
Abstract
Introduction: Combined oxidative phosphorylation deficiency 20 (COXPD20) is a mitochondrial respiratory chain complex (RC) disorder, caused by disease-causing variants in the VARS2 gene, which encodes a mitochondrial aminoacyl-tRNA synthetase. Here we describe a patient with fatal mitochondrial encephalopathy caused by a homozygous VARS2 gene missense variant.
Case Report: We report the case of a girl, the first child of non-consanguineous and healthy parents, born from an uneventful term pregnancy, who presented, in the neonatal period, major hypotonia and microcephaly. At 4 months of age she showed poor eye contact, nystagmus, global psychomotor development delay and failure to thrive, without dysmorphic features. Focal seizures started at 24 months which evolved to a severe epileptic encephalopathy and finally to super refractory status epilepticus, leading to her death at 28 months of age. Etiologic investigation encompassing metabolic and genetic causes failed to disclose a diagnosis. Post-mortem exome sequencing allowed the identification of a pathogenic variant in VARS2 gene in the homozygous state (c.1100C > T, p.Thr367Ile) in the patient, inherited from her heterozygous parents, leading to the diagnosis of COXPD2.
Conclusion: To the best of our knowledge, this is the fifth case described in the literature of a child with disease-causing variant in VARS2. With this report we expand the knowledge about the phenotype associated with this very rare mitochondrial defect, further emphasizing the use of exome sequencing as a very powerful diagnostic tool.
Communicated by: Daniela Karall
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Amino acyl
- Exome sequencing
- Mitochondrial diseases
- Mitochondrial encephalopathy
- RNA
- Severe epileptic encephalopathy
- Transfer
- VARS2 gene
Introduction
Defects in the mitochondrial respiratory chain (RC) have emerged as the most common cause of childhood and adult neurometabolic disease, with an estimated prevalence of 1 per 5,000 live births (Taylor et al. 2014). RC is a multiheteromeric enzymatic structure that performs oxidative phosphorylation (OXPHOS), a fundamental reaction that supplies ∼90% of the energy used by mammalian cells (Diodato et al. 2014; Baertling et al. 2017). Signs and symptoms of the disorder may develop at any time throughout the patient’s life, often occurring in association with neurological impairment and frequently resulting in chronic disability and premature death (Taylor et al. 2014).
The RC consists of five complexes, composed by ∼85 structural proteins, 13 of which are encoded by mtDNA, whereas the others are encoded by nuclear genes. Aside from mitochondrial DNA-encoded ribosomal and transfer RNAs (rRNAs and tRNAs), the mitochondrial translation apparatus consists of more than 100 proteins, encoded by nuclear genes, translated by cytosolic ribosomes and imported into the mitochondria matrix. These include mitochondrial aminoacyl-tRNA-synthetases (mt-aaRSs), ribosomal structural and assembly proteins, tRNA modifying and methylating enzymes, and several initiation, elongation, and termination factors of mitochondrial translation (Taylor et al. 2014; Diodato et al. 2014; Baertling et al. 2017; Valente et al. 2007). An increasing number of mitochondrial translation disorders caused by causative variants in genes encoding mt-aaRSs, which catalyze the ligation of specific amino acids to their cognate tRNAs, have been discovered (Diodato et al. 2014).
Combined oxidative phosphorylation deficiency 20 (COXPD20 – OMIM #615917) is a disorder of the RC, caused by homozygous or compound heterozygous variants in the VARS2 gene (OMIM #612802) located on chromosome 6p21 (https://www.omim.org/entry/615917). The VARS2 gene encodes a mt-aaRS, which catalyzes the attachment of valine to tRNA(Val) for mitochondrial translation, designed as valyl tRNA-synthetase. Variants in this gene are associated with early-onset mitochondrial encephalopathies (Taylor et al. 2014; Online Mendelian Inheritance in Man 2017, https://www.omim.org/entry/615917).
Four patients with COXPD20 have been previously described. However, limited clinical information was provided, leaving the phenotype of VARS2 deficiency largely uncharacterized (Taylor et al. 2014; Diodato et al. 2014; Baertling et al. 2017; Pronicka et al. 2016; Mikol and Polivka 2005).
Here we report the fifth case of COXPD20, identified by exome sequencing (ES), caused by a homozygous variant in VARS2 in a patient with fatal mitochondrial encephalopathy. We present detailed clinical data to better characterize the phenotypic spectrum associated with this disease.
Case Report
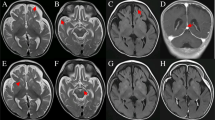
We report the clinical description of a child, first daughter of Portuguese, non-consanguineous and healthy parents, who was born after an uneventful full-term pregnancy, by cesarean delivery due to pelvic presentation, with APGAR scores of 9 and 10, although small for the gestational age (weight: 2,755 g, 10th percentile, height 45.5 cm, 3th percentile and head circumference 32 cm, 5th percentile). Expanded newborn metabolic screening was normal. Since the neonatal period, she presented microcephaly, severe global hypotonia, progressive feedings difficulties, and failure to thrive. At 4 months, severe global psychomotor development delay was reported, with head lag, poor eye contact, and pendular nystagmus. Dysmorphic features were absent. At 6 months, a brain magnetic resonance imaging (MRI) showed global atrophy and a small glioepithelial cyst associated with left hippocampal molding. Renal and hepatic function tests, creatine kinase, lactate dehydrogenase, ammonia, total homocysteine, pH, and lactic acid were normal. Abdominal ultrasound was also normal. Otorhinolaryngology and cardiac evaluations (including echocardiogram) were unremarkable. Ophthalmologic evaluation revealed visual evoked potentials reduced amplitude with preserved latency, with no other ocular abnormalities. First metabolic evaluation disclosed lactate 5.35 mmol/L (normal 0.63–2.44), pyruvate 71 μmol/L (normal 54.1–119.9), L/P ratio 75 (normal 10–25), plasma aminoacid profile with a slight elevation of alanine (464 μmol/L; normal 236–400) and proline (330 μmol/L; normal 100–280) and a normal organic acids profile. Despite the normalization of lactate value, mitochondrial etiology was suspected. The glucose loading test didn’t reveal hyperlactacidemia (fast lactate 2.56; fast pyruvate 115; fast L/P 22). After 2 g/kg glucose: lactate 1.98, pyruvate 122 and L/P ratio 16). Brain MRI was repeated at 12 months of age, revealing global atrophy and symmetrical diffuse T2 hyperintensity of the supra and infratentorial white matter (Fig. 1). Array comparative genomic hybridization (array-CGH) did not show copy-number changes. Deltoid muscle biopsy performed at 18 months revealed a histology with minimal and uncharacteristic changes, without ragged-red fibers. The biochemical study of respiratory chain complexes by spectrophotometry revealed an elevation of citrate synthase (CS) 215.6 nmol/min/mg NCP (non-collagenic proteins) (normal 85–180) with normal activity of complexes I–IV: 20.4 nmol/min/mg NCP/CS (normal 8.8–30.8), 13.4 nmol/min/mg NCP/CS (normal 12–35), 39.5 nmol/min/mg NCP/CS (normal 22.2–62.2), and 13.5 nmol/min/mg NCP/CS (normal 11.5–34.5), respectively; complex II + III 2.6 nmol/min/mg NCP/CS (normal 2.6–12).

(a–c) Axial T2 weighted images of the patient at age of 1 year. (a) Bilateral hypersignal of the cerebellar white matter. (b) Neuroglial cyst with left hippocampal molding. (c) Bilateral and symmetrical supratentorial white matter involvement. (d–f) Axial T1 weighted images of the patient at age of 24 months. (d) Loss of volume of cerebellum with enlargement of cerebellar folia and small size of left inferior cerebellar hemisphere. (e) Neuroglial cyst with left hippocampal molding. (f) More prominent decrease of supratentorial brain volume with loss of periventricular white matter. (g–i) Axial T2 weighted images of the patient at age of 26 months. (g, h) More severe and diffuse loss of brain volume than in the previous scan; sequelae lesions with volume loss of inferior left cerebellar hemisphere and left parieto-occipital cortical encephaloclastic lesion; (i) Bilateral and symmetrical supratentorial periventricular and subcortical white matter involvement
At 24 months of age she had global psychomotor delay, with absent language, severe hypotonia without the ability to sit independently and spastic tetraparesis; pendular nystagmus was less evident. At this stage, focal seizures began, characterized by staring and tonic extension with clonic movements of the right superior limb, during sleep and while waking up, lasting less than 60 s. During video-electroencephalogram monitoring, a convulsive event was registered, as well as a left parietal focus with spike-waves extending to the middle line. Sodium valproate (20 mg/kg/day) was initiated, with partial control of seizures (maximum dose of 40 mg/kg/day). Given the association of nystagmus, hypotonia, and epilepsy with fast rhythms in EEG, the hypothesis of infantile neuroaxonal dystrophy was considered and skin biopsy was performed, showing no axonal spheroids or any other abnormalities.
Due to the recurrence of seizures with right head deviation and right limb fencing posture, carbamazepine (maximum dose 20 mg/kg/day) and clobazam (2.5 mg per day) were sequentially added on. Despite these antiepileptic drugs, the clinical status evolved to an epileptic encephalopathy with regression and admission to a pediatric department at the age of 26 months. EEG showed severe diffuse encephalopathy with slowing and focal left parietal epileptic activity with middle line involvement. At that time, metabolic reevaluation including LCR study was performed: plasma lactate 2.72 mmol/L, pyruvate 124 μmol/L and L/P ratio of 22; LCR lactate 1.45 mmol/L (normal 1.2–2.1), pyruvate 90 μmol/L (normal 59.2–89.2) and L/P 16 (normal 10–25). Plasma aminoacid profile showed a slight elevation of alanine 502 μmol/L (100–400), proline 544 μmol/L (118–230) and glycine 340 μmol/L (40–290). LCR aminoacids were normal, as urinary organic acids. Seizures progressed into convulsive status epilepticus (CSE) and led to admission in the Pediatric Intensive Care Unit (PICU). During hospitalization, multiple combinations of drugs were tried in order to try to control super refractory SE, namely midazolam, thiopental (maximum 10 mg/kg/h), methylprednisolone, ketamine, lidocaine (4 mg/kg/h), pyridoxine (22 mg/kg/day), propofol (maximum 8.9 mg/kg/h), lamotrigine (maximum 100 mg/day), topiramate, vigabatrin, levetiracetam, felbamate (50 mg 12/12 h), and lacosamide (4.5 mg 12/12 h). Ketogenic diet was tried but ketosis was never achieved. Brain MRI was repeated, revealing worsening of the previous global atrophy. Continuous EEG monitoring disclosed continuous polyspike or spike and wave discharges, with generalized distribution and right hemisphere lateralization, with periodic epileptiform discharges – a pattern of electrographic generalized status epilepticus. Mechanical ventilation was initiated followed by tracheostomy due to vocal cords’ paresis. At this time a diagnostic panel for epileptic encephalopathies revealed two intronic variants in KCNB1 and RNASEH2C genes, both inherited from her asymptomatic father.
By 28 months she died in the PICU, due to cardio-respiratory arrest in the context of a severe epileptic encephalopathy. The anatomopathological report revealed an encephalon with severe neuronal loss and spongiosis.
Two years after the child’s death, ES was performed using the SureSelect Human All Exon kit and a HiSeq sequencer (Illumina). For data analysis, a custom validated pipeline was applied using FastQC and QualiMap for quality control, BWA for alignment (GRCh37), GATK HaplotypeCaller for variant calling, and Ensembl VEP and GEMINI for annotation. Exome data was then filtered by focusing on variants that resulted in a change at the protein level and, if known in the databases, its minor allele frequency (MAF) had to be below 1%. All filtered variants were further analyzed using Alamut software (Interactive Biosoftware) for pathogenicity prediction, previous description in affected individuals using the Human Gene Mutation Database (HGMD) and frequency was confirmed at the Genome Aggregation Database (gnomAD). This pipeline allowed the identification of a homozygous c.1100C > T substitution at VARS2 that replaces a highly conserved threonine by an isoleucine at codon 367 (p.Thr367Ile), that was subsequently confirmed, as well as both parents carrier status, by Sanger sequencing.
Discussion
Combined oxidative phosphorylation deficiency 20 (COXPD20) is a very rare mitochondrial respiratory chain complex (RC) disorder, caused by disease-causing variants in the VARS2 gene. Variants in this gene have been recently identified as the cause of mitochondrial encephalopathy in four individuals. Our report widens the clinical phenotype related to VARS2 deficiency. The presently described patient initially presented hypotonia, global psychomotor development delay, spastic tetraparesis and pendular nystagmus and later developed epileptic encephalopathy.
Mitochondrial diseases (MD) have emerged as a common cause of metabolic inherited diseases, but their diagnosis remains challenging due to clinical heterogeneity and constantly expanding number of causative genes (Taylor et al. 2014; Diodato et al. 2014; Valente et al. 2007). These disorders are characterized by morphological and/or functional mitochondrial abnormalities (Mikol and Polivka 2005) and the molecular mechanism potentially involves different gene products affecting mtDNA replication and expression, including aaRS2 (Taylor et al. 2014). However, most mutations in aaRS2 genes have been reported in single, or in just a few cases, which hampers the establishment of definitive genotype–phenotype correlations. In the study performed by Pronicka et al. (2016), most of the neonates with causative variants identified were born from the first pregnancy of healthy unrelated parents. Our patient, the first daughter of healthy parents, was carrying the c.1100C > T, p.Thr367Ile pathogenic variant, compatible with an autosomal recessive inheritance. In a study performed by Pronicka et al. (2016), 29 neonates with MD died before the establishment of a molecular diagnosis and half of the deaths occurred in the early neonatal period. In our case, the diagnosis was established 2 years after death and 4 years after the onset of the symptoms. Molecular confirmation of the clinical diagnosis allows adequate genetic counseling and the possibility of a prenatal diagnosis in future pregnancies.
Aside from our report, four additional patients with VARS2 variants have been described (Tables 1 and 2). Taylor et al. (2014) reported a 10-year-old boy who presented in the first year of life with muscle weakness and hypotonia and later developed central nervous system disease, including progressive external ophthalmoplegia, ptosis, and ataxia, without family history of similar disease. Biochemical cells studies showed a deficiency of mitochondrial respiratory complexes I and IV (Diodato et al. 2014). This patient was a compound heterozygous for VARS2 variants c.1135G > A (p.Ala379Thr); c.1877C > A (p.Ala626Asp) (Taylor et al. 2014).
Diodato et al. (2014) reported a single patient with a homozygous missense variant c.1100C > T, (p.Thr367Ile) in VARS2. The patient was a boy with delayed psychomotor development, facial dysmorphism, and microcephaly that became apparent soon after birth. At 4 years of age, he developed seizures and myoclonic jerks. Brain MRI showed hyperintense lesions in the periventricular region, insulae, and right frontotemporal cortex. Muscle biopsy was histologically normal, but muscle homogenate showed isolated complex I deficiency (25% residual activity). Patient fibroblasts showed no enzymatic defects, but oxygen consumption was impaired, suggesting defective mitochondrial respiration (Diodato et al. 2014). Our case presents the same variant reported by Diodato et al. (2014) and has similar clinical manifestations; however, in our patient RC study was normal. Pronicka et al. (2016) described a neonate, born at a gestational age of 40 weeks with a birth weight of 3,420 g, who presented with stridor, lactic acidosis, and cardiomyopathy in the early neonatal period who was a compound heterozygous for the disease-causing variants: c.1100C > T (p.Thr367Ile), c.1490G > A (p.Arg497His) (Baertling et al. 2017; Pronicka et al. 2016).
Finally, Baertling et al. (2017) reported a case of a neonate presenting mitochondrial encephalopathy with severe lactic acidosis, hypertrophic cardiomyopathy, epilepsy, and abnormalities on brain imaging including corpus callosum and cerebellum hypoplasia, as well as a massive lactate peak on MR-spectroscopy. As in the previous case, disease-causing variants in compound heterozygosity were documented, one of them a novel variant: c.601C > T p.Arg201Trp, c.1100C > T (p.Thr367Ile) (Baertling et al. 2017).
It was also noted the high prevalence of the causative variant c.1100C > T, p.(Thr367Ile) identified in 3 out of 4 of the previous reports (Table 1). Hypotonia was present in all patients and most of them also showed microcephaly, failure to thrive, global developmental delay, and seizures. Pendular nystagmus and spastic tetraparesis observed in our patient were not previously described (Table 2).
Interestingly, specific MRI patterns seem to be associated with some patients with aaRS2 variants (e.g., DARS2, EARS2, RARS2) (Diodato et al. 2014), although it is not possible to establish any pattern for VARS2. In the patient described by Diodato et al., MRI showed hyperintense lesions involving mainly the insulae and frontotemporal right cortex. In our case, MRI showed global atrophy and diffuse and symmetrical T2 hyperintensity of the supra and infratentorial white matter. Relevant histological modifications in muscle are ragged-red fibers with or without cytochrome C oxidase (COX) activity (Mikol and Polivka 2005). Diodato et al. (2014) reported a mosaic for COX-positive fibers in muscle biopsy, although our patient showed no abnormalities in muscle histology. The neuropathological finding in patients with mitochondrial encephalomyopathies is not specific and consists of spongiosis, neuronal loss, focal necrosis, capillary proliferation, and mineral deposits (Mikol and Polivka 2005). In our case, the anatomopathological report revealed an encephalon with severe lesions, with atrophy and spongiosis, that can be correlated with microcephaly as well as with a highly refractory epileptic encephalopathy.
It is important to notice that mutations in the VARS2 gene should be considered in the differential diagnosis of mitochondrial encephalopathies. However, additional clinical reports are required to further define the phenotypic spectrum and the disease course associated with VARS2 deficiency and to clarify whether MRI patterns and histology are specific or not. ES has led to an exponential increase in the identification of causative genes in MD (Pronicka et al. 2016; Wortmann et al. 2015).
In conclusion, variants in nuclear genes causing mitochondrial translation defects represent a new potentially broad field of MD. Causative variants in VARS2 have been recently identified as the cause of mitochondrial encephalopathy in four individuals. Here we describe the fifth patient and expand the phenotypic spectrum of COXDP20. We also confirmed the relevance of ES as molecular diagnostic test for MD, mostly in single patients presenting with heterogeneous clinical syndromes but with biochemical markers suggesting a “mitochondrial signature.”
References
Baertling F, Alhaddad B, Seibt A et al (2017) Neonatal encephalocardiomyopathy caused by mutations in VARS2. Metab Brain Dis 32(1):267–270
Diodato D, Melchionda L, Haack TB et al (2014) VARS2 and TARS2 mutations in patients with mitochondrial encephalomyopathies. Hum Mutat 35(8):983–989
Mikol J, Polivka M (2005) Mitochondrial encephalomyopathies. Ann Pathol 25(4):282–291
Online Mendelian Inheritance in Man. Combined oxidative phosphorylation deficiency 20; coxpd20. June 2017. https://www.omim.org/entry/615917
Pronicka E, Piekutowska-Abramczuk D, Ciara E et al (2016) New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med 14(1):174
Taylor RW, Pyle A, Griffin H et al (2014) Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312(1):68–77
Valente L, Tiranti V, Marsano RM et al (2007) Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet 80(1):44–58
Wortmann SB, Koolen DA, Smeitink JA, Van den Heuvel L, Rodenburg RJ (2015) Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Metab Dis 38(3):437–443
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendices
Take-Home Message
Combined oxidative phosphorylation deficiency 20 (COXPD20) is a very rare mitochondrial respiratory chain complex disorder, caused by disease-causing variants in the VARS2 gene. Our report extends the phenotypical spectrum of VARS2-related disorders and emphasizes Exome Sequencing ability to identify variants in nuclear gene in patients with suspected mitochondrial disease.
Contributions of Individual Authors
Sandra Pereira: analysis of clinical data and drafting the article.
Mariana Adrião: analysis of clinical data and drafting the article.
Margarida Ayres Basto: acquisition and interpretation of radiological data.
Mafalda Sampaio: analysis of clinical data and critical revision of the article.
Esmeralda Rodrigues: analysis of metabolic data and critical revision of the article.
Laura Vilarinho: acquisition, analysis, and interpretation of metabolic data and critical revision of the article.
Elisa Leão Teles: analysis of metabolic data and critical revision of the article.
Isabel Alonso: acquisition, analysis, and interpretation of genetic data and critical revision of the article.
Miguel Leão: final review and approval of the version to be published.
Conflict of Interest
Sandra Pereira, Mariana Adrião, Margarida Ayres, Mafalda Sampaio, Esmeralda Rodrigues, Laura Vilarinho, Elisa Leão Teles, Isabel Alonso, and Miguel Leão declare that they have no conflict of interest.
Details of Funding
The content of the article has not been influenced by the sponsors.
Details of Ethics Approval
Research in accordance with the Declaration of Helsinki and the International Medical Research.
A Patient Consent Statement
Written informed consent to publication was obtained.
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
Copyright information
© 2018 Society for the Study of Inborn Errors of Metabolism (SSIEM)
About this chapter

Cite this chapter
Pereira, S. et al. (2018). Mitochondrial Encephalopathy: First Portuguese Report of a VARS2 Causative Variant. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 42. JIMD Reports, vol 42. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2018_89
Download citation
DOI: https://doi.org/10.1007/8904_2018_89
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-58364-7
Online ISBN: 978-3-662-58365-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)