
Abstract
Fibroblast growth factors (FGF) have pleiotropic roles in human development and metabolism, and FGF signaling through FGF receptors (FGFRs) has been implicated in a wide range of cancers. Extensive pre-clinical and clinical studies are currently underway to elucidate the therapeutic possibilities: monoclonal antibodies, ligand traps, heparanoids, and kinase inhibitors all have potential for the treatment of FGFR-driven cancers.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others

Keywords
- Brivanib
- Dovitinib
- FGF receptor (FGFR)
- Fibroblast growth factor (FGF)
- Aberrant FGF
- Signaling
- A-loop tyrosine phosphorylation
- Core homology region
- CRKL
- ELISA
- Endocrine FGFs
- Exon 8 and exon 9
- FGFR binding specificity
- FGFR dimerization
- FGFR1–3 genes
- FRS2α, 3
- Immunohistochemical staining
- Mechanisms
- Overexpression
- Paracrine-acting FGF subfamilies
- Preclinical studies
- Prognostic markers
- Pyruvate kinase
- Role in cancer
- Single nucleotide polymorphisms (SNPs)
- Somatic mutations
- Subfamilies
- Therapy
- Palifermin
Target: FGF, FGFR
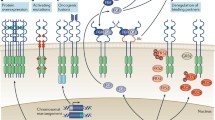
Fibroblast growth factor (FGF) signaling plays pleiotropic roles in human development and metabolism. Based on primary sequence, structural similarity, and phylogenetic analysis, the 18 human FGFs (FGF1–FGF10 and FGF16–FGF23) are grouped into five paracrine subfamilies and one endocrine subfamily. The paracrine subfamilies include the FGF1 subfamily comprising FGF1 and 2; the FGF7 subfamily comprising FGF3, 7, 10, and 22; the FGF4 subfamily comprising FGF4, 5, and 6; the FGF8 subfamily comprising FGF8, 17, and 18; and the FGF9 subfamily comprising FGF9, 16, and 20. The endocrine-acting FGF19 subfamily comprises FGF19, 21, and 23. The paracrine-acting FGF subfamilies play essential roles in spermatogenesis, mesoderm induction, somitogenesis, organogenesis, and pattern formation, whereas members of the FGF19 subfamily signal in an endocrine fashion to regulate major metabolic processes including glucose, lipid, cholesterol, and bile acid metabolism and serum phosphate/vitamin D homeostasis (Kuro-o 2008; Martin 1998; Ornitz 2005; Yu and White 2005).
The core homology region of FGFs (approximately 120 amino acids long) adopts a β-trefoil fold consisting of 12 antiparallel β-strands (β1–β12) that arrange into 3 sets of 4-stranded b-sheets in paracrine FGFs. Endocrine FGFs , however, lack the β11 strand and as a result have an atypical trefoil fold. The globular β-trefoil core domain is flanked by highly divergent N- and C-terminal tails. All FGFs bind heparan sulfate (HS) albeit with differing affinities. The HS binding site (HBS) in FGFs is composed of residues from the β1 to β2 loop and from the region between β10 and β12. Paracrine FGFs have substantial affinity for HS and therefore can only act locally, whereas the weak affinity of the FGF19 subfamily members allows them to avoid entrapment in the extracellular matrix (ECM) and enter blood circulation (Beenken and Mohammadi 2012; Mohammadi et al. 2005a).
FGFs carry out their diverse actions by binding and activating the FGF receptor (FGFR) subfamily of receptor tyrosine kinases encoded by four genes in humans (FGFR1–4). FGFR1–3 genes are composed of 19 exons, whereas FGFR4 gene contains 18 exons. The prototypical FGFR is composed of three extracellular immunoglobulin domains (D1–D3) connected by flexible linker sequences, a transmembrane domain, and an intracellular conserved tyrosine kinase domain. Structural studies have shown that ligand binding requires both D2 and D3 domains. Like FGFs, FGFRs are also HS-binding proteins. HBS in FGFRs is located in D2 and is composed of basic residues that collectively localize onto one of the b-sheets of this domain. The D1 and D1–D2 linker are dispensable for ligand binding and in fact suppress FGF and HS binding affinity of the D2–D3 region. In FGFR1–3, exon 8 (known as “IIIb”) and exon 9 (known as “IIIc”) code for the second half of D3 and are spliced in a mutually exclusive fashion to the common exon 7 (known as “IIIa”) that encodes the first half of D3. This splicing event is tissue specific and results in the expression of epithelial “b” isoforms (FGFR1b–FGFR3b) or mesenchymal “c” isoforms (FGFR1c–FGFR3c) thereby expanding the number of principal FGFRs to seven, namely, FGFR1c, FGFR1b, FGFR2c, FGFR2b, FGFR3c, FGFR3b, and FGFR4 (Beenken and Mohammadi 2009; Johnson et al. 1991).
FGF–FGFR binding specificity /promiscuity is critical in FGF signaling and is principally dictated by primary sequence differences between the 18 FGFs and the 17 principal FGFRs. Tissue-specific alternative splicing in the D3 domain of FGFR1–3 is the main mechanism in the regulation of FGF–FGFR binding specificity. Generally, paracrine FGF subfamilies also exhibit tissue-specific expression patterns and are expressed in either epithelial or mesenchymal compartments. The epithelially expressed FGFs typically show specificity for FGFRc isoforms expressed in the mesenchyme and vice versa, resulting in the establishment of an epithelial–mesenchymal signaling loop (Beenken and Mohammadi 2011). It is well documented that FGF7 and FGF10, which are expressed exclusively in the mesenchyme, specifically activate FGFR2b to mediate the epithelial–mesenchymal signaling required for the development of multiple organs and glands including lung, thyroid, pituitary, lachrymal, and salivary glands. In contrast, the members of the FGF4, FGF8, and FGF9 subfamilies are expressed in the epithelium and activate the mesenchymal FGFRc isoforms to govern patterning and morphogenesis of multiple tissues and organs, including the brain, lung, heart, kidney, eye, limb, and ear (Beenken and Mohammadi 2009). For instance, FGF8b binds FGFR1c–FGFR3c and FGFR4 but does not recognize “b” isoforms. FGF2 binds with comparable high affinity to both FGFR1c and FGFR2c but does not bind the remaining five FGFRs. FGF1 overrides the specificity barrier set by alternative splicing and binds equally well to both “b” and “c” isoforms of FGFRs. To date, crystal structures of eight FGF–FGFR complexes have been published including FGF1–FGFR1c (PDB ID: 1EVT), FGF1–FGFR2c (PDB ID: 1DJS), FGF1–FGFR3c (PDB ID: 1RY7), FGF1–FGFR2b (PDB ID: 3OJM), FGF2–FGFR1c (PDB ID: 1CVS), FGF2–FGFR2c (PDB ID: 1EV2), FGF8–FGFR2c (PDB ID: 2FDB), and FGF10–FGFR2b (PDB ID: 1NUN). Structural data show that the D3 alternative splicing alters the primary sequences of key FGF binding sites in D3 including the bC’–bE and bF–bG loops and bF and bG strands to narrow the ligand binding specificity of FGFRb isoforms to mesenchymally expressed FGFs and that of FGFRc isoforms to epithelially expressed FGFs. The structural data also show that the specificity/promiscuity profile of a given FGF is principally dictated by the primary sequence of its N-terminal region. The structural data have begun to illuminate the shared primary sequence and secondary structural elements within the N-termini of members of a given FGF subfamily that explain overlapping FGFR binding specificity/promiscuity profile of the subfamily (Goetz and Mohammadi 2013).
A wealth of genetic studies in mice and flies and cell-based studies has established that paracrine FGF–FGFR signaling is HS dependent. Recent data show that HS controls the diffusion of paracrine FGFs and hence shapes the morphogenetic gradients in the extracellular matrix. Aside from controlling the diffusion of FGFs, HS impinges on paracrine FGF signaling through many other mechanisms as well, including coordination/stabilization of FGF–FGFR binding and dimerization, providing thermal stability and protecting against proteolytic degradation, acting as a storage reservoir for ligand, and limiting the dimensionality of FGF (Beenken and Mohammadi 2009).
HS-assisted FGF–FGFR dimerization is a key event for signal transmission across the plasma membrane by paracrine FGFs. The symmetric model of FGF–FGFR dimerization bears a 2:2:2 FGF–FGFR–HS stoichiometry in which multivalent protein–protein contacts between the two FGF–FGFR halves are the main driving force of dimerization and HS facilitates these protein–protein contacts (Mohammadi et al. 2005b). The FGFRs, located in the center of the dimer, interact directly via the membrane-proximal end of D2. The FGFs, located at either side of the centrally located FGFRs, interact with both receptors through primary and secondary receptor binding sites. On the membrane distal end of the 2:2:2 FGF–FGFR–HS symmetric dimer, the spatially separate HS binding sites of two FGFs and of two receptor D2 domains merge into one large HS-binding canyon, into which two HS oligosaccharides bind. By simultaneously engaging the HS binding sites of FGF and receptor D2 domains in the canyon, HS fortifies both the primary FGF–FGFR interface and the dimer interface that consists of both direct receptor–receptor and secondary ligand–receptor contacts. The nonreducing end of the oligosaccharide is tucked between the two receptor D2 domains, while the reducing end interacts with HS binding site of the ligand. On average, each oligosaccharide engages in about 30 hydrogen bonds with FGF and FGFR. The binding of HS does not cause significant conformational changes to occur in either the FGF ligand or receptor. Because the endocrine-acting FGF19 subfamily members have extremely low HS affinity, HS is incapable of enhancing endocrine FGF–FGFR binding and dimerization. To overcome this deficiency, endocrine FGFs rely on α-/β-Klotho coreceptors which form binary complexes with the cognate FGFRs of endocrine FGFs to increase the affinity of FGFR for endocrine FGFs and induce FGFR dimerization (Beenken and Mohammadi 2012).
Biology of the Target
HS- or Klotho-dependent dimerization of the FGFR extracellular domains juxtaposes the cytoplasmic kinase domains allowing them to transphosphorylate each other on A-loop tyrosines. A-loop tyrosine phosphorylation elevates the intrinsic kinase activity of FGFR kinase by stabilizing the active conformation of the kinase. A-loop phosphorylation is then followed by phosphorylation on tyrosines in the C-tail, kinase insert, and juxtamembrane regions. Among many downstream signaling pathways that FGFR kinase activation triggers are RAS–MAPK, PI hydrolysis/PKC/Ca2+, PI3K–AKT, and RAC1/CDC42 signaling pathways (Dailey et al. 2005; Eswarakumar et al. 2005).
Phosphorylation of an FGFR-invariant tyrosine (Y766 in FGFR1) at the C-tail of FGFR creates a binding site for the SH2 domain of PLCγ (also known as FRS1) and is required for PLCγ phosphorylation and activation. PLCγ recruitment serves two purposes: (i) it facilitates phosphorylation of PLCγ to increase its enzymatic activity, and (ii) it brings PLCγ to the vicinity of its substrate PIP2 in the plasma membrane. Hydrolysis of PIP2 generates two second messengers: IP3 and DAG that stimulate Ca2+ release from intracellular stores and PKC activation, respectively. Activated PKC then activates the MAPK pathway in a Ras-independent manner by phosphorylating and activating Raf (Schlessinger 2000).
In contrast to PLCγ, CRKL is an adaptor protein that lacks intrinsic enzymatic activity. Recruitment of CRKL to the phosphorylated tyrosine in the juxtamembrane region of FGFR1 and FGFR2 leads to translocation of associated Rac1/Cdc42 to the plasma membrane. These G-proteins act through their effector protein, PAK, to activate the MAPK pathway by phosphorylating Raf1 and Mek1, leading to changes in cytoskeletal reorganization and cell motility (Seo et al. 2009).
FRS2α is another major adaptor protein for FGFRs that, unlike PLCγ and CRKL, associates constitutively (receptor tyrosine phosphorylation independent) with the juxtamembrane region of FGFR. Phosphorylation of FRS2α by the A-loop phosphorylated (activated) FGFR generates docking sites for the SH2 domains of the adaptor protein GRB2 and the phosphatase Shp2. Grb2 is constitutively associated with SOS, Cbl, and Gab1. Since SOS is a guanine nucleotide exchange factor (GEF) for Ras, Grb2–SOS activates the RAS–MAPK pathway. Grb2–Cbl mediates FRS2 degradation, since Cbl is an E3 ubiquitin ligase. Finally, the PI3K–AKT pathway is activated by Grb2–Gab1 (Gotoh 2008).
Target Assessment
Quantitative PCR is used to measure transcripts of FGFs and FGFRs in excised tissues. Serum and urine levels of FGF can be measured using ELISA . Immunohistochemical staining is also commonly used to detect the presence of FGF and FGFR proteins in tumor tissues.
Role of the Target in Cancer
Rank: 7 – clear role in cancer but not yet a primary therapeutic target.
Uncontrolled FGF signaling can be strongly oncogenic as it can promote not only cell proliferation and migration but also neoangiogenesis, as originally shown by Klagsbrun through studies in the 1970s and 1980s on what was then known as tumor angiogenesis factor (TAF). There is ample evidence for the involvement of deregulated FGF signaling in human cancer. FGF signaling can be deregulated through a variety of mechanisms, including receptor mutations leading to constitutive activation or loss of ligand binding specificity, transcriptional upregulation of ligands and/or receptors leading to autocrine signaling, and genetic translocations generating constitutively active FGFR fusion proteins. Aberrant FGF signaling is best known for causing craniosynostosis and dwarfism syndromes such as Apert’s syndrome (AS), Pfeiffer’s syndrome (PS), and achondroplasia (ACH) (Wilkie 2005). Interestingly, many of the germ line mutations in FGFRs associated with skeletal disorders also occur as somatic mutations in cancer. The FGFR2 S252W and N549K mutations that cause AS and PS, respectively, are also detected in endometrial cancers. Mutations of the analogous N546 in FGFR1 and N535 in FGFR4 are detected in glioblastomas and rhabdomyosarcomas, respectively. A mutation of FGFR2 W290C associated with PS has been found in lung carcinomas. Mutations of K650 in the A-loop of FGFR3 kinase are responsible for severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) as well as thanatophoric dysplasia types I and II (TDI, TDII). This residue is also frequently mutated in bladder and cervical cancers and in multiple myeloma. Mutations at the K650 codon in FGFR3 leading to thanatophoric dysplasia have been identified in spermatocytic seminomas, and these mutations increase in prevalence in sperm DNA as paternal age increases. FGFR3 G380R mutation, the most common cause of achondroplasia, is also seen in bladder cancer. This mutation leads to gain of function by promoting both receptor dimerization and receptor recycling, thereby impairing efficient receptor degradation. Many of these FGFR mutations have been structurally characterized and have been shown to lead to ligand-dependent or ligand-independent gain of function by enhancing ligand–receptor affinity, overriding ligand binding specificity, or relieving FGFR kinase autoinhibiton (Beenken and Mohammadi 2011).
There is also a long list of other FGFR somatic mutations /alterations detected in cancers that do not occur in skeletal disorders. In 8p11 myeloproliferative syndrome (EMS), a hematologic cancer, FGFR1 kinase is constitutively activated by being fused to eight different dimerizing/oligomerizing domains, including the zinc finger gene ZNF198 and BCR. Interestingly, blocking the recruitment of PLCg-1 to the FGFR1 kinase fusion proteins by mutating Y766 in the PLCg-1 binding site of FGFR1 attenuates EMS, suggesting a role for PI hydrolysis/PKC/Ca2+ signaling in the progression of this cancer. A subset of glioblastomas harbor oncogenic chromosomal translocations that fuse in frame the tyrosine kinase domains of FGFR1 or FGFR3 to the transforming acidic coiled-coil (TACC) domain TACC1 or TACC3. The FGFR3–TACC3 fusion occurs in bladder cancer as well. Additionally, FGFR1 kinase domain gain-of-function mutations are seen in glioblastomas, and malignant prostate cells have elevated levels of FGFR1 expression. Oncogenic t(4:14) rearrangements of FGFR3 have been described in multiple myeloma. Translocations of FGFR3 are also seen in peripheral T-cell lymphomas. FGFR4 mutations are found in rhabdomyosarcomas and correlate with more aggressive cancer. These mutations, including V550E and V550L, promote receptor autophosphorylation and constitutive signaling (Beenken and Mohammadi 2009).
Overexpression of both FGFs and FGFRs has long been implicated in cancer. FGF1 overexpression in ovarian tumors is associated with poor survival. FGF1, 2, 6, 7, 8, and 9 are found to be overexpressed in prostate cancers. Overexpressed FGF3 is seen in breast cancers as is FGF2. Overexpressed FGF8 has been detected in 50% of in situ prostate tumors and 80% of advanced prostate cancers, and FGF8 is also overexpressed in breast cancer. Overexpression of FGF5 has been recorded in esophagus, colon, prostate, and lung cancers as well as in melanoma. FGF10 is overexpressed in breast cancers. FGF18 is overexpressed in colon cancer as is FGF19. Hepatocellular carcinomas show overexpression of FGF2, 8, 17, and 18. Decreased expression of Sprouty proteins, major cytoplasmic negative regulators of FGF signaling, is observed in breast and prostate cancers (Turner and Grose 2010).
FGFR1 overexpression is seen in ovarian cancer, bladder cancer, oral squamous carcinoma, prostate cancer, squamous cell lung cancer, small and non-small cell lung cancer, breast cancer, and rhabdomyosarcoma. In ~10% of gastric cancers, gene amplification leads to increased FGFR2 expression which correlates with a poor prognosis. FGFR2 is also overexpressed in about 10% of human endometrial carcinomas and in triple-negative breast cancers. An FGFR2 variant with a C-terminal truncation is expressed in cancer cell lines. This truncation attenuates receptor endocytosis, leading to increased levels of cell surface receptor and accompanying signaling. FGFR2 overexpression enables FGF7-dependent stimulation of gastric cancer growth. Autocrine signaling can occur when mesenchymal isoforms of FGFR are misexpressed in epithelial tissues. For instance, a switch from FGFR2b to FGFR2c in bladder cancers signals a change to a more highly invasive bladder or prostate cancer (Knights and Cook 2010).
The mechanisms by which FGF–FGFR signaling leads to cancer are being continuously explored. One interesting recent development in cancer biology has been the association of FGF signaling with the Warburg effect (Hitosugi et al. 2009). The Warburg effect describes the phenomenon that cancer cells have greater uptake of glucose compared to normal cells and preferentially engage in glycolysis, even in the presence of oxygen. Pyruvate kinase (PK) is a rate-limiting enzyme in glycolysis and catalyzes the conversion of phosphoenolpyruvate to pyruvate. Of the four isoenzymes of PK (M1, M2, L, and R), PKM2 is found mainly in malignant cells. In normal physiology, pyruvate is subsequently converted to acetyl-CoA by pyruvate dehydrogenase A1 (PDHA1) and then enters the Krebs cycle, and only under hypoxic conditions will pyruvate be converted to lactate by lactate dehydrogenase A (LDH-A). In cancer cells, however, pyruvate is converted to lactate in both hypoxic and oxidative environments. This physiology of the Warburg effect is oncogenic, possibly because it assists rapid cell division by supplying an increased amount of basic building blocks like nucleic and amino acids through the upregulation of glycolysis. Interestingly, FGFR1 is implicated in mediating the Warburg effect by numerous mechanisms, including regulating pyruvate production, preventing pyruvate from entering oxidative metabolism, and increasing the conversion of pyruvate to lactate. FGFR1 directly tyrosine phosphorylates PKM2 to inhibit its activity. Additionally, PDH kinase 1 (PDHK1), a mitochondrial Ser/Thr kinase and an inhibitor of PDHA1, is activated by FGFR1-mediated tyrosine phosphorylation and promotes cancer cell growth. FGFR1 also tyrosine phosphorylates LDH-A, thereby increasing its activity and improving its binding to its substrate, NADH (Fan et al. 2011).
FGF signaling has also been shown to confer loss of cell polarity and increased migratory phenotypes upon cancer cells by inhibiting epithelial–mesenchymal transition (EMT). For instance, pathological FGF signaling can lead to prostate carcinogenesis via EMT. Overexpression of FGF10 in prostatic mesenchyme leads to upregulation of androgen receptor expression in the adjacent epithelium and transforms the epithelium into well-differentiated prostate adenocarcinoma. Interestingly, dominant-negative FGFR1 is able to revert the induced cancer back to normal epithelium. Inducible expression of FGFR1 also leads to development of prostate adenocarcinoma through EMT and is associated with increased Sox9 expression, a known regulator of EMT. Deactivating inducible FGFR1 signaling led to the regression of prostatic intraepithelial neoplasia and slowed progression of adenocarcinoma (Yilmaz and Christofori 2009).
The list of mechanisms by which FGF signaling contributes to tumorigenesis keeps expanding. FGF1 and FGF2 are released when tumor cells decay in the necrotic center of tumor and act as an impetus for neoangiogenesis, with melanomas being an example of this process. By implanting xenografts of prostate cancer bone metastases from humans into mice, FGF9 signaling was found to have a role in mediating the progression of bone metastases in prostate cancer. Neutralizing antibody to FGF9 reduced the size of the bone tumors that developed from the xenografts. Another FGF9 subfamily member, FGF20, was found to be necessary for maintaining the mitogenic state of β-catenin-transformed rat kidney epithelial cells, since FGF20 siRNA interfered with β-catenin-mediated growth in these cells (Beenken and Mohammadi 2009).
High-Level Overview
Diagnostic, Prognostic, Predictive
Single nucleotide polymorphisms (SNPs) in FGFR2 are associated with breast cancers carrying the BRCA2 mutation. These SNPs are postulated to increase affinity for transcription factors, causing increased FGFR2 expression. Eighty percent of superficial papillary bladder tumors harbor gain-of-function FGFR3 mutations, and thus, FGFR3 mutations are being considered as a marker for non-muscle-invasive tumors. Detection of FGFR3 mutant proteins in urine has been shown to be a marker of tumor recurrence (Miyake et al. 2010).
FGFRs are beginning to be appreciated as prognostic markers for cancer. The G388R mutation in the transmembrane domain of FGFR4 is associated with prostate cancer progression, more aggressive colon cancer, and also predicts a poor prognosis in head and neck squamous cell carcinomas and gastric cancer. The mutation has been shown to slow down receptor internalization resulting in increased cell surface expression of FGFR4 and accompanied sustained signaling. In addition, the expression of the mutated FGFR4 induces cell migration and has also been found to confer resistance to chemotherapy. Interfering with FGFR4 signaling with an antibody resensitized cells to chemotherapy (Beenken and Mohammadi 2009).
Therapeutic
Currently, only one FGF is being used as a therapy for cancer patients. Recombinant N-terminally truncated FGF7, known as palifermin , is FDA approved for the alleviation of radiation and chemotherapy-induced mucositis in cancer patients undergoing bone marrow transplant (Spielberger et al. 2004). By administering palifermin for 3 days prior to chemotherapy and then for 3 days following hematopoietic stem cell transplant, palifermin reduced patients’ use of opioids, reduced the median duration of mucositis from 9 to 6 days, and reduced the incidence of severe mucositis from 62% to 20%. The improvement in quality of life provided by this drug is significant, since some patients were enabled to continue oral feeding during their cancer therapy who otherwise would have been prevented from doing so by severe mucositis. No significant side effects from palifermin have been documented. Palifermin primarily acts by inducing increased epithelial cell proliferation. The new epithelium that is induced can persist for up to 1 week following a dose of palifermin. Other proposed mechanisms of FGF7 action include upregulating Nrf2 that activates genes encoding antioxidant enzymes. FGF7 may also favorably impact the course of mucositis by reducing the Th1/Th2 ratio of cytokines and by reducing TNF-a and IFN-γ through its induction of IL-13.
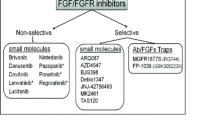
Presently, no drugs that exclusively target FGFRs are being used in cancer therapy, but sunitinib, a broad-spectrum receptor tyrosine kinase inhibitor with coverage of FGFRs but whose activity is primarily mediated through inhibition of VEGF, PDGF, and KIT pathways, is FDA approved for treatment of GI stromal tumors, renal cell carcinomas, and pancreatic and neuroendocrine tumors. There are currently over 100 active trials evaluating the activity of sunitinib against various cancers.
Preclinical Summary
In in vitro experiments, targeting FGFR signaling has been shown to slow down the growth of multiple myeloma, bladder cancer, glioblastomas, and lung and colon cancer. Expression of a kinase-dead dominant-negative version of FGFR3c but not FGFR3b led to apoptosis in colorectal cancer cells, highlighting the specificity of FGFR signaling in carcinogenesis. The two inhibitors that have long been used in the laboratory to inhibit FGFRs for in vitro experiments, SU5402 and PD173074, have had significant issues with toxicity in vivo. Numerous new receptor kinase inhibitors are in the pipeline. For instance, ponatinib is a pan-BCR–ABL and pan-FGFR inhibitor that, in addition to having promise for the treatment of imatinib-resistant CML, is able to induce apoptosis of cells from 8p11 myeloproliferative syndrome patients by reducing phosphorylation of FGFR1 fusion proteins and can improve survival in mice transplanted with FGFR1 fusion kinase-expressing leukemia/lymphoma cell lines (Knights and Cook 2010). AZ12908010 is a compound with FGFR1-3 selective inhibition that suppresses myeloma, urothelial, breast, and gastric cancer cell lines. LY2874455 is a pan-FGFR kinase inhibitor that functions by reversibly competing for ATP. LY2874455 inhibits FGF-induced MAPK signaling in vivo in murine heart tissue and also reduces tumor growth in xenografts of urinary tract cancer, gastric cancer, and multiple myeloma. Inhibition of FGFR2 and FRS2 phosphorylation by LY2874455 in gastric cancer xenografts underlays the reduction in tumor growth. FIIN-1, discovered at the Dana–Farber Cancer Institute, was developed by analysis of the co-crystal structure of PD173074 with FGFR1, and it is the first selective and irreversible pan-FGFR inhibitor. It functions by binding a cysteine in the ATP binding site. FIIN-1 inhibits inducible FGFR1 activation in vitro and has antiproliferative activity against a wide range of tumor cell lines. Inhibition of FGFR3 by PD173074 reduces growth of UCC. It also was able to reduce cell growth in endometrial cancer cell lines expressing FGFR2 with kinase-activating mutations (N549K, K649N) as well as induce apoptosis in HER-2-positive breast cancer cell lines.
Monoclonal antibodies are of considerable value for cancers overexpressing certain FGFRs or FGFs or for cancers harboring FGFR extracellular domain mutations. Monoclonal antibodies directed against FGF8 and FGF19 have reduced tumor growth in mouse models of prostate cancer and hepatocellular cancer, respectively. An antibody against FGF8 has induced regression of established tumors in mouse models of breast cancer. An antibody against FGF2 has inhibited tumor cell proliferation in preclinical studies of melanoma, and monoclonal antibodies against FGFR3, such as R3Mab and PRO-001, have shown antiproliferative and cytotoxic properties in mouse models of bladder cancer and MM, respectively (Qing et al. 2009).
The research for monoclonal antibodies against FGF19 is of particular interest. FGFR4 signaling is required for hepatocarcinogenesis, since transgenic FGF19 mice that develop hepatocellular carcinoma fail to do so when bred with FGFR4 knockout mice (French et al. 2012), and an anti-FGFR4 monoclonal antibody was shown to inhibit FGFR4 signaling and tumor growth in vivo. Given a direct link between FGFR4 and liver tumorigenesis, this research is proof of principle that FGFR4 is a worthwhile therapeutic target. Importantly, FGF19 is specifically overexpressed in hepatocellular carcinomas (HCCs) containing the 11q13.3 amplicon, and FGF19 mediates its effects on tumor growth via b-catenin signaling (Sawey et al. 2011). Anti-FGF19 antibody 1A6 was able to inhibit 50% of cell lines harboring the 11q13.3 amplicon, but none of the HCC cell lines lacking the amplicon, suggesting that FGF biologic therapies will have their greatest impact when carefully targeted using genetic data. FGF19 signaling has also been implicated in colon cancer, and preclinical research is underway in this field. Colon cancers with activated pregnane X receptor (PXR) have aggressive characteristics of tissue invasion, metastasis, and cell growth, and this pathophysiology is mediated by PXR’s activation of the FGF19 promoter (Wang et al. 2011). Anti-FGF19 antibody inhibits the aggressive phenotype of colon cancer seen with activated PXR. These results raise the option of targeting the FGF19–FGFR4-β-Klotho pathway to inhibit tumor growth.
Clinical Summary
Compounds that have advanced farthest in clinical trials and are closest to therapeutic use tend to also inhibit RTKs other than FGFRs and usually to an even greater degree. These drugs include brivanib , dovitinib (formerly CHIR-258), and BIBF1120 that have been promising in their ability to inhibit VEGF-independent and VEGF-dependent angiogenesis, and their eventual use in the clinic is anticipated. Even though they broadly inhibit many RTKs, the side effects from these drugs are less severe than those from FGFR-specific drugs, since the efficacious dose is lower. Combining knockdown of FGFRs with knockdown of other RTKs in combination with radiotherapy is a possible alternative to avoid the significant side effects of full FGFR inhibition. As one example of this class of drugs, dovitinib is an inhibitor of FGFR and VEGFR, and a phase I/II dose-escalation study in patients with advanced melanoma showed a reasonable safety profile, with primary side effects of nausea, fatigue, and diarrhea. Twenty-six percent of patients had stable disease after 8 weeks of treatment, and 53% continued to have progressive grade III or IV disease (Kim et al. 2011). Most notable is brivanib, an RTK inhibitor selective for VEGFR and FGFR that has been evaluated in several phase III trials. In the BRISK-FL study, it was compared against sorafenib as first-line therapy for unresectable HCC but did not meet criteria for non-inferiority as median overall survival was 9.5 months for brivanib compared to 9.9 months for sorafenib. In the AGITG CO.20 trial, brivanib was added to the anti-EGF Ab cetuximab to treat chemo-refractory colorectal cancer, but it did not significantly increase overall survival relative to cetuximab alone and also increased toxicity.
Anticipated Results
Due to the high degree of homology between ATP-binding pockets of RTK domains, compounds that preferentially inhibit one receptor subfamily tend to cross-inhibit other subfamilies as well, so the development of FGFR-specific inhibitors for clinical application has been challenging. However, there are several FGFR-specific inhibitors currently under investigation. AZD4547 is being tested in clinical trials for its efficacy against breast cancers that overexpress FGFR1 and are estrogen receptor positive, with results pending. BGJ398, another kinase inhibitor, is going to be studied in clinical trials in patients with solid tumors where FGFR1 or FGFR2 has been amplified or there is a mutated FGFR3. The main downside of FGFR-specific inhibitors is that the high doses needed for therapeutic inhibition end up leading to serious side effects, such as deregulation of calcium and phosphate metabolism and tissue calcification due to inhibition of FGF23’s hormonal functions (Knights and Cook 2010).
Several heparanoids that antagonize the ability of heparan sulfate to promote FGF–FGFR binding and signaling have been investigated in clinical trials. Among the most well known is suramin, a polysulfated naphthylurea. Although phase I/II trials showed some benefit in bladder, kidney, and prostate cancers, phase III trials have failed to demonstrate a gain in survival through suramin administration. Other heparanoids such as PI-88 (muparfostat) are still being evaluated in clinical trials, but it has yet to have a dosing schedule established that avoids significant hematologic toxicity. Thalidomide is a small molecule that inhibits angiogenesis including FGF2-induced angiogenesis (Beenken and Mohammadi 2009).
One strategy to target overexpression of FGFs in certain cancers is through the use of ligand traps that sequester FGFs. FP-1039 is a ligand trap consisting of the extracellular FGFR1c domain fused to the Fc of IgG and is being used in clinical studies to examine its efficacy against advanced or recurrent cancers (clinicaltrials.gov: NCT00687505). This can enable titrating factor levels to physiologic levels rather than completely abolishing the signal. A soluble form of FGFR found in breast cancers may eventually also be used for this purpose (Ezzat et al. 2001).
All of the above approaches – including monoclonal antibodies, kinase inhibitors, heparanoids, and ligand traps – hold promise for the treatment of FGFR-driven cancers. Clinical trial results for all these potential therapeutics are highly anticipated, and the field of FGF–FGFR signaling will be further stimulated once some of these therapeutics start to be used in the clinic.
References
Beenken A, Mohammadi M. The FGF family: biology, phathophysiology, and therapy. Nat Rev Drug Discov. 2009;8:235–53.
Beenken A, Mohammadi M. The molecular bases for FGF receptor activation in craniosynostosis and dwarfism syndromes. In: Muenke M, Kress W, Collmann H, Solomon BD, editors. Craniosynostoses: molecular genetics, principles of diagnosis, and treatment. Basel: Karger; 2011;45–57.
Beenken A, Mohammadi M. The structural biology of the FGF19 subfamily. In: Kuro-o M, editor. Endocrine FGFs and Klothos. Austin, Texas: Landes Bioscience; Adv Exp Med Biol. 2012;728:1–24.
Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:233–47.
Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–49.
Ezzat S, Zheng L, Yu S, Asa SL. A soluble dominant negative fibroblast growth factor receptor 4 isoform in human MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2001;287:60–5.
Fan J, Hitosugi T, Chung TW, Xie J, Ge Q, Gu TL, Polakiewicz RD, Chen GZ, Boggon TJ, Lonial S, Khuri FR, Kang S, Chen J. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol Cell Biol. 2011;31:4938–50.
French DM, Lin BC, Wang M, Adams C, Shek T, Hotzel K, Bolon B, Ferrando R, Blackmore C, Schroeder K, Rodriguez LA, Hristopoulos M, Venook R, Ashkenazi A, Desnoyers LR. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS One. 2012;7:e36713.
Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol. 2013;14:166–80.
Gotoh N. Regulation of growth factor signaling by FRS2 family docking/scaffold adaptor proteins. Cancer Sci. 2008;99:1319–25.
Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu TL, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J, Chen J. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2, ra73.
Johnson DE, Lu J, Chen H, Werner S, Williams LT. The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol Cell Biol. 1991;11:4627–34.
Kim KB, Chesney J, Robinson D, Gardner H, Shi MM, Kirkwood JM. Phase I/II and pharmacodynamic study of dovitinib (TKI258), an inhibitor of fibroblast growth factor receptors and VEGF receptors, in patients with advanced melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2011;17:7451–61.
Knights V, Cook SJ. De-regulated FGF receptors as therapeutic targets in cancer. Pharmacol Ther. 2010;125:105–17.
Kuro-o M. Endocrine FGFs and Klothos: emerging concepts. Trends Endocrinol Metab. 2008;19:239–45.
Martin GR. The roles of FGFs in the early development of vertebrate limbs. Genes Dev. 1998;12:1571–86.
Miyake M, Sugano K, Sugino H, Imai K, Matsumoto E, Maeda K, Fukuzono S, Ichikawa H, Kawashima K, Hirabayashi K, Kodama T, Fujimoto H, Kakizoe T, Kanai Y, Fujimoto K, Hirao Y. Fibroblast growth factor receptor 3 mutation in voided urine is a useful diagnostic marker and significant indicator of tumor recurrence in non-muscle invasive bladder cancer. Cancer Sci. 2010;101:250–8.
Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005a;16:107–37.
Mohammadi M, Olsen SK, Goetz R. A protein canyon in the FGF-FGF receptor dimer selects from an à la carte menu of heparan sulfate motifs. Curr Opin Struct Biol. 2005b;15:506–16.
Ornitz DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–13.
Qing J, Du X, Chen Y, Chan P, Li H, Wu P, Marsters S, Stawicki S, Tien J, Totpal K, Ross S, Stinson S, Dornan D, French D, Wang QR, Stephan JP, Wu Y, Wiesmann C, Ashkenazi A. Antibody-based targeting of FGFR3 in bladder carcinoma and t(4;14)-positive multiple myeloma in mice. J Clin Invest. 2009;119:1216–29.
Sawey ET, Chanrion M, Cai C, Wu G, Zhang J, Zender L, Zhao A, Busuttil RW, Yee H, Stein L, French DM, Finn RS, Lowe SW, Powers S. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell. 2011;19:347–58.
Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25.
Seo JH, Suenaga A, Hatakeyama M, Taiji M, Imamoto A. Structural and functional basis of a role for CRKL in a fibroblast growth factor 8-induced feed-forward loop. Mol Cell Biol. 2009;29:3076–87.
Spielberger R, Stiff P, Bensinger W, Gentile T, Weisdorf D, Kewalramani T, Shea T, Yanovich S, Hansen K, Noga S, McCarty J, LeMaistre CF, Sung EC, Blazar BR, Elhardt D, Chen MG, Emmanouilides C. Palifermin for oral mucositis after intensive therapy for hematologic cancers. N Engl J Med. 2004;351:2590–8.
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–29.
Wang H, Venkatesh M, Li H, Goetz R, Mukherjee S, Biswas A, Zhu L, Kaubisch A, Wang L, Pullman J, Whitney K, Kuro-o M, Roig AI, Shay JW, Mohammadi M, Mani S. Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. J Clin Invest. 2011;121:3220–32.
Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203.
Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33.
Yu X, White KE. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev. 2005;16:221–32.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media New York
About this entry
Cite this entry
Mohammadi, M., Beenken, A. (2017). FGF-FGFR Signaling in Cancer. In: Marshall, J. (eds) Cancer Therapeutic Targets. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-0717-2_19
Download citation
DOI: https://doi.org/10.1007/978-1-4419-0717-2_19
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-0716-5
Online ISBN: 978-1-4419-0717-2
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences