
Abstract
Nanoscale systems have emerged in the past two decades as attractive platforms for delivering nucleic acids in vivo while performing other therapeutic or diagnostic functions, though their full potential for improving human health has yet to be realized in the clinic. Bioengineering techniques have been crucial for modifying and optimizing synthetic and viral delivery systems to include drugs, imaging agents and targeting moieties as well as reducing toxicity effects and increasing delivery efficiency and specificity. Directed delivery technologies can complement these nanoscale systems to localize therapy in vivo. The use of nucleic acid analogs can also enhance therapeutic efficacy under ideal circumstances. This chapter will review some of the recent developments in RNA and DNA delivery research with a focus on progress toward human therapies, the challenges that have been encountered, and the engineering approaches that have been employed. In addition to on-going work on the optimization of delivery systems, three challenging areas are identified: (1) the development of heterogeneous, three-dimensional microenvironments for testing delivery systems, (2) imaging approaches to understand the dynamic interactions of systems from administration through delivery in the human population, and (3) development and translation of directed technologies capable of enhancing delivery in a clinical setting and producing a sustained therapeutic effect.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The ability to deliver nucleic acids to specific sites within the body is a significant engineering problem, but one that, when fully addressed, will radically change how diseases and conditions are treated. Only a small fraction of the discoveries made in the laboratory have progressed to clinical trials because of challenges in developing efficient, sensitive, and specific delivery systems. Ideally, a delivery system is taken up only by the targeted cells and does not cause unwanted side effects, the nucleic acid cargo is not degraded before reaching its intracellular target, and there is sufficient delivery to produce a sustained therapeutic effect. However, in a complex, heterogeneous human population, all of these characteristics are hard to achieve reproducibility.
Of the more than 1,800 gene therapy clinical trials started by the end of 2012, more than two-thirds have used viral vectors, but fewer than 5 % have progressed beyond the equivalent of phase II of the US Food and Drug Administration (FDA) regulatory process. Two-thirds of the studies have been carried out in the United States and a similar fraction has focused on cancer-related indications [1]. The number of small interfering ribonucleic acids (siRNA) clinical trials is more modest at around 30 studies, although for this type of nucleic acid, the use of synthetic vectors outnumbers the use of viral vectors [2].
Synthetic and viral delivery systems offer contrasting advantages. In general terms, viral vectors promise excellent performance characteristics and make better use of multiple biological systems but have significant safety concerns, whereas synthetic vectors have better safety profiles, but much lower efficiency. Much of the work over the past two decades has focused on addressing these safety and efficacy concerns and optimizing one or two steps of what is a multi-step delivery process. However, beyond discovery-driven research and individual step optimization, it is clear that by taking a systems approach to delivery, there are multiple paths to more widespread clinical reality with safe and effective multifunctional nanoscale delivery systems tailored to individual conditions and therapeutic agents.
Delivery systems are not absolutely required for deoxyribonucleic acid (DNA) to generate a therapeutic effect in vivo. Local delivery of naked DNA into skeletal muscle [3], heart muscle [4], and into tumors [5] is sufficient to induce a response. Indeed, naked or plasmid DNAs (pDNA) are the third most common delivery approach used in gene therapy clinical trials and are three times more common than liposome systems [1]. However, for RNA interference (RNAi) therapies, nucleases will degrade up to 70 % of naked siRNA molecules within minutes of systemic administration [6], requiring chemical modification or a protective carrier for efficient use. To increase their efficiency and effectiveness, they need to be protected from capture and degradation before reaching their target, to have their anionic backbone shielded for efficient cell uptake, and to be targeted to cells of interest without provoking an immune response or increasing systemic clearance. Although at a very early stage, the potential to rationally design and engineer multifunctional vectors is particularly exciting and a step toward making nucleic-acid-based therapy a routine clinical tool.
The growing availability of tools to engineer delivery systems at the nanometer level, commensurate with the persistence length of nucleic acids, has led to a deeper understanding of how the unique properties of matter can be exploited in this size range, as well as enhancing in vivo delivery. The size, shape, charge, composition, mechanical stiffness, and surface functionalization of delivery systems influence both the physiological and cellular barriers to delivery. For example, particles above 200 nm in size are susceptible to phagocytosis in the reticular endothelial system, and particles below 100 nm can get caught in Kupffer cells, while particles below 5 nm are removed by glomerular filtration. Particles below 40 nm can freely diffuse in a cell’s cytoplasm, while below 5 nm, they can cross the capillary endothelium and particles below 1 nm in size can translocate the cell membrane. Most nanoscale delivery systems fall in the 5–200 nm range and, after systemic administration, circulate in the blood stream until cleared or moved into the extracellular space. If they come in contact with a target cell, they can enter the cell through a number of mechanisms. For example, if they enter by endocytosis, the nucleic acids need to make a timely escape before degradation in a lysosome.
All of these interactions can be tuned by engineering the delivery system’s physical, chemical, and biological characteristics. An ideal multifunctional nanoscale delivery system efficiently delivers its nucleic acid cargo to the desired site and effectively releases the cargo at the desired time, while performing additional functions, such as simultaneous drug delivery, providing imaging contrast, or environmental sensing. For efficient and effective delivery, the system should include site-specific targeting and the ability to disrupt the normal cell pathways to deliver its nucleic acid payload. While doing this, the delivery system and payload must be carefully tuned not to stimulate the immune system, create toxicity, or be captured or degraded by the circulatory system. In this chapter, we will review recent research developments in both viral and nonviral vectors, focusing on progress toward the clinical application of these nanoscale delivery systems.
2 Delivery Systems
The delivery of nucleic acids in vivo requires a safe, stable, sensitive, and specific approach. In order to prevent degradation, to reduce side effects, and promote efficient delivery, nucleic acids can be encapsulated, bound, or complexed into nanometer-sized constructs. The physical and chemical properties of these constructs can be tuned to change clearance rates, uptake processes, and intracellular fate and to account for delivery method. Although systemic injection is the most common delivery method, physical methods such as stereotactic injection and ballistic delivery as well as the use of optical, magnetic, electric, and ultrasound fields to generate force and disrupt the cell membrane can be used to enhance uptake. Effective delivery of nucleic acids can also be achieved by topical administration [7], nasal delivery [8], and direct injection into the brain [9].
2.1 Viral Delivery Systems for Nucleic Acids
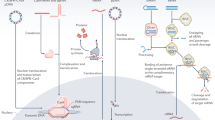
Viruses are efficient vehicles for delivering nucleic acids to a wide range of organisms. Viruses have evolved natural mechanisms to enter their host cells and are capable of delivering kilobase-long nucleic acid sequences to a wide range of target cells. These are major advantages in vivo, where the goal is to deliver the nucleic acids to specific targets without causing significant harm to the host. As such, viral vectors have been designed to be stable, achieve efficient transduction, and have high viral titer and sustained expression of the heterologous nucleic acids of interest (transgenes). Figure 18.1 illustrates general schemes for the design of three commonly used viral vectors. Despite the many advantages of viral delivery vectors, they are known to be capable of producing several adverse effects, which include immune reactions, oncogene activation, and nonspecific cellular tropism [10].

Viral vector designs
2.1.1 Retroviral Vectors
Retroviruses are enveloped RNA viruses of 80–130 nm in diameter with a genome size of 8–11 kilobases (kb) [11]. Retroviral genomes are made up of gag, pol, env, and 5′ and 3′ long terminal repeats (LTR). Retroviral vectors are rendered replication defective by deleting the structural, envelope, and enzymatic genes, and instead, the desired transgenes are inserted [12]. These replication-defective vectors, or proviruses, are incapable of continuing to spread after the initial infection of the target cells. The initial round of vector replication is dependent on several cis acting elements from the viral genome, including a promoter and polyadenylation signal, reverse transcription signal, transfer RNA (tRNA), primer binding site (PBS), and a polyurine tract, to initiate complementary DNA (cDNA) synthesis. Cellular tRNA binds to the PBS to initiate reverse transcription. Vectors containing the transgene are then packaged into viral particles as directed by a packaging signal (Ψ), PBS, and LTRs. Helper viruses, or plasmids, that carry the viral genes gag/pol and env, but lacking the packaging signal ψ [13], are required to express the viral proteins needed to produce vector-containing infectious viral particles. The helper function can also be provided by transfecting helper plasmids into cell cultures to produce helper cells [14]. The process of expressing the transgene in the retroviral vectors and delivering them to the target cells is called transduction [13].
Entry of the viral vectors into targeted cells is an essential first step in viral-vector-based gene delivery. Gaining entry into cells involves the interaction of the viral envelope with the host cell receptor. For lentiviral vectors, which are derived from human immunodeficiency virus type 1 (HIV1) [15], this involves a two-stage process [16]. Initially, the glycoprotein 120 (gp120) subunit of the envelope protein interacts with the target cell receptor CD4. The resulting conformational changes in gp120 allow binding with the co-receptors CXCR4 or CCR5, which belong to the chemokine family [17, 18]. Subsequently, another envelope protein subunit, gp41, signals fusion that is then followed by the release of viral capsid into the cytoplasm of the target cells. In addition to viral entry, the envelope plays an important role in determining cellular tropism by a technique called pseudotyping, where a viral vector is combined with foreign envelope proteins. Cellular tropism enhances the range of susceptible cells for the viral vectors. The lentiviral vector envelope can be substituted by the vesicular stomatitis virus G glycoprotein (VSV-G). This pseudotyping strategy has been shown not only to increase the range of possible target cell types, but also to improve stability and increase viral titers [13, 19]. Gammaretroviral vectors, e.g., murine leukemia viral vectors, transduce only dividing cells [20] and have a narrow range of cells that can have genetic material transferred to them. However, these vectors have ready access to the host genome because the nuclear membrane is removed during cell division. In contrast, lentiviral vectors transduce a wide variety of cell types, both dividing and nondividing cells such as neurons and B and T cells, albeit with varying degree of transduction efficiency [19, 21, 22].
A distinguishing feature of retroviruses is their ability to integrate into the host genome, resulting in sustained expression of the transgene. However, integration presents challenges in vector design if the vector is inserted at a site where the regulatory elements in the vector result in transcriptional activation of oncogenes [23]. Such insertional mutagenesis was the underlying cause for the adverse events in an early human gene transfer trial involving patients with severe combined immunodeficiency defect (SCID) [24]. Some patients involved in this trial subsequently developed T-cell leukemia. For lentiviral vectors, the risk of insertional mutagenesis can be reduced by designing self-inactivation (SIN) vectors that lack the viral transcriptional control elements, promoter/enhancer, and, thus, reduce the possibility of activating an oncogene located adjacent to the vector integration site [25]. Strategies have also been developed to target specific chromosomal insertion sites for the transgenes [26]. Research has also led to approaches to avoid integration. Because integration requires integrase attachment on the LTRs, mutating the integrase gene or modifying the attachment sequences of the LTRs may eliminate integration [27]. The transgene can also be expressed as episomal DNA without integration into the host genome. Such expression can occur for a relatively long duration in nondividing cells, such as retina cells [28, 29], or transiently in dividing cells.
Gammaretroviral vectors can accommodate up to 7 kb sequences [30, 31], while lentiviral vectors can accommodate larger transgenes, up to 10 kb [25]. In addition to the length of the inserted sequences, vector design must also consider stability issues. Retroviral vectors have half-lives of a few hours at 37 °C and up to a few months at −80 °C. A contributing factor to the instability is the loss of function of the envelope protein. Another factor to consider is viral titer. Lentiviral vectors can be produced at 107 transducing units/ml. The titer can be increased to 109–1010 transducing units/ml by ultracentrifugation [25]. However, titers may decrease when larger transgenes are inserted.
The first human gene transfer was conducted in 1989, as an immunotherapy to treat patients with advanced melanoma, using retroviral vectors and tumor-infiltrating lymphocytes [32, 33]. Targeted gene therapy began in 1990 with the adenosine deaminase gene for treatment of SCID [34]. Since then, numerous disorders have been treated by gene therapy [35]. They include inherited genetic disorders that involve autosomal X-linked recessive single genes or some autosomal dominant genes, and some acquired diseases, such as cancer, vascular diseases, neurodegenerative disorders, and inflammatory diseases. The first clinical trial using lentiviral vectors was approved in 2002 [36]. Currently several clinical trials are underway in which lentiviral vectors have been used to: (1) treat HIV infection, (2) transduce neuronal cells of the central nervous system for the treatment of Parkinson’s disease [37], and (3) deliver beta-globin gene for beta-thalassemia treatment [38].
There are safety concerns with the use of retroviral vectors for gene delivery. Both gammaretroviral and lentiviral vectors have the potential to illicit an immune response from the host. Immune reactions toward the viral vector result in rejection of all expressing cells. However, it should be noted that immune response is a beneficial outcome if lentiviral vectors are used to deliver genetic vaccines to treat HIV infection. Another serious safety issue is homologous recombination, which occurs when the packaging virus recombines with the vector to produce replication competent viruses. These viruses can produce harmful infections, and additionally gammaretroviruses can also cause cancer if an oncogene is activated by insertional mutagenesis [39]. In contrast, there is no evidence that lentiviral vectors result in oncogene activation through insertional mutagenesis. Another concern is the spread of the vector beyond the intended target tissue, which may cause persistent unwanted biological activity or unpredictable responses. All of these concerns have increased caution in the use of retroviral vectors although their advantages can be overwhelmingly attractive.
2.1.2 Adeno-Associated Viral Vectors
Adeno-associated virus (AAV) is a small, single-stranded DNA virus of 4,681 nucleotides (nt). The wild-type (wt) genome is made up of two genes, rep and cap, that encode four replication proteins and three capsid proteins, respectively. The three capsid proteins, Vp1, Vp2, and Vp3, are produced from the same open reading frame, but from differential splicing (Vp1) and alternative translational start sites (Vp2 and Vp3, respectively) [40]. Vp3 is the most abundant subunit in the virion and interacts with the host cell receptor. Recognition of Vp3 by the receptor determines cellular tropism of the virus. A phospholipase domain, essential for viral infectivity, has been identified in the unique N-terminus of Vp1 [41, 42]. The functional significance of Vp2 remains to be resolved. The viral genome is flanked on either side by 145-bp inverted terminal repeats (ITR) [43]. With the deletion of most of the viral genes, transgenes can be inserted into the cis 145-bp ITRs, to create a recombinant AAV (rAAV). The transgene can be expressed in the transduced cells without integration into the host cell genome and persists as episomal DNA [44]. The small genome size poses a limitation for AAV vector in delivering large transgenes. However, recent studies have shown delivery of genomes up to 6 kb, although delivery of these larger payloads was less efficient [43, 45]. Typically, transgenes of up to 5 kb are delivered. Strategies have been developed to improve efficiency, for example by splitting the vectors, with each vector containing approximately half of the transgene within the same cell. This approach, however, is still limited by viral packaging capacity. Additionally, cells have to be infected with different viral particles to achieve full transgene expression.
Transcapsidation is an approach to improve the packaging capacity, increase tissue tropisms, and transduction efficiency, where more than 100 different capsids from different serotypes can be exchanged to produce dozens of rAAV containing the same genome [45]. AAV serotype 2 is the best-studied AAV and was the first one used for gene transfer. However, vectors derived from alternative serotypes, e.g., 1, 4, 5, and 6 have been packaged with the same vector genome, but different viral capsids to improve efficiency and tropisms [46, 47].
rAAVs have been used in gene therapies in human muscle, liver, lung, central nervous system [48], and recently in the retina [49]. In a gene therapy trial of Parkinson’s patients, there was an improvement in the Unified Parkinson’s Disease Rating Scale (UPDRS) after 6 months for patients who received the glutamic acid decarboxylase (GAD) gene carried by an AAV2 vector that was delivered to the subthalamic nucleus [50]. rAAV2 vector carrying the gene encoding retinal pigment epithelium-specific 65-kilodalton protein (RPE65) has been used in a gene therapy trial to treat severe retinal dystrophy [51]. These results show promise, but further clinical trials are needed to demonstrate clinical significance.
2.2 Nonviral Delivery Systems
There has been a four-decade long history of nonviral delivery system development, prompted by on-going safety concerns associated with viral vectors. DNA transfection protocols emerged in the late 1970s, and liposome-based gene delivery strategies were first reported in the 1980s, though the field really took off in the late 1990s with the discovery of siRNA which required the use of a delivery system in vivo. Nonviral delivery systems are attractive because they typically have lower immunogenicity, lower toxicity, and their production can be easily scaled for widespread clinical use; but they also have disadvantages. These disadvantages lead to lower efficiency at each stage of the delivery process, with an overall efficiency <0.1 %.
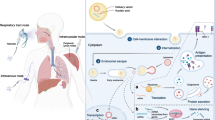
There are a number of basic engineering considerations in the design of all synthetic vectors. They should be nontoxic, nonimmunogenic, and biodegradable, protect their nucleic acid cargo against degradation, and efficiently deliver their cargo to both the cells of interest and the desired intracellular target. Optimizing the efficiency of nonviral delivery has been the focus of much research, which includes understanding the best targeting approach for selecting the cells of interest, how the stability of formulations change after administration, and how to get efficient endosomal release. Some of the barriers to delivery are illustrated in Fig. 18.2 and include: (1) formation of nucleic acid complex, (2) entry of the complex into the cells of interest, (3) endosomal escape of the nucleic acids, (4) dissociation of the complex, and (5) transport of the nucleic acid to site of action [52]. In contrast to viral vectors, synthetic vectors are poorly optimized to take advantage of existing cellular architecture. In particular, they cannot control endosomal release, have low diffusion rate, and are unable to take advantage of active transport mechanisms. Ideally for DNA, delivery is perinuclear, and for siRNA, it is targeting of the RNA-induced silencing complex (RISC). However, the rate-limiting steps of delivery are not fully understood, and there is not a good mechanistic understanding of how to rationally optimize loading rate.

Systemic and cellular barriers to delivery in vivo
A further challenge has been that the optimization of synthetic vectors for efficient in vitro cell culture delivery rarely translates to similar in vivo results. Monolayers of selected cells, in a carefully controlled environment, are not representative of the complex in vivo environment. The in vivo environment is a three-dimensional heterogeneous structure in an extracellular matrix, complete with enzymes, different cell morphologies, and a circulatory and immune system that cannot be easily replicated in vitro. In the worst case, some parameters can be optimized in vitro based on misleading effects which do not occur in vivo. In cell culture, size can be an advantage due to sedimentation efficiency, but in vivo smaller (less than 40 nm) particles are favored because of faster diffusion rates [53]. Another example is that positively charged vectors are beneficial in vitro for binding nucleic acids and enhancing uptake by interacting with the negatively charged cell membrane. However in vivo, negatively charged serum will bind to the vector, significantly reducing effectiveness. Some of the common designs for synthetic nanoscale delivery systems are illustrated in Fig. 18.3 and described in more detail in the following sections.

Synthetic delivery system designs
2.2.1 Bioconjugation
Bioconjugation is a technique for improving delivery by covalently linking nucleic acids to bioactive targeting agents. Bioconjugation to lipids, sugars, polyethylene glycol (PEG), and peptides is in principle more attractive than delivery with cationic liposomes and cationic polymers due to the advantages of smaller-size and enhanced pharmacokinetics. They easily clear systemic circulation and, thus, can be useful for targeting oligonucleotides to cells that are not in direct contact with the vasculature such as hepatocytes. Bioconjugates of oligonucleotides have been used to deliver oligonucleotides to treat liver fibrosis [54]. Lipids, which are common targeting agents, have also been conjugated with nucleic acids for delivery of DNA, antisense oligonucleotides, and siRNA. Development of bioconjugated therapeutic drugs is more advanced; for example, site-specific anticancer drugs have been designed to consist of the hydrophobic drug linked to the nucleoside analog to form an amphiphilic bioconjugate. After cell uptake, the inactive bioconjugates or prodrugs are activated only at the target site by pH change or enzymatic cleavage.
2.2.2 Cell-Penetrating Peptides
Cell-penetrating peptides (CCP) are short peptides, typically arginine or lysine rich, that enhance cell uptake of an attached cargo [55]. Because of their cationic nature, they bind to the anionic glycan moieties of the extracellular matrix through electrostatic interactions [56]. Their ability to penetrate the cell membrane has been exploited to deliver large molecules, such as drugs, into cells. The positively charged CPPs are also useful for delivering negatively charged DNA or siRNA via electrostatic interactions. However, electrostatic interactions require excess CPPs, in approximately a 10:1 ratio, to maintain an overall positive charge in order to bind to glycans. A major shortcoming of CPPs as a drug delivery vehicle is their lack of specificity, but approaches have been developed to overcome this shortcoming and enhance target cell specificity. For neutral molecules, such as antisense oligomers and peptide nucleic acids (PNA), CPPs can be used as a delivery vehicle by covalent chemical conjugation. There are inherent disadvantages to using this conjugation process because the cargo may be altered as a result. Alternatively, as a way to combat this disadvantage, CPPs can form a complex with PNA, via complementary sequence hybridization, and the resulting conjugate is then used to deliver antisense oligomers to the cells.
Major families of CPPs studied in the published literature to date include: (1) penetratin, a drosophila antennapedia-derived peptide, (2) Tat peptide derived from HIV, and (3) transportan peptides [57]. These CPPs have been used to deliver therapeutic peptides that target tumor tissues by receptor-mediated endocytosis [58]. For example, the N-terminus of elastin-like polypeptides that are responsive to thermal control can be fused to CPPs. Tumor cells are specifically and selectively heated to 40–43 °C by microwave, radio-frequency, or high-intensity focused ultrasound. At these elevated temperatures, the thermally responsive elastin-like polypeptide delivery complex along with the therapeutic peptides and are taken up by tumor cells.
2.2.3 Liposomes
Liposomes are artificial vesicles composed of a lipid bilayer, typically in the size range of 50 nm to several microns, which self-assemble to entrap a liquid core. The surface chemistry, size, and charge of the liposome can be easily tuned through different preparation methods, and in general, they have good biocompatibility and pharmacokinetic profiles for a range of cargos. Liposomes can be fabricated with multiple concentric bilayers, with a single bilayer enclosing a liquid core, or they can form a solid or a nanostructured lipid nanoparticle [59]. Lipid nanoparticles can be fabricated down to 50 nm in size, and, as long as the formation of a crystalline structure is prevented through a blend of liquid and solid phases, there is efficient cargo loading and improved stability. However, the lipophilic core of solid particles is in general not attractive for the delivery of nucleic acids alone.
Liposomes have gained the most attention among the nonviral delivery systems because of their flexibility, biocompatibility, and tunability as well as their low toxicity, immunogenicity, and biodegradability in vivo. Two major engineering advances have helped push liposomal drug delivery products to commercial and clinical success: (1) the modification of lipids to bypass the reticular endothelial system and promote targeting, and (2) efficient cargo loading processes.
Since their first use for delivery of nucleic acids, many different types of liposomes have been developed and tested, with cationic lipids emerging as a preferred choice, often in combination with other “helper” lipids. The positively charged head groups of cationic lipids form lipoplexes with negatively charged nucleic acids, giving up to 100 % loading efficiency under the right concentrations and ratios. Endosomal escape is also primarily mediated by the cationic lipids that are believed to destabilize the endosome by forming cationic–anionic pairs with anionic lipids of the endosome membrane. This process can be enhanced by engineering the geometry of the cationic lipids and the choice of helper lipids [60].
Liposome-based delivery systems have a number of advantages, including the ability to transport large pieces of DNA, and a degree of enzyme protection in the lipoplex. Other advantages include low immunogenicity, they can be modified easily to target specific cells and their production can be scaled easily and relatively inexpensively. These advantages are offset by a number of limitations, including low overall delivery efficiency, challenges in encapsulating mixed cargoes, burst release of cargo rather than controlled release, poor storage stability, and lack of controlled release in the intracellular region of interest. For DNA delivery, ideally, this release is in the perinuclear space and the DNA can be trafficked across the nuclear envelope for efficient delivery, while for siRNA it would be close to the RISC.
Engineering of the liposome surface or the encapsulated space can address some of these limitations and increase functionality. Surface modification with PEG can provide a degree of stealth and increase circulation half-life [61], while incorporating antibodies, peptides, aptamers, affibodies, and vitamins can increase targeting efficiency [62], though it remains to be seen whether targeting robustly results in improved efficiency in humans. Mixing in helper lipids can improve fusogenicity by promoting fusion and by causing membrane destabilization [63]. Furthermore, the use of environmental triggers can increase control [64]. For example, lyso-lecithin formulations, which are thermosensitive, have progressed to clinical trials for treatment of breast and liver cancer. Photosensitive liposomes are being tested in animals and magnetosensitive liposomes are in preliminary development [59]. Environmental sensitivity can be added through enzyme-cleavable lipids and pH-sensitive lipids, both of which destabilize the liposome. Use of bioactive lipids, such as ceramide, has also been explored for tumor targeting in murine models [65]. A recent review covers many of the challenges and opportunities related to the surface chemistry of liposomes [59].
Despite nearly 50 years of research, attractive properties and many in vivo demonstrations, few liposome delivery systems have reached the market. To illustrate this point, liposomes have great potential as vehicles for DNA and peptide-based vaccines because their formulation can be adapted to protect multiple cargoes, target specific tissues, perform an immunostimulating and adjuvant role, and tuned for both humoral and cellular immune responses [66]. However, liposomes are weaker on commercially significant parameters like shelf-stability, cost-effectiveness, and the ability to scale up production reproducibly. Solid and nanostructured lipid particles that compose different lipids potentially do not share all of these weaknesses, but introduce other concerns like aggregation or coagulation.
Several engineering challenges remain for the more widespread use of liposomal delivery. Many of the studies to optimize delivery have been based on empirical observations; however, with the increasing availability of proteomic tools to understand the arrangement of transportation complexes and super-resolution microscopy to study trafficking events, a more mechanistic understanding is increasingly possible [67].
2.2.4 Polymers
Polymer nanoparticles can be synthetized easily in a wide range of sizes from 10 nm to several microns, and in a range of designs, including spherical and core–shell structures. Many families of polymers have been explored for nucleic acid delivery, including synthetic and natural polymers [68]. In principle, there are three ways in which polymer nanoparticles can be used for delivery: (1) by direct conjugation of the therapeutic agent with the polymer, (2) associating the agent with polymer through an electrostatic or hydrophobic interaction, or (3) by encapsulation of the agent. The most common route for nucleic acid delivery is by the formation of association complexes between the anionic backbone of the nucleic acid and a cationic polymer. The use of high buffering capacity moieties like polyanimes, so-called proton sponges, or the use of pH-sensitive components in the range pH 5–7 can also be used to aid endosomal escape to increase efficiency once captured. Furthermore, the addition of hydrophilic polymers like PEG can provide steric stability and minimize interactions in the physiological environment and reduce immune stimulation. This has driven researchers to explore heteropolymers and polymer-liposome complexes to identify efficient nonimmunogenic formations and has led to more than 35 combinations being tested so far in vivo [68].
The low molecular weight polycationic polymers, such as the polyethylene imines (PEI), are among the most commonly studied because of their small size, good association with nucleic acids, their high buffering capacity, and well-controlled chemistry. PEI/nucleic acid complexes (polyplexes) have good stability and can complex well with both siRNA and DNA, but their unmodified use is limited by stability and toxicity issues [69]. Polyarginine [70] and polylysine [71] are both lower toxicity alternatives but require additional modification for efficient in vivo use.
Naturally occurring polymers including albumin, collagen, gelatin, and chitosan have also been considered as delivery vehicles [72]. The degradation of these polymers in the presence of particular enzymes makes them attractive for controlled release. Chitosan is among the best studied and, with modification, may be attractive for oral delivery [73].
Advances in polymer chemistry have led to a number of designs incorporating functional components for enhancing efficiency and functionality, including redoxable di-sulfides, pH labile linkers, polythioketals sensitive to reactive oxygen species, and pH-sensitive hydrazone linkers [68]. The sensitivity of these smart polymers is likely to broaden over the next decade to include a wider range of physicochemical characteristics and biological processes. For controlled release of multiple therapeutic agents, the porosity of polymer nanoparticles can be tuned by formulation and manufacturing and has led to a resurgence of interest in biodegradeable polymers like the synthetic copolymer polylactide–polyglycolide (PLGA), which can be mixed with PEI for delivery of siRNA in vivo [74]. Polymeric micelles, hydrogel nanoparticles, and other protein-based nanoparticles have also been explored for drug delivery [75] and when used in combination with cationic polymers may offer some advantages for multifunctional delivery.
2.2.5 Dendrimers
Dendrimers are highly branched three-dimensional synthetic macromolecules, which can be produced with well-defined sizes in the range of 1–10 nm and with low dispersity. Higher generations of dendrimers resulting in the size range of 5–20 nm are favored for in vivo work because of efficient kidney filtering below 5 nm and high positive charge densities potentially creating toxicity issues and endocytosis efficiency drops as the size approaches 100 nm [76]. Their globular structure allows loading of nucleic acids and other cargos onto the outside surface via covalent bonding or electrostatic interactions to form dendriplexes, often through the use of amino groups grafted onto the end of the dendrimer branches. Poly(amidoamine) (PAMAM) dendrimers are the best studied, although they require chemical modification to reduce toxicity in vivo [77].
The small size of dendrimers makes them attractive for targeting the brain and deep tissues [76]. There have been some demonstrations of their use to deliver DNA [78] and siRNA [79] in vivo, although in vivo performance does not always match in vitro promise. By optimizing the choice of generation and surface chemistry, dendrimers do offer a potentially interesting route to cross the blood–brain barrier and deliver nucleic acid therapies to the brain.
2.2.6 Nanoparticles
The unique properties of materials in the nanometer to micron size range have provoked a lot of interest among physical and life scientists. For life scientists in particular, five broad classes of particles have emerged as the most studied because of their properties: (1) small particles of iron oxide, (2) quantum dots, (3) silica particles, (4) gold particles, and (5) carbon nanoparticles. The ability to design and engineer these particles to modulate their interactions with biological molecules, to carry a therapeutic cargo, and to possess characteristics favorable for imaging have led to optimism that nanometer-sized platforms can be used as multifunctional theranostics [80]. However, concerns about cytotoxicity and organ accumulation have dampened enthusiasm for the in vivo use of some of these materials.
Nucleic acids can be electrostatically immobilized on the surface of nanoparticles and protected from enzyme degradation using cationic polymers, such as the polyethylenimines or polyallylamines functionalized onto the surface of the particle. Functionalization with a mixture of polymers can also provide a degree of biocompatibility and stability in vivo. Much of the research work so far on nanoparticles has focused on in vitro and rodent studies, although to progress to human studies will require a more judicious choice of surface decoration and a better understanding of the kinetics of these particles and their nucleic acid cargoes. Some of the multifunctional imaging and delivery combinations for nanoparticles are illustrated in Table 18.1 and described in more detail in the following sections.
2.2.6.1 Small Particles of Iron Oxide Nanoparticles
Small particles of iron oxide nanoparticles (SPIONs) are nanometer-sized particles of superparamagnetic magnetite and maghemite. These properties make them attractive for T2 magnetic resonance imaging (MRI), for targeting using a magnetic field and for hyperthermia. The biocompatibility and excellent in vivo imaging characteristics have led to widespread use of SPIONs for diagnostic imaging and for decorating then with different moieties, such as dyes, polymers, and peptides. Although there have been concerns about the off-target impact of these particles, a noteworthy study compared uptake of SPIONs decorated with a near-infrared dye, siRNA, and membrane translocation peptides in a mice tumor model using in vivo MRI and ex vivo optical imaging and found that there was no indication of an inflammation or cytotoxicity response [81]. Plasma DNA can also be delivered in vivo using SPIONs and imaged using MRI, fluorescence, and transmission electron microscopy (TEM) [82]. The multifunctional nature of SPIONs and their flexible surface chemistry has enabled target refinement and improvements in stability, leading to higher efficiency, as well as the delivery of a combination of anticancer drugs and siRNA in vivo [83].
2.2.6.2 Quantum Dots
At nanometer scales, semiconductor materials can possess very attractive photophysical properties, including tunable fluorescence, resistance to photobleaching, and long-term stability. The hydrophobic surface can be functionalized with polymers and decorated with peptides for targeted nucleic acid delivery in live cells [84]. Concerns about toxicity and efficiency and challenges to image deep tissues have hindered progress toward more routine use of quantum dots in vivo. In vivo work with quantum dots has focused on conjugation with antibodies for diagnostic purposes and for drug delivery [85, 86], but there may be a resurgence of interest with smaller, less toxic polymer quantum dot hybrids [87].
2.2.6.3 Silica Particles
In contrast to the other nanoparticles, silica does not offer unique imaging characteristics in the nanometer-size range, but does have excellent biocompatibility, stability and can be fabricated inexpensively into a range of shapes with different porosities, providing both interior and surface space to carry a therapeutic cargo. Some recent examples of progress in this field include a demonstration that second-generation polyamidoamine dendrimers covalently attached to the surface of 250 nm mesoporous silica particles provide protection for complexed pDNA and can successfully transfect HeLa cells in vitro [88]. Silica particles decorated with the cationic polymer polyethylenimine and loaded with siRNA have also been used to knock down EGFP expression in PANC-1 cells in vitro [89]. The porous region has been used to extend functionality beyond nucleic acid delivery to include chemotherapy agents [90], and anti-malarials [91]. The use of mesoporous silica particles to co-deliver of doxorubicin and siRNA to silence P-glycoprotein exporters in a xenograft breast cancer mouse model has recently been shown to provide synergistic tumor growth inhibition [92].
2.2.6.4 Gold Nanoparticles
The excellent chemical, physical, and optical properties of gold in the nanometer range, as well as easy synthesis of a variety of shapes and configurations has led to a long interest in gold-based synthetic vectors. Cationic polymers covalently attached to gold particles were found to be an efficient vehicle for pDNA transfection of COS-7 cells in vitro [93], and knockdown enhanced green fluorescence protein in endothelial cells in vitro using antisense oligonucleotides [94]. Increasingly, complicated designs have been developed to enhance efficiency and prolong effectiveness of siRNA using multiple layers [95], giving 70 % gene silencing of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in a mouse model for more than 10 days post-injection [96].
2.2.6.5 Carbon Nanoparticles
Carbon has many unusual physical and chemical properties in the nanometer range and can form several different structures which are of interest as delivery vehicles. Three of these structures, nanodiamonds, carbon nanotubes (CNT), and graphene sheets have attractive features and have been explored as synthetic vectors over the past decade.
The increasing availability, innate photoluminescence, characteristic Raman signal, and good biocompatibility of nanometer-sized (~5 nm) diamonds makes them attractive for gene delivery [97–99]. In vitro delivery of siRNA into Ewing sarcoma cells has been demonstrated [99] and the attractive optical properties had led to early in vivo studies in Caenorhabditis elegans [100], although the kinetics of the functionalized nanodiamonds have not yet been fully characterized in vivo.
CNT have the ability to enter a range of cell types by passive diffusion and act as a vector [101]. A lot of effort has gone into optimizing surface modification of the CNT to enhance efficiency and biocompatibility. Hybrid cationic polymer-CNT have been demonstrated to be effective for delivering siRNA in mice at doses of <1 mg/kg with relatively high clearance [102].
Graphene sheets have high surface area and mechanical strength as well as exceptional conductivity and attractive functionalization chemistry. Oxidizing graphene followed by functionalization has emerged as an early solution to its hydrophobic nature and has been used as a siRNA vector for in vitro experiments [98]. The strong near-infrared absorption of graphene oxide has been used to demonstrate a photothermal effect in a mouse tumor model [103], and it has been used for delivery of doxorubicin in vivo [104], though more data are required to understand cellular uptake mechanisms, biodistribution, and toxicity.
2.3 Directed Delivery
Complementary to the carrier-mediated viral and nonviral systems described previously, delivery of nucleic acids can be enhanced locally using a range of forces, or delivered locally using a number of noninvasive and minimally invasive techniques. Local delivery or the use of fields to target regions is attractive to avoid initial systemic clearance processes, enhance cargo concentration in the region of the cells of interest, and avoid side effects associated with large doses. However, disruption of the normal physiology of the local region because of these delivery methods may have unintended consequences, including generating an inflammatory response, altering lymphatic flow and perturbation of physiological functions. Ideally, the goal for directed delivery is to combine both regional and cell-specific delivery, to minimize disturbance of neighboring cells.
2.3.1 Hydrodynamic Delivery
Hydrodynamic delivery, or the use of a rapid injection of fluid to deliver force in a noncompressible environment, can disrupt physical barriers, such as endothelial layers and cell membranes, to enable efficient delivery to parenchymal cells. In less than a decade, this technique progressed from first demonstration in a mouse model to use in a human clinical trial, in part because it is a simple, effective, and versatile approach and can be repeated multiple times [105]. This approach has been used for both DNA and RNA delivery to a range of tissues including liver, kidney, skeletal muscle, and myocardium, as well as tumors, through veins, arteries, and ducts. While effective in rodents, scaling hydrodynamic delivery to humans has been challenging because of concerns about the induction of irregularities in cardiac function, localized increase in blood pressure, expansion in organ size, and structural deformation, with the effects lasting up to several days in animals [105]. A number of refinements have been developed including the use of catheters, balloons, computer-controlled injectors [106], and image-guidance [107], which may address some of these challenges.
2.3.2 Ballistic Delivery
The ballistic delivery of nucleic acids attached to dense particles is an attractive approach for dermal targets because it is more efficient and less invasive than injection. Gene guns, using a burst of helium to give nucleic-acid-coated gold or tungsten particles enough momentum to penetrate physical barriers, emerged in the 1980s as an alternative transfection tool. For in vivo applications, the particles have enough momentum to breach the stratum corneum of the skin and have been used for vaccine delivery, but have also been used to delivery pDNA to mouse skeletal muscle cells [108]. The approach has a number of challenges, which include the risk of inflammation and damage in the target region, limited particle choices, limited nucleic acid loading capacity, indiscriminate and poor uniformity of the delivery, and sensitivity to a wide range of environmental factors. Although particle-mediated vaccine delivery systems have been tested in humans [109], it remains unclear whether cost and complexity will be barriers to wider use. However, engineering advances such as contoured shock tubes and better vaccine preservation techniques, and the lure of dose-sparing and more controlled immune response are continuing to drive interest beyond clinical studies toward regulatory approval [110]. Two other dermal delivery methods have emerged for vaccine delivery, microneedles and liquid jet injectors, both of which are reviewed in detail by Kis et al. [110].
2.3.3 Electric Field-Assisted Delivery
Electroporation is an extremely effective and simple technique for increasing the permeability of cells for nucleic acid delivery that has been used for more than 30 years. A number of techniques have been developed for electroporation in vivo and this technique has been used to successfully transfect liver, skin, tumor, and skeletal muscle cells [108]. Electroporation has been commonly used in DNA vaccine studies and enhanced efficacy up to 1,000-fold overnaked DNA intramuscular injections in animal models [111]. At least 10 phase I or phase II human clinical studies have been conducted with promising results [112], though with some significant adverse effects also reported that include subjects reporting pain and bleeding at the injection site [113]. At an earlier stage of development are minimally invasive electroporation devices for transdermal delivery [114] and multifunctional systems [115] that have the potential to address some of the concerns with naked DNA delivery by intramuscular injection and invasive electroporation.
2.3.4 Ultrasound-Assisted Delivery
Ultrasound is routinely used for in vivo imaging, although it is also known to have a therapeutic effect at higher intensities because of mechanical stimulation or disruption. Microbubbles of gas can also undergo oscillations under ultrasound stimulation disrupting the membrane and increasing the permeability of neighboring cells. This sonoporation approach has been studied for more than 25 years although all the mechanisms involved in internalization and other effects on the cell are not well used. To avoid tissue damage, relatively high frequencies (~1 MHz) and low-to-moderate mechanical indices are used, which is sufficient to cause stable cavitation of the bubbles at lower intensities and inertial cavitation at higher intensities. The 500–5,000 nm bubbles are typically composed of gas cores of a high molecular weight hydrophobic gas and a shell containing phospholipids, surfactants, targeting moieties, and nucleic acids. In vivo delivery studies have focused on organs routinely imaged by ultrasound, including successful delivery in heart, skeletal muscle, and kidney, as well as the pancreas, liver, and the central nervous system, either through direct injection into the organ or into the blood stream [116]. Both siRNA [117] and pDNA [118] have been delivered in vivo with delivery limited to the focal region of the noninvasive ultrasound. Ultrasound-mediated delivery bypasses the endocytic pathway, although there is little to no control about where the nucleic acids will enter the cell. Some concerns have been raised about efficiency [119] and that microbubbles may exacerbate underlying hypotensive reactions in some patients [120], suggesting that further optimization is required to move beyond early animal studies [121].
2.3.5 Magnetic Field-Assisted Delivery
The magnitude and gradient of external magnetic fields can be used to target superparamagnetic particles in vivo, as well as provide a basis for MRI, spectroscopy, and hysteretic heating. However, achieving strong enough field gradients to generate sufficient force on submicron particles is very challenging in regions other than the extremities. A local magnetizable structure can enhance the field gradient, such as implantable stents [122]. Much of the in vivo work with magnetic targeting has focused on drug and cell delivery work, although more generally solid, lipid-based, and microbubble magnetic vectors have been used to deliver DNA [123] and siRNA [81]. Some of the challenges to be addressed include concerns about toxicity, particularly at higher concentrations, aggregation of the particles, and overall efficiency of the nucleic acid delivery to target cells. A recent review has comprehensively described the progress and prospects for magnetically enhanced delivery [124].
2.3.6 Optically Assisted Delivery
Arguably, the least developed method for membrane pore generation is the use of optical fields, although it was first demonstrated nearly 30 years ago. An optical field can generate a pore through thermal heating with continuous wave lasers, while at femtosecond timescales, disruption can occur through the generation of low-density plasma and intermediate pulses can generate bubbles and thermoelastic stresses [125]. Experimental results suggest that shorter pulses result in better viability, though not necessarily an improvement in efficiency, and they have been used to deliver DNA [126] and messenger RNA (mRNA) [127] in vitro. In vivo studies are more challenging by the need for local optical access; however, two approaches have been demonstrated: (1) the transfection of zebrafish blastomeres with pDNA [128] and (2) a microendoscope with an axicon tip [129]. Other optical properties have also explored for nucleic acid delivery. Particles driven by laser-induced plasma jets have been demonstrated [130], however many engineering challenges still exist and scientists have yet to find an efficient, effective and reproducible way to deliver nucleic acids in vivo using optical fields.
3 Therapeutic Components
Nucleic acids and their analogs have been increasingly used as therapeutic agents over the past 40 years. The development of a host of tools for engineering monomers as well as polymeric sequences that have favorable physical, chemical, or biological characteristics has helped drive nucleic acid therapies toward being practical and reproducible in the clinic. The desired type of therapeutic effect drives the choice of nucleic acid, which along with delivery considerations narrows down the choice of analog. DNA, RNA, and nucleic acid analogs have all been explored extensively as therapeutic cargos for nanoscale delivery systems. The chemical structure of DNA, RNA, and three of analogs are illustrated in Fig. 18.4 and described in more detail in the following sections.

Nucleic acids and analogs
3.1 DNA-Based Therapeutics
pDNA have been used extensively as a therapeutic agent and delivered in a large number of different ways. pDNA can be systemically delivered with low uptake efficiency, or by a number of physical mechanisms, including electroporation, gene gun transfer, ultrasound, or by forming chemical complexes with cationic polymers and lipids [131, 132]. Uptake is thought to be primarily accomplished by endocytosis [133] and once inside, pDNA escapes from lysosomal degradation.
For treatment of muscle diseases, naked pDNA containing the transgenes can be delivered to muscles by either intramuscular injection [134] or intravascular injection by a procedure known as hydrodynamic limb vein injection [135, 136]. This procedure involves the use of a tourniquet to temporarily isolate an area in a peripheral vein or artery of a limb from normal blood flow. This is followed by injection of the pDNA into the vessel in the anterograde direction. The pDNA is delivered in a large volume of saline so as to facilitate extravasation of the pDNA from the vasculature into the muscle tissue. Naked pDNA is susceptible to nuclease degradation, which limits the serum half-life to approximately 10 min [137]. The rapid clearance is mitigated by delivering an excessively high number of copies of the transgene. Transgene expression in a plasmid is achieved by placing it under transcriptional control of an appropriate promoter/enhancer. The expression is not as efficient as viral vectors that are integrated into the host genome. However, in nondividing cells, pDNA transgene expression has been shown to persist for years after direct intramuscular injection [134, 138]. A major advantage of naked pDNA over viral delivery of transgene is that the former is nonimmunogenic.
In addition to injection, pDNA can also be delivered using bacteria. Production of pDNA in bacteria involves the use of specific selection markers, such as an antibiotic resistance gene to identify bacterial colonies that are transfected. However, given the safety concerns with antibiotic-resistant bacterial strains, alternative antibiotic-free strategies have been developed which may be a more practical way of delivering pDNA in humans [139].
3.2 RNA-Based Therapeutics and Delivery Systems
This section describes a range of RNA molecules that can be used as research tools in drug discovery or therapeutic application: aptamers, ribozymes, antisense, and siRNA. These are powerful tools to silence gene expression, but they have to get into the cells and bind to the complementary genes for silencing. For in vivo use, these therapeutics must overcome barriers of limited stability, poor cellular uptake, unfavorable subcellular trafficking, lack of targeting, nonspecific tissue distribution, and susceptibility to nuclease degradation.
3.2.1 Aptamers
Aptamers are single-stranded DNA or RNA oligonucleotides of 20–80 bases that are generated in vitro from random libraries of nucleic acids [140]. Because they are single-strand, they exist in many three-dimensional shapes including hairpin-like monomers, duplexes, triplexes, or quadruplexes [141–145]. The basic technology for screening and selecting aptamers was developed two decades ago. This process is called Systematic Evolution of Ligands by Exponential enrichment (SELEX) [146–148]. However, it was soon discovered that aptamer specificity and binding affinity can be improved with modification of the basic SELEX screening. For example, procedures involving the immobilization of fluorescence-labeled targets (FlyMag-SELEX) or the oligonucleotides (Capture-SELEX) on magnetic beads have been developed to improve selection of aptamers that target organic molecules or pharmaceuticals [149, 150]. Highly specific aptamers have been selected with the modified SELEX for use in biosensors and assays for detecting drug targets, antibiotic, or pharmaceuticals. Aptamers have also been isolated by a Cell-SELEX procedure that target cell surface markers for therapeutic and diagnostic purposes.
Aptamers are versatile molecules and can bind to a range of targets including small molecules such as adenosine triphosphate (ATP) or macromolecules such as proteins or microorganisms [151, 152]. Aptamer binding has been likened to antibodies in that they are highly specific and bind with high affinity. For these reasons, aptamers have been used in basic research to investigate protein interactions and in clinical applications as therapeutics or as vehicles to deliver drugs. Aptamer-mediated drug delivery holds promise as it minimizes off-target side effects. In clinical applications, aptamers have the added advantages of low immunogenicity in contrast to antibodies and storage stability. Intracellular stability against nuclease activities can be achieved by chemical modifications involving primarily the sugar moieties, the nitrogenous base, and replacing the phosphate backbone with phosphorothionate [153]. The 2′-ribose can be modified by adding methyl- or amino groups [154, 155]. The nitrogenous base can also be substituted with uracil derivatives and other pyrimidines [156].
Currently, there are six RNA-based aptamers that have been clinically evaluated. A vascular epithelial growth factor (VEGF)-specific RNA-modified aptamer (Macugen by Pfizer/Eyetech) has been approved by the FDA for treating age-related macular degeneration [157–160]. A clinical trial sponsored by Regado Bioscience is evaluating REG1 as part of a dual-aptamer therapy for acute coronary syndrome [161]. Kang and his colleagues identified two aptamers that specifically recognize cell surface membrane proteins expressed on glioblastoma cell lines. These results suggest that aptamers may be promising cancer therapeutics.
3.2.2 Ribozymes
The seminal research of Thomas Cech showed that RNA is capable of enzymatic activities for which he was awarded the Nobel Prize in 1989 [162]. The term ribozyme refers to antisense RNA molecules that have catalytic activity. Biologically active ribozymes share either the “hammerhead” or “hairpin” motifs that reflect their secondary structures. Most ribozymes self-catalyze as evidenced by splicing of introns and subsequent ligation [163]. They recognize specific nucleotide sequences in the catalytic sites. Hammerhead ribozymes cleave RNA at UA, UC, or UU whereas hairpin ribozymes cleave CUG sequences [164, 165]. Ribozymes have been engineered and synthesized to improve site-specific enzymatic activities and stability while maintaining the hammerhead and hairpin motifs. Automated solid-phase RNA synthesis [166] has been successfully used to produce synthetic ribozyme analogs that are more stable and are amenable to labeling with fluorescent tags, radioisotopes (for NMR studies), and other groups to improve biological activity and resistance to RNAse degradation. The scope, utility, and intracellular stability can be enhanced by increasing the secondary structure such as stem–loop on either side of the ribozymes. Chemically modified ribozymes have been produced that act as riboswitches in biosensor technology [167, 168].
Clinical applications of ribozymes include use as antivirals or gene therapies. Studies with HeLa cells transfected with hammerhead ribozymes targeting the gag transcripts showed reduced gag expression [169, 170]. Similarly, a hammerhead ribozyme targeting the 5′ leader sequence showed reduced HIV1 replication in T-cell lines [169]. Targeting the leader sequence (with either hammerhead or hairpin ribozymes) has been demonstrated to prevent HIV1 from establishing infection [170–172]. The latter approach is promising for vaccine development. Immunotherapies for infectious diseases, such as HIV1 infection, have been developed by using the infected individuals own immune cells such as CD4, CD8, CD34, or antigen presenting cells that were transduced with ribozyme carried on a murine retroviral vector [173]. However, the use of ribozymes for HIV1 gene therapy is challenging because the virus mutates at a high rate such that the cleavage sequences could be disrupted.
3.2.3 Small Interfering RNA
siRNA was discovered by Professors Andrew Fire and Craig Mello who were awarded the Nobel prize in 1996 for their seminal research [174]. They discovered that long, double-stranded RNA, introduced into cells either by viral pathogens, such as HIV1 or jumping genes, can be cut into short 20–25 nucleotide pieces by an enzyme called Dicer. These short double-stranded RNAs bind to several proteins to form the RISC. The RISC contains a helicase module that unwinds the two strands of siRNA to form the single-stranded short RNA or siRNA. The siRNA binds to the complementary mRNA, which is cleaved by the endonuclease activity in RISC and silenced from protein expression [158]. The sequence-specific binding of siRNA to turn off or turn down gene expression is a powerful approach to: (1) identify gene function, (2) regulate gene expression, (3) determine drug targets, and (4) develop therapeutics for disease. Despite these promising approaches, siRNA development for therapeutic purposes is fraught with challenges. The full therapeutic potential of siRNA has yet to be fully realized because of the barriers to delivery and creating a sustained therapeutic effect, including shielding the negatively charged backbone, preventing nuclease degradation, promoting uptake only by cells of interest, and ensuring endosomal escape at the optimum time. The susceptibility of siRNA to nuclease degradation [175, 176] limits their serum half-life [137]. The intracellular stability can be improved with chemical modification of the oligonucleotides. Another challenge with therapeutic uses is that the intracellular siRNA concentration decreases with each cell division.
siRNA delivery systems have been an active area of research. Various nanotechnology approaches have been used to improve delivery to tumor cells in animal models. For example, Cho et al. reported an innovative nanoparticle construct that expresses siRNA in vivo by conjugating to the particle surface target ligands and double-stranded DNA nanocassettes containing a promoter and a shRNA gene [177]. Other approaches include the use of plasmids and viral vectors to deliver siRNA into both dividing and nondividing cells, stem cells, zygotes, and their differentiating progeny [178]. Another delivery option is Lipofectamine 2000, which is a cationic liposome, that has been used in a complex with siRNA to transfect mammalian cells [55, 179]. However, its toxicity and low transfection efficiency in certain types of cells limited its usefulness. Other approaches include nanodelivery systems and CCP, as discussed earlier.
It is a little more than a decade since the first demonstration of RNA inference in vivo [180] and great strides have been made in understanding and optimizing delivery. siRNA for silencing disease-associated mRNA transcripts has been successfully used as an antiviral for HIV1 susceptible cells [181, 182]. siRNA has also been shown to be capable of knocking down targets relevant to many diseases, including ovarian cancer [183], cirrhosis [184], and hypercholesterolemia [185]. siRNA therapeutics have progressed rapidly into clinical trials, with more than 14 diseases being targeted by more than 20 therapeutics in more than 30 clinical trials. A review, in 2011, noted that 8 of 9 intravenous siRNA therapeutics used a synthetic carrier: four were composed of cationic liposomes, one of anionic liposomes, two were polymeric, and one was naked siRNA [2]. Furthermore, localized delivery of siRNA by intravitreal injection, intralesional injection, and topical application has been tested in clinical trials as well as one study on oral delivery of E coli, which produce shRNA for treatment of familial adenomatous polyposis. There have also been three trials studying autologous cell therapy using siRNA.
3.3 Nucleic Acid Analogs
The modification of the phosphodiester backbone, sugar ring, or nucleobase can introduce new functions or enhance the performance of nucleic acids. Over the past two decades, an increasing variety of analogs have been synthesized to complement the dideoxynucleotides used in sequencing. The most widely studied are the backbone analogs, including PNA, locked nucleic acids (LNA), and morpholinos, as well as fluorescent base analogs such as 2-animopurine (2-AP). Differences in the conformational structure, as a result of these analog bases, such as the sugar ring pucker, can result in very different antisense activity [186] and have led to expanding interest in engineering modified oligonucleotides for gene silencing in vivo [187].
3.3.1 Peptide Nucleic Acids
PNAs are stable, neutral charge DNA analogs first developed in the 1980s with a peptide-based backbone, but maintaining the nucleobases to preserve base-pairing rules. They bind DNA and RNA with high affinity and have a number of interesting properties, including resistance to enzyme degradation, double-stranded DNA invasion, triplex formation, and act via a translation inhibition mechanism [187]. With charge neutrality, water solubility is length-dependent and there is much stronger binding to DNA/RNA because of the lack of charge repulsion, as well as the potential for co-delivery with hydrophobic drugs. PNA can persist in the cytoplasm for at least 48 h and binds more quickly to negatively supercoiled DNA, making it attractive for in vivo therapeutic use. Recently, PGLA nanoparticles were used to systemically delivery PNA and DNA to human cells in non-obese diabetic (NOD) mice and demonstrate site-specific gene editing [188]. Site-specific intradermal delivery of PNA, coupled to a cell penetration peptide, to keratinocytes has also been demonstrated [189]. PNA is also attractive for fluorescence in situ hybridization (FISH) because it can bind under unfavorable conditions. It also has been incorporated into FDA-approved vitro molecular diagnostic tools, and coupled to imaging agents to investigate mRNA expression in vivo [190], suggesting a route to multifunctional delivery in vivo.
3.3.2 Locked Nucleic Acids
LNA is a chemically modified RNA analog first described in 1998 with high affinity for complementary RNA or DNA and has been used to improve in vitro methods including microarray profiling, allele-specific polymerase chain reaction (PCR) and FISH [191]. The nucleosides are locked in a North sugar confirmation, making them useful in anti-miRNA, antisense, and siRNA applications, while having improved nuclease resistance and potentially lower immuno-stimulation [187]. Several derivatives of LNA have been developed to further improve performance and enable further chemical modification, and two LNA-based therapeutics have progressed to human clinical trials [192]. The exclusive licensing of LNA to Exiqon A/S and nontrivial synthesis may be barriers to more widespread research and development of LNA-based multifunctional approaches.
3.3.3 Morpholinos
Morpholinos are modified oligonucleotides with an uncharged substitute for the phosphodiester linkages and the furanose sugars, developed in the mid-1980s. Their preparation is a more cost-effective transformation of ribonucleosides, and assembly efficiency is high through the morpholine nitrogen. They show similar or increased affinity and nuclease resistance and can be used as translational inhibitors [187]. Antisense mopholino oligonucleotides have been delivered in vivo to a splice-reporter mouse model using a dendritic transporter resulting in the expression of splice-corrected GFP [193]. Some concerns have been raised about off-target effects and the assessment of efficacy [194], although these are addressable through well-conceived control experiments.
4 Challenges and Opportunities for Multifunctional Delivery in Vivo
The broad challenge for multifunctional nanoscale delivery systems is identification of the most robust method to deliver a targeted therapeutic dose of nucleic acids and other components efficiently in vivo with minimal invasiveness, while minimizing side effects and maximizing clinically actionable information.
Table 18.2 highlights some of the challenges and solutions that have identified so far. However, many challenges remain, as highlighted in the previous sections of this chapter. Each of the components, carrier, nucleic acid cargo, and functional components can be optimized independently and readily developed under in vitro circumstances, although it is challenging to make the leap to system optimization for the complex and less predictable environment of human therapies.
One ongoing area of intense work is the optimization of vectors. For synthetic vectors, the primary challenge is to enhance uptake efficiency and control intracellular fate through the use of ligands to activate signaling cascades and nuclear targeting, without increasing immunogenicity. This will require a deeper understanding of the mechanisms underlying the RISC, nuclear transport, active transport of particles in the cytoplasm, routes to deep tissue penetration, such as the exosome pathways. While viral vectors have much higher efficiency, their challenges are around reducing side effects, such as oncogene activation, immunogenicity, adequate viral titers, production, length of the transgene, and the range of cell targets. These challenges are active areas of research and understanding the the virology underpinning these vectors has been crucial in overcoming the barriers.
Another area where there is still room for development is the optimization of the composition of vectors during development of nanoscale delivery systems by utilizing realistic three-dimensional heterogeneous microenvironments. A better understanding of the physical and chemical environments in living organs, combined with the engineering of cell culture, using microfabricated structures, has enabled the generation of complex microenvironments that more closely mimic the physiological environment in vivo beyond the conventional two-dimensional culture systems [195].
A third area where there is a need for deep understanding is the dynamic interaction of the delivery systems with the entry site, the extracellular matrix, and circulatory system, as well as how these interactions modify the therapeutic effect in a heterogeneous human population. Viral vectors enter cells via membrane receptors and therefore serve as experimental systems because mutations can be introduced in the receptors leading to altered downstream intracellular events. The emergence of molecular imaging is a promising direction for helping to unravel these interactions using MRI, positron emission tomography (PET), single-photon emission computed tomography (SPECT), ultrasound (US), or optical imaging moieties (some of which were described earlier in this chapter) and enables imaging theranostics as a process for achieving precision medicine [196]. In vivo imaging agents that are sensitive to their environment, bioswitches that turn on/off the delivery of payloads, and biomarker analysis, will help provide new methods for characterizing toxicity, reduce off-target effects, and minimize immune stimulation, as well as optimize deep tissue delivery and circulation time. More detailed analysis of the overall in vivo efficiency across multiple model systems is required to understand the kinetics of circulation, degradation, internalization, and intracellular fate of nucleic acid therapies, and the relative contributions of helper ligands.
A final area where engineering will play a significant role is in the development of targeted delivery of nanoscale systems. The use of minimally invasive delivery systems and fields to localize delivery are likely to grow to address challenges like sustained delivery, off-target effects, and maximizing therapeutic effect with minimizing dose.
The most challenging question is the long-term impact of multifunctional nanoscale delivery systems on clinical care. With naked nucleic acid delivery and simple delivery systems already in clinical trials, the value proposition for multifunction delivery systems in current clinical practice is unclear, particularly if the multifunctional components are designed for real-time monitoring and optimization of care. The current clinical workflow model is based on quality control and careful planning of treatment in advance to minimize errors, but also limiting personalized analysis to static or slowly varying biomarkers. This makes it very difficult to detect immune responses and off-target effects or to modify dose based on whether a therapeutic response is observed. Building information from the increased functionality of these nanoscale delivery systems into the clinical workflow will help protect and improve health as well as expand the knowledge based in medical and associated sciences.
References
Edelstein M (2012) Gene therapy clinical trials worldwide. J Gene Med. http://www.abedia.com/wiley/vectors.php. Accessed 21 Jan 2013
Burnett JC, Rossi JJ, Tiemann K (2011) Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J 6:1130–1146. doi:10.1002/biot.201100054
Jiao S, Williams P, Berg R et al (1992) Direct gene transfer into nonhuman primate myofibers in vivo. Hum Gene Ther 3:21–33. doi:10.1089/hum.1992.3.1-21
Ardehali A, Fyfe A, Laks H et al (1995) Direct gene transfer into donor hearts at the time of harvest. J Thorac Cardiovasc Surg 109:716–720. doi:10.1016/S0022-5223(95)70353-5
Vile R, Hart I (1993) In vitro and in vivo targeting of gene expression to melanoma cells. Cancer Res 53:962–967
Khatri N, Rathi M, Baradia D et al (2012) In vivo delivery aspects of miRNA, shRNA and siRNA. Crit Rev Ther Drug Carrier Syst 29:487–527
Inoue T, Sugimoto M, Sakurai T et al (2007) Modulation of scratching behavior by silencing an endogenous cyclooxygenase-1 gene in the skin through the administration of siRNA. J Gene Med 9:994–1001. doi:10.1002/jgm.1091
Bitko V, Musiyenko A, Shulyayeva O, Barik S (2005) Inhibition of respiratory viruses by nasally administered siRNA. Nat Med 11:50–55. doi:10.1038/nm1164
DiFiglia M, Sena-Esteves M, Chase K et al (2007) Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci USA 104:17204–17209. doi:10.1073/pnas.0708285104
Sinn PL, Anthony RM, McCray PB Jr (2011) Genetic therapies for cystic fibrosis lung disease. Hum Mol Genet 20:R79–R86. doi:10.1093/hmg/ddr104
Kamimura K, Suda T, Zhang G, Liu D (2011) Advances in gene delivery systems. Pharmaceut Med 25:293–306. doi:10.2165/11594020-000000000-00000
Maetzig T, Galla M, Baum C, Schambach A (2011) Gammaretroviral vectors: biology, technology and application. Viruses 3:677–713. doi:10.3390/v3060677
Coffin JMJM, Hughes SHSH, Varmus HEHE (1997) Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Hu WS, Pathak VK (2000) Design of retroviral vectors and helper cells for gene therapy. Pharmacol Rev 52:493–511
Lewis P, Hensel M, Emerman M (1992) Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J 11:3053–3058
Lee C-L, Dang J, Joo K-I, Wang P (2011) Engineered lentiviral vectors pseudotyped with a CD4 receptor and a fusogenic protein can target cells expressing HIV-1 envelope proteins. Virus Res 160:340–350. doi:10.1016/j.virusres.2011.07.010
Dimitrov DS (1997) How do viruses enter cells? The HIV coreceptors teach us a lesson of complexity. Cell 91:721–730
Moore JP (1997) Coreceptors: implications for HIV pathogenesis and therapy. Science 276:51–52
Cockrell AS, Kafri T (2007) Gene delivery by lentivirus vectors. Mol Biotechnol 36:184–204
Miller DG, Adam MA, Miller AD (1990) Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol 10:4239–4242
Durand S, Cimarelli A (2011) The inside out of lentiviral vectors. Viruses 3:132–159. doi:10.3390/v3020132
Pfeifer A, Ikawa M, Dayn Y, Verma IM (2002) Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proc Natl Acad Sci USA 99:2140–2145. doi:10.1073/pnas.251682798
Bahrami S, Pedersen FS (2009) Viral technology for delivery of nucleic acids. In: Jorgensen L, Nielsen HM (eds) Delivery technologies for biopharmaceuticals. Wiley, Chichester, pp 93–112
Cavazzana-Calvo M (2000) Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 288:669–672. doi:10.1126/science.288.5466.669
Mátrai J, Chuah MKL, VandenDriessche T (2010) Recent advances in lentiviral vector development and applications. Mol Ther 18:477–490. doi:10.1038/mt.2009.319
Izmiryan A, Basmaciogullari S, Henry A et al (2011) Efficient gene targeting mediated by a lentiviral vector-associated meganuclease. Nucleic Acids Res 39:7610–7619. doi:10.1093/nar/gkr524
Sarkis C, Philippe S, Mallet J, Serguera C (2008) Non-integrating lentiviral vectors. Curr Gene Ther 8:430–437
Nightingale SJ, Hollis RP, Pepper KA et al (2006) Transient gene expression by nonintegrating lentiviral vectors. Mol Ther 13:1121–1132. doi:10.1016/j.ymthe.2006.01.008
Yáñez-Muñoz RJ, Balaggan KS, MacNeil A et al (2006) Effective gene therapy with nonintegrating lentiviral vectors. Nat Med 12:348–353. doi:10.1038/nm1365
Barquinero J, Eixarch H, Pérez-Melgosa M (2004) Retroviral vectors: new applications for an old tool. Gene Ther 11(Suppl 1):S3–S9. doi:10.1038/sj.gt.3302363
Daniel R, Smith JA (2008) Integration site selection by retroviral vectors: molecular mechanism and clinical consequences. Hum Gene Ther 19:557–568. doi:10.1089/hum.2007.148
Culver K, Cornetta K, Morgan R et al (1991) Lymphocytes as cellular vehicles for gene therapy in mouse and man. Proc Natl Acad Sci USA 88:3155–3159
Rosenberg SA, Aebersold P, Cornetta K et al (1990) Gene transfer into humans–immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med 323:570–578. doi:10.1056/NEJM199008303230904
Blaese RM, Culver KW, Miller AD et al (1995) T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science 270:475–480
Stone D (2010) Novel viral vector systems for gene therapy. Viruses 2:1002–1007. doi:10.3390/v2041002
D’Costa J, Mansfield SG, Humeau LM (2009) Lentiviral vectors in clinical trials: current status. Curr Opin Mol Ther 11:554–564
Schambach A, Baum C (2008) Clinical application of lentiviral vectors: concepts and practice. Curr Gene Ther 8:474–482
Bank A, Dorazio R, Leboulch P (2005) A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Ann N Y Acad Sci 1054:308–316. doi:10.1196/annals.1345.007
Pedersen FS, Pyrz M, Duch M (2011) Retroviral replication. Encyclopedia of Life Sciences. doi:10.1002/9780470015902.a0000430.pub3
Muralidhar S, Becerra SP, Rose JA (1994) Site-directed mutagenesis of adeno-associated virus type 2 structural protein initiation codons: effects on regulation of synthesis and biological activity. J Virol 68:170–176
Girod A, Wobus CE, Zádori Z et al (2002) The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J Gen Virol 83:973–978
Zádori Z, Szelei J, Lacoste MC et al (2001) A viral phospholipase A2 is required for parvovirus infectivity. Dev Cell 1:291–302
Grieger JC, Samulski RJ (2005) Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J Virol 79:9933–9944. doi:10.1128/JVI.79.15.9933-9944.2005
McCarty DM, Young SM Jr, Samulski RJ (2004) Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet 38:819–845. doi:10.1146/annurev.genet.37.110801.143717
Allocca M, Doria M, Petrillo M et al (2008) Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest 118:1955–1964. doi:10.1172/JCI34316
Gao G, Vandenberghe LH, Wilson JM (2005) New recombinant serotypes of AAV vectors. Curr Gene Ther 5:285–297
Taymans J-M, Vandenberghe LH, Haute CVD et al (2007) Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum Gene Ther 18:195–206. doi:10.1089/hum.2006.178
Carter BJ (2005) Adeno-associated virus vectors in clinical trials. Hum Gene Ther 16:541–550. doi:10.1089/hum.2005.16.541
Bennett J (2006) Commentary: an aye for eye gene therapy. Hum Gene Ther 17:177–179. doi:10.1089/hum.2006.17.177
LeWitt PA, Rezai AR, Leehey MA et al (2011) AAV2-GAD gene therapy for advanced Parkinson’s disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol 10:309–319. doi:10.1016/S1474-4422(11)70039-4
Bainbridge JWB, Smith AJ, Barker SS et al (2008) Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 358:2231–2239. doi:10.1056/NEJMoa0802268
Zabner J, Fasbender AJ, Moninger T et al (1995) Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem 270:18997–19007
Ng CP, Pun SH (2008) A perfusable 3D cell-matrix tissue culture chamber for in situ evaluation of nanoparticle vehicle penetration and transport. Biotechnol Bioeng 99:1490–1501. doi:10.1002/bit.21698
Ye Z, Houssein HSH, Mahato RI (2007) Bioconjugation of oligonucleotides for treating liver fibrosis. Oligonucleotides 17:349–404. doi:10.1089/oli.2007.0097
Mo RH, Zaro JL, Shen W-C (2012) Comparison of cationic and amphipathic cell penetrating peptides for siRNA delivery and efficacy. Mol Pharm 9:299–309. doi:10.1021/mp200481g
Shiraishi T, Nielsen PE (2011) Peptide nucleic acid (PNA) cell penetrating peptide (CPP) conjugates as carriers for cellular delivery of antisense oligomers. Artif DNA PNA XNA 2:90–99
Sanders WS, Johnston CI, Bridges SM et al (2011) Prediction of cell penetrating peptides by support vector machines. PLoS Comput Biol 7:e1002101. doi:10.1371/journal.pcbi.1002101
Bidwell GL 3rd, Raucher D (2010) Cell penetrating elastin-like polypeptides for therapeutic peptide delivery. Adv Drug Deliv Rev 62:1486–1496. doi:10.1016/j.addr.2010.05.003
Puri A, Loomis K, Smith B et al (2009) Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit Rev Ther Drug Carrier Syst 26:523–580
Semple SC, Akinc A, Chen J et al (2010) Rational design of cationic lipids for siRNA delivery. Nat Biotechnol 28:172–176. doi:10.1038/nbt.1602
Chapter 17—Engineering cationic liposome s… [Methods Enzymol 464:343–454 (2009). doi:10.1016/S0076-6879(09)64017-9]—PubMed—NCBI. http://www.ncbi.nlm.nih.gov/pubmed/19903563. Accessed 4 Feb 2013
Kenjo E, Asai T, Yonenaga N et al (2013) Systemic delivery of small interfering RNA by use of targeted polycation liposomes for cancer therapy. Biol Pharm Bull 36:287–291
Zuhorn IS, Bakowsky U, Polushkin E et al (2005) Nonbilayer phase of lipoplex-membrane mixture determines endosomal escape of genetic cargo and transfection efficiency. Mol Ther 11:801–810. doi:10.1016/j.ymthe.2004.12.018
Guo X, Szoka FC Jr (2003) Chemical approaches to triggerable lipid vesicles for drug and gene delivery. Acc Chem Res 36:335–341. doi:10.1021/ar9703241
Stover TC, Sharma A, Robertson GP, Kester M (2005) Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin Cancer Res 11:3465–3474. doi:10.1158/1078-0432.CCR-04-1770
Giddam AK, Zaman M, Skwarczynski M, Toth I (2012) Liposome-based delivery system for vaccine candidates: constructing an effective formulation. Nanomedicine (Lond) 7:1877–1893. doi:10.2217/nnm.12.157
Lam AP, Dean DA (2010) Progress and prospects: nuclear import of nonviral vectors. Gene Ther 17:439–447. doi:10.1038/gt.2010.31
Wu Z-W, Chien C-T, Liu C-Y et al (2012) Recent progress in copolymer-mediated siRNA delivery. J Drug Target 20:551–560. doi:10.3109/1061186X.2012.699057
Lee S-Y, Huh MS, Lee S et al (2010) Stability and cellular uptake of polymerized siRNA (poly-siRNA)/polyethylenimine (PEI) complexes for efficient gene silencing. J Control Release 141:339–346. doi:10.1016/j.jconrel.2009.10.007
Kim SW, Kim NY, Choi YB et al (2010) RNA interference in vitro and in vivo using an arginine peptide/siRNA complex system. J Control Release 143:335–343. doi:10.1016/j.jconrel.2010.01.009
Guo J, Cheng WP, Gu J et al (2012) Systemic delivery of therapeutic small interfering RNA using a pH-triggered amphiphilic poly-l-lysine nanocarrier to suppress prostate cancer growth in mice. Eur J Pharm Sci 45:521–532. doi:10.1016/j.ejps.2011.11.024
Panyam J, Labhasetwar V (2003) Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev 55:329–347
He C, Yin L, Tang C, Yin C (2013) Multifunctional polymeric nanoparticles for oral delivery of TNF-α siRNA to macrophages. Biomaterials. doi:10.1016/j.biomaterials.2013.01.033
Woodrow KA, Cu Y, Booth CJ et al (2009) Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat Mater 8:526–533. doi:10.1038/nmat2444
Bamrungsap S, Zhao Z, Chen T et al (2012) Nanotechnology in therapeutics: a focus on nanoparticles as a drug delivery system. Nanomedicine (Lond) 7:1253–1271. doi:10.2217/nnm.12.87
Pérez-Martínez FC, Ocaña AV, Pérez-Carrión MD, Ceña V (2012) Dendrimers as vectors for genetic material delivery to the nervous system. Curr Med Chem 19:5101–5108
Albertazzi L, Gherardini L, Brondi M et al (2013) In vivo distribution and toxicity of PAMAM dendrimers in the central nervous system depend on their surface chemistry. Mol Pharm 10:249–260. doi:10.1021/mp300391v
Liu Y, Huang R, Han L et al (2009) Brain-targeting gene delivery and cellular internalization mechanisms for modified rabies virus glycoprotein RVG29 nanoparticles. Biomaterials 30:4195–4202. doi:10.1016/j.biomaterials.2009.02.051
Yu T, Liu X, Bolcato-Bellemin A-L et al (2012) An amphiphilic dendrimer for effective delivery of small interfering RNA and gene silencing in vitro and in vivo. Angew Chem Int Ed Engl 51:8478–8484. doi:10.1002/anie.201203920
Xie J, Lee S, Chen X (2010) Nanoparticle-based theranostic agents. Adv Drug Deliv Rev 62:1064–1079. doi:10.1016/j.addr.2010.07.009
Medarova Z, Pham W, Farrar C et al (2007) In vivo imaging of siRNA delivery and silencing in tumors. Nat Med 13:372–377. doi:10.1038/nm1486
Cormode DP, Skajaa GO, Delshad A et al (2011) A versatile and tunable coating strategy allows control of nanocrystal delivery to cell types in the liver. Bioconjug Chem 22:353–361. doi:10.1021/bc1003179
Taratula O, Garbuzenko O, Savla R et al (2011) Multifunctional nanomedicine platform for cancer specific delivery of siRNA by superparamagnetic iron oxide nanoparticles-dendrimer complexes. Curr Drug Deliv 8:59–69
Derfus AM, Chen AA, Min D-H et al (2007) Targeted quantum dot conjugates for siRNA delivery. Bioconjug Chem 18:1391–1396. doi:10.1021/bc060367e
Gao X, Cui Y, Levenson RM et al (2004) In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol 22:969–976. doi:10.1038/nbt994
Zrazhevskiy P, Sena M, Gao X (2010) Designing multifunctional quantum dots for bioimaging, detection, and drug delivery. Chem Soc Rev 39:4326–4354. doi:10.1039/b915139g
Zhang P, Liu W (2010) ZnO QD@PMAA-co-PDMAEMA nonviral vector for plasmid DNA delivery and bioimaging. Biomaterials 31:3087–3094. doi:10.1016/j.biomaterials.2010.01.007
Radu DR, Lai C-Y, Jeftinija K et al (2004) A polyamidoamine dendrimer-capped mesoporous silica nanosphere-based gene transfection reagent. J Am Chem Soc 126:13216–13217. doi:10.1021/ja046275m
Hom C, Lu J, Liong M et al (2010) Mesoporous silica nanoparticles facilitate delivery of siRNA to shutdown signaling pathways in mammalian cells. Small 6:1185–1190. doi:10.1002/smll.200901966
Meng H, Liong M, Xia T et al (2010) Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano 4:4539–4550. doi:10.1021/nn100690m
Bhattarai SR, Muthuswamy E, Wani A et al (2010) Enhanced gene and siRNA delivery by polycation-modified mesoporous silica nanoparticles loaded with chloroquine. Pharm Res 27:2556–2568. doi:10.1007/s11095-010-0245-0
Meng H, Mai WX, Zhang H et al (2013) Codelivery of an optimal drug/siRNA combination using mesoporous silica nanoparticles to overcome drug resistance in breast cancer in vitro and in vivo. ACS Nano. doi:10.1021/nn3044066
Thomas M, Klibanov AM (2003) Conjugation to gold nanoparticles enhances polyethylenimine’s transfer of plasmid DNA into mammalian cells. Proc Natl Acad Sci USA 100:9138–9143. doi:10.1073/pnas.1233634100
Rosi NL, Giljohann DA, Thaxton CS et al (2006) Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science 312:1027–1030. doi:10.1126/science.1125559
Guo S, Huang Y, Jiang Q et al (2010) Enhanced gene delivery and siRNA silencing by gold nanoparticles coated with charge-reversal polyelectrolyte. ACS Nano 4:5505–5511. doi:10.1021/nn101638u
Bonoiu AC, Bergey EJ, Ding H et al (2011) Gold nanorod–siRNA induces efficient in vivo gene silencing in the rat hippocampus. Nanomedicine (Lond) 6:617–630. doi:10.2217/nnm.11.20
Zhang X-Q, Chen M, Lam R et al (2009) Polymer-functionalized nanodiamond platforms as vehicles for gene delivery. ACS Nano 3:2609–2616. doi:10.1021/nn900865g
Zhang L, Lu Z, Zhao Q et al (2011) Enhanced chemotherapy efficacy by sequential delivery of siRNA and anticancer drugs using PEI-grafted graphene oxide. Small 7:460–464. doi:10.1002/smll.201001522
Alhaddad A, Adam M-P, Botsoa J et al (2011) Nanodiamond as a vector for siRNA delivery to Ewing sarcoma cells. Small 7:3087–3095. doi:10.1002/smll.201101193
Mohan N, Chen C-S, Hsieh H–H et al (2010) In vivo imaging and toxicity assessments of fluorescent nanodiamonds in caenorhabditis elegans. Nano Lett 10:3692–3699. doi:10.1021/nl1021909
Pantarotto D, Singh R, McCarthy D et al (2004) Functionalized carbon nanotubes for plasmid DNA gene delivery. Angew Chem Int Ed Engl 43:5242–5246. doi:10.1002/anie.200460437
McCarroll J, Baigude H, Yang C-S, Rana TM (2010) Nanotubes functionalized with lipids and natural amino acid dendrimers: a new strategy to create nanomaterials for delivering systemic RNAi. Bioconjug Chem 21:56–63. doi:10.1021/bc900296z
Yang K, Zhang S, Zhang G et al (2010) Graphene in mice: ultrahigh in vivo tumor uptake and efficient photothermal therapy. Nano Lett 10:3318–3323. doi:10.1021/nl100996u
Zhang W, Guo Z, Huang D et al (2011) Synergistic effect of chemo-photothermal therapy using PEGylated graphene oxide. Biomaterials 32:8555–8561. doi:10.1016/j.biomaterials.2011.07.071
Suda T, Liu D (2007) Hydrodynamic gene delivery: its principles and applications. Mol Ther 15:2063–2069. doi:10.1038/sj.mt.6300314
Suda T, Suda K, Liu D (2008) Computer-assisted hydrodynamic gene delivery. Mol Ther 16:1098–1104. doi:10.1038/mt.2008.66
Kamimura K, Zhang G, Liu D (2010) Image-guided, intravascular hydrodynamic gene delivery to skeletal muscle in pigs. Mol Ther 18:93–100. doi:10.1038/mt.2009.206
Mellott AJ, Forrest ML, Detamore MS (2012) Physical non-viral gene delivery methods for tissue engineering. Ann Biomed Eng. doi:10.1007/s10439-012-0678-1
Fuller DH, Loudon P, Schmaljohn C (2006) Preclinical and clinical progress of particle-mediated DNA vaccines for infectious diseases. Methods 40:86–97. doi:10.1016/j.ymeth.2006.05.022
Kis EE, Winter G, Myschik J (2012) Devices for intradermal vaccination. Vaccine 30:523–538. doi:10.1016/j.vaccine.2011.11.020
Sardesai NY, Weiner DB (2011) Electroporation delivery of DNA vaccines: prospects for success. Curr Opin Immunol 23:421–429. doi:10.1016/j.coi.2011.03.008
Littel-van Van Drunen, den Hurk S, Hannaman D (2010) Electroporation for DNA immunization: clinical application. Expert Rev Vaccines 9:503–517. doi:10.1586/erv.10.42
Daud AI, DeConti RC, Andrews S et al (2008) Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol 26:5896–5903. doi:10.1200/JCO.2007.15.6794
Broderick KE, Shen X, Soderholm J et al (2011) Prototype development and preclinical immunogenicity analysis of a novel minimally invasive electroporation device. Gene Ther 18:258–265. doi:10.1038/gt.2010.137
Yamaoka A, Guan X, Takemoto S et al (2010) Development of a novel Hsp70-based DNA vaccine as a multifunctional antigen delivery system. J Control Release 142:411–415. doi:10.1016/j.jconrel.2009.11.005
Suzuki R, Oda Y, Utoguchi N, Maruyama K (2011) Progress in the development of ultrasound-mediated gene delivery systems utilizing nano- and microbubbles. J Control Release 149:36–41. doi:10.1016/j.jconrel.2010.05.009
He Y, Bi Y, Hua Y et al (2011) Ultrasound microbubble-mediated delivery of the siRNAs targeting MDR1 reduces drug resistance of yolk sac carcinoma L2 cells. J Exp Clin Cancer Res 30:104. doi:10.1186/1756-9966-30-104
Raju BI, Leyvi E, Seip R et al (2013) Enhanced gene expression of systemically administered plasmid DNA in the liver with therapeutic ultrasound and microbubbles. IEEE Trans Ultrason Ferroelectr Freq Control 60:88–96
Liu Y, Yan J, Prausnitz MR (2012) Can ultrasound enable efficient intracellular uptake of molecules? A retrospective literature review and analysis. Ultrasound Med Biol 38:876–888. doi:10.1016/j.ultrasmedbio.2012.01.006
Cosgrove D, Harvey C (2009) Clinical uses of microbubbles in diagnosis and treatment. Med Biol Eng Comput 47:813–826. doi:10.1007/s11517-009-0434-3
Alzaraa A, Gravante G, Chung WY et al (2012) Targeted microbubbles in the experimental and clinical setting. Am J Surg 204:355–366. doi:10.1016/j.amjsurg.2011.10.024
Chorny M, Fishbein I, Yellen BB et al (2010) Targeting stents with local delivery of paclitaxel-loaded magnetic nanoparticles using uniform fields. Proc Natl Acad Sci USA 107:8346–8351. doi:10.1073/pnas.0909506107
Zheng X, Lu J, Deng L et al (2009) Preparation and characterization of magnetic cationic liposome in gene delivery. Int J Pharm 366:211–217. doi:10.1016/j.ijpharm.2008.09.019
Plank C, Zelphati O, Mykhaylyk O (2011) Magnetically enhanced nucleic acid delivery. Ten years of magnetofection-progress and prospects. Adv Drug Deliv Rev 63:1300–1331. doi:10.1016/j.addr.2011.08.002
Vogel A, Noack J, Hüttman G, Paltauf G (2005) Mechanisms of femtosecond laser nanosurgery of cells and tissues. Applied Physics B 81:1015–1047. doi:10.1007/s00340-005-2036-6
Tirlapur UK, König K (2002) Cell biology: Targeted transfection by femtosecond laser. Nature 418:290–291. doi:10.1038/418290a
Barrett LE, Sul J-Y, Takano H et al (2006) Region-directed phototransfection reveals the functional significance of a dendritically synthesized transcription factor. Nat Methods 3:455–460. doi:10.1038/nmeth885
Kohli V, Robles V, Cancela ML et al (2007) An alternative method for delivering exogenous material into developing zebrafish embryos. Biotechnol Bioeng 98:1230–1241. doi:10.1002/bit.21564
Tsampoula X, Taguchi K, Cižmár T et al (2008) Fibre based cellular transfection. Opt Express 16:17007. doi:10.1364/OE.16.017007
Menezes V, Mathew Y, Takayama K et al (2012) Laser plasma jet driven microparticles for DNA/drug delivery. PLoS ONE 7:e50823. doi:10.1371/journal.pone.0050823
Kawakami S, Higuchi Y, Hashida M (2008) Nonviral approaches for targeted delivery of plasmid DNA and oligonucleotide. J Pharm Sci 97:726–745. doi:10.1002/jps.21024
Passineau MJ, Zourelias L, Machen L et al (2010) Ultrasound-assisted non-viral gene transfer to the salivary glands. Gene Ther 17:1318–1324. doi:10.1038/gt.2010.86
Al-Dosari MS, Gao X (2009) Nonviral gene delivery: principle, limitations, and recent progress. AAPS J 11:671–681. doi:10.1208/s12248-009-9143-y
Wolff JA, Ludtke JJ, Acsadi G et al (1992) Long-term persistence of plasmid DNA and foreign gene expression in mouse muscle. Hum Mol Genet 1:363–369
Hagstrom JE, Hegge J, Zhang G et al (2004) A facile nonviral method for delivering genes and siRNAs to skeletal muscle of mammalian limbs. Mol Ther 10:386–398. doi:10.1016/j.ymthe.2004.05.004
Sebestyén MG, Hegge JO, Noble MA et al (2007) Progress toward a nonviral gene therapy protocol for the treatment of anemia. Hum Gene Ther 18:269–285. doi:10.1089/hum.2006.186
Kawabata K, Takakura Y, Hashida M (1995) The fate of plasmid DNA after intravenous injection in mice: involvement of scavenger receptors in its hepatic uptake. Pharm Res 12:825–830
Wang M, Orsini C, Casanova D et al (2001) MUSEAP, a novel reporter gene for the study of long-term gene expression in immunocompetent mice. Gene 279:99–108
Vandermeulen G, Marie C, Scherman D, Préat V (2011) New generation of plasmid backbones devoid of antibiotic resistance marker for gene therapy trials. Mol Ther 19:1942–1949. doi:10.1038/mt.2011.182
Kanwar JR, Roy K, Kanwar RK (2011) Chimeric aptamers in cancer cell-targeted drug delivery. Crit Rev Biochem Mol Biol 46:459–477. doi:10.3109/10409238.2011.614592
Bouchard PR, Hutabarat RM, Thompson KM (2010) Discovery and development of therapeutic aptamers. Annu Rev Pharmacol Toxicol 50:237–257. doi:10.1146/annurev.pharmtox.010909.105547
Huang D-B, Vu D, Cassiday LA et al (2003) Crystal structure of NF-kappaB (p50)2 complexed to a high-affinity RNA aptamer. Proc Natl Acad Sci USA 100:9268–9273. doi:10.1073/pnas.1632011100
Mashima T, Matsugami A, Nishikawa F et al (2009) Unique quadruplex structure and interaction of an RNA aptamer against bovine prion protein. Nucleic Acids Res 37:6249–6258. doi:10.1093/nar/gkp647
Phan AT, Kuryavyi V, Darnell JC et al (2011) Structure-function studies of FMRP RGG peptide recognition of an RNA duplex-quadruplex junction. Nat Struct Mol Biol 18:796–804. doi:10.1038/nsmb.2064
Sussman D, Wilson C (2000) A water channel in the core of the vitamin B(12) RNA aptamer. Structure 8:719–727
Ellington AD, Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346:818–822. doi:10.1038/346818a0
Ellington AD, Szostak JW (1992) Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 355:850–852. doi:10.1038/355850a0
Tuerk C, Gold L (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510
Nutiu R, Li Y (2005) In vitro selection of structure-switching signaling aptamers. Angew Chem Int Ed Engl 44:1061–1065. doi:10.1002/anie.200461848
Stoltenburg R, Nikolaus N, Strehlitz B (2012) Capture-SELEX: selection of DNA aptamers for aminoglycoside antibiotics. J Anal Methods Chem 2012:415697. doi:10.1155/2012/415697
Binning JM, Leung DW, Amarasinghe GK (2012) Aptamers in virology: recent advances and challenges. Front Microbiol 3:29. doi:10.3389/fmicb.2012.00029
Kang D, Wang J, Zhang W et al (2012) Selection of DNA aptamers against glioblastoma cells with high affinity and specificity. PLoS ONE 7:e42731. doi:10.1371/journal.pone.0042731
Eckstein F, Gish G (1989) Phosphorothioates in molecular biology. Trends Biochem Sci 14:97–100
Green LS, Jellinek D, Bell C et al (1995) Nuclease-resistant nucleic acid ligands to vascular permeability factor/vascular endothelial growth factor. Chem Biol 2:683–695
Ruckman J, Green LS, Beeson J et al (1998) 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J Biol Chem 273:20556–20567
Lin Y, Nieuwlandt D, Magallanez A et al (1996) High-affinity and specific recognition of human thyroid stimulating hormone (hTSH) by in vitro-selected 2′-amino-modified RNA. Nucleic Acids Res 24:3407–3414
Keefe AD, Pai S, Ellington A (2010) Aptamers as therapeutics. Nat Rev Drug Discov 9:537–550. doi:10.1038/nrd3141
Khar RK, Jain GK, Warsi MH et al (2010) Nano-vectors for the ocular delivery of nucleic acid-based therapeutics. Indian J Pharm Sci 72:675–688. doi:10.4103/0250-474X.84575
Sanghvi YS (2011) A status update of modified oligonucleotides for chemotherapeutics applications. Curr Protoc Nucleic Acid Chem Chap 4: Unit 4.1.1–22. Doi: 10.1002/0471142700.nc0401s46
Thiel KW, Giangrande PH (2009) Therapeutic applications of DNA and RNA aptamers. Oligonucleotides 19:209–222. doi:10.1089/oli.2009.0199
Cohen MG, Purdy DA, Rossi JS et al (2010) First clinical application of an actively reversible direct factor IXa inhibitor as an anticoagulation strategy in patients undergoing percutaneous coronary intervention. Circulation 122:614–622. doi:10.1161/CIRCULATIONAHA.109.927756
Altman S, Cech TR (2013) The nobel prize in chemistry 1989. http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1989/. Accessed 4 Apr 2013
Scherer LJ, Rossi JJ (2003) Approaches for the sequence-specific knockdown of mRNA. Nat Biotechnol 21:1457–1465. doi:10.1038/nbt915
Burke JM (1996) Hairpin ribozyme: current status and future prospects. Biochem Soc Trans 24:608–615
Usman N, Beigelman L, McSwiggen JA (1996) Hammerhead ribozyme engineering. Curr Opin Struct Biol 6:527–533
El-Sagheer AH, Brown T (2010) New strategy for the synthesis of chemically modified RNA constructs exemplified by hairpin and hammerhead ribozymes. Proc Natl Acad Sci USA 107:15329–15334. doi:10.1073/pnas.1006447107
Liang JC, Bloom RJ, Smolke CD (2011) Engineering biological systems with synthetic RNA molecules. Mol Cell 43:915–926. doi:10.1016/j.molcel.2011.08.023
Wieland M, Berschneider B, Erlacher MD, Hartig JS (2010) Aptazyme-mediated regulation of 16S ribosomal RNA. Chem Biol 17:236–242. doi:10.1016/j.chembiol.2010.02.012
Sarver N, Cantin EM, Chang PS et al (1990) Ribozymes as potential anti-HIV-1 therapeutic agents. Science 247:1222–1225
Yu M, Ojwang J, Yamada O et al (1993) A hairpin ribozyme inhibits expression of diverse strains of human immunodeficiency virus type 1. Proc Natl Acad Sci USA 90:6340–6344
Dropulić B, Lin NH, Martin MA, Jeang KT (1992) Functional characterization of a U5 ribozyme: intracellular suppression of human immunodeficiency virus type 1 expression. J Virol 66:1432–1441
Ojwang JO, Hampel A, Looney DJ et al (1992) Inhibition of human immunodeficiency virus type 1 expression by a hairpin ribozyme. Proc Natl Acad Sci USA 89:10802–10806
Wong-Staal F, Poeschla EM, Looney DJ (1998) A controlled, phase 1 clinical trial to evaluate the safety and effects in HIV-1 infected humans of autologous lymphocytes transduced with a ribozyme that cleaves HIV-1 RNA. Hum Gene Ther 9:2407–2425. doi:10.1089/hum.1998.9.16-2407
(2013) Press release: the 2006 nobel prize in physiology or medicine. http://www.nobelprize.org/nobel_prizes/medicine/laureates/2006/press.html. Accessed 12 Apr 2013
Pecot CV, Calin GA, Coleman RL et al (2011) RNA interference in the clinic: challenges and future directions. Nat Rev Cancer 11:59–67. doi:10.1038/nrc2966
Sibley CR, Seow Y, Wood MJA (2010) Novel RNA-based strategies for therapeutic gene silencing. Mol Ther 18:466–476. doi:10.1038/mt.2009.306
Cho Y-S, Lee GY, Sajja HK et al (2013) Targeted delivery of siRNA-generating DNA nanocassettes using multifunctional nanoparticles. Small. doi:10.1002/smll.201201973
Wall NR, Shi Y (2003) Small RNA: can RNA interference be exploited for therapy? Lancet 362:1401–1403. doi:10.1016/S0140-6736(03)14637-5
Dalby B, Cates S, Harris A et al (2004) Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications. Methods 33:95–103. doi:10.1016/j.ymeth.2003.11.023
McCaffrey AP, Meuse L, Pham T-TT et al (2002) RNA interference in adult mice. Nature 418:38–39. doi:10.1038/418038a
Liu YP, Vink MA, Westerink J-T et al (2010) Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 16:1328–1339. doi:10.1261/rna.1887910
Van den Haute C, Eggermont K, Nuttin B et al (2003) Lentiviral vector-mediated delivery of short hairpin RNA results in persistent knockdown of gene expression in mouse brain. Hum Gene Ther 14:1799–1807. doi:10.1089/104303403322611809
Halder J, Kamat AA, Landen CN Jr et al (2006) Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clin Cancer Res 12:4916–4924. doi:10.1158/1078-0432.CCR-06-0021
Sato Y, Murase K, Kato J et al (2008) Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol 26:431–442. doi:10.1038/nbt1396
Frank-Kamenetsky M, Grefhorst A, Anderson NN et al (2008) Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci USA 105:11915–11920. doi:10.1073/pnas.0805434105
Marquez VE, Siddiqui MA, Ezzitouni A et al (1996) Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides? J Med Chem 39:3739–3747. doi:10.1021/jm960306+
Deleavey GF, Damha MJ (2012) Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol 19:937–954. doi:10.1016/j.chembiol.2012.07.011
McNeer NA, Schleifman EB, Cuthbert A et al (2012) Systemic delivery of triplex-forming PNA and donor DNA by nanoparticles mediates site-specific genome editing of human hematopoietic cells in vivo. Gene Ther. doi:10.1038/gt.2012.82
Rogers FA, Hu R-H, Milstone LM (2013) Local delivery of gene-modifying triplex-forming molecules to the epidermis. J Invest Dermatol 133:685–691. doi:10.1038/jid.2012.351
Chakrabarti A, Zhang K, Aruva MR et al (2007) Radiohybridization PET imaging of KRAS G12D mRNA expression in human pancreas cancer xenografts with [(64)Cu]DO3A-peptide nucleic acid-peptide nanoparticles. Cancer Biol Ther 6:948–956
Karkare S, Bhatnagar D (2006) Promising nucleic acid analogs and mimics: characteristic features and applications of PNA, LNA, and morpholino. Appl Microbiol Biotechnol 71:575–586. doi:10.1007/s00253-006-0434-2
Campbell MA, Wengel J (2011) Locked vs. unlocked nucleic acids (LNA vs. UNA): contrasting structures work towards common therapeutic goals. Chem Soc Rev 40:5680–5689. doi:10.1039/c1cs15048k
Li Y-F, Morcos PA (2008) Design and synthesis of dendritic molecular transporter that achieves efficient in vivo delivery of morpholino antisense oligo. Bioconjug Chem 19:1464–1470. doi:10.1021/bc8001437
Eisen JS, Smith JC (2008) Controlling morpholino experiments: don’t stop making antisense. Development 135:1735–1743. doi:10.1242/dev.001115
Huh D, Hamilton GA, Ingber DE (2011) From 3D cell culture to organs-on-chips. Trends Cell Biol 21:745–754. doi:10.1016/j.tcb.2011.09.005
Kelkar SS, Reineke TM (2011) Theranostics: combining imaging and therapy. Bioconjug Chem 22:1879–1903. doi:10.1021/bc200151q
Acknowledgments
The authors are indebted to Drs. William Heetderks and Antonio Sastre for their critical review and to Ms. Christine Rogers for her assistance with preparing the manuscript. Figure 26.4 Used with permission and modified from Biochimica et Biophysica Acta (BBA)—Proteins and Proteomics, 1697, Yoon S. Cho-Chung, Antisense protein kinase A RIα-induced tumor reversion: portrait of a microarray, 71–79, Copyright 2004, with permission from Elsevier.
Conflict of Interest
The authors declare that they have no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag London
About this chapter
Cite this chapter
Conroy, R., Seto, B. (2014). Multifunctional Nanoscale Delivery Systems for Nucleic Acids. In: Cai, W. (eds) Engineering in Translational Medicine. Springer, London. https://doi.org/10.1007/978-1-4471-4372-7_18
Download citation
DOI: https://doi.org/10.1007/978-1-4471-4372-7_18
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-4371-0
Online ISBN: 978-1-4471-4372-7
eBook Packages: EngineeringEngineering (R0)