
Abstract
Symbiont microbial communities play important roles in animal biology and are thus considered integral components of metazoan organisms, including parasitic worms (helminths). Nevertheless, the study of helminth microbiomes has thus far been largely overlooked, and symbiotic relationships between helminths and their microbiomes have been only investigated in selected parasitic worms. Over the past decade, advances in next-generation sequencing technologies, coupled with their increased affordability, have spurred investigations of helminth-associated microbial communities aiming at enhancing current understanding of their fundamental biology and physiology, as well as of host–microbe interactions. Using the blood fluke Schistosoma mansoni as a key example of parasitic worms with complex life cycles involving multiple hosts, in this chapter we (1) provide an overview of protocols for sample collection and (2) outline an example workflow to characterize worm-associated microbial communities using high-throughput sequencing technologies and bioinformatics analyses of large-scale sequence data.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others
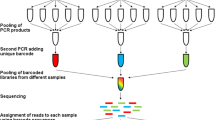

References
Dheilly NM, Martínez Martínez J, Rosario K et al (2019) Parasite microbiome project: grand challenges. PLoS Pathog 15(10):e1008028
Formenti F, Cortés A, Brindley PJ et al (2020) A bug’s life: delving into the challenges of helminth microbiome studies. PLoS Negl Trop Dis 14(9):e0008446
Jenkins TP, Brindley PJ, Gasser RB et al (2019) Helminth microbiomes—a hidden treasure trove? Trends Parasitol 35(1):13–22
Sinnathamby G, Henderson G, Umair S et al (2018) The bacterial community associated with the sheep gastrointestinal nematode parasite Haemonchus contortus. PLoS One 13(2):e0192164
White EC, Houlden A, Bancroft AJ et al (2018) Manipulation of host and parasite microbiotas: survival strategies during chronic nematode infection. Sci Adv 4(3):eaap7399
Hogan G, Walker S, Turnbull F et al (2019) Microbiome analysis as a platform R&D tool for parasitic nematode disease management. ISME J 13(11):2664–2680
Jorge F, Dheilly NM, Poulin R (2020) Persistence of a Core microbiome through the ontogeny of a multi-host parasite. Front Microbiol 11:954
Bouchery T, Lefoulon E, Karadjian G et al (2013) The symbiotic role of Wolbachia in Onchocercidae and its impact on filariasis. Clin Microbiol Infect 19(2):131–140
Slatko BE, Taylor MJ, Foster JM (2010) The Wolbachia endosymbiont as an anti-filarial nematode target. Symbiosis 51(1):55–65
Cortés A, Rooney J, Bartley DJ et al (2020) Helminths, hosts, and their microbiota: new avenues for managing gastrointestinal helminthiases in ruminants. Expert Rev Anti-Infect Ther 18(10):977–985
Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST (2011) Bayesian community-wide culture-independent microbial source tracking. Nat Methods 8(9):761–3
Pérez-Cobas AE, Gómez-Valero L, Buchrieser C (2020) Metagenomic approaches in microbial ecology: an update on whole-genome and marker gene sequencing analyses. Microb Genom 6(8):mgen000409
Tringe SG, Hugenholtz P (2008) A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol 11(5):442–446
Case RJ, Boucher Y, Dahllof I et al (2007) Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl Environ Microbiol 73(1):278–288
Colley DG, Bustinduy AL, Secor WE et al (2014) Human schistosomiasis. Lancet 383(9936):2253–2264
Klindworth A, Pruesse E, Schweer T et al (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41(1):e1
Mann VH, Morales ME, Rinaldi G et al (2010) Culture for genetic manipulation of developmental stages of Schistosoma mansoni. Parasitology 137(3):451–462
Mouahid G, Rognon A, de Carvalho AR et al (2018) Transplantation of schistosome sporocysts between host snails: a video guide. Wellcome Open Res 3:3
Illumina (2013) Metagenomic sequencing library preparation. Preparing 16S ribosomal RNA gene amplicons for the Illumina MiSeq system
Bolyen E, Rideout JR, Dillon MR et al (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37(8):852–857
Callahan BJ, McMurdie PJ, Rosen MJ et al (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13(7):581–583
Quast C, Pruesse E, Yilmaz P et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(Database issue):D590–D596
Robeson II MS, O’Rourke DR, Kaehler BD, Ziemski M, Dillon MR, Foster JT, Bokulich NA. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. bioRxiv. https://doi.org/10.1101/2020.10.05.326504
Werner JJ, Koren O, Hugenholtz P et al (2012) Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J 6(1):94–103
Glassing A, Dowd SE, Galandiuk S et al (2016) Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog 8:24
Karstens L, Asquith M, Davin S et al (2019) Controlling for contaminants in low-biomass 16S rRNA gene sequencing experiments. mSystems 4(4):e00290–e00219
Claassen S, du Toit E, Kaba M, Moodley C, Zar HJ, Nicol MP (2013) A comparison of the efficiency of five different commercial DNA extraction kits for extraction of DNA from faecal samples. J Microbiol Methods 94(2):103–110. https://doi.org/10.1016/j.mimet.2013.05.008
Zakrzewski M, Proietti C, Ellis JJ et al (2017) Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 33(5):782–783
Fouhy F, Clooney AG, Stanton C et al (2016) 16S rRNA gene sequencing of mock microbial populations—impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol 16(1):123
Yang B, Wang Y, Qian PY (2016) Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinform 17:135
Teng F, Darveekaran Nair SS, Zhu P et al (2018) Impact of DNA extraction method and targeted 16S-rRNA hypervariable region on oral microbiota profiling. Sci Rep 8(1):16321
Salter SJ, Cox MJ, Turek EM et al (2014) Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12:87
Eisenhofer R, Minich JJ, Marotz C et al (2019) Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol 27(2):105–117
Thatcher SA (2015) DNA/RNA preparation for molecular detection. Clin Chem 61(1):89–99
Pollock J, Glendinning L, Wisedchanwet T et al (2018) The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Appl Environ Microbiol 84(7)
Olson ND, Morrow JB (2012) DNA extract characterization process for microbial detection methods development and validation. BMC Res Notes 5:668
Giacomin P, Zakrzewski M, Jenkins TP et al (2016) Changes in duodenal tissue-associated microbiota following hookworm infection and consecutive gluten challenges in humans with coeliac disease. Sci Rep 6:36797
Witzke MC, Gullic A, Yang P et al (2020) Influence of PCR cycle number on 16S rRNA gene amplicon sequencing of low biomass samples. J Microbiol Methods 176:106033
Illumina (2020) How much PhiX spike-in is recommended when sequencing low diversity libraries on Illumina platforms?
Janssen S, McDonald D, González A et al (2018) Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 3(3):e00021–e00018
Illumina (2017) MiSeq® Reporter Software Guide
Amir A, McDonald D, Navas-Molina JA et al (2017) Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2(2):e00191–e00116
Bokulich NA, Kaehler BD, Rideout JR et al (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6(1):90
DeSantis TZ, Hugenholtz P, Larsen N et al (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Glockner FO, Yilmaz P, Quast C et al (2017) 25 years of serving the community with ribosomal RNA gene reference databases and tools. J Biotechnol 261:169–176
Kim D, Hofstaedter CE, Zhao C et al (2017) Optimizing methods and dodging pitfalls in microbiome research. Microbiome 5(1):52
Willner D, Daly J, Whiley D et al (2012) Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS One 7(4):e34605
Bittinger K, Charlson ES, Loy E et al (2014) Improved characterization of medically relevant fungi in the human respiratory tract using next-generation sequencing. Genome Biol 15(10):487
Jervis-Bardy J, Leong LE, Marri S et al (2015) Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome 3:19
Davis NM, Proctor DM, Holmes SP et al (2018) Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6(1):226
Finotello F, Mastrorilli E, Di Camillo B (2018) Measuring the diversity of the human microbiota with targeted next-generation sequencing. Brief Bioinform 19(4):679–692
Lloyd-Price J, Abu-Ali G, Huttenhower C (2016) The healthy human microbiome. Genome Med 8(1):51
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550
Fernandes AD, Reid JN, Macklaim JM et al (2014) Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2:15
Mandal S, Van Treuren W, White RA et al (2015) Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis 26:27663
Segata N, Izard J, Waldron L et al (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12(6):R60
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
1 Electronic Supplementary Material
Schistosoma mansoni free-swimming developmental stages (i.e., cercariae and miracidia) collected for helminth microbiota profiling. Cercariae were obtained from experimentally infected Biomphalaria glabrata, whereas miracidia were hatched from eggs isolated from the liver of mice experimentally infected with parasite cercariae (MP4 218651 kb)
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Formenti, F., Rinaldi, G., Cantacessi, C., Cortés, A. (2021). Helminth Microbiota Profiling Using Bacterial 16S rRNA Gene Amplicon Sequencing: From Sampling to Sequence Data Mining. In: de Pablos, L.M., Sotillo, J. (eds) Parasite Genomics. Methods in Molecular Biology, vol 2369. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1681-9_15
Download citation
DOI: https://doi.org/10.1007/978-1-0716-1681-9_15
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-1680-2
Online ISBN: 978-1-0716-1681-9
eBook Packages: Springer Protocols