
Abstract
Hemochromatosis is characterized by iron overload secondary to mutations in the hemochromatosis gene (HFE), which codes HFE. The current hemochromatosis classification is type 1 HFE-hemochromatosis (HFE-related), type 2 Hemochromatosis (non-HFE-related), and type 3 mutated transferrin receptor. The most common HFE mutations are p.C282Y, p.H63D, and p.S65C. Most patients with hemochromatosis are homozygous for C282Y.
Iron homeostasis requires a normal function of hepcidin, a hormone made in the liver, and a normal functioning HFE. The interaction between hepcidin and ferroportin (FPN) regulates the amount of circulating iron.
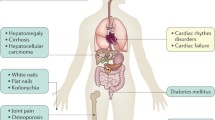
The classical manifestations of hemochromatosis are cirrhosis, and its complications, including hepatocellular carcinoma, hyperpigmentation, and diabetes. Men are more frequently affected by progressive iron overload than women.
Patients with an abnormal liver profile in association with either transferrin saturation of at least 45% or high serum ferritin, or both, should have hemochromatosis genetic testing.
Iron produces liver injury because of its association with hepatic comorbidities and other toxic elements that stimulate liver fibrosis. The treatment of iron overload is the removal of iron by phlebotomy. Patients homozygous for C282Y with serum ferritin greater than 300 ng/ml for men, and more than 200 ng/mL in women and a transferrin saturation equal or greater than 45% are recommended to have phlebotomy aimed at serum ferritin of 50 to 100 micrograms/L, and hemoglobin between 11-12 g/dl.
Liver transplantation is associated with normalization of hepcidin and absence of iron accumulation in the graft, and it is an option for patients with decompensated liver disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Hepcidin
- Ferroportin
- Hemochromatosis
- Nonhemochromatosis mutations
- Iron
- Hepatocellular carcinoma
- Arthropathy
- Alcohol
- Hemochromatosis gene mutations
- hyperpigmentation
A 26-year-old man was admitted to Behavioral Health for detoxification from heroin, which he had been using for 5 years. He had a history of polysubstance abuse since age fifteen. His physical exam was normal. His hepatic panel was notable for high serum activity of aspartate (AST) and alanine (ALT) amino transaminases, 128 IU/L (normal range: 10–40) and 437 IU/L (normal range: 10–45) IU/L, respectively, and alkaline phosphatase (AP) of 180 IU/L (normal range: 40–120), and normal serum concentration of bilirubin and albumin, and normal prothrombin time. The liver sonogram revealed hepatomegaly. Liver disease workup was notable for the presence of hepatitis C RNA, consistently documented, suggestive of chronic hepatitis C virus infection, a serum ferritin concentration of 4545 ng/ml (normal range 15–150) with a transferrin saturation of 96%. Hemochromatosis gene testing revealed two copies of C282Y consistent with primary hemochromatosis. The patient was referred to genetics and to the hepatology clinic to treat chronic hepatitis C and iron overload.
Hemochromatosis is a disease characterized by iron overload [1]. It results from mutations in the hemochromatosis gene (HFE), which codes HFE. There are non-HFE mutations that result in iron overload [1]. Hemochromatosis has been classified according to the gene where the mutation has been identified [1] (Table 11.1).
Hepcidin, a hormone made in the liver, controls iron absorption, which requires a normal functioning HFE [2]. The interaction between hepcidin and ferroportin (FPN) regulates the amount of iron in the bloodstream [3,4,5]. The development of progressive iron overload is more common in men than in women. It appears to be influenced by other genetic factors, the environment, blood loss, and alcohol consumption.
Iron Homeostasis
Iron is an essential element in human beings. Iron participates in energy generation reactions within the cytochrome p450 enzymatic group; when iron is complexed with protoporphyrins it forms heme, the major oxygen-binding molecule. Iron is transported in the circulation by transferrin, by haptoglobin in association with hemoglobin, and is stored in ferritin. Ferritin is a spherical protein comprised of 24 subunits of heavy or light types. The ferritin concentration reflects iron stores, although it is also an acute phase reactant. Ferrous iron is delivered to ferritin by Poly (rC)-binding protein 1 (PCBP1), a cytosolic iron chaperone, to be stored [6]; iron is released from ferritin, as documented in vitro, by lysosomal proteolysis [7].
Iron’s only source to human beings is the diet. Dietary non-heme iron is absorbed through the apical membrane of the enterocyte in the ferrous form (Fe ++), which originates from the reduction of ferric iron (Fe+++) by duodenal cytochrome b reductase 1 [8]; other ferric reductases may also be involved. Ferrous iron enters from the intestinal lumen through the brush border into the cytoplasm of the enterocyte through the divalent metal ion transporter-1 (DMT1) [9], whereas heme iron (i.e., from meat), which is more easily absorbed than ferrous iron, enters the enterocyte via the heme carrier protein-1 (HCP-1), which was subsequently identified as the folate transporter [10], formally named solute carrier family 46 (SLC46A1). Ferrous iron is transported across the basolateral membrane of the enterocyte by FPN [5]; it is oxidized to ferric iron by hephaestin, a multi-copper oxidase located in the basolateral membrane, and binds to transferrin in the blood [11]. Alternatively, in the enterocyte, iron is incorporated into ferritin at the conversion of ferrous into ferric iron by the ferroxidase activity of the H chain of ferritin. Regardless of the iron’s source entering the enterocyte, the exit to the blood is via the same route, i.e., FPN [3,4,5].
FPN is the only known exporter of iron [3,4,5]. It is expressed on the basolateral surface of the duodenal enterocytes [5, 12], membranes of macrophages [12], the sinusoidal surface of hepatocytes [13], and the placental surface facing the fetal circulation [4]. FPN is encoded by two tissue-specific differentially spliced transcripts, FPN1A and FPN1B, the latter being strongly expressed in the duodenum and erythroid cell precursors, and which differ between each other in the presence of 5’IRE (iron response element) in the former, and its absence in the latter, not being repressed in iron deficiency states [14].
The export of iron depends on copper-containing ferroxidases, including ceruloplasmin, hephaestin, and possibly zyklopen, which oxidize ferrous iron by the use of molecular oxygen [11, 15, 16]. Ceruloplasmin deficiency impairs iron absorption and its release from macrophages and is associated with liver and brain iron accumulation [15, 17]. The role of ferroxidases in the export of iron is proposed to be the facilitation of efflux of reduced iron by oxidizing it to its ferric form, thus allowing its uptake by apotransferrin and decreasing the concentration of ferrous iron on the cell surface, and maintaining a ferrous iron gradient to the extracellular face of FPN, which drives iron transport [18].
Iron travels bound to transferrin and is taken up by almost all cells by the transferrin receptor 1 (TFR1), to which diferric transferrin binds, being internalized by endosomes, where iron is released by endosomal acidification. Iron regulatory proteins 1 and 2 (IRP1 and IRP2) are cytoplasmic proteins that play a role in iron homeostasis by regulating ferritin translation and TfR1 mRNA stability, thus regulating the receptor expression central for iron uptake into the cell [19]. Ferric iron is reduced by STEASP3 and transported through the endosomal membrane by DMT1 [20]. In this form, iron is utilized by the cells or stored in ferritin. Apotransferrin (i.e., transferrin without iron) bound to TFR1 is recycled from the endosome to the surface of the cells where it is released from the bond at a neutral pH, making the receptor available to bind additional holotransferrin (i.e., diferric transferrin) [21]. The hepatocytes also express transferrin receptor 2 (TFR2); however, its affinity for holotransferrin is lower than that for TfR1, and is reported not to play a major role in the uptake of iron bound to transferrin, according to animal experiments [22].
Iron also circulates in relatively small amounts bound to proteins other than transferrin, i.e., nontransferrin bound iron (NTBI); however, when transferrin saturation increases, this type of iron also increases, being of pathological importance in iron overload conditions like hemochromatosis. NTBI is toxic and cleared by the hepatocytes by the transporters DMT1 [23] and Zrt-, Irt-like protein 14 (Zip14) [24].
Macrophages phagocytize senescent erythrocytes, digesting them and their hemoglobin, and releasing heme, which is subsequently degraded by the inducible oxygenase-1 (HO-1), making iron available for cytoplasmic storage, or export to plasma, also by the same route that iron leaves the enterocyte, i.e., via FPN. During hemolysis, heme may be exported in intact form by heme transporters bound to hemopexin, a plasma heme carrier, as an alternative pathway [18].
Hepcidin, a protein made in the liver, controls iron homeostasis in human beings [2]. The iron not transferred to plasma is stored in macrophages, hepatocytes, and possibly placenta. The iron not transferred from enterocytes to the plasma is lost by the sloughing intestinal epithelial cells every few days. In addition, iron is lost during menstruation.
Hepcidin
The concentration of iron in human beings tends to be constant. It was reported from a study from the United States that in men, the distribution of iron stores is 9.82 ± 2.82 mg/kg and 5.5 ± 3.35 mg/kg in 93% of women, with the remaining 7% being deficient by 3.87 ± 3.23 mg/kg [25]. In iron deficiency, iron absorption increases, and in association with parenteral iron administration, it decreases [26]. It is the hormone hepcidin that controls iron homeostasis [2]. Hepcidin, encoded by HAMP in chromosome 19, is a peptide hormone produced by the hepatocytes. It is a negative regulator of iron absorption that exerts its function by binding to FPN, encoded by FPN1 (SLC40A1). The binding of hepcidin to FPN leads to the internalization of the latter, with its final degradation, limiting the absorption of iron by the enterocyte, and its recycling by the macrophages to maintain a homeostatic transferrin saturation [27]. Low concentrations of hepcidin are associated with increased iron absorption versus high hepcidin concentrations, which are associated with low iron absorption [2].
Hepcidin is generated from an 84-amino acid prepropeptide that contains an NH2-terminal 24-amino acid signal sequence cleaved to yield prohepcidin, which is cleaved by furin-like prohormone convertases at the COOH-terminal peptide bond after a polybasic sequence [28]. It circulates in plasma mostly free, with minimal binding to albumin and alpha 2-macroglobulin [29, 30]. Up to the residue nine of the terminal amino acids of hepcidin, including a single thiol cysteine, is required for its function [28, 31, 32], i.e., control of the expression of FPN, the iron exporter, by mediating its endocytosis and proteolysis by lysosomes, for which ubiquitination of lysines in the cytoplasmic loop of FPN may be required [33], and thus regulating the amount of iron that is delivered to plasma and all iron consuming tissues [27]. In regard to FPN, the extracellular FPN residue, C326, is essential for hepcidin binding [31]; in addition, some aromatic side chains, including phenylalanine and tyrosine, in the interacting segments of hepcidin and FPN, have been identified experimentally as important elements for their interaction [32].
The regulation of hepcidin is transcriptional and the major regulators of hepcidin synthesis are iron bound to transferrin (iron-transferrin), iron stores, inflammation, and erythroid activity [34]. It is reported that bone morphogenic proteins (BMP) receptors [35], likely Alk2 or Alk3 [36], as type I subunits, and ActRIIA, as type II subunits [37], and the SMAD pathway are pivotal in the regulation of hepcidin, the promoter of which has several BMP response elements. Hemojuvelin (HJV), a protein linked to glycophosphatidylinositol, is a coreceptor of BMP required for its activation in the context of iron homeostasis [38].
Extracellular iron sensors seem to be involved in iron homeostasis; however, the mechanisms have not been completely elucidated and may include transferrin receptor types 1 and 2 (TfR1 and TfR2), holotransferrin, and membrane serine protease mariptase-2 (TMPRSS6): (i) the disruption of transferrin receptor type 2 (TfR2) is associated with a loss of extracellular iron sensing [39, 40], (ii) holotransferrin and the protein involved in hemochromatosis, HFE, compete for binding with the TfR1, which has been interpreted to suggest that the HFE-TfR1 complex may be another sensor for holotransferrin, independent from TfR2, or via its interaction with TfR2 and with HJV [41], and (iii): TMPRSS6 cleaves BMP, an HJV agonist, thus inactivating the hepcidin-BMP signaling.
In regard to the effect of iron stores on hepcidin expression, it was documented that TfR2, HJV, BMP6, and HFE are involved in the response of hepcidin to acute iron overload but not in chronic iron overloading or BMP-6 expression [42]. Conditions that also regulate hepcidin include erythropoiesis [26], inflammation [43], and liver injury [44,45,46].
In high iron concentrations, the highly saturated transferrin stabilizes TfR2, disrupting the interaction between the HFE protein and TfR1 leading to increased BMP-6 secretion from nonparenchymal liver cells, which facilitates the formation of a complex comprised of BMP receptor/BMP6/HJV /neogenin/TfR2/HFE that induces the expression of hepcidin [47,48,49]. An increase in iron increases HAMP expression, and hence an increase in hepcidin, which prevents the transport of iron from the enterocyte and macrophage via FPN. Excess iron and inflammation both induce HAMP expression, which, in turn, results in decreased iron absorption and diminished iron release from macrophages.
Low iron hepatic concentrations decrease BMP6 secretion, which hampers the BMP signaling pathway, reducing the expression of hepcidin. In the presence of inflammation, interleukin 6 and activin B are induced in the liver, which activates hepcidin’s transcription via the STAT3/JAK2 pathway and BMP signaling pathway, respectively.
A decrease in hepcidin may result from mutations in the HFE or TfR2 genes, leading to hemochromatosis in the adult. Mutations in the HJV or HAMP genes lead to juvenile hemochromatosis. Juvenile hemochromatosis is an autosomal recessive disorder resulting in iron overload at an early age, and high transferrin saturation, and ferritin concentrations [50].
A deficiency in hepcidin results in increased expression of ferroportin (FPN) at the cellular surface, mostly of enterocytes, and macrophages, which leads to an increase in the export of iron. Serum iron concentrations and transferrin saturation then increase, leading to increased iron uptake by the liver, pancreas, and heart resulting in iron overload, which characterizes hemochromatosis [1].
Mutations in FPN are also associated with iron overload due to resistance to hepcidin. A77D, V162del, and G490D mutations inhibit FPN activity, i.e., loss of function, associated with increased iron deposition in Kupffer cells, and normal transferrin saturation. The Y64N and C326Y variants are resistant to the inhibition by hepcidin. The N144D and N144H are partially resistant, i.e., gain of function, associated with high transferrin saturation, and increased deposition of iron in the liver parenchyma [51, 52].
Iron Mediated Injury
It has been considered that iron, in its ferrous state, interacts with H2O2 to generate the highly injurious hydroxyl radical [53, 54]; it has been stated, however, that these experiments were not conducted in proper models of iron overload, nor experimental conditions analogous to the pathophysiology of iron overload in human beings [55]. Iron alone appears to be associated with limited hepatic inflammation and fibrosis but, in association with other toxic elements or hepatic comorbidities, and by stimulating hepatic stellate cells combined with cofactors to be identified, may drive liver injury and consequent cirrhosis [55].
Genetics
The hemochromatosis gene (HFE (H for high and FE for iron)) is located on the short arm of chromosome 6 (6p22.1), within the extended HLA class I region [56]. HFE has six exons, with histone genes on each side; at least nine alternatively spliced forms have been documented. The product of HFE is HFE, a membrane associated beta 2-microglobulin protein, comprised of an alpha chain encoded by HFE and a beta 2-microglobulin, as the beta chain.
The most common mutations in HFE are substitutions of: (i) cysteine for tyrosine at position 282 (p.C282Y), (ii) histidine for aspartate at position 63 (p.H63D), and (iii) serine for cysteine at position 65 (p.S65C). C282Y is the most common mutation, considered to have originated in Northern Europe, with a frequency of one in 200 individuals [57]; most patients with hemochromatosis are homozygous for C282Y. H63D is geographically distributed more widely than C282Y [58]. Approximately 4% of patients with hemochromatosis are compound heterozygous for the mutations, i.e., C282Y/H63D. The evidence of iron overload in subjects with the S65C mutation is not strong; however, in combination with C282Y, there can be a phenotypic expression of the disease [59].
Experiments conducted in COS-7 cells revealed that HFE protein is expressed on the cell surface and that it binds beta 2-microglobulin; however, these characteristics disappear in the mutated C282Y, which was documented to remain in the endoplasmic reticulum and in the middle compartment of the Golgi apparatus, which does not process the protein normally, subsequently undergoing accelerated degradation [60]. The H63D and S65C mutations alter the α1 binding groove but do not prevent HFE presentation on cell surfaces.
Mutations can increase the phenotypic expression of C282Y in homozygosity in genes that concern iron homeostasis. The mutations pR59G, pG71D, or pR56X in the HAMP gene [61] and by the mutations S105L, E302K, N372D, R335Q L101P, and G320V in the HJV gene [62], both in the heterozygote state, in patients with homozygosity for C282Y result in marked iron overload, manifested relatively early [61]. Single nucleotide polymorphisms (SNP) may also influence the expression of C282Y homozygosity, including SNP in the haptoglobin (Hp) [63] and BMP2 genes [64]; however, the findings could not be reproduced [65].
Large genome-wide association studies (GWAS) identified three variants in serum transferrin, rs3811647, rs1799852, and rs2280673, in addition to the HFE C282Y mutation, which explained approximately 40% of the genetic variation in serum transferrin [66]. A subsequent GWAS confirmed rs3811647 in the TF gene as the only SNP associated with iron homeostasis through serum transferrin and iron concentrations [67]. Eleven genome-wide-significant loci that included HFE, SLC40A1, TF, TFR2, TFRC, TMPRSS6, and the newly identified ABO, ARNTL, FADS2, NAT2, TEX14 were documented, with SNPs at ARNTL, TF, and TFR2 that affect iron indices in patients homozygous for C282Y [68]. By the same methodology, the proprotein convertase subtilisin/kexin type 7 (PCSK7) rs236918 C allele was identified as a risk factor in patients homozygous for C282Y. These studies have helped to explain the variability of the C282Y homozygous phenotypic expression [69].
Epidemiology
In 1990, important studies from Australia documented the prevalence of iron overload in patients with hemochromatosis. In homozygous, it was reported as 0.36% in a group of 1968 subjects, mostly Caucasian, translated as 1 in 300 subjects; a transferrin saturation greater than 45% along with high serum ferritin had a predictive value for accurate diagnosis of 64%, finding that suggested the use of this finding in screening programs [70].
Subsequent studies reported the frequency of the C282Y mutation in the general population to be 6.2% from a group of 127,613 participants included in an individual patient meta-analysis from 36 studies [71], four of which were from the United States, which documented from: (i) 100 randomly selected individuals, a prevalence of the C282Y homozygosity of 1% in Caucasians, and that of C282Y heterozygosity 8% in Caucasians, 3% in Hispanics, and 2% in African Americans, and of H63D homozygosity 4% in Caucasians and 1% in Hispanics, and heterozygosity in 24% in Caucasians, 15% in Hispanics, and 3.5% in African Americans [72], (ii) 10,198 adult subjects a frequency of the C282Y mutation of 0.063, of H63D 0.152 and of 0.016 for S65C mutation in whites [73], (iii) 5171 participants in the Third National Health and Nutrition Examination Survey conducted from 1992 to 1994 an estimated prevalence of the C282Y gene mutation of 0.26% (95% confidence interval (CI), 0.12%–0.49%), and 1.89% (95% CI, 1.48%–2.43%) for H63D homozygosity, with compound heterozygosity being 1.97% (95% CI, 1.54%–2.49%); an estimated prevalence of C282Y heterozygosity of 9.54% in non-Hispanic whites, 2.33% in non-Hispanic blacks, and 2.75% in Mexican-Americans [74], and (iv) 4865 subjects a frequency of C282Y of 0.0507 in whites, and 0.0067 in African Americans, and for H63D, 0.1512 in whites, and 0.0263 in African Americans [75]. It was concluded from the meta-analysis that a genotype frequency for C282Y could be calculated to be 0.38%, i.e., 1 in 260 subjects, with a wide geographic variation. However, it was documented from 41,038 subjects, from whom 152 were homozygous for the mutation, that only one patient had signs and symptoms suggestive of hemochromatosis, although a history of hepatitis and abnormal serum activity of serum aspartate aminotransferase, not correlated with iron burden or age, was more common in the subjects with the mutation than in those without it, suggesting that the penetrance of the mutation was low, with less than 1% of homozygous developing the clinical disease [76]. The biochemical expression of C282Y, i.e., high serum ferritin concentration and high transferrin saturation, is reported as 75% for men and 50% for women, as reported from studies that evaluated subjects of European descent [77, 78].
A meta-analysis that included 2802 European heritage subjects with hemochromatosis documented the proportion of homozygosity for C282Y as 80.6% and compound heterozygosity for C282Y and H63D as 5.3%. In the control subjects, the reported proportion of C282Y homozygosity was 0.6%, and compound heterozygosity was 1.3% [71].
In contrast to C282Y, H63D has less geographic variation, with a reported average allelic frequency of 14%. S65C, which can be associated with iron overload in association with C282Y, was documented to have a frequency of approximately 0.5%, most commonly found in Brittany, France [71].
From a study that included what was reported as a racially diverse group comprised of 99,711 subjects, 299 were documented as homozygous for the C282Y mutation, with an estimated prevalence of 0.44% in non-Hispanic whites, 0.11% in Native Americans, 0.027% in Hispanics, 0.014% in blacks, 0.012 in Pacific Islanders, and very uncommon in Asians, with a proportion of 0.000039% [77].
Natural History
The natural history of hemochromatosis was documented from a group of patients with variable HFE genotypes known as the HealthIron cohort from a total of 31,192 of Northern European extraction subjects recruited from 1990 to 1994 in Australia. The study of serum iron indices in the 203 patients identified as homozygous for HFE C282Y and followed for a mean of 12 years documented that the predicted probability of having serum ferritin greater than 1000 mcg/ml was between 13% and 35% for men, and 16% to 22% for women. In patients with normal serum ferritin at baseline, the prediction to go over 1000 mcg/ml at follow up was less than 15% without any therapeutic interventions. These data were interpreted to indicate that sufficient iron to place patients at risk for complications would be accumulated by age 55. In addition, it was concluded that in men and not in women, a transferrin saturation greater than 95% at a mean age of 55 years increased the probability of having high serum ferritin by age 65 [79]. It was also concluded from the same group of subjects that 28.4% of men homozygous for C282Y had documented iron overload, versus 1.2% for women. There was an increase in reported symptoms, including fatigue and “use of arthritis medicine,” and of liver disease in men versus women [78]; this difference was interpreted to result from blood loss from menstruation [78, 79]. One hundred and eighty patients, 84 of whom were men, were identified as compound heterozygotes, i.e., C282Y/H63D, compared with 330 control subjects, 149 of whom were men in regard to iron studies, which revealed significantly increased serum concentration of serum ferritin and transferrin saturation in compound heterozygotes regardless of the gender, as compared to the normal genotype. Men and women, compound heterozygotes, had similar comorbidities related to iron overload as those with the wild type, with only one of 82 men and zero of 95 women with documented iron overload disease. These data were interpreted to indicate that although they may have abnormal iron indices in patients with compound heterozygosity, disease from iron overload is rare [80].
SNP have been identified in genes associated with iron metabolism, including TF, the gene that codes transferrin [78], and DCYTB, which codes cytochrome b reductase 1 [81] that likely modify the variable expression of iron overload disease [81]. In addition, an SNP in the gene TGF-beta1, which codes tumor growth factor-beta 1, was identified as associated with cirrhosis in patients with hemochromatosis [82]. Genetic factors also seem to protect from progressive disease, as reported by an increase in the survival of patients homozygous for C282Y identified as carrying HLA-A*03, B*14 [83].
Exogenous factors that include excessive alcohol use [84, 85], and hepatic comorbidities that include fatty liver [86], and chronic hepatitis C viral infection [87], have also been reported in association with an increase in the clinical expression of genetic hemochromatosis by accelerating liver injury from iron overload. Conversely, the C282Y mutation was reported in 44% of 27 patients with porphyria cutanea tarda (PCT) versus 12% in a control group of 12 patients; this finding has suggested that the mutation may be a triggering factor for the manifestations of the PTC [88].
Clinical Manifestations
The classic clinical manifestations of hemochromatosis are cirrhosis, hyperpigmentation, and diabetes. They tend to become apparent in the fourth decade of life by which 15 to 40 g of iron have accumulated in the body.
Four states have been described in the disease: (I) genetic mutation and increased serum transferrin saturation, in some cases, (II) iron overload not associated with symptoms, (III) iron overload associated with symptoms of early disease, e.g., fatigue, and (IV) iron overload in association with end-organ damage including cirrhosis.
From a study conducted in Australia that included 251 patients with hemochromatosis enrolled from 1947 to 1991, most patients reported symptoms independent from the presence of cirrhosis [89]. Fatigue has been reported as the most common symptom in hemochromatosis [78, 90, 91]; however, most patients are asymptomatic at the time of diagnosis. Symptoms have been reported in association with serum ferritin greater than 1000 microg/L [1].
From a group of 251 patients [79], hepatomegaly was the most common finding in physical examination (81%), followed by hyperpigmentation (72%), body hair loss (16%), splenomegaly (10%), with less than 10% of patients having peripheral edema, jaundice, and ascites. Seven percent of men had gynecomastia. Except for hyperpigmentation and gynecomastia, all signs were significantly more common in patients with than in those without cirrhosis [79].
Serum activity of AST and ALT was abnormal in 60 to greater than 65% of patients [89, 92], especially in the presence of cirrhosis [92].
Cardiac complications from hemochromatosis were documented in 10% of patients with the disease in a study that investigated the relationship between iron overload, age, and clinical symptoms [90], with electrocardiogram changes being reported in 35% of patients in another study, found significantly more common in patients with cirrhosis (46%) than in those without (21%) [89].
Bronze diabetes was the name for hemochromatosis, diabetes being one of the most recognized complications of this disease. A quantitative immunohistochemical and ultrastructural study of human pancreas in hemochromatosis documented that in patients with the disease iron overload was prominent in the exocrine tissue, there was a reduced number of immunoreactive B cells, to which iron deposit was restricted, in association with a loss of their endocrine granules; the findings were reported to be different from those reported in type I and II diabetes and suggested a role of iron in the pathogenesis of diabetes in hemochromatosis [93]. The prevalence of diabetes in a study that included 410 patients with hemochromatosis from Canada and France was 14%, with a documented statistical association with iron overload [90]. It has been subsequently reported that the frequency of diabetes in patients with hemochromatosis was not higher in patients homozygous for C282Y versus those without HFE mutations [76, 77]; the screening nature of these studies might have decreased the prevalence of diabetes by not including patients with known hemochromatosis who might have had this complication of the disease [76, 77]. Also, the increased diabetes prevalence in the general population was proposed as a reason for the absence of diabetes as a more prevalent condition in patients with hemochromatosis than in those without the disease [78]. The interpretation of the risk of diabetes in hemochromatosis seems to be evolving as factors including increased iron entry into beta cells by mechanisms that include transferrin receptors, and DMT1, decreased insulin secretion due to pancreatic atrophy, islet cells inflammation, injury of beta cells, and genetic predisposition are identified [94].
Hypogonadotropic hypogonadism is a complication of hemochromatosis, which seems to be decreasing in incidence because of screening measures for hemochromatosis. The prevalence of hypogonadism in men was 14.6% from 1983 and 1995 [95, 96]. Hypogonadism significantly decreased to 3% from 1996 to 2003, [95], and to 6.4%, in a study that included subjects diagnosed from 1983 to the first half of the 2000 decade, with a reported prevalence of 5.2% in women [96]; however, the prevalence of hypogonadism in the latter referenced study was 89% in patients with cirrhosis and 33% in those with diabetes [96]. A study that assessed pituitary function in idiopathic hemochromatosis in 36 men documented that the pituitary deficiency associated with the disease is gonadotrophic [97]; however, another study reported abnormalities in both pituitary and end-organ function [98]. Impotence was documented in 40% of men [90]. A study that used mailed questionnaires and that had the Dutch Hemochromatosis Association as a source documented that some aspect of sexual function was impaired in 7% to 39% of the 69 participants, findings not different from that reported from studies from the general population [99]. The pathogenesis of hypopituitarism as a cause of hypogonadism in regard to iron overload has not been elucidated. In regard to testicular function, iron is required for spermatogenesis; it is considered that iron transport through the seminiferous tubules’ basal membrane is independent and protects against iron overload associated with hemochromatosis [100]. Thus, a pituitary dysfunction that affects the hypophyseal–gonadal axis seems to be favored, although direct iron damage has not been unequivocally excluded [101].
The arthropathy associated with hemochromatosis is more common in men than in women and prevalent between 40 and 55 years of age; the most commonly involved joints are hips, knees, first carpometacarpal (CMC), proximal interphalangeal (PIP), distal PIP, second and third metacarpophalangeal (MCP), and ankles [102]. With a specialty clinic in rheumatology from where 62 patients were included and the UK Haemochromatosis Society, with 470 patient survey respondents, as sources, a study reported that a diagnosis of hemochromatosis was made in the fifth decade of life, with 77% and 76% of subjects, respectively, having reported symptoms related to joints for 8 years prior to the diagnosis, with the involvement of the MCP in 38.5% of patients, and ankles in 29.5%, followed by knees, hips, and PIP joints; at the time of clinical assessment or submission of the questionnaire for the study, the most commonly affected joints were PIP in 64.5% of subjects, knees in 64%, ankles in 61%, and MCP in 60%, in the group seen at the specialty clinic, versus CMC in 59%, wrists in 52%, PEP in 47%, MCP in 46%, knee in 42%, and ankle in 35% in the self-reporting group. It was concluded from this study that symptoms from arthropathy predate the diagnosis of hemochromatosis by almost 10 years and that arthropathy of MCP joints and ankles at a relatively early age in the absence of trauma, rare in osteoarthritis, is an indication to exclude hemochromatosis [103].
A web-based cross-sectional study conducted in Australia in patients with hemochromatosis that included the assessment of the quality of life [104] revealed that patients who reported arthritis in association with the disease had lower mean utility (0.52: F(1,198) = 10.854, p = 0.001), 0.66 as the reference value, utility being “...a measure of the strength of preference for a particular health state ‘that, when combined with life years gained (LYG),’…the outcome reflects both morbidity and mortality: quality adjusted life years (QALYs)”[105]. This finding is consistent with the prior study that revealed that, although cirrhosis was the most important complication affecting survival, arthritis was the most important complication affecting the quality of life [106].
The pathogenesis of hemochromatosis related arthropathy has not been elucidated, although iron has been considered a relevant etiologic factor [107]. A study that examined the synovium and arthropathy radiographic changes in 27 patients with hemochromatosis documented the most prevalent findings as villus formation and iron deposition within synovial intimal cells [116]. Arthropathy did not correlate with histology but was present in 26% of cases with the above reported histological findings but did correlate with total body iron as per the differential ferrioxamine test [108]. Other histological studies of the synovium from patients with hemochromatosis have documented infiltration with mononuclear cells and lymphocytes, neovascularization, and hyperplasia, findings reported as prominent in association with hemosiderin deposition; although the type of cellular invasion in hemochromatosis was reported as similar to osteoarthritis, the neutrophil invasion was more pronounced in the former than in the latter, and it may contribute to the rapid progressive arthropathy of hemochromatosis [109]. Arthropathy is not reversed or its progression halted by iron removal [107, 108].
The prevalence of osteoporosis in patients with cirrhosis and hemochromatosis was 25% to 78% [110, 111], and osteopenia was 41% [112]. In a study that included 38 men with hemochromatosis, hypogonadism was documented in the minority of patients, and femoral neck bone mineral density was decreased in association with an increased degree of hepatic iron concentrations similar in patients with and without cirrhosis. These data were interpreted to suggest that factors, including iron itself, other than cirrhosis and hypogonadism, are involved in the pathogenesis of bone disease in hemochromatosis [111]. From a study that included 87 patients it was reported that osteoporosis and hypogonadism were associated with bone disease, findings related to the degree of iron overload [112]. An association between iron overload and osteoporosis was reported in a study of 304 patients with hemochromatosis, 23% of whom had been diagnosed with the bone complication. Also, wrist and vertebral fractures were prevalent in the latter group [113]. One mechanism in the development of bone disease in hemochromatosis involved the facilitation of osteoclasts’ differentiation by iron [114], and the deterrence of the osteogenic differentiation of multipotent mesenchymal stem cells, from which osteoblasts derive themselves, and which are inhibited by iron, the latter by the inhibition of the osteogenic transcription factor runt-related transcription factor 2 (Runx2) [115]. Iron removal via phlebotomy was reported in association with improvement in bone density [116].
The most recognized dermatological manifestation of hemochromatosis is hyperpigmentation, characterized by a brown or gray slate color, most prominent on sun exposed areas. A retrospective study that included 22 patients documented hyperpigmentation in seventeen (77%) [117]; however, this study’s retrospective nature may have overestimated the prevalence. Skin biopsies documented siderosis in eccrine sweat glands reported as specific for the disease, and melanin accumulation, in the epidermis’ basal layers [118], the former associated with a decrease after phlebotomy but not the latter. In addition, ichthyosis and koilonychias are reported in patients with the disease [119].
Laboratory Tests
The hallmark of hemochromatosis is high serum ferritin concentrations and transferrin saturation. Serum ferritin can be normal in the early stages of the disease. The increase in serum ferritin can result from a reaction to an acute event, i.e., ferritin being an acute phase reactant, and from hepatic comorbidities including alcoholic liver disease and chronic hepatitis C; thus, alone, it is not diagnostic of hemochromatosis. Transferrin saturation is the earliest laboratory evidence of hemochromatosis [120, 121]. It was documented that an unsaturated iron binding capacity equal or less than 25 micromoles/L confirmed C282Y homozygosity with 83% sensitivity and 99% specificity, indicating similar performance to transferrin saturation in the identification of patients with hemochromatosis [122]. The guidelines from the American College of Gastroenterology (ACG) recommend diagnostic workup that includes serum ferritin, iron, transferrin saturation, and unsaturated iron binding capacity [123]. The guidelines indicate that patients with hepatitis, suggested by serum activity of transaminases greater than 35 IU/ml with normal transferrin saturation and serum ferritin concentrations do not have hemochromatosis. Patients with hepatitis in association with either transferrin saturation of at least 45% or high serum ferritin, or both, should have hemochromatosis genetic testing. The guidelines recommend that: (i) patients who are homozygous for C282Y should have phlebotomy, (ii) for homozygous for C282Y with serum ferritin greater 1000 mg/dl or hepatitis, a liver biopsy can be considered to rule out cirrhosis. Serum ferritin concentration equal or greater than 1000 microg/L, a platelet count equal or lower than 200 × 10(9)/L, and serum activity of AST greater than the upper limit of normal were associated with the diagnosis of cirrhosis in 77% of patients in a study from Canada, and in 90% of patients in a study from France, findings that have been used to avoid a liver biopsy [124]. Serum hyaluronidase was reported to identify cirrhosis in patients with hemochromatosis and serum ferritin concentrations greater than 1000 micrograms/L [125]; however, in another study, the concentration of hyaluronic acid did not accurately predict severe fibrosis. The use of this marker in cirrhosis has not been incorporated into standard practice. For patients who are compound heterozygotes (i.e., C282Y/H63D), and who have non-C282Y hemochromatosis, evaluation of hepatic iron concentration should be done by liver biopsy or by magnetic resonance imaging (MRI); if increased tissue iron is documented, phlebotomy should be started if ferritin is greater than 1000 mg/dl [123].
Elevated serum ferritin concentration in association with high serum transferrin saturation can also result from mutations in hemojuvelin, hepcidin, and TfR2 genes, in contrast to patients with ferroportin disease with gain of function [126] or aceruloplasminemia [15, 17] who have high serum ferritin concentrations but normal transferrin saturation. In this context, hepatic iron concentration assessment by liver biopsy or MRI is recommended; hepatic iron measure greater than 150 micromoles/g is consistent with ferroportin disease or aceruloplasminemia, between 50 and 150 micromoles/g with conditions of dysmetabolic iron overload syndrome [127], and less or equal to 50, is consistent with L ferritin disease [128]. The ACG guidelines suggest not to proceed with genetic testing in patients with iron indices suggestive of iron overload who are negative for the C282Y and H63D mutations [123].
In a study comprised of 182 patients with hemochromatosis from the United States, liver cirrhosis was absent in patients who are homozygous for C282Y and in compound heterozygous for C282Y/H63D whose serum ferritin was less than 1000 microgram/L; in combination with hepatitis, the probability of cirrhosis was 72%, and in patients with serum ferritin concentrations less than 1000 micrograms/L, it was 7.4%. These findings have suggested that a liver biopsy is not paramount in evaluating patients with hemochromatosis, as inferences regarding indications for phlebotomy and screening for hepatocellular carcinoma (HCC) can be made from non-invasive laboratory findings [124, 129].
The ACG practice guidelines suggest a non-invasive test, i.e., a non-contrast enhanced MRI in conjunction with the software used for the estimation of hepatic iron concentration (i.e., MRI T2*) in patients being investigated for iron overload in the absence of C282Y homozygosity. The use of transient elastography has not been sufficiently explored in patients with iron overload. One hundred and three consecutive subjects, 57 with hemochromatosis and 46 as control subjects, were prospectively evaluated and documented as having similar fibrosis measures, 5.20 kPa in the disease group and 4.9 kPa in the control group; however, 22.8% of patients had values greater than 7.1 kPa, suggesting significant fibrosis, in contrast to no subjects from the control group. There was no correlation reported between these measures and serum ferritin concentration. These findings were considered preliminary, identifying the need for additional studies [130]. A follow-up study from 2005 to 2013 included 77 patients homozygous for C282Y, with either serum ferritin greater than 1000 micrograms/L, who had hepatitis, or both who had a liver biopsy and transient elastography done. The transient elastography measure was 17.2 kPa in patients with severe fibrosis (F3-F4), comprised of 19.5% of the group, versus 4.9 kPa in patients without advanced fibrosis, with the ability to predict fibrosis stage in 77% of the patients accurately when the lower and upper thresholds of 6.4 kPa, no fibrosis, and 13.9 kPa, severe fibrosis, were used; however, intermediate kPa values were not useful in fibrosis determination. Validation studies have been recommended prior to inclusion in general practice [131]. A liver biopsy is the reliable method to determine the stage of fibrosis and to diagnose cirrhosis, and it is suggested as an option if this information is sought [123].
Early diagnosis allows for treatment in a timely fashion to prevent complications from the disease; in this regard, counseling and screening of family members must be followed. Population screening is not recommended; thus, the task of family screening falls on the clinician making the diagnosis. A study included 672 asymptomatic patients homozygous for C282Y identified as part of liver disease workup or screening after the diagnosis of a relative. In addition, a group of untreated patients who were homozygous for the mutation were followed for 24 years. Histologically, the amount of liver iron and fibrosis degree was similar in both groups, with diseases known to be associated with hemochromatosis more commonly documented in individuals identified during a liver disease workup, who were also older than those without symptoms. Fifty-six percent of liver samples from men had iron overload graded from 2 to 4, fibrosis staged from 2 to 4 in 18.4%, and cirrhosis in 5.6%; in women, the proportions were 34.5%, 18.8%, and 1.9% for the findings listed above. Hepatic iron concentration correlated with fibrosis and cirrhosis, with a 7.5 fold decrease in mean fibrosis score associated with phlebotomy. Of note, patients with cirrhosis did not report any symptoms. Thus, homozygous patients with C282Y identified by family screening or indicated liver disease workup had the same degree of iron and fibrosis. Importantly, both decreased substantially in association with phlebotomy [132]. In the absence of screening guidelines, it is reiterated that workup for iron overload conditions in patients with an abnormal liver profile is important. Genetic testing in children is not recommended before age 18. However, if there is a mutation in a parent, checking for mutations in the other parent allows inferring the children’s genetic characteristics. If one parent is negative, the children are obligate heterozygotes, not at risk for developing clinical complications from the disease [133]. The exception in children concerns those with serum ferritin concentrations of 1000 micrograms/L seen in juvenile hemochromatosis and other uncommon forms of iron overload, which do require investigation when the condition is identified [134].
Histology
Perls’ Prussian blue stain is used to assess hemosiderin deposition in the liver. Early stages of iron overload in homozygous hemochromatosis are characterized by hemosiderin as small granules in hepatocytes in the periportal area in a “cruciate pattern”. The iron stores tend to remain in zone one, with decreased expression in the perivenular zones. Fibrosis also develops from the portal tracts and subsequently evolves into irregular fibrosis and cirrhosis. Cirrhosis from hemochromatosis is described as exhibiting high amounts of iron stores in hepatocytes that outline the nodules with partial acini preservation. Cirrhosis in hemochromatosis is micronodular. In the late stages of the disease, after iron has accumulated itself in non-treated patients, iron can appear in Kupffer cells lining the sinusoids [135]. In addition, areas devoid or with minimal parenchymal iron accumulation appear surrounded by heavily hemosiderin laden hepatocytes in an appearance reminiscent of glades in the forest; the significance of these structures is not known [136]; however, it was reported that iron free foci are preneoplastic lesions and that, in a patient with hemochromatosis, may herald the development of HCC, and hence are an indication for screening for HCC [137].
In heterozygotes, iron overload can be modest; when notable, the hemosiderin appears in the hepatocytes in the portal areas and zone 1 of the liver acini. Fibrosis tends to be absent, and Kupffer cells are devoid of iron [135].
In association with iron depletion, iron stores disappear from the liver. It is documented that the changes in the iron expression in association with phlebotomy exhibit a pattern that is the reverse of the pattern of accumulation, i.e., the last cells noted to have large iron granules are in the portal areas, and zone 1 [135]. Decreased fibrosis has been reported in association with iron depletion over time, but regression from F4 was documented in a minority of patients [89].
Treatment
The treatment of iron overload is the removal of iron by phlebotomy [123]. Survival of patients with hemochromatosis is decreased in comparison to that of individuals without the disease. Over 8.1 +/− 6.8 years (range, 0–31 years), seventeen patients from the group of 85 with hemochromatosis had died, with cirrhosis increasing the risk of death by 5.5 times over that of patients without cirrhosis. Patients who were not cirrhotic at the time of diagnosis had similar estimated survival to those from age- and sex-matched members of the normal population. These results were interpreted to indicate that the diagnosis and treatment of hemochromatosis prior to the development of cirrhosis could result in an improved long-term survival, similar to that of the general population; thus, it identified cirrhosis as the complication that marked the natural history of this disease associated with decreased survival in patients with hemochromatosis [138].
From a study that included 251 patients with hemochromatosis followed up for 14.1 +/− 6.8 years, the survival was reported as expected in patients without cirrhosis or diabetes or in patients diagnosed between 1982 and 1991, a period during which the identification of patients with hemochromatosis increased, as compared to prior decades. Survival was reduced in relation to iron overload. The number of deaths from HCC, cirrhosis, and diabetes was higher than expected, and an association between HCC and cirrhosis versus iron removed was reported. This important follow-up study documented that the prognosis of patients with hemochromatosis and the development of its complications depend on the amount and duration of exposure to excess iron and, thus, emphasized that early diagnosis and treatment prevent complications from iron overload [89]. The reader is referred to the excellent editorial [139] related to the publication cited above [89].
A study that included 672 asymptomatic homozygous for C282Y subjects screened because of family history or during general health assessments revealed a similar degree of hepatic iron and fibrosis, which was prevalent, and reversed in association with phlebotomy, except in cases of cirrhosis. This study further highlighted the importance of early diagnosis and treatment prior to cirrhosis development [132].
Clinical guidelines recommend starting treatment in C282Y homozygous subjects who have serum ferritin greater than 300 ng/ml for men, and more than 200 ng/mL in women and a transferrin saturation equal or greater than 45%. Five hundred (500) ml are removed once or twice a week, to decrease serum ferritin to 50 to 100 micrograms/L, with periodic phlebotomies to maintain the serum ferritin at that level, and hemoglobin between 11 and 12 g/dl. Compound heterozygotes, C282Y/H63D, or homozygous for H63D [140] tend not to develop iron overload in the absence of comorbidities including excessive alcohol drinking and chronic hepatitis C; in cases of uncertainty, a liver biopsy to determine hepatic iron index can be done to determine if phlebotomy is indicated [141]. A liver biopsy also allows for the calculation of hepatic iron index (HII) to differentiate C282Y from compound heterozygotes and as a prognostic factor for cirrhosis development. HII is calculated by dividing the hepatic iron content in micromoles per gram of dry liver weight by the patient age in years, with an index equal or greater than 1.9 and/or 71 micromoles/g dry liver weight distinguishing homozygous from compound heterozygotes and secondary iron overload [142].
In regard to fibrosis in association with phlebotomy, in a study that defined regression of fibrosis as a decrease in at least two METAVIR units, comprised of 36 patients homozygous for C282Y, regression was documented in nine of thirteen patients (69%) with baseline F3, and in eight of 23 (35%) with baseline F4 [143]. Although removal of iron may decrease the degree of fibrosis, patients with cirrhosis should continue to undergo screening tests for HCC, as the risk of this complication remains. Also in association with phlebotomy, a consensus report documented that “weakness,” which may be similar to fatigue, was unchanged in 40% of patients, and arthralgias, unchanged in 50%, and even worsened in 20% [144].
In a study of survival and causes of death in cirrhotic and in non-cirrhotic patients with primary hemochromatosis that included 163 patients diagnosed between 1959 and 1983 and followed for an average of 10 years, the cumulative survival was calculated as 92% at 5 years, 76% at ten, 59% at 15, and 49% at 20; inadequate depletion of iron during the first eighteen months of therapy was associated with a significant decreased survival [89], supporting the recommendation of iron depletion in patients who meet criteria for such, as described [123]. In a hereditary hemochromatosis database 95 cirrhotic were identified. Sixty had undergone genetic testing, 87% of whom were homozygous for C282Y. The documented cumulative survival at one year was 88%, at five, 69%, and at 20, 56%. Advanced disease, determined by the Child-Pugh score, and HCC, which tended to be diagnosed between the ages of 48 and 79 years, were associated with death, on multivariate analysis. It was concluded that early diagnosis and treatment to prevent the development of cirrhosis may reduce the incidence of HCC in patients with hemochromatosis [145]. A study from France that included 1085 patients documented the overall mortality of patients with hemochromatosis similar to the general population; however, deaths from liver disease were increased in the disease group, with death occurring in all the subjects between the ages of 53 and 81 years, and no difference between life span and diagnosis of hemochromatosis. Serum ferritin, however, was higher in patients who died from liver disease than in patients who died from other causes, 3816 ± 1932 μg/L and 2417 ± 3723 μg/L (p = 0.11), respectively. Iron depletion of at least 10 grams was not associated with a mortality rate different from the general population; however, removing 2 to 20 g of iron was associated with a standardized mortality rate (SMR) lower than that of the general population. Diabetes and arthropathy were not associated with increased mortality in patients with hemochromatosis. However, patients with cardiomyopathy of any kind had increased mortality with deaths from the cardiovascular disease being reported as fewer in patients with serum ferritin less than 1000 micrograms/L, and in those who had iron depletion characterized as low, i.e., 2 to 10 g total. The group of patients who died from liver disease was comprised of men, in the majority, who had mean serum ferritin of 3816 ± 1932 μg/L; however, excessive alcohol intake was documented in ten of 27 (37%); in this group, 20 of 27 (74%) died from complications of liver cancer, which represented an SMR of 15.96 (CI: 9.75–24.65). Cox proportional hazards models estimated that age of at least 56 years, diabetes, alcohol use, and liver fibrosis, at least F3, were independently associated with a risk of death, all of which carried a hazard ratio of at least 3. Of note, nine patients in the study group died from extrahepatic cancers compared to the 21.81 expected in the general population. Patients with serum ferritin levels less than 1000 microgram/L had lower mortality from extrahepatic malignancies than the general population [146].
Normalization of serum ferritin due to iron depletion was associated with fatigue improvement in patients with hereditary hemochromatosis and elevated serum ferritin levels [147]. A randomized, participant—blinded, placebo controlled trial conducted in Australia between 2012 and 2016 included patients aged 18 to 70 with hemochromatosis, homozygous for C282Y, to study the effect of iron depletion by erythrocytapheresis versus a sham intervention (i.e., plasmapheresis) performed every three weeks. Fatigue was the primary outcome, in association with a decrease in ferritin, which was aimed to decrease to less than 300 micrograms/L. At baseline, the serum ferritin was from 300–1000 μg/L, and the transferrin saturation was high. There was a significant improvement in the Modified Fatigue Impact Scale scores in association with erythrocytapheresis in the 50 patients who completed the study from the original 54 versus that of the 44 from the original 50 who had been randomized to the sham procedure; in addition, there was a significant improvement in the cognitive subcomponent of the tool in association with the therapeutic intervention versus the sham intervention, but not in the physical or psychosocial subcomponents [147]. This study indicated that iron depletion in patients with serum ferritin between 300 and 1000 micrograms/L should be considered.
Chelators are alternative therapies if phlebotomy is not tolerated or feasible. Deferasirox is a chelating agent approved for the treatment of iron overload. In a dose escalation trial in a group of patients homozygous for HFE-related hemochromatosis the daily administration of this drug for 48 weeks was associated with a decrease in the median serum ferritin concentrations by 63.5%, 74.8%, and 74.1% at doses of 5, 10, and 15 mg/kg, respectively, and to less than 250 ng/mL in all the doses studied. Significant side effects were increased ALT greater than three times the upper limit of normal in a few patients, and increased serum creatinine, both more common in the high than in the low dose. A second study, prospective and open label in ten patients with hemochromatosis in whom phlebotomy had been inadequate, or not tolerated, deferasirox at doses from 5 to 15 mg/kg per day was associated with a significant decrease in median serum ferritin, mean transferrin saturation, median iron concentration, and mean serum activity of ALT; in this study, the drug was not associated with increased serum creatinine [148]. Deferoxamine and deferiprone are chelators approved to treat secondary iron overload.
Erythrocytapheresis is a procedure through which red cells are exchanged [149]. A study that included 38 patients homozygous for C282Y newly diagnosed randomized to receive erythrocytapheresis versus phlebotomy documented that significantly fewer treatment sessions of erythrocytapheresis were required to reach target of serum ferritin concentration of 50 micrograms/L, nine versus 27 for phlebotomy [150]. A subsequent randomized crossover trial compared the number of sessions needed to reach a serum ferritin of 50 micrograms/L after one year of treatment with erythrocytapheresis versus phlebotomies, with subsequent crossover to the alternate treatment documented that significantly more sessions were necessary for phlebotomy versus erythrocytapheresis, 3.3 vs. 1.9 (mean difference, 1.4; 95% confidence interval, 1.1–1.7), with the time between treatment sessions 2.3 times longer for erythrocytapheresis than for phlebotomy. Although quality of life by the SF-36 was not significantly different in association with the two treatment modalities, joint swelling, an important complication of hemochromatosis, was substantially more common during the period of phlebotomy treatment versus that of erythrocytapheresis, which was the preferred treatment of the majority of patients. Erythrocytapheresis was more expensive than phlebotomy [151].
Proton pump inhibitors (PPI) decrease iron absorption; it has been reported that the use of PPI in patients with hemochromatosis required a reduced number of phlebotomy sessions to reach treatment targets. From a randomized controlled trial that included 30 homozygous for C282Y who underwent phlebotomies when serum ferritin was greater than 100 micrograms/L, it was documented that patients on PPIs required significantly fewer sessions than those not on PPIs, which suggested that PPIs can be used in combination with iron depletion therapy [152]; PPIs alone are not recommended as a treatment for iron overload in hemochromatosis [123].
It is recommended that patients being investigated for iron overload get a hormonal evaluation that includes testosterone, follicle stimulating hormone, luteinizing hormone, GnRH testing, and pituitary MRI, regardless of clinical manifestations. In patients younger than 40 years, weekly phlebotomy is proposed as treatment of hypogonadism combined with testosterone or gonadotropin treatment in subjects older than 40 years; sperm cryopreservation is suggested [101].
Liver Transplantation
Liver transplantation in patients with hemochromatosis is associated with normalization of hepcidin, thus preventing iron overload in the graft [153]. The indication for liver transplantation in patients with hemochromatosis is decompensated liver disease or hepatocellular carcinoma [123].
The survival post liver transplant in a study that included 260 patients with decompensated liver disease from iron overload and recruited from several centers in the United States was documented as 64%, 48%, and 34% at one, three, and 5 years, respectively, in homozygous for C282Y, which was lower than that for heterozygotes that included H63D. Iron overload not from hemochromatosis was associated with a significant decreased survival versus that of the patients transplanted for other causes with an odds ratio of 1.4 (95% CI:1.15–1.61, p < 0.001) [154]. Subsequently, by the use of the United Network for Organ Sharing as a source, the survival of patients transplanted for hemochromatosis between 1990 and 1996 was documented as 79.1%, 71.8%, and 64.6%, at one, three, and 5 years, respectively. The survival improved for patients transplanted for hemochromatosis between 1997 and 2006, being 86.1%, 80.8%, and 77.3% as one, three, and 5 years, which was similar to that of patients transplanted for other conditions, with a hazard ratio for death of 0.89 (95% CI: 0.65–1.22) [155]. However, a retrospective review that included 22 patients with hemochromatosis transplanted for end-stage liver disease, for HCC, or for subacute liver failure documented a patient and graft survival of 80.7%, and 74%, at one and 5 years, respectively, with several deaths occurring over a median period of 46 months post transplantation from multiorgan failure or recurrence of HCC; bacterial infections were the most common causes of complications, and iron removal prior to transplantation was reported to be associated with a decrease in cardiac complications and bacterial infections [156]. Thus, although there is a documented improvement in post transplant survival in patients with hemochromatosis, a complication that depends on the pathogenesis of the disease, i.e., iron overload, does have an impact on post surgical comorbidities. The finding that iron removal was associated with a decrease in infection rate support that early intervention is associated with improved general and post liver transplantation outcomes.
Diet changes include avoidance of iron supplements in any of their forms, vitamin C supplements because it increases iron absorption, alcohol, and raw fish and shellfish [157], the latter because of transmission of pathogens that thrive in iron laden conditions (e.g., Listeria monocytogenes, Yersinia enterocolitica, Escherichia coli, and Vibrio vulnificus) and because of impaired immune function in association with iron overload [158,159,160,161].
Treatment of Other Genetic Iron Overload Conditions
In ferroportin disease, loss of function, serum ferritin is substantially increased. Iron depletion is the treatment to keep the serum ferritin between 50 and 100 micrograms/L; however, it is not well tolerated in general. Low transferrin saturation and anemia tend to develop quickly despite high serum ferritin. Chelation and erythropoietin have been reported to be a therapeutic option.
In aceruloplasminemia, phlebotomy is poorly tolerated; thus, chelation is the therapeutic alternative.
In juvenile hemochromatosis, the cardiac function from iron load may be seriously impaired for which aggressive and urgent iron removal may be necessary.
Hepcidin deficiency or resistance to the effects of hepcidin on FPN is associated with iron overload, the focus of this chapter. Mutations in TfR2 [162, 163], HJV [38], transferrin, and hepcidin [164] are associated with iron overload,
Ferroportin disease has high penetrance and results from a mutation with the autosomal dominant transmission in the SLC40A1 gene, which encodes ferroportin1/IREG1/MTP1 a product expressed in macrophages, enterocytes, and hepatocytes. This mutation results in decreased expression or decreased ability to transport iron. Phenotypically, it is characterized by high serum ferritin concentrations in association with low or normal transferrin saturation, accumulation of iron mostly in macrophages, anemia, and poor tolerability to phlebotomy [126]. Another form of ferroportin disease is characterized by loss of function, associated with hyperferritinemia, high transferrin saturation, and iron accumulation in the hepatocyte but low incidence of liver fibrosis and cirrhosis [165].
Potential Pharmacological Treatment of Hepcidin-Ferroportin Disease
A decrease in hepcidin is associated with iron overload. A phase II clinical trial of LJPC-401, a synthetic human hepcidin, (ClinicalTrials.gov Identifier: NCT03395704), for the treatment of iron overload in adult patients with hereditary hemochromatosis was associated with a statistically significant reduction in change in transferrin saturation, the primary efficacy endpoint of the study, and frequency of phlebotomies, a key secondary efficacy endpoint of the study [166].
A decrease in iron absorption or export from duodenal cells into the circulation would decrease iron accumulation. Polyphenols have iron chelating properties and are present in the human diet. It was reported from an animal study that quercetin, a polyphenol, was associated with iron chelation in the intestinal lumen of rats. In the long term, it was associated with the regulation of FPN expression that resulted in a decrease of iron export [167]. A clinical trial on the Effects of Polyphenols on Iron Absorption in Iron Overload Disorders (POLYFER) is listed in ClinicalTrials.gov Identifier: NCT03453918; no published results were identified.
References
Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016 Aug 13;388(10045):706–16.
Nicolas G, Viatte L, Bennoun M, Beaumont C, Kahn A, Vaulont S. Hepcidin, a new iron regulatory peptide. Blood Cells Mol Dis. 2002 Nov-Dec;29(3):327–35.
Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000 Jun 30;275(26):19906–12.
Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000 Feb 17;403(6771):776–81.
McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000 Feb;5(2):299–309.
Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008 May 30;320(5880):1207–10.
Kidane TZ, Sauble E, Linder MC. Release of iron from ferritin requires lysosomal activity. Am J Physiol Cell Physiol. 2006 Sep;291(3):C445–55.
McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001 Mar 2;291(5509):1755.
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997 Jul 31;388(6641):482–8.
Nakai Y, Inoue K, Abe N, Hatakeyama M, Ohta KY, Otagiri M, et al. Functional characterization of human proton-coupled folate transporter/heme carrier protein 1 heterologously expressed in mammalian cells as a folate transporter. J Pharmacol Exp Ther. 2007 Aug;322(2):469–76.
Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999 Feb;21(2):195–9.
Canonne-Hergaux F, Donovan A, Delaby C, Wang HJ, Gros P. Comparative studies of duodenal and macrophage ferroportin proteins. Am J Physiol Gastrointest Liver Physiol. 2006 Jan;290(1):G156–63.
Ramey G, Deschemin JC, Durel B, Canonne-Hergaux F, Nicolas G, Vaulont S. Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica. 2010 Mar;95(3):501–4.
Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009 May;9(5):461–73.
Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci U S A. 1999 Sep 14;96(19):10812–7.
Cherukuri S, Potla R, Sarkar J, Nurko S, Harris ZL, Fox PL. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab. 2005 Nov;2(5):309–19.
Harris ZL, Takahashi Y, Miyajima H, Serizawa M, MacGillivray RT, Gitlin JD. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci U S A. 1995 Mar 28;92(7):2539–43.
Ganz T. Systemic iron homeostasis. Physiol Rev. 2013 Oct;93(4):1721–41.
Kuhn LC. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics: Integrated Biometal Sci. 2015 Feb;7(2):232–43.
Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005 Nov;37(11):1264–9.
Chua AC, Graham RM, Trinder D, Olynyk JK. The regulation of cellular iron metabolism. Crit Rev Clin Lab Sci. 2007;44(5–6):413–59.
Chua AC, Delima RD, Morgan EH, Herbison CE, Tirnitz-Parker JE, Graham RM, et al. Iron uptake from plasma transferrin by a transferrin receptor 2 mutant mouse model of haemochromatosis. J Hepatol. 2010 Mar;52(3):425–31.
Shindo M, Torimoto Y, Saito H, Motomura W, Ikuta K, Sato K, et al. Functional role of DMT1 in transferrin-independent iron uptake by human hepatocyte and hepatocellular carcinoma cell, HLF. Hepatol Res. 2006 Jul;35(3):152–62.
Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc Natl Acad Sci U S A. 2006 Sep 12;103(37):13612–7.
Cook JD, Flowers CH, Skikne BS. The quantitative assessment of body iron. Blood. 2003 May 1;101(9):3359–64.
Finch C. Regulators of iron balance in humans. Blood. 1994 Sep 15;84(6):1697–702.
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004 Dec 17;306(5704):2090–3.
Nemeth E, Preza GC, Jung CL, Kaplan J, Waring AJ, Ganz T. The N-terminus of hepcidin is essential for its interaction with ferroportin: structure-function study. Blood. 2006 Jan 1;107(1):328–33.
Krause A, Neitz S, Magert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000 Sep 1;480(2–3):147–50.
Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001 Mar 16;276(11):7806–10.
Fernandes A, Preza GC, Phung Y, De Domenico I, Kaplan J, Ganz T, et al. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood. 2009 Jul 9;114(2):437–43.
Preza GC, Ruchala P, Pinon R, Ramos E, Qiao B, Peralta MA, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011 Dec;121(12):4880–8.
Qiao B, Sugianto P, Fung E, Del-Castillo-Rueda A, Moran-Jimenez MJ, Ganz T, et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012 Jun 6;15(6):918–24.
Ganz T, Olbina G, Girelli D, Nemeth E, Westerman M. Immunoassay for human serum hepcidin. Blood. 2008 Nov 15;112(10):4292–7.
Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006 May;38(5):531–9.
Steinbicker AU, Bartnikas TB, Lohmeyer LK, Leyton P, Mayeur C, Kao SM, et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood. 2011 Oct 13;118(15):4224–30.
Xia Y, Babitt JL, Sidis Y, Chung RT, Lin HY. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood. 2008 May 15;111(10):5195–204.
Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004 Jan;36(1):77–82.
Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006 Sep 29;281(39):28494–8.
Johnson MB, Chen J, Murchison N, Green FA, Enns CA. Transferrin receptor 2: evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol Biol Cell. 2007 Mar;18(3):743–54.
D'Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J Hepatol. 2012 Nov;57(5):1052–60.
Ramos E, Kautz L, Rodriguez R, Hansen M, Gabayan V, Ginzburg Y, et al. Evidence for distinct pathways of hepcidin regulation by acute and chronic iron loading in mice. Hepatology. 2011 Apr;53(4):1333–41.
Maes K, Nemeth E, Roodman GD, Huston A, Esteve F, Freytes C, et al. In anemia of multiple myeloma, hepcidin is induced by increased bone morphogenetic protein 2. Blood. 2010 Nov 4;116(18):3635–44.
Fujita N, Sugimoto R, Takeo M, Urawa N, Mifuji R, Tanaka H, et al. Hepcidin expression in the liver: relatively low level in patients with chronic hepatitis C. Mol Med. 2007 Jan-Feb;13(1–2):97–104.
Girelli D, Pasino M, Goodnough JB, Nemeth E, Guido M, Castagna A, et al. Reduced serum hepcidin levels in patients with chronic hepatitis C. J Hepatol. 2009 Nov;51(5):845–52.
Harrison-Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, Clemens D, et al. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006 Aug 11;281(32):22974–82.
Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005 Aug;115(8):2187–91.
Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009 Apr;41(4):478–81.
Andriopoulos B Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009 Apr;41(4):482–7.
Camaschella C. Juvenile haemochromatosis. Baillieres Clin Gastroenterol. 1998 Jun;12(2):227–35.
Drakesmith H, Schimanski LM, Ormerod E, Merryweather-Clarke AT, Viprakasit V, Edwards JP, et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood. 2005 Aug 1;106(3):1092–7.
Schimanski LM, Drakesmith H, Merryweather-Clarke AT, Viprakasit V, Edwards JP, Sweetland E, et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood. 2005 May 15;105(10):4096–102.
Figueiredo MS, Baffa O, Barbieri Neto J, Zago MA. Liver injury and generation of hydroxyl free radicals in experimental secondary hemochromatosis. Res Exp Med. 1993;193(1):27–37.
Kadiiska MB, Burkitt MJ, Xiang QH, Mason RP. Iron supplementation generates hydroxyl radical in vivo. An ESR spin-trapping investigation. J Clin Invest. 1995 Sep;96(3):1653–7.
Bloomer SA, Brown KE. Iron-induced liver injury: a critical reappraisal. Int J Mol Sci. 2019 Apr;30:20(9).
Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996 Aug;13(4):399–408.
Bacon BR, Olynyk JK, Brunt EM, Britton RS, Wolff RK. HFE genotype in patients with hemochromatosis and other liver diseases. Ann Intern Med. 1999 Jun 15;130(12):953–62.
Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ. Global prevalence of putative haemochromatosis mutations. J Med Genet. 1997 Apr;34(4):275–8.
Asberg A, Thorstensen K, Hveem K, Bjerve KS. Hereditary hemochromatosis: the clinical significance of the S65C mutation. Genet Test. 2002 Spring;6(1):59–62.
Waheed A, Parkkila S, Zhou XY, Tomatsu S, Tsuchihashi Z, Feder JN, et al. Hereditary hemochromatosis: effects of C282Y and H63D mutations on association with beta2-microglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7 cells. Proc Natl Acad Sci U S A. 1997 Nov 11;94(23):12384–9.
Jacolot S, Le Gac G, Scotet V, Quere I, Mura C, Ferec C. HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood. 2004 Apr 1;103(7):2835–40.
Le Gac G, Scotet V, Ka C, Gourlaouen I, Bryckaert L, Jacolot S, et al. The recently identified type 2A juvenile haemochromatosis gene (HJV), a second candidate modifier of the C282Y homozygous phenotype. Hum Mol Genet. 2004 Sep 1;13(17):1913–8.
Van Vlierberghe H, Langlois M, Delanghe J, Horsmans Y, Michielsen P, Henrion J, et al. Haptoglobin phenotype 2-2 overrepresentation in Cys282Tyr hemochromatotic patients. J Hepatol. 2001 Dec;35(6):707–11.
Milet J, Dehais V, Bourgain C, Jouanolle AM, Mosser A, Perrin M, et al. Common variants in the BMP2, BMP4, and HJV genes of the hepcidin regulation pathway modulate HFE hemochromatosis penetrance. Am J Hum Genet. 2007 Oct;81(4):799–807.
Milet J, Le Gac G, Scotet V, Gourlaouen I, Theze C, Mosser J, et al. A common SNP near BMP2 is associated with severity of the iron burden in HFE p.C282Y homozygous patients: a follow-up study. Blood Cells Mol Dis. 2010 Jan 15;44(1):34–7.
Benyamin B, McRae AF, Zhu G, Gordon S, Henders AK, Palotie A, et al. Variants in TF and HFE explain approximately 40% of genetic variation in serum-transferrin levels. Am J Hum Genet. 2009 Jan;84(1):60–5.
de Tayrac M, Roth MP, Jouanolle AM, Coppin H, le Gac G, Piperno A, et al. Genome-wide association study identifies TF as a significant modifier gene of iron metabolism in HFE hemochromatosis. J Hepatol. 2015 Mar;62(3):664–72.
Benyamin B, Esko T, Ried JS, Radhakrishnan A, Vermeulen SH, Traglia M, et al. Novel loci affecting iron homeostasis and their effects in individuals at risk for hemochromatosis. Nat Commun. 2014 Oct 29;5:4926.
Stickel F, Buch S, Zoller H, Hultcrantz R, Gallati S, Osterreicher C, et al. Evaluation of genome-wide loci of iron metabolism in hereditary hemochromatosis identifies PCSK7 as a host risk factor of liver cirrhosis. Hum Mol Genet. 2014 Jul 15;23(14):3883–90.
Leggett BA, Halliday JW, Brown NN, Bryant S, Powell LW. Prevalence of haemochromatosis amongst asymptomatic Australians. Br J Haematol. 1990 Apr;74(4):525–30.
European Association For The Study Of The L. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010 Jul;53(1):3–22.
Marshall DS, Linfert DR, Tsongalis GJ. Prevalence of the C282Y and H63D polymorphisms in a multi-ethnic control population. Int J Mol Med. 1999 Oct;4(4):389–93.
Beutler E, Felitti V, Gelbart T, Ho N. The effect of HFE genotypes on measurements of iron overload in patients attending a health appraisal clinic. Ann Intern Med. 2000 Sep 5;133(5):329–37.
Steinberg KK, Cogswell ME, Chang JC, Caudill SP, McQuillan GM, Bowman BA, et al. Prevalence of C282Y and H63D mutations in the hemochromatosis (HFE) gene in the United States. JAMA. 2001 May 2;285(17):2216–22.
Phatak PD, Ryan DH, Cappuccio J, Oakes D, Braggins C, Provenzano K, et al. Prevalence and penetrance of HFE mutations in 4865 unselected primary care patients. Blood Cells Mol Dis. 2002 Jul-Aug;29(1):41–7.
Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T. Penetrance of 845G--> a (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002 Jan 19;359(9302):211–8.
Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005 Apr 28;352(17):1769–7.
Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008 Jan 17;358(3):221–30.
Gurrin LC, Osborne NJ, Constantine CC, McLaren CE, English DR, Gertig DM, et al. The natural history of serum iron indices for HFE C282Y homozygosity associated with hereditary hemochromatosis. Gastroenterology. 2008 Dec;135(6):1945–52.
Gurrin LC, Bertalli NA, Dalton GW, Osborne NJ, Constantine CC, McLaren CE, et al. HFE C282Y/H63D compound heterozygotes are at low risk of hemochromatosis-related morbidity. Hepatology. 2009 Jul;50(1):94–101.
Constantine CC, Anderson GJ, Vulpe CD, McLaren CE, Bahlo M, Yeap HL, et al. A novel association between a SNP in CYBRD1 and serum ferritin levels in a cohort study of HFE hereditary haemochromatosis. Br J Haematol. 2009 Oct;147(1):140–9.
Osterreicher CH, Datz C, Stickel F, Hellerbrand C, Penz M, Hofer H, et al. TGF-beta1 codon 25 gene polymorphism is associated with cirrhosis in patients with hereditary hemochromatosis. Cytokine. 2005 Jul 21;31(2):142–8.
Barton JC, Barton JC, Acton RT. Longer survival associated with HLA-A*03, B*14 among 212 hemochromatosis probands with HFE C282Y homozygosity and HLA-A and -B typing and haplotyping. Eur J Haematol. 2010 Nov;85(5):439–47.
Fletcher LM, Dixon JL, Purdie DM, Powell LW, Crawford DH. Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology. 2002 Feb;122(2):281–9.
Adams PC, Agnew S. Alcoholism in hereditary hemochromatosis revisited: prevalence and clinical consequences among homozygous siblings. Hepatology. 1996 Apr;23(4):724–7.
Powell EE, Ali A, Clouston AD, Dixon JL, Lincoln DJ, Purdie DM, et al. Steatosis is a cofactor in liver injury in hemochromatosis. Gastroenterology. 2005 Dec;129(6):1937–43.
Diwakaran HH, Befeler AS, Britton RS, Brunt EM, Bacon BR. Accelerated hepatic fibrosis in patients with combined hereditary hemochromatosis and chronic hepatitis C infection. J Hepatol. 2002 May;36(5):687–91.
Stuart KA, Busfield F, Jazwinska EC, Gibson P, Butterworth LA, Cooksley WG, et al. The C282Y mutation in the haemochromatosis gene (HFE) and hepatitis C virus infection are independent cofactors for porphyria cutanea tarda in Australian patients. J Hepatol. 1998 Mar;28(3):404–9.
Niederau C, Fischer R, Purschel A, Stremmel W, Haussinger D, Strohmeyer G. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology. 1996 Apr;110(4):1107–19.
Adams PC, Deugnier Y, Moirand R, Brissot P. The relationship between iron overload, clinical symptoms, and age in 410 patients with genetic hemochromatosis. Hepatology. 1997 Jan;25(1):162–6.
Delatycki MB, Allen KJ, Nisselle AE, Collins V, Metcalfe S, du Sart D, et al. Use of community genetic screening to prevent HFE-associated hereditary haemochromatosis. Lancet. 2005 Jul 23-29;366(9482):314–6.
Lin E, Adams PC. Biochemical liver profile in hemochromatosis. A survey of 100 patients. J Clin Gastroenterol. 1991 Jun;13(3):316–20.
Rahier J, Loozen S, Goebbels RM, Abrahem M. The haemochromatotic human pancreas: a quantitative immunohistochemical and ultrastructural study. Diabetologia. 1987 Jan;30(1):5–12.
Barton JC, Acton RT. Diabetes in HFE hemochromatosis. J Diabetes Res. 2017;2017:9826930.
Walsh CH, Wright AD, Williams JW, Holder G. A study of pituitary function in patients with idiopathic hemochromatosis. J Clin Endocrinol Metab. 1976 Oct;43(4):866–72.
McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab. 2005 Apr;90(4):2451–5.
Charbonnel B, Chupin M, Le Grand A, Guillon J. Pituitary function in idiopathic haemochromatosis: hormonal study in 36 male patients. Acta Endocrinol. 1981 Oct;98(2):178–83.
McNeil LW, McKee LC Jr, Lorber D, Rabin D. The endocrine manifestations of hemochromatosis. Am J Med Sci. 1983 May-Jun;285(3):7–13.
Van Deursen C, Delaere K, Tenkate J. Hemochromatosis and sexual dysfunction. Int J Impot Res. 2003 Dec;15(6):430–2.
Leichtmann-Bardoogo Y, Cohen LA, Weiss A, Marohn B, Schubert S, Meinhardt A, et al. Compartmentalization and regulation of iron metabolism proteins protect male germ cells from iron overload. Am J Physiol Endocrinol Metab. 2012 Jun 15;302(12):E1519–30.
El Osta R, Grandpre N, Monnin N, Hubert J, Koscinski I. Hypogonadotropic hypogonadism in men with hereditary hemochromatosis. Basic Clin Androl. 2017;27:13.
Kiely PD. Haemochromatosis arthropathy–a conundrum of the Celtic curse. J R Coll Physicians Edinb. 2018 Sep;48(3):233–8.
Richardson A, Prideaux A, Kiely P. Haemochromatosis: unexplained metacarpophalangeal or ankle arthropathy should prompt diagnostic tests: findings from two UK observational cohort studies. Scand J Rheumatol. 2017 Jan;46(1):69–74.
Hawthorne G, Richardson J, Osborne R. The assessment of quality of life (AQoL) instrument: a psychometric measure of health-related quality of life. Qual Life Res Int J Qual Life Asp Treat Care Rehab. 1999 May;8(3):209–24.
de Graaff B, Neil A, Sanderson K, Yee KC, Palmer AJ. Quality of life utility values for hereditary haemochromatosis in Australia. Health Qual Life Outcomes. 2016 Feb 29;14:31.
Adams PC, Speechley M. The effect of arthritis on the quality of life in hereditary hemochromatosis. J Rheumatol. 1996 Apr;23(4):707–10.
Ines LS, da Silva JA, Malcata AB, Porto AL. Arthropathy of genetic hemochromatosis: a major and distinctive manifestation of the disease. Clin Exp Rheumatol. 2001 Jan-Feb;19(1):98–102.
Walker RJ, Dymock IW, Ansell ID, Hamilton EB, Williams R. Synovial biopsy in haemochromatosis arthropathy. Histological findings and iron deposition in relation to total body iron overload. Ann Rheum Dis. 1972 Mar;31(2):98–102.
Heiland GR, Aigner E, Dallos T, Sahinbegovic E, Krenn V, Thaler C, et al. Synovial immunopathology in haemochromatosis arthropathy. Ann Rheum Dis. 2010 Jun;69(6):1214–9.
Sinigaglia L, Fargion S, Fracanzani AL, Binelli L, Battafarano N, Varenna M, et al. Bone and joint involvement in genetic hemochromatosis: role of cirrhosis and iron overload. J Rheumatol. 1997 Sep;24(9):1809–13.
Guggenbuhl P, Deugnier Y, Boisdet JF, Rolland Y, Perdriger A, Pawlotsky Y, et al. Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporos Int. 2005 Dec;16(12):1809–14.
Valenti L, Varenna M, Fracanzani AL, Rossi V, Fargion S, Sinigaglia L. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos Int. 2009 Apr;20(4):549–55.
Richette P, Ottaviani S, Vicaut E, Bardin T. Musculoskeletal complications of hereditary hemochromatosis: a case-control study. J Rheumatol. 2010 Oct;37(10):2145–50.
Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, et al. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. 2009 Mar;15(3):259–66.
Balogh E, Tolnai E, Nagy B Jr, Nagy B, Balla G, Balla J, et al. Iron overload inhibits osteogenic commitment and differentiation of mesenchymal stem cells via the induction of ferritin. Biochim Biophys Acta. 2016 Sep;1862(9):1640–9.
Handzlik-Orlik G, Holecki M, Wilczynski K, Dulawa J. Osteoporosis in liver disease: pathogenesis and management. Ther Adv Endocrinol Metab. 2016 Jun;7(3):128–35.
Parkash O, Akram M. Hereditary hemochromatosis. J College Phys Surg Pakistan: JCPSP. 2015 Sep;25(9):644–7.
Schwartz RA. Dermatologic manifestations of hemochromatosis. Medscape. 2019 April;22:2019.
Chevrant-Breton J, Simon M, Bourel M, Ferrand B. Cutaneous manifestations of idiopathic hemochromatosis. Study of 100 cases. Arch Dermatol. 1977 Feb;113(2):161–5.
McLaren CE, McLachlan GJ, Halliday JW, Webb SI, Leggett BA, Jazwinska EC, et al. Distribution of transferrin saturation in an Australian population: relevance to the early diagnosis of hemochromatosis. Gastroenterology. 1998 Mar;114(3):543–9.
Olynyk JK, Cullen DJ, Aquilia S, Rossi E, Summerville L, Powell LW. A population-based study of the clinical expression of the hemochromatosis gene. N Engl J Med. 1999 Sep 2;341(10):718–24.
Adams PC, Bhayana V. Unsaturated iron-binding capacity: a screening test for C282Y hemochromatosis? Clin Chem. 2000 Nov;46(11):1870–1.
Kowdley KV, Brown KE, Ahn J, Sundaram V. ACG clinical guideline: hereditary hemochromatosis. Am J Gastroenterol. 2019 Aug;114(8):1202–18.
Beaton M, Guyader D, Deugnier Y, Moirand R, Chakrabarti S, Adams P. Noninvasive prediction of cirrhosis in C282Y-linked hemochromatosis. Hepatology. 2002 Sep;36(3):673–8.
Crawford DH, Murphy TL, Ramm LE, Fletcher LM, Clouston AD, Anderson GJ, et al. Serum hyaluronic acid with serum ferritin accurately predicts cirrhosis and reduces the need for liver biopsy in C282Y hemochromatosis. Hepatology. 2009 Feb;49(2):418–25.
Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004 Jan-Feb;32(1):131–8.
Deugnier Y, Bardou-Jacquet E, Laine F. Dysmetabolic iron overload syndrome (DIOS). Presse Med. 2017 Dec;46(12 Pt 2):e306–e11.
Cadenas B, Fita-Torro J, Bermudez-Cortes M, Hernandez-Rodriguez I, Fuster JL, Llinares ME, et al. L-ferritin: one gene, five diseases; from hereditary Hyperferritinemia to Hypoferritinemia-report of new cases. Pharmaceuticals. 2019 Jan;23:12(1).
Morrison ED, Brandhagen DJ, Phatak PD, Barton JC, Krawitt EL, El-Serag HB, et al. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med. 2003 Apr 15;138(8):627–33.
Adhoute X, Foucher J, Laharie D, Terrebonne E, Vergniol J, Castera L, et al. Diagnosis of liver fibrosis using FibroScan and other noninvasive methods in patients with hemochromatosis: a prospective study. Gastroenterol Clin Biol. 2008 Feb;32(2):180–7.
Legros L, Bardou-Jacquet E, Latournerie M, Guillygomarc'h A, Turlin B, Le Lan C, et al. Non-invasive assessment of liver fibrosis in C282Y homozygous HFE hemochromatosis. Liver Int. 2015 Jun;35(6):1731–8.
Powell LW, Dixon JL, Ramm GA, Purdie DM, Lincoln DJ, Anderson GJ, et al. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med. 2006 Feb 13;166(3):294–301.
Adams PC. Implications of genotyping of spouses to limit investigation of children in genetic hemochromatosis. Clin Genet. 1998 Mar;53(3):176–8.
Delatycki MB, Powell LW, Allen KJ. Hereditary hemochromatosis genetic testing of at-risk children: what is the appropriate age? Genet Test. 2004 Summer;8(2):98–103.
Searle J, Kerr JFR, Halliday JW, Powell LW. Iron storage disease. In: MacSween PPA RNM, Scheuer PJ, Burt AD, Portmann BC, editors. Pathology of the liver. Edinburgh: Churchill Livingstone; 1994. p. 219–42.
Powell LW, Kerr JF. The pathology of the liver in hemochromatosis. Pathobiol Annu. 1975;5:317–37.
Deugnier YM, Charalambous P, Le Quilleuc D, Turlin B, Searle J, Brissot P, et al. Preneoplastic significance of hepatic iron-free foci in genetic hemochromatosis: a study of 185 patients. Hepatology. 1993 Dec;18(6):1363–9.
Adams PC, Speechley M, Kertesz AE. Long-term survival analysis in hereditary hemochromatosis. Gastroenterology. 1991 Aug;101(2):368–72.
Powell LW. Hemochromatosis: the impact of early diagnosis and therapy. Gastroenterology. 1996 Apr;110(4):1304–7.
Gochee PA, Powell LW, Cullen DJ, Du Sart D, Rossi E, Olynyk JK. A population-based study of the biochemical and clinical expression of the H63D hemochromatosis mutation. Gastroenterology. 2002 Mar;122(3):646–51.
Walsh A, Dixon JL, Ramm GA, Hewett DG, Lincoln DJ, Anderson GJ, et al. The clinical relevance of compound heterozygosity for the C282Y and H63D substitutions in hemochromatosis. Clin Gastroenterol Hepatol. 2006 Nov;4(11):1403–10.
Summers KM, Halliday JW, Powell LW. Identification of homozygous hemochromatosis subjects by measurement of hepatic iron index. Hepatology. 1990 Jul;12(1):20–5.
Falize L, Guillygomarc'h A, Perrin M, Laine F, Guyader D, Brissot P, et al. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology. 2006 Aug;44(2):472–7.
Adams P, Brissot P, Powell LW. EASL international consensus conference on haemochromatosis. J Hepatol. 2000 Sep;33(3):485–504.
Beaton MD, Adams PC. Prognostic factors and survival in patients with hereditary hemochromatosis and cirrhosis. Can J Gastroenterol. 2006 Apr;20(4):257–60.
Bardou-Jacquet E, Morcet J, Manet G, Laine F, Perrin M, Jouanolle AM, et al. Decreased cardiovascular and extrahepatic cancer-related mortality in treated patients with mild HFE hemochromatosis. J Hepatol. 2015 Mar;62(3):682–9.
Ong SY, Gurrin LC, Dolling L, Dixon J, Nicoll AJ, Wolthuizen M, et al. Reduction of body iron in HFE-related haemochromatosis and moderate iron overload (mi-iron): a multicentre, participant-blinded, randomised controlled trial. Lancet Haematol. 2017 Dec;4(12):e607–e14.
Cancado R, Melo MR, de Moraes BR, Santos PC, Guerra-Shinohara EM, Chiattone C, et al. Deferasirox in patients with iron overload secondary to hereditary hemochromatosis: results of a 1-yr phase 2 study. Eur J Haematol. 2015 Dec;95(6):545–50.
Ullrich H, Fischer R, Grosse R, Kordes U, Schubert C, Altstadt B, et al. Erythrocytapheresis: do not forget a useful therapy! Transfus Med Hemother. 2008;35(1):24–30.
Rombout-Sestrienkova E, Nieman FH, Essers BA, van Noord PA, Janssen MC, van Deursen CT, et al. Erythrocytapheresis versus phlebotomy in the initial treatment of HFE hemochromatosis patients: results from a randomized trial. Transfusion. 2012 Mar;52(3):470–7.
Rombout-Sestrienkova E, Winkens B, Essers BA, Nieman FH, Noord PA, Janssen MC, et al. Erythrocytapheresis versus phlebotomy in the maintenance treatment of HFE hemochromatosis patients: results from a randomized crossover trial. Transfusion. 2016 Jan;56(1):261–70.
Vanclooster A, van Deursen C, Jaspers R, Cassiman D, Koek G. Proton pump inhibitors decrease phlebotomy need in HFE hemochromatosis: double-blind randomized placebo-controlled trial. Gastroenterology. 2017 Sep;153(3):678–80 e2.
Bardou-Jacquet E, Philip J, Lorho R, Ropert M, Latournerie M, Houssel-Debry P, et al. Liver transplantation normalizes serum hepcidin level and cures iron metabolism alterations in HFE hemochromatosis. Hepatology. 2014 Mar;59(3):839–47.
Kowdley KV, Brandhagen DJ, Gish RG, Bass NM, Weinstein J, Schilsky ML, et al. Survival after liver transplantation in patients with hepatic iron overload: the national hemochromatosis transplant registry. Gastroenterology. 2005 Aug;129(2):494–503.
Yu L, Ioannou GN. Survival of liver transplant recipients with hemochromatosis in the United States. Gastroenterology. 2007 Aug;133(2):489–95.
Dar FS, Faraj W, Zaman MB, Bartlett A, Bomford A, O'Sullivan A, et al. Outcome of liver transplantation in hereditary hemochromatosis. Transplant Int. 2009 Jul;22(7):717–24.
Aranda N, Viteri FE, Montserrat C, Arija V. Effects of C282Y, H63D, and S65C HFE gene mutations, diet, and life-style factors on iron status in a general Mediterranean population from Tarragona. Spain Ann Hematol. 2010 Aug;89(8):767–73.
Waterlot Y, Cantinieaux B, Hariga-Muller C, De Maertelaere-Laurent E, Vanherweghem JL, Fondu P. Impaired phagocytic activity of neutrophils in patients receiving haemodialysis: the critical role of iron overload. Br Med J. 1985 Aug 24;291(6494):501–4.
de Sousa M. Immune cell functions in iron overload. Clin Exp Immunol. 1989 Jan;75(1):1–6.
Pinto JP, Dias V, Zoller H, Porto G, Carmo H, Carvalho F, et al. Hepcidin messenger RNA expression in human lymphocytes. Immunology. 2010 Jun;130(2):217–30.
Porto G, Cruz E, Teles MJ, de Sousa M. HFE related hemochromatosis: uncovering the inextricable link between iron homeostasis and the immunological system. Pharmaceuticals. 2019 Aug;22:12(3).
Girelli D, Trombini P, Busti F, Campostrini N, Sandri M, Pelucchi S, et al. A time course of hepcidin response to iron challenge in patients with HFE and TFR2 hemochromatosis. Haematologica. 2011 Apr;96(4):500–6.
Worthen CA, Enns CA. The role of hepatic transferrin receptor 2 in the regulation of iron homeostasis in the body. Front Pharmacol. 2014;5:34.
Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003 Jan;33(1):21–2.
Mayr R, Janecke AR, Schranz M, Griffiths WJ, Vogel W, Pietrangelo A, et al. Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol. 2010 Nov;53(5):941–9.
Jolla L. La Jolla Pharmaceutical Company Announces Positive Results from Pre-Specified Interim Analysis of Phase 2 Study of LJPC-401 in Patients with Hereditary Hemochromatosis 2019 [11/05/2019].
Lesjak M, Hoque R, Balesaria S, Skinner V, Debnam ES, Srai SK, et al. Quercetin inhibits intestinal iron absorption and ferroportin transporter expression in vivo and in vitro. PLoS One. 2014;9(7):e102900.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer-Verlag London Ltd., part of Springer Nature
About this chapter

Cite this chapter
Bergasa, N.V. (2022). Hemochromatosis. In: Bergasa, N.V. (eds) Clinical Cases in Hepatology. Springer, London. https://doi.org/10.1007/978-1-4471-4715-2_11
Download citation
DOI: https://doi.org/10.1007/978-1-4471-4715-2_11
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-4714-5
Online ISBN: 978-1-4471-4715-2
eBook Packages: MedicineMedicine (R0)