oxygen reduction properties in acid medium of Fe/N/C catalysts [116], we have shown that those obtained after high-temperature pyrolysis in NH3 of ironII acetate adsorbed on nonporous carbon black were practically unable to reduce H2O2 electrochemically in H2SO4 solution at pH 1. Therefore, we do not believe that the catalytic sites in Fe/N/C would follow the oxygen reducing path described by Eqs. (10.9)–(10.12), but rather a path described by Eqs. (10.3)–(10.6), which is also illustrated by the turnover along the left arrow in Fig. 10.25. Essentially, the same conclusions about the oxygen reduction path were reached by Chlistunoff for a pyrolyzed Fe/PANI catalyst (where PANI = polyaniline), with the exception that he also claimed that his results indicated that a relatively strong interaction between O2 and a ferric site may be possible, adding another starting point for ORR, besides the already known ferrous site indicated by a solid circle in Fig. 10.25 [138].
Although there are some disagreements between various groups about the exact reaction path during ORR, all groups agree on the fact that the first electron transfer (Eqs. 10.3 or 10.9) is the rate-limiting step in the O2 reduction reaction in acidic medium [136, 137, 139]. As such, the first reduction step of O2 on L4FeII site is responsible for the observed Tafel slopes of about 60 mV/decade (at 25 °C) or about 70 mV/decade (at 80 °C) which are usually observed for O2 reduction on Pt (see for instance Table 3.4 in ref. [21] and also the slope of 61 mV/decade determined over a current range of nearly three decades in Fig. 3 of [8]).
10.6.2 O2 Reduction on N-Doped Carbons
While it may be relatively easy to understand the interaction between O2 (a diradical in its fundamental state) and the 3dz2 orbital of the L4FeII catalytic site (see the Energy Scheme in Fig. 10.21), it is not so easy to understand the interaction mechanism and reaction path followed during the reduction reaction of O2 on N-doped carbons in acid medium. Several calculations were performed by various groups to determine the precise origin of the reduction capabilities and the oxygen reduction mechanism on these N-based catalysts. They will now be reviewed.
In their first quantum calculations of the ORR properties of N-doped graphite [140], Anderson and his collaborators found that carbon radical sites were formed adjacent to substitutional N in graphite and were active for O2 electroreduction to H2O2 via an adsorbed OOH intermediate. They also found that substitutional N atoms that were at a distance from graphite sheet edges (graphitic N of type N3 in Fig. 10.4) led to more ORR activity than those closer to the edge (graphitic N of type N2 in Fig. 10.4). In a subsequent publication [141], they focused on the properties of pyridinic nitrogen atoms at the graphite edge and found that, at potentials greater than 0.3 V (when N atoms were not hydrogenated), they were not active toward ORR because their OOH bonds were too weak. At potentials below 0.3 V, the hydrogenation of these pyridinic N made them radical centers. Because the latter bonded too strongly to OOH, they were not active either.
Later, in studying the properties of two pyridinic N atoms substituting for adjacent edge CH groups (–N–C–N–) [142], Anderson and his colleagues discovered that, at potentials below 1.70 V, one of these N atoms would be in NH form and that the adsorption of possible oxygen intermediates on these edge pyridinic nitrogen atoms would be blocked after an initial reduction leading to O bonded to one N and H bonded to the other one (–NO–C–NH–). However, this edge structure, with its unpaired electron, can bond OOH to the carbon bridging the pyridinic N atoms, with a bond strength that led to a two-electron reduction to H2O2. This overall reaction took place according to the following equations:
If two carbon atoms in a graphene layer were each substituted with a nitrogen atom, the latter would be of the pyridinic type because their substitution for the carbon on the graphene edge is energetically favorable. However, it is more likely to find these pyridinic N atoms far apart from each other because the (–N–C–N–) configuration is not energetically favorable [143]. The latter (–N–C–N–) configuration described by Anderson and his colleagues is therefore unlikely and leaves us once again with pyridinic-type nitrogen without ORR activity. However, Ikeda and his colleagues [144] confirmed that graphitic N of type 2 in Fig. 10.4 are capable of reducing oxygen as long as they are on a zigzag edge of the graphene plane. If the analogous (–C–N–C–) configuration is realized in an armchair edge, O2 adsorption does not give rise to stable structures.
Thus far, all N-doped carbons appear to only be able to reduce oxygen on graphitic-type nitrogen atoms, and this reduction only leads to the production of H2O2. However, experimentally, it is known that metal-free N-doped carbons are capable of reducing O2 to water with an apparent transfer of 4e−, as reported by various research groups covered in this chapter. It is also clear from Fig. 3 in ref. [116], for instance, that similar limiting current densities are reached at the same rotation rate (from 100 to 1,600 rpm) for ORR on a metal-free N-doped carbon, a Fe/N/C or a Pt/C electrocatalyst.
The first report of the reduction of oxygen to water on a graphene layer doped with nitrogen of the pyridinic type was published in 2011 [145]. Here the N-containing graphene sheet is modeled as C45NH20 as shown in Fig. 10.26a. Each atom is numbered and a spin density (upper number) and atomic charge (lower number) were calculated for each atom. All carbon or nitrogen atoms on the edge are terminated by hydrogen atoms. The ORR process was simulated beginning with a first electron transfer. In acidic environment, O2 can adsorb H+ to form H+–O–O [146] and this intermediate can be reduced at the electrode according to Eq. (10.15):

(a) Charge distribution and spin density distribution on the N-graphene with pyridine structure (C45NH20). Each atom is identified by a number. The fractions next to each atom represents the spin density value (numerator) and charge density value (denominator); (b) Spin density distribution on the electron density isovalue plane; the most negative values are in red, the most positive in blue. Spin and charge density values for C37 are (0.235/0.084), N (0.144/−0.690), and C9 (0.194/0.150) (according to Fig. 4 in ref. [145]; reprinted with permission of The American Chemical Society)
In the next step, OOH adsorbs on a carbon near the pyridinic N atom on the graphene sheet. The adsorbed OOH species is represented as *OOH. There are two possible reaction paths described as follows:
Reaction path 1
Reaction path 2
Energetically, it was found that reaction path 1 was more favorable than reaction path 2.
The above reaction does not happen on graphene; only on N-doped graphene. One of the reasons is that HOMO-LUMO gap decreases by a factor of two compared to pure graphene (2.7 eV), after nitrogen atom substitution. Hence, the chemical reactivity of N-doped graphene is significantly improved because the electrons are more easily excited from valence band to conduction band when the band gap is smaller. The other reason is that nitrogen doping introduces an unpaired electron which causes spin and charge densities to appear in the N-doped graphene model as shown in Fig. 10.26a. Figure 10.26b illustrates the change in spin density introduced on individual atoms in the graphene layer. As spin densities are the largest on C37 and C9, these atoms predominantly bond OOH as *OOH in the equations of reaction path 1. Therefore, the occurrence of a large spin density on these carbon atoms transforms them into catalytic sites for ORR.
Another recent work [147] proposes an interesting mechanism for a 4e− reduction of O2 on N-doped graphite of type 2 along the zigzag edge (see Fig. 10.4). This mechanism is illustrated in Fig. 10.27. A notable feature in this mechanism is state III with *OOH on the catalytic site. In this state, due to the higher strength of the C–O vs. the O–O bond, *OOH is easily dissociated into *O and OH−. The remaining *O becoming a carbonyl group, cleaves the C–N bond in step 3a to form a stable CHO group and the broken-bonded nitrogen atom becomes pyridinic in IV. According to Jung and his colleagues [147], this new type of active site that switches between pyridinic and graphitic types of nitrogen atoms may reconcile the experimental controversy as to whether the pyridinic, graphitic, or both types of nitrogen atoms are the ORR-active sites for N-doped graphene materials.

10.7 Conclusions
Despite decades of research on Fe- or Co-based electrocatalysts for the oxygen reduction reaction (ORR) in acidic medium, such as that in PEM fuel cells, the role of the metal is still one that raises a great deal of controversy. Consequently, the nature of the catalytic site in these non-noble metal ORR catalysts is still a topic of debate. One camp within the scientific community believes that the metal is an integral and electrochemically active part of the catalytic site, while the other believes that the metal is merely a chemical catalyst for the formation of special oxygen-reducing N-doped carbon structures.
From this work, we conclude that the three models for the nature of the active ORR sites in non-noble metal catalysts, proposed and advocated by van Veen, Yeager, and Wiesener during the 1980s, all have merit because there appears to be more than a single type of active ORR site in Fe/N/C (or Co/N/C) catalysts in acidic medium. Indeed, FeN4/C active sites, proposed by van Veen and having a D1 Mössbauer signature, are present in Fe/N/C catalysts. Other sites, referred to as N–FeN2 + 2/C, having a D3 Mössbauer signature, are also present in the same catalysts. The latter is derived from C–N x –Me, the model proposed by Yeager for the catalytic site, in which the transition metal ion M (Fe or Co) bonds to the carbon support by coordination through pyridinic nitrogen substituting for carbon in the support.
Our group has shown that C–N x –Fe is only the precursor of the true catalytic site, which is formed after a high-temperature pyrolysis step. We initially referred to this catalytic site as FeN2/C. Complementary experiments relating to the influence of the carbon support on the activity of Fe/N/C catalysts led us to conclude that (1) FeN2/C, whose structure was obviously incomplete, was in fact FeN2 + 2/C and (2) this site is hosted in the slit-shaped micropores of the carbon support. Our most recent Mössbauer experiments revealed yet more details about the exact structure. It appears that FeN2 + 2/C may exist either with or without an axial coordination of the iron ion in the site to a supplementary pyridinic-type nitrogen atom located below the FeN2 + 2/C site. This supplementary coordination pulls the iron ion out of its FeN2 + 2 coordination plane and changes its FeII spin from S = 1 to S = 2, modifying the relative energy level of the 3d orbitals of FeII and the electronic population of its 3dz2 orbital, which binds to O2 in the first step of the oxygen reduction reaction. N–FeN2 + 2/C with its S = 2 spin is ORR active, while FeN2 + 2/C with its S = 1 spin is inactive. Finally, it is also very likely that the CNx sites proposed by Wiesener are also present in Fe/N/C catalysts since their total nitrogen content always significantly outnumbers the active-site-related Fe ion content.
In Fe/N/C catalysts obtained from iron acetate as the iron precursor, we have shown that (1) the catalytic activity follows the relative concentration of N–FeN2 + 2/C-type catalytic sites, as measured by Mössbauer spectroscopy and neutron activation analysis of the Fe content, and (2) the catalytic activity also follows of the relative intensity of the FeN2C4 + ions as measured by ToF-SIMS experiments. From these two observations, and the fact that (3) individual iron atoms can be detected in these catalysts surrounded by nitrogen atoms and (4) the Fe-containing site can be poisoned by KCN in alkaline medium, it was deduced that iron is indeed active in these sites and that among the three types of catalytic sites present in these catalysts (N–FeN2 + 2/C, FeN4/C, and CNx), N–FeN2 + 2/C are the most active (when they are coupled with a neighboring protonated nitrogen atom). Iron-based active sites are also acid resistant.
ToF-SIMS results obtained for catalysts made with ClFeTMPP as the iron precursor in similarly made catalysts show that FeN4/C is the major type of catalytic site present. However, the two other active sites are also present in these catalysts, N–FeN2 + 2/C being here in smaller proportion. The opposite is true when the iron precursor is iron acetate.
As far as CNx sites are concerned, they may indeed be the only type of site in the catalyst when the latter is obtained from a nitrogen precursor and carbon support only, i.e., without involving any strategic metal like Fe or Co in the synthesis procedure. The ORR mass activity of these CNx catalysts is about 50–100 times lower, at 0.8 V vs. RHE in the kinetic region of the ORR current-potential curve, than that of catalysts produced in the presence of a strategic metal like Fe or Co. When at least one of these metals is present during the synthesis, it appears highly probable that the supplementary activity imparted to these catalysts may well be explained by the presence of some Fe–Nx (or Co–Nx) active sites, given that even very small contents of Fe (or Co), i.e., well below 0.1 wt% (1,000 ppm), in the catalysts precursor may significantly increase the catalytic activity. Such small strategic metal contents are often considered negligible by many researcher groups who use several wt% Fe (and/or Co) in their synthesis but deny any active role of the metal in the ORR properties of these catalysts.
Finally, it is relatively easy to understand how iron ions may be involved in every step of the oxygen reduction reaction in acidic medium because its 3dz2 orbital is able to accept one electron of the oxygen diradical molecule. On the other hand, it is not so easy to understand, without performing quantum calculations, how O2 interacts with N-doped carbon. According to the sequence of reactions published in the literature about the activity of CNx catalytic sites, it appears that doping carbon with nitrogen atoms by substitution for carbon in a graphene layer modifies the electronic charge and the spin of each neighboring carbon atom, enabling some of these carbon atoms to interact with O2. However, it is still not completely clear what type of nitrogen atom substitution, i.e., pyridinic or graphitic type, in graphene layers results in the best CNx-based catalysts for ORR.
References
Kerr RA (2011) Peak oil production may already be here. Science 331(6024):1510–1511
Chow J, Kopp RJ, Portney PR (2003) Energy resources and global development. Science 302(5650):1528–1531
Cho A (2010) Energy’s tricky tradeoffs. Science 329(5993):786–787
Olah G, Prakash GKS, Goeppert A (2011) Anthropogenic chemical carbon cycle for a sustainable future. J Am Chem Soc 133:12881–12898
http://www1.eere.energy.gov/hydrogenandfuelcells/mypp/pdfs/fuel_cells.pdf
Gasteiger H, Kocha SS, Sompalli B, Wagner FT (2005) Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl Catal B Environ 56:9–35
Ishihara A, Ohgi Y, Matsuzawa K, Mitsushima S, Ota KI (2010) Progress in non-precious metal oxide-based cathode for polymer electrolyte fuel cells. Electrochim Acta 55:8005–8012
Lefèvre M, Proietti E, Jaouen F, Dodelet JP (2009) Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 324(5923):71–74
Wu G, More KL, Johnston CM, Zelenay P (2011) High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron and cobalt. Science 332(6027):443–447
Proietti E, Jaouen F, Lefèvre M, Larouche N, Tian J, Herranz J, Dodelet JP (2011) Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells. Nat Commun 2:416
Jahnke H, Schönborn M, Zimmermann G (1976) Organic dyestuffs as catalysts for fuel cells. Top Curr Chem 61:133–181
Bagotzky VS, Tarasevich MR, Radyushkina KA, Levina OE, Andrusyova SI (1977) Electrocatalysis of the oxygen reduction process on metal chelates in acid electrolyte. J Power Sources 2:233–240
Bouwkamp-Wijnoltz AL, Visscher W, van Veen JAR, Boellaard E, Van der Kraan AM, Tang SC (2002) On active-site heterogeneity in pyrolyzed carbon supported iron porphyrin catalysts for the electrochemical reduction of oxygen: an in-situ Mössbauer study. J Phys Chem B 106:12993–13001
van Veen JAR, Colijn HA, Van Baar JF (1988) On the effect of heat-treatment on the structure of carbon-supported metalloporphyrins and phthalocyanines. Electrochim Acta 33:801–804
Yeager E (1984) Electrocatalysis for O2 reduction. Electrochim Acta 1984(29):1527–1537
Scherson DA, Tanaka A, Gupta SL, Tryk D, Fierro C, Holze R, Yeager EB (1986) Transition metal macrocycles supported on high area carbon: pyrolysis-mass spectrometry studies. Electrochim Acta 31:1247–1258
Tanaka A, Gupta SL, Tryk D, Fierro C, Yeager EB, Scherson, DA (1992) Electrochemical and spectroscopic aspects of heat-treated transition metal macrocycles as electrocatalysts for oxygen reduction. In: Scherson D, Tryk D, Daroux M, Xing X (eds) Proceedings of the symposium “on structural effects in electrocatalysis and oxygen electrochemistry”, The Electrochemical Society, Pennington, NJ, pp 555–572
Wiesener K (1986) N4-chelates as electrocatalysts for cathodic oxygen reduction. Electrochim Acta 31:1073–1078
Franke R, Ohms D, Wiesener KJ (1989) Investigation of the influence of thermal treatment on the properties of carbon materials modified by N4-chelates for the reduction of oxygen in acidic media. J Electroanal Chem 260:63–73
Gupta S, Tryk D, Bae I, Aldred W, Yeager E (1989) Heat-treated polyacrylonitrile-based catalysts for oxygen electroreduction. J Appl Electrochem 19:19–27
Dodelet JP (2006) Oxygen reduction in PEM fuel cell conditions: heat-treated non-precious metal-N4 macrocycles and beyond. In: Zagal JH, Bedioui F, Dodelet JP (eds) N4-macrocyclic metal complexes. Springer, New York, pp 83–147, Chapter 3
Bouwkamp-Wijnoltz AL, Visscher W, vanVeen JAR, Tang SC (1999) Electrochemical reduction of oxygen: an alternative method to prepare active CON4 catalysts. Electrochim Acta 45:379–386
Gouerec P, Biloul A, Contamin O, Scarbeck G, Savy M, Riga J, Weng LT, Bertrand P (1997) Oxygen reduction in acid media catalyzed by heat-treated cobalt tetraazaannulene supported on an active charcoal: correlation between the performance after longevity tests and the active site configuration as seen by XPS and ToF-SIMS. J Electroanal Chem 422:61–75
Lefèvre M, Dodelet JP, Bertrand P (2000) O2 reduction in PEM fuel cells: activity and active site structural information for catalysts obtained by the pyrolysis at high temperature of Fe precursors. J Phys Chem B 104:11238–11247
Murakami M, Iijima S, Yoshimura S (1986) Morphology and structure of a one dimensional graphite polymer, poly-per-naphthalene. J Appl Phys 60:3856–3863
He P, Lefèvre M, Faubert G, Dodelet JP (1999) Oxygen reduction catalysts for polymer electrolyte fuel cells from the pyrolysis of various transition metal acetates adsorbed on 3,4,9,10-perylenetetracarboxylic dianhydride. J New Mater Electrochem Syst 2:243–251
Faubert G, Côté R, Dodelet JP, Lefèvre M, Bertrand P (1999) Oxygen reduction catalysts for polymer electrolyte fuel cells from the pyrolysis of FeII acetate adsorbed on 3,4,9,10-perylenetetracarboxylic dianhydride. Electrochim Acta 44:2589–2603
Casanovas J, Manelicart J, Rubio J, Illas F, Jiménez Mateos JM (1996) Origin of the large N1s binding energy in X-ray photoelectron spectra of calcined carbonaceous materials. J Am Chem Soc 118:8071–8076
Jiménez-Mateos J, Fierro JLG (1996) X-ray photoelectron spectroscopic study of petroleum fuel cokes. Surf Interface Anal 24:223–236
Lefèvre M, Dodelet JP, Bertrand P (2002) Molecular oxygen reduction in PEM fuel cells: evidence for the simultaneous presence of two active sites in Fe-based catalysts. J Phys Chem B 106:8705–8713
Herranz J, Lefèvre M, Larouche N, Stansfield B, Dodelet JP (2007) Step-by-step synthesis of non-noble electrocatalysts for O2 reduction under proton exchange membrane fuel cell conditions. J Phys Chem C 111:19033–19042
Bron M, Radnik J, Fieber-Herdmann M, Bogdanoff P, Fiechter SJ (2002) EXAFS, XPS and electrochemical studies on oxygen reduction catalysts obtained by heat-treatment of iron phenanthroline complexes supported on high surface area carbon black. J Electroanal Chem 535:113–119
Jaouen F, Marcotte S, Dodelet JP, Lindberg G (2003) Oxygen reduction catalysts for polymer electrolyte fuel cells from the pyrolysis of iron acetate adsorbed on various carbon supports. J Phys Chem B 107:1376–1386
Kinoshita K (1988) Carbon, electrochemical and physicochemical properties. Wiley, New York, pp 3–12
Hess WH, Herd CR (1993) Microstructure, morphology and general physical properties. In: Donnet JB, Bansal RC, Wang MJ (eds) Carbon black, 2nd edn. Dekker, New York, pp 89–173, Chapter 3
Johnson GE, Decker WA, Forney AJ, Field JH (1968) Hydrogen cyanide produced from coal and ammonia. Ind Eng Chem Process Des Dev 7:137–143
Sherwood TK, Gilligand ER, Ing SW (1960) Hydrogen cyanide: synthesis from its elements and from ammonia and carbon. Ind Eng Chem 52:601–604
Griffiths DM, Standing HA (1966) Thermodynamic aspects of the reactions of carbon and coal at high temperatures. In: Given PH (ed) Coal science. American Chemical Society, Washington, DC, pp 666–676, Chapter 42
Jaouen F, Dodelet JP (2007) Non-noble electrocatalysts for O2 reduction: how does heat-treatment affect their activity and structure? Part I. Model for carbon black gasification by NH3: parametric calibration and electrochemical validation. J Phys Chem C 111:5963–5970
Jaouen F, Serventi AM, Lefèvre M, Dodelet JP, Bertrand P (2007) Non-noble electrocatalysts for O2 reduction: how does heat-treatment affect their activity and structure? Part II. Structural changes observed by electron microscopy, Raman, and mass spectroscopy. J Phys Chem C 111:5971–5976
Charreteur F, Jaouen F, Ruggeri S, Dodelet JP (2008) Fe/N/C non-precious catalysts for PEM fuel cells: influence of the structural parameters of pristine commercial carbon blacks on their activity for oxygen reduction. Electrochim Acta 53:2925–2938
Sadezky A, Muckenhuber H, Grothe H, Niessner R, Pöschl U (2005) Raman microscopy of soot and related carbonaceous materials: spectral analysis and structural information. Carbon 43:1731–1742
Katagiri G, Ishida H, Ishitani H (1988) Raman spectra of graphite edge planes. Carbon 26:565–571
Jaouen F, Lefèvre M, Dodelet JP, Cai M (2006) Heat-treated Fe/N/C catalysts for O2 electroreduction: are active sites hosted in micropores? J Phys Chem B 110:5553–5558
Ravikovitch PI, Vishnyakov A, Russo R, Neimark AV (2000) Unified approach to pore size characterization of microporous carbonaceous materials from N2, Ar, and CO2 adsorption isotherms. Langmuir 16:2311–2320
Jaouen F, Herranz J, Lefèvre M, Dodelet JP, Kramm UI, Herrman I, Bogdanoff P, Maruyama J, Nagaoka T, Garsuch A, Dahn J, Olson T, Pylypenko S, Atanassov P, Ustinov EA (2009) Cross-laboratory experimental study of non-noble metal electrocatalysts for the oxygen reduction reaction. Appl Mater Interfaces 1:1623–1639
Yuasa M, Yamaguchi A, Itsuki H, Tanaka K, Yamamoto M, Oyaizu K (2005) Modifying carbon particles with polypyrrole for adsorption of cobalt ions as electrocatalytic site for oxygen reduction. Chem Mater 17:4278–4281
Maruyama J, Abe I (2007) Fuel cell cathode catalyst with Heme-like structure formed from nitrogen of glycine and iron. J Electrochem Soc 154:B297–B304
Bron M, Fiechter S, Bogdanoff P, Tributsch H (2002) Thermogravimetry/spectrometry investigations on the formation of oxygen reduction catalysts for PEM fuel cells on the basis of heat-treated iron phenanthroline complexes. Fuel Cells 2:127–142
Schulenburg H, Stankov S, Schünemann V, Radnik J, Dorbrandt I, Fiechter S, Bogdanoff P, Tributsch HJ (2003) Catalysts for the oxygen reduction from heat-treated iron (III) tetramethoxyphenylporphyrin chloride: structure and stability of active sites. J Phys Chem B 107:9034–9041
Maldonado S, Stevenson KJ (2004) Direct preparation of carbon nanofiber electrodes via pyrolysis of iron(II) phthalocyanine: electrocatalytic aspects of oxygen reduction. J Phys Chem B 108:11375–11383
Maldonado S, Stevenson KJ (2005) Influence of nitrogen doping on oxygen reduction electrocatalysis at carbon nanofiber electrodes. J Phys Chem B 109:4707–4716
Maldonado S, Morin S, Stevenson KJ (2006) Structure, composition, and chemical reactivity of carbon nanotubes by selective nitrogen doping. Carbon 44:1429–1437
Lyon JL, Stevenson KJ (2007) Anomalous electrochemical dissolution and passivation of iron growth catalysts in carbon nanotubes. Langmuir 23:11311–11318
Wiggins-Camacho JD, Stevenson KJ (2009) Effect of nitrogen concentration on capacitance, density of states, electronic, conductivity, and morphology of N-doped carbon nanotube electrodes. J Phys Chem C 113:19082–19090
Wiggins-Camacho JD, Stevenson KJ (2011) Indirect electrocatalytic degradation of cyanide at nitrogen-doped carbon nanotube electrodes. Environ Sci Technol 45:3650–3656
Ramaswamy N, Mukerjee S (2011) Influence of inner-and outer-sphere electron transfer mechanisms during electrocatalysis of oxygen reduction in alkaline media. J Phys Chem C 115:18015–18026
Gara M, Compton RG (2011) Activity of carbon electrodes towards oxygen reduction in acid: a comparative study. New J Chem 35:2647–2652
Marcotte S, Villers S, Guillet N, Roué L, Dodelet JP (2004) Electroreduction of oxygen on Co-based catalysts: determination of the parameters affecting the two-electron transfer reaction in an acid medium. Electrochim Acta 50:179–188
Meng H, Jaouen F, Proietti E, Lefèvre M, Dodelet JP (2009) pH effect on oxygen reduction activity of Fe-based electrocatalysts. Electrochem Commun 11:1986–1989
Herranz J, Jaouen F, Dodelet JP (2009) Electrochemical evidence of two types of active sites for oxygen reduction in Fe-based catalysts. ECS Trans 25:117–128
Matter PH, Zhang L, Ozkan U (2006) The role of nanostructure in nitrogen-containing carbon catalysts for the oxygen reduction reaction. J Catal 239:83–96
Matter PH, Ozkan U (2006) Non-metal catalysts for dioxygen reduction in an acid electrolyte. Catal Lett 109:115–123
Matter PH, Wang E, Arias M, Biddinger EJ, Ozkan U (2006) Oxygen reduction reaction catalysts prepared from acetonitrile pyrolysis over alumina-supported metal particles. J Phys Chem B 110:18374–18384
Matter PH, Wang E, Ozkan U (2006) Preparation of nanostructured nitrogen-containing carbon catalysts for the oxygen reduction reaction from SiO2- and MgO-supported metal particles. J Catal 243:395–403
Matter PH, Wang E, Arias M, Biddinger EJ, Ozkan U (2007) Oxygen reduction reaction activity and surface properties of nanostructures nitrogen-containing carbon. J Mol Catal A Chem 264:73–81
Matter PH, Wang E, Millet JMM, Ozkan U (2007) Characterization of the iron phase in CNx-based oxygen reduction reaction catalysts. J Phys Chem C 111:1444–1450
Biddinger E, Ozkan U (2007) Methanol tolerance of CNx oxygen reduction catalysts. Top Catal 46:339–348
Biddinger E, von Deak D, Ozkan US (2009) Nitrogen-containing carbon nanostructures as oxygen reduction catalysts. Top Catal 52:1566–1574
Biddinger EJ, Knapke DS, von Deak D, Ozkan US (2010) Effect of sulfur as a growth promoter for CNx nanostructures as PEM and DMFC ORR catalysts. Appl Catal B Environ 96:72–82
Woods MP, Biddinger EJ, Matter PH, Mirkelamoglu B, Ozkan US (2010) Correlation between oxygen reduction reaction and oxidative dehydrogenation activities over nanostructures carbon catalysts. Catal Lett 136:1–8
von Deak D, Biddinger EJ, Luthman KA, Ozkan US (2010) The effect of phosphorus in nitrogen-containing carbon nanostructures on oxygen reduction in PEM fuel cells. Carbon 48:3635–3658
Biddinger EJ, Ozkan US (2010) Role of graphitic edge plane exposure in carbon nanostructures for oxygen reduction reaction. J Phys Chem C 114:15306–15314
Bao X, von Deak D, Biddinger EJ, Ozkan US, Haddad CM (2010) A computational exploration of the oxygen reduction reaction over a carbon catalyst containing a phosphinate functional group. Chem Commun 46:8621–8623
von Deak D, Biddinger EJ, Ozkan U (2011) Carbon corrosion characteristics of CNx nanostructures in acidic media and implications for ORR performance. J Appl Electrochem 41:757–763
Biddinger EJ, von Deak D, Singh D, Marsh H, Tan B, Knapke DS, Ozkan US (2011) Examination of catalyst loading effect on the selectivity of CNx and Pt/VC ORR catalysts using RRDE. J Electrochem Soc 158:B402–B409
von Deak D, Singh D, Biddinger EJ, King JC, Bayram B, Miller JT, Ozkan US (2012) Investigation of sulfur poisoning of CNx oxygen reduction catalysts for PEM fuel cells. J Catal 285:145–151
Birry L, Zagal JH, Dodelet JP (2010) Does CO poison Fe-based catalysts for ORR? Electrochem Commun 12:628–631
Nallathambi V, Wu G, Subramanian NP, Kumaraguru SP, Lee JW, Popov BN (2007) Highly active carbon composite electrocatalysts for PEM fuel cells. ECS Trans 11:241–247
Li X, Liu L, Lee JW, Popov BN (2008) Development of tellurium-modified carbon catalysts for oxygen reduction reaction in PEM fuel cells. J Power Sources 182:18–23
Nallathambi V, Li X, Lee JW, Popov BN (2008) Development of nitrogen-modified carbon-based catalysts for oxygen reduction in PEM fuel cells. ECS Trans 16:405–417
Subramanian NP, Li X, Nallathambi V, Kumaraguru SP, Mercado HC, Wu G, Lee JW, Popov BN (2009) Nitrogen-modified carbon-based catalysts for oxygen reduction reaction in polymer electrolyte membrane fuel cells. J Power Sources 188:38–44
Nallathambi V, Lee JW, Kumaraguru SP, Wu G, Popov BN (2008) Development of high performance carbon composite catalysts for oxygen reduction in PEM proton exchange membrane fuel cells. J Power Sources 183:34–42
Liu G, Li X, Popov BN (2009) Stability study of nitrogen-modified carbon composite catalysts for oxygen reduction reaction in polymer electrolyte membrane fuel cells. ECS Trans 25:1251–1259
Liu G, Li X, Ganesan P, Popov BN (2009) Development of non-precious metal oxygen-reduction catalysts for PEM fuel cells based on N-doped ordered porous carbon. Appl Catal B Environ 93:156–165
Jaouen F, Dodelet JP (2007) Average turn-over frequency of O2 electro-reduction for Fe/N/C and Co/N/C catalysts in PEFCs. Electrochim Acta 52:5975–5984
Liu G, Li X, Ganesan P, Popov BN (2010) Studies of oxygen reduction reaction active sites and stability of nitrogen-modified carbon composite catalysts for PEM fuel cells. Electrochim Acta 55:2853–2858
Liu G, Li X, Lee JW, Popov BN (2011) A review of the development of nitrogen-modified carbon catalysts for oxygen reduction at USC. Catal Sci Technol 1:207–217
Li G, Liu G, Popov BN (2010) Activity and stability of non-precious metal catalysts for oxygen reduction in acid and alkaline electrolytes. J Power Sources 195:6373–6378
Ozaki JI, Tanifuji SI, Kimura N, Furuichi A, Oya A (2006) Enhancement of oxygen reduction activity by carbonization of furan resin in the presence of phthalocyanines. Carbon 44:1298–1352
Ozaki JI, Kimura N, Tomonori A, Oya A (2007) Preparation and oxygen reduction activity of BN-doped carbons. Carbon 45:1847–1853
Niwa H, Horiba K, Harada Y, Oshima M, Ikeda T, Terakura K, Ozaki JI, Miyata S (2009) X-ray absorption analysis of nitrogen contribution to oxygen reduction reaction in carbon alloy cathode catalysts for polymer electrolyte fuel cells. J Power Sources 187:93–97
Chokai M, Taniguchi M, Moriya S, Matsubayashi K, Shinoda T, Nabae Y, Kuroki S, Hayakawa T, Kakimoto MA, Ozaki JI, Miyata S (2010) Preparation of carbon alloy catalysts for polymer electrolyte fuel cell from nitrogen-containing rigid-rod polymers. J Power Sources 195:5947–5951
Kobayashi R, Ozaki JI (2009) Novel N-doped carbon cathode catalyst for polymer electrolyte membrane fuel cells formed on carbon black. Chem Lett 38:396–397
Ozaki JI, Tanifuji SI, Furuichi A, Yabutsuka K (2010) Enhancement of oxygen reduction activity of nanoshell carbons by introducing nitrogen atoms from metal phthalocyanines. Electrochim Acta 55:1864–1871
Niwa H, Kobayashi M, Horiba K, Harada Y, Oshima M, Terakura K, Ikeda T, Koshigoe Y, Ozaki JI, Miyata S, Ueda S, Yamashita Y, Yoshikawa H, Kobayashi K (2011) X-ray photoemission spectroscopy analysis of N-containing carbon-based cathode catalysts for polymer electrolyte fuel cells. J Power Sources 196:1006–1011
Kobayashi M, Niwa H, Harada Y, Horiba K, Oshima M, Ofuchi H, Terakura K, Ikeda T, Koshigoe Y, Ozaki JI, Miyata S, Ueda S, Yamashita Y, Yosikawa H, Kobayashi K (2011) Role of residual transition metal atoms in oxygen reduction reaction in cobalt phthalocyanine-based carbon cathode catalysts for polymer electrolyte fuel cell. J Power Sources 196:8346–8351
Wang X, Hou Z, Ikeda T, Huang SF, Terakura K, Boero M, Oshima M, Kakimoto MA, Miyata S (2011) Selective nitrogen doping in graphene: enhanced catalytic activity for the oxygen reduction reaction. Phys Rev B 84:245434-1–245434-7
Nabae Y, Moriya S, Matsubayashi K, Lyth SM, Malon M, Wu L, Islam NM, Koshigoe Y, Kuroki S, Kakimoto MA, Miyata S, Ozaki JI (2010) The role of Fe species in the pyrolysis of Fe phthalocyanine and phenolic resin for preparation of carbon-based cathode catalysts. Carbon 48:2613–2624
Wu L, Nabae Y, Moriya S, Matsubayashi K, Islam NM, Kuroki S, Kakimoto MA, Ozaki JI, Miyata S (2010) Pt-free cathode catalysts prepared via multi-step pyrolysis of Fe phthalocyanine and phenolic resin for fuel cells. Chem Commun 46:6377–6379
Lyth SM, Nabae Y, Moriya S, Kuroki S, Kakimoto MA, Ozaki JI, Miyata S (2009) Carbon nitride as a nonprecious catalyst for electrochemical oxygen reduction. J Phys Chem C 113:20148–20151
Lyth SM, Nabae Y, Islam NM, Kuroki S, Kakimoto M, Miyata S (2011) Electrochemical oxygen reduction activity of carbon nitride supported on carbon black. J Electrochem Soc 158:B194–B201
Iwazaki T, Obinata R, Sugimoto W, Takasu Y (2009) High oxygen reduction activity of silk derived activated carbon. Electrochem Commun 11:376–378
Iwasaki T, Yang H, Obinata R, Sugimoto W, Takasu Y (2010) Oxygen-reduction activity of silk-derived carbons. J Power Sources 195:5840–5847
Rao CV, Cabrera CR, Ishikawa Y (2010) In search of the active site in nitrogen-doped carbon nanotube electrodes for the oxygen reduction reaction. J Phys Chem Lett 1:2622–2627
Yu D, Zhang Q, Dai L (2010) Highly efficient metal-free growth of nitrogen-doped single-walled carbon nanotubes on plasma-etched substrates for oxygen reduction. J Am Chem Soc 132:15127–15129
Shrestha S, Mustain WE (2010) Properties of nitrogen-functionalized ordered mesoporous carbon prepared using polypyrrole precursor. J Electrochem Soc 157:B1665–B1672
Wang X, Lee JS, Zhu Q, Liu J, Wang Y, Dai S (2010) Ammonia-treated ordered mesoporous carbons as catalytic materials for oxygen reduction reaction. Chem Mater 22:2178–2180
Kundu S, Nagaiah TC, Xia W, Wang Y, Van Dommele S, Bitter JH, Santa M, Grundmeier G, Bron M, Schuhmann W, Muhler M (2009) Electrocatalytic activity and stability of nitrogen-containing nanotubes in the oxygen reduction reaction. J Phys Chem C 113:14302–14310
Xiong W, Du F, Liu Y, Perez A, Supp M, Ramakrishnan S, Dai L, Jiang L (2010) 3-D carbon nanotube structures used as high performance catalyst for oxygen reduction reaction. J Am Chem Soc 132:15839–15841
Choi CH, Lee SY, Park SH, Woo SI (2011) Highly active N-doped-CNT grafted on Fe/C prepared by pyrolysis of dicyandiamide on Fe2O3/C for electrochemical oxygen reduction reaction. Appl Catal B Environ 103:362–368
Geng D, Liu H, Chen Y, Li R, Sun X, Ye S, Knights S (2011) Non-noble metal oxygen reduction electrocatalysts based on carbon nanotubes with controlled nitrogen contents. J Power Sources 196:1795–1801
Oh HS, Oh JG, Lee WH, Kim HJ, Kim H (2011) The influence of the structural properties of carbon on the oxygen reduction reaction of nitrogen modified carbon based catalysts. Int J Hydrogen Energy 36:8181–8186
Oh HS, Oh JG, Roh B, Hwang I, Kim H (2011) Development of highly active and stable non-precious oxygen reduction catalysts for PEM fuel cells using polypyrrole and chelating agent. Electrochem Commun 13:879–881
Jin H, Zhang H, Zhong H, Zhang J (2011) Nitrogen-doped carbon xerogel: a novel carbon-based electrocatalyst for oxygen reduction reaction in proton exchange membrane (PEM) fuel cells. Energy Environ Sci 4:3389–3394
Jaouen F, Dodelet JP (2009) O2 reduction mechanism on non-noble metal catalysts for PEM fuel cells. Part I: Experimental rates for O2 electroreduction, H2O2 electroreduction, and H2O2 disproportionation. J Phys Chem C 113:15422–15432
Kramm U, Herranz J, Larouche N, Arruda TM, Lefèvre M, Jaouen F, Bogdanoff P, Fiechter S, Abs-Wurmbach I, Mukerjee S, Dodelet JP (2012) Structure of the catalytic sites in Fe/N/C-catalysts for O2-reduction in PEM fuel cells. Phys Chem Chem Phys 14:11673–11688
Walker FA, Simonis U (2006) Iron porphyrin chemistry. In: Scott RA (ed) Encyclopedia of inorganic chemistry. Wiley Online Library. doi: 10.1002/0470862106.ia111
Zagal JH, Paez MA, Silva JF (2006) Fundamental aspects on the catalytic activity of metallomacrocyclics for the electrochemical reduction of O2. In: Zagal JH, Bedioui F, Dodelet JP (eds) N4-macrocyclic metal complexes. Springer, New York, pp 41–82, Chapter 2
Koslowski UI, Abs-Wurmbach I, Fiechter S, Bogdanoff P (2008) Nature of the catalytic centers of porphyrin-based electrocatalysts for ORR: a correlation of kinetic current density with the site density of Fe-N4 centers. J Phys Chem C 112:15356–15366
Kramm UI, Abs-Wurmbach I, Herrmann-Geppert I, Radnik J, Fiechter S, Bogdanoff P (2011) Influence of the electron-density of FeN4-centers towards the catalytic activity of pyrolyzed FeTMPPCl-based ORR-electrocatalysts. J Electrochem Soc 158:B69–B78
Melendres CA (1980) Mössbauer and Raman spectra of carbon-supported iron phthalocyanine. J Phys Chem 84:1936–1939
Kuzmann E, Nath A, Chechersky V, Li S, Wei Y, Chen X, Li J, Homonnay Z, Gal M, Garg VK, Klencsar Z, Vertes A (2002) Mössbauer study of oxygenated iron-phthalocyanines, a precursor of magnetic storage material. Hyperfine Interact 139/140:631–639
Schulz CE, Hu C, Scheidt WR (2006) On spin Hamiltonian fits for Mössbauer spectra of high-spin Fe(II) porphyrinate systems. Hyperfine Interact 170:55–60
Sams JR, Tsin TB (1979) Mössbauer spectroscopy of iron porphyrins. In: Dolphin D (ed) The porphyrins. Volume IV. Physical chemistry, part B. Academic, New York, pp 425–478, Chapter 9
Collman JP, Gagne R, Reed CA, Halbert TR, Lang G, Robinson WT (1975) “Picket fence porphyrins” synthetic model for oxygen binding hemoproteins. J Am Chem Soc 97:1427–1439
Collman JP, Hoard JL, Kim N, Lang G, Reed CA (1975) Synthesis, stereochemistry, and structure-related properties of α, β, γ, δ-tetraphenylporphinatoiron (II). J Am Chem Soc 97:2676–2681
Herranz J, Jaouen F, Lefèvre M, Kramm UI, Proietti E, Dodelet JP, Bogdanoff P, Fiechter S, Abs-Wurmbach I, Bertrand P, Arruda TM, Mukerjee S (2011) Unveiling N-protonation and anion-binding effects on Fe/N/C catalysts for O2 reduction in proton exchange-membrane fuel cells. J Phys Chem C 115:16087–16097
Li Y, Zhou W, Wang H, Xie L, Liang Y, Wei F, Idrobo JC, Pennycook J, Dai H (2012) An oxygen reduction electrocatalyst based on carbon nanotube-graphene complexes. Nat Nanotechnol 7:394–400
Thorum M, Hankett JM, Gewirth AA (2011) Poisoning the oxygen reduction reaction on carbon-supported Fe and Cu electrocatalysts: evidence for metal-centered activity. J Phys Chem Lett 2:295–298
Gupta S, Fierro C, Yeager E (1991) The effect of cyanide on the electrochemical properties of transition metal macrocycles for oxygen reduction in alkaline solutions. J Electroanal Chem 306:239–250
Burgess J, Twigg MV (2006) Iron: inorganic & coordination chemistry. In: King RB (ed) Encyclopedia of inorganic chemistry. Wiley, New York
Li W, Yu A, Higgins DC, Llanos BG, Chen Z (2010) Biologically inspired highly durable iron phthalocyanine catalysts for oxygen reduction reaction in polymer electrolyte membrane fuel cells. J Am Chem Soc 132:17056–17058
Waldron K, Rutherford JC, Ford D, Robinson NJ (2009) Metalloproteins and metal sensing. Nature 460:823–830
Boulatov R (2006) Billion-year old oxygen cathode that actually works: respiratory oxygen reduction and its biomimetic analogs. In: Zagal JH, Bedioui F, Dodelet JP (eds) N4-macrocyclic metal complexes. Springer, New York, pp 41–82, Chapter 1
Anderson AB, Sidik RA (2004) Oxygen reduction on FeII and FeIII coordinated to N4 chelates. Reversible potentials for the intermediate steps from quantum theory. J Phys Chem B 108:5031–5035
Bouwkamp-Wijnoltz AL, Visscher W, Van Veen JAR (1998) The selectivity of oxygen reduction by pyrolyzed iron porphyrin supported on carbon. Electrochim Acta 43:3141–3152
Chlistunoff J (2011) RRDE and voltammetric study of ORR on pyrolyzed Fe/polyaniline catalysts. On the origin of variable Tafel slopes. J Phys Chem C 115:6496–6507
Van Veen JAR, Van Baar JF, Kroese CJ, Coolegem JGF, De Wit N, Colijn HA (1981) Oxygen reduction on transition metal porphyrins in acid electrolyte: I. Activity. Ber Bunsenges Phys Chem 85:693–700
Sidik RA, Anderson AB, Subramanian NP, Kumaraguru SP, Popov BN (2006) O2 reduction on graphite and nitrogen-doped graphite: experiment and theory. J Phys Chem B 110:1787–1793
Vayner E, Anderson AB (2007) Theoretical predictions concerning oxygen reduction in nitride graphite edges and a cobalt center bonded to them. J Phys Chem C 111:9330–9336
Kurak KA, Anderson AB (2009) Nitrogen-treated graphite and oxygen reduction on pyridinic edge sites. J Phys Chem C 113:6730–6734
Huang SF, Terakura K, Ozaki T, Ikeda T, Boero M, Oshima M, Ozaki JI, Miyata S (2009) First-principles calculations of the electronic properties of graphene clusters doped with nitrogen and boron: analysis of catalytic activity for the oxygen reduction reaction. Phys Rev B 80:235410-1–235410-4
Ikeda T, Boero M, Huang SF, Terakura K, Oshima M, Ozaki JI (2008) Carbon alloy catalysts: active sites for oxygen reduction reaction. J Phys Chem C 112:14706–14709
Zhang L, Xia Z (2011) Mechanism of oxygen reduction reaction on nitrogen-doped graphene for fuel cells. J Phys Chem C 115:11170–11176
Wang Y, Balbuena PB (2005) Ab initio molecular dynamics simulations of the oxygen reduction reaction on a Pt(111) surface in the presence of hydrated hydronium (H3O)+(H2O)2: direct or series pathway? J Phys Chem B 109:14896–14907
Kim H, Lee K, Woo SI, Jung Y (2011) On the mechanism of enhanced oxygen reduction reaction in nitrogen-doped graphene nanoribbons. Phys Chem Chem Phys 13:17505–17510
Acknowledgments
I am very grateful to NSERC, General Motors of Canada, and MDEIE (Gouvernement du Québec) for providing research funds in support of my work on non-noble metal catalysts for ORR in PEM fuel cells at INRS for more than 20 years. I would like to thank the numerous students, postdoctoral fellows, and research associates that have worked in my group in this field. In particular, I wish to thank Eric Proietti and Regis Chenitz for their help in the editing and final presentation of this chapter as well as Ulrike Kramm and Frédéric Jaouen for providing comments.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag London
About this chapter
Cite this chapter
Dodelet, JP. (2013). The Controversial Role of the Metal in Fe- or Co-Based Electrocatalysts for the Oxygen Reduction Reaction in Acid Medium. In: Shao, M. (eds) Electrocatalysis in Fuel Cells. Lecture Notes in Energy, vol 9. Springer, London. https://doi.org/10.1007/978-1-4471-4911-8_10
Download citation
DOI: https://doi.org/10.1007/978-1-4471-4911-8_10
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-4910-1
Online ISBN: 978-1-4471-4911-8
eBook Packages: EnergyEnergy (R0)