
Abstract
Fabry disease (FD) is an X-linked, lysosomal storage disease. Mutations in the gene coding for alpha-galactosidase A lead to globotriaosylceramide (Gb-3) accumulation in lysosomes and in placenta and umbilical cord. Impact of FD and treatment with enzyme replacement (ERT) on foetal development is undisclosed.
A 38-year-old primigravida with FD (G85N) is reported. She has 50% reduced alpha-galactosidase A activity and elevated plasma and urine-Gb-3. She was severely affected with ischaemic stroke at age 23, hypertension, albuminuria and moderately reduced renal function. ERT was initiated at age 23 years in 2001 and continued during spontaneous pregnancy at age 38. In third trimester she developed moderate-to-severe pre-eclampsia, successfully managed by methyldopa. Chorion villus sampling revealed a male foetus without the maternal gene mutation. Planned Caesarean section was performed without complications at gestational age week 38 + 6, delivering a healthy boy. Histopathological placental examination showed no sign of Gb-3 accumulation. Literature survey disclosed a total of 12 cases, 8 were treated with ERT during pregnancy and 5 infants inherited the family mutation. All outcomes were successful. In the six cases with available placental histopathological examination, Gb-3 accumulation was only seen on the foetal side if the foetus had the inherited mutation.
In conclusion, the present case, describing the first data from a severely affected FD patient receiving ERT during pregnancy complicated by pre-eclampsia, together with all other published cases, has emphasized that ERT is safe during pregnancy and resulting in successful foetal outcome; despite this, ERT is by the health authorities advised against during pregnancy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Alpha-galactosidase A enzyme deficiency
- Enzyme replacement therapy
- Fabry disease
- Lysosomal storage disease
- Placental examination
- Pre-eclampsia
- Pregnancy
Introduction
Fabry disease (FD, OMIM # 301500) is an X-linked, lysosomal storage disease. Mutations in the galactosidase alpha gene cause missing or reduced activity of the alpha-galactosidase A enzyme, leading to storage of globotriaosylceramide (Gb-3) and related glycosphingolipids in lysosomes (Brady 1967; Kint 1970; Bishop et al. 1988). In female patients with FD, phenotypic expression is variable due to X-chromosome inactivation and residual enzyme activity (Echevarria et al. 2016), varying from nearly unaffected to full phenotypic disease similar to males (Macdermot et al. 2001; Whybra et al. 2001; Wilcox et al. 2008). Gb-3 accumulation has been found in the placenta and umbilical cord during pregnancy with a male foetus with a mutation in the α-galactosidase A gene (Thurberg and Politei 2012), leading to considerations of potential impact on foetal well-being during pregnancy. No randomized clinical trials have been performed in pregnant patients with FD, and the efficacy and safety of enzyme replacement therapy (ERT) on pregnancy and foetal development have not been established. The present case describes the first data from a severely affected, ERT-receiving FD patient during pregnancy complicated by pre-eclampsia. The present paper also provides a review of the literature, since cases of patients with FD during pregnancy are rare, and currently treatment is done on individual, experimental basis. Controlled clinical trials or even large single-centre cohort presentations are unlikely to be performed on this rare topic; hence, documentation of treatment and outcome in all individual patients with FD during pregnancy is important to gather global information.
Case
Clinical Findings
We present a case of a 38-year-old primigravida with FD, diagnosed at age 2 years, with a missense mutation G85N, reduced alpha-galactosidase A activity to 50% of normal lower limit (10 nmol/h/mg protein, reference range: 20–65) and elevated plasma-Gb-3 and urine-Gb-3 of 6.5 μmol/L (reference range, 1.6–3.3) and 13.9 mol Gb-3/mol sphingomyelin (reference range, <0.3) in 2001 before ERT, respectively. Phenotypically, she was severely affected with ischaemic stroke, hypertension, albuminuria and moderately reduced renal function at age 23 years and transient ischaemic attack at age 32. She has been treated with ERT since 2001 (as the first patient in Denmark) as well as acetylsalicylic acid 75 mg/day and enalapril 10 mg/day. Cardiac involvement is limited to short PQ interval.
At age 37, the patient initiated in vitro fertilization with donor sperm and planned pre-implantation genetic diagnosis. However, spontaneous pregnancy was achieved at age 38. The patient consented to chorion villus sampling at gestational age of 10 + 5 weeks and genetic analysis revealed that the male foetus had not inherited the maternal gene mutation.
During pregnancy, enalapril was switched to labetalol 100 mg × 3/day, while acetylic acid (75 mg/day) and ERT (agalsidase-beta, 1 mg/kg, every other week) were continued. In the third trimester, blood pressure and albuminuria increased while renal function decreased, consistent with moderate-to-severe pre-eclampsia (Fig. 1). The condition was successfully managed by methyldopa (initially 250 mg × 3/day increasing to 500 mg × 3/day). Eclampsia did not develop. Mild haemolysis, slightly elevated liver enzymes and platelets low in the normal range were observed; however, criteria for HELLP syndrome (i.e. haemolysis, elevated liver enzymes and low platelet count) were not met.

Blood pressure, urine albumin-creatinine ratio and eGFR during pregnancy and 1 month after delivery; Systolic blood pressure (mmHg) (upper black dots), diastolic blood pressure (mmHg) (lower black dots); eGFR (mL/min/1.73 m2) (punctuated black line); Urine albumin-to-creatinine ratio (mg/g) (punctuated black line with triangles); Arrows indicate start and end of pregnancy, respectively
Planned Caesarean section without complications was performed at gestational age week 38 + 6, delivering a healthy boy without FD. Weight, length and head circumference at birth were 2,675 g, 47 cm and 34 cm, respectively. Apgar score was 10/1, 10/5 and 10/10.
One year after delivery the renal function and proteinuria was unchanged compared to pre-pregnancy values.
Material and Methods
Placenta
Tissues: Term placenta was obtained from an uncomplicated normal pregnancy from a non-Fabry person to compare with the placenta from the Fabry patient. Tissue for immunohistochemical analyses was fixed in 4% formaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) by immersion for 2 h followed by further processing.
Antibodies: Polyclonal rabbit antihuman α-Gal A was kindly provided by Genzyme Corp. (Framingham, MA, USA). For immunohistochemical light microscopy analysis, visualization was performed using horse radish peroxidase (HRP)-conjugated secondary goat anti-rabbit antibody (Dako, P044801-2, Glostrup, Denmark) (1:200).
Immunohistochemistry: Placental tissue for light microscopic investigations was dehydrated in graded alcohols and embedded in paraffin. Paraffin sections of 2 μm were cut on a Leica RM 2165 microtome (Leica, Ballerup, Denmark) and processed as previously described (Vinge et al. 2010). Briefly, sections were heated and placed in xylene overnight, prior to rehydration in graded alcohols. Rehydrated sections were heated in Tris-EGTA buffer for antigen retrieval in a microwave oven for approximately 20 min, cooled and permeabilized with 0.05% saponin (1% bovine serum albumin (BSA), 0.2% gelatine, 0.05% saponin in 0.01 M PBS) and blocked for endogenous peroxidase activity before incubation with primary antibodies. Sections were incubated with the primary antibody in 0.01 M PBS, 0.1% BSA and 0.02 M NaN3, followed by incubation with HRP-conjugated secondary antibody. Peroxidase labelling was visualized by incubation with diaminobenzidine and 0.03% H2O2 for 10 min. Sections were counterstained with Meier’s haematoxylin stain and examined in a Leica DMR (Leica, Wetzlar, Germany) microscope equipped with a Leica DFC320 camera (Leica, Wetzlar, Germany) and processed using Adobe Photoshop 8.0 software.
Review of Literature
A literature search was done in PubMed using search criteria Fabry disease AND pregnancy AND live birth AND/OR placental histology and including only full papers (i.e. not abstracts) written in English language.
Results
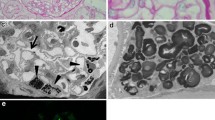
Immunohistochemical detection of alpha-galactosidase A in human placenta showed a distinct granular labelling for α-galactosidase A in the foetal placenta including syncytiotrophoblast cells, in endothelial cells and in interstitial cells. There was no difference in labelling between the Fabry patient (Fig. 2a) and the control (Fig. 2b).

Immunohistochemical labelling for α-galactosidase A in term placenta from the Fabry patient (a) and from control (b). Labelling (brown colour) was seen in vascular endothelial cells (arrows), in interstitial cells (arrowheads) and in syncytiotrophoblast cells (S). Bars, 20 μm
The literature review, and including our case, documented 13 cases of Fabry pregnancies resulting in live births, 6 of which had been treated with agalsidase-beta, 3 with agalsidase-alpha, and 4 without ERT (Tables 1, 2 and 3). All pregnancy outcomes were positive independent of ERT and genetic affection of the foetus. Histopathological examination of the placental tissue disclosed Gb-3 accumulation in the maternal side of the placenta in both ERT-treated and untreated mothers, while Gb-3 accumulation could be seen on both maternal and foetal side in two cases where the foetus had inherited the family mutation.
Discussion
ERT with agalsidase-alpha or agalsidase-beta is recommended for female patients with verified FD and symptoms. All ERT-treated females were symptomatic before pregnancy in the included cases. The pregnant FD females have followed general guidelines for treatment during pregnancy, although a closer monitoring has been preferred. ERT dosage of either agalsidase-alpha or agalsidase-beta has remained unchanged during pregnancy. Included cases have not presented complications related to ERT or negative effect of ERT on foetal outcome. However, in one ERT-treated case (8%), a foetus showed signs of growth restrain the last 2 weeks before birth, amniorrhexis occurred in week 36, and due to pathological cardiotocographic monitoring, acute Caesarean section was performed, delivering a healthy boy.
There are no cases describing progression of FD during pregnancy of patients receiving ERT. Contrarily, in one case (Kalkum et al. 2009, Case 1) there were described increasing acroparaesthesias and fatigue after discontinuation of ERT early in pregnancy, why ERT was re-introduced at week 14 (Kalkum et al. 2009). In another case (Kalkum et al. 2009, Case 2) the patient was treated solely with ERT, while carbamazepine and gabapentin (for neuropathic pain) and irbesartan (for hypertension) were discontinued according to general guidelines. Interestingly blood pressure remained stable during pregnancy, and the patient did not experience neuropathic pain (Kalkum et al. 2009). In a case by Germain et al. (2010), the pregnant female had severe proteinuria and massive Gb-3 storage in podocytes as well as focal/segmental glomerulosclerosis prior to pregnancy. Treatment with ACE and ARB was discontinued as recommended, and patient continued ERT as monotherapy without worsening of proteinuria during pregnancy (Germain et al. 2010).
Data on complications during pregnancy of FD patients is scarce. One retrospective survey study reported data from 41 women (102 pregnancies) and found no life-threatening complications but a higher rate of proteinuria and hypertension, while pre-eclampsia, gestational diabetes, premature births, miscarriage and intrauterine death were not found more commonly in FD patients vs the general population (Holmes and Laney 2015). In the study 10% (4/41) of females were treated with ERT, and none had complications (Holmes and Laney 2015). Gestational diabetes is seen in 2–10% of the general population, and pre-eclampsia is seen in 3% of the general population (Holmes and Laney 2015). In the present study, a numerically higher percentage of pre-eclampsia was observed 8%, but sample size is small (1/13), and in a larger series with 102 FD pregnancies, 5% had pre-eclampsia (not significantly different from the general population). Holmes and Laney did not discuss severity and treatment of complications during pregnancy. The current case was treated with enalapril due to proteinuria prior to the pregnancy. As guidelines suggest, ACE inhibitors are to be avoided during pregnancy (Vest and Cho 2012), and the patient was successfully switched to labetalol. Furthermore, as pre-eclampsia developed, the current case was uptitrated in methyldopa according to guidelines of non-Fabry patients and was well-managed. The present review did not disclose life-threatening complications during pregnancy in FD patients.
Four published cases present data for FD pregnancies without ERT. In the first case (Bouwman et al. 2010, Case A), the patient was asymptomatic at age 39, with an uneventful pregnancy which resulted in a healthy boy without the family mutation and without any accumulation of Gb-3 in the placenta (Bouwman et al. 2010). In the second case (Vedder et al. 2006, Case A; Bouwman et al. 2010, Case C), a mildly affected 23-year-old female with acroparaesthesias as only symptom was not receiving ERT. Pregnancy and birth disclosed no complications, but the male infant inherited the family mutation, and there was seen Gb-3 accumulation in both the maternal and placental side of the placenta (Vedder et al. 2006; Bouwman et al. 2010). Parent et al. (2010) presented a symptomatic patient at age 36, with acroparaesthesias, abdominal cramp, diarrhoea, hypohidrosis, hearing loss and tinnitus. ERT was discontinued at gestational week 4 due to concerns of potential risks. Pregnancy and birth were uncomplicated, giving a healthy boy without the family mutation. Placental examination showed Gb-3 storage inclusion, predominantly on the maternal side. Interestingly, the patient reported diminished acroparaesthesias and gastrointestinal symptoms during pregnancy, which returned to pre-pregnancy intensity 6 weeks after delivery (Parent et al. 2010). In a case with a non-Fabry mother (having a child with a hemizygote FD father) giving an obligate carrier infant girl, pregnancy was uncomplicated. Birth was complicated by foetal distress leading to vacuum extraction, but a healthy girl was delivered. No Gb-3 storage was seen in the placenta (Vedder et al. 2006).
In summary, in non-ERT patients (n = 4), no progression or complications were observed during pregnancy, while the ERT patients (n = 9) did not disclose progression of FD, but two pregnancies were complicated by gestational diabetes and hyperthyroidism and pre-eclampsia. Direct comparison between ERT- and non-ERT-treated females will be difficult as there is bias by indication as ERT-treated females per indication are (more) symptomatic. Gb-3 storage was seen in the placenta of one female receiving ERT on both the maternal and foetal side (the foetus had inherited the family mutation). In pregnancy of FD females without ERT, Gb-3 storage was seen in the placenta, at least in the two symptomatic patients, and storage on the foetal side was seen if the foetus had inherited the family mutation. In the placenta of the obligate carrier female, no storage was seen which might be due to residual endogenous enzyme production.
That Gb-3 accumulation can occur prenatally was shown in 1985 in a 22-week foetus (Tsutsumi et al. 1985), where the cornea was examined biochemically and histopathologically. The α-galactosidase activity in the cornea was very low compared with that of the normal control. Histopathological examination demonstrated presence of intracytoplasmic lamellar bodies surrounded by a single membrane in the epithelial cells. The lamellar bodies were thought to result from abnormal accumulation of ceramide trihexoside. It was concluded that ceramide trihexoside had already begun to accumulate in the epithelial cells in mid-trimester gestation (Tsutsumi et al. 1985). As discussed by Bouwman et al. (2010), the accumulation of Gb-3 in placental tissue differs as is seen in the included cases. The pathophysiology is still not fully elaborated, but accumulation seems to depend on both maternal and foetal characteristics, e.g. disease status, severity, enzyme activity levels and ERT (Bouwman et al. 2010). It is still unknown if ERT can cross the placental barrier and clear foetal placental tissue from Gb-3. Accumulation of Gb-3 has been described in both maternal and foetal placental tissue despite ERT treatment in one case (Thurberg and Politei 2012). The present case cannot contribute to answer this question as the foetus was unaffected and therefore had endogenous enzyme production.
The present case represents the sixth case of agalsidase-beta treatment during pregnancy and brings the total of ERT-treated pregnancies to nine and confirms as shown earlier (Bouwman et al. 2010; Germain et al. 2010; Politei 2010; Tasci and Bicik 2015; Kalkum et al. 2009; Parent et al. 2010) that the treatment seems to be safe for mother and foetus throughout pregnancy.
ERT is used in comparable rare lysosomal storage disorders, e.g. Pompe disease, mucopolysaccharidoses (MPS) and Gaucher disease. However, as in FD, ERT is not approved during pregnancy due to insufficient data. Kłos et al. (2017) published a review on ERT during pregnancy of Pompe disease patients (n = 5) and found it to be seemingly safe with no complications for neither mother nor infant. Contrarily, in one patient there was observed disease progression, while ERT was withheld during pregnancy (Kłos et al. 2017). A recent case series of MPS patients during pregnancy showed a general tendency for both pregnancy and delivery being of high-risk (Stewart et al. 2016). ERT was given in four cases with no adverse effect. All infants developed normally regardless of ERT status during pregnancy (Stewart et al. 2016). In Gaucher disease, ERT during pregnancy has not yet been found associated with any adverse effects (Elstein et al. 2004). Outcomes were similar for ERT-treated and untreated females, and all included infants were healthy and developed according to expectations. The study emphasized that Gaucher females who need ERT should be continued during pregnancy (Elstein et al. 2004).
The present review is the first to put together all clinical information on published Fabry pregnancies. Our case has emphasized that even in a severely affected female with FD combined with severe complication to pregnancy, safe and successful outcome is possible. However, strict monitoring is recommended. Furthermore, neither maternal disease severity nor treatment with ERT seems to affect foetal outcome if the patient is well-managed. General guidelines of treating pre-eclampsia and other pregnancy-related complications should be followed.
Regulatory authorities advise against ERT during pregnancy in FD as well as other rare lysosomal storage diseases. Controlled clinical trials or even large single-centre cohort presentations are unlikely to be performed on this rare topic; hence, documentation of treatment and outcome in all individual patients with FD during pregnancy is important. Interestingly, the medical community seems to have chosen ERT during pregnancy despite the warning from regulatory authorities. It seems to be time to reconsider whether it is prudent and due time to change the advice against ERT in Fabry pregnancies, since no adverse pregnancy outcomes have been published.
References
Bishop DF, Kornreich R, Desnick RJ (1988) Structural organization of the human a-galactosidase A gene: further evidence for the absence of a 3′ untranslated region. Proc Natl Acad Sci U S A 85:3903–3907
Bouwman MG, Hollak CEM, van den Bergh Weerman MA et al (2010) Analysis of placental tissue in Fabry disease with and without enzyme replacement therapy. Placenta 31:344–346. https://doi.org/10.1016/j.placenta.2010.02.004
Brady R (1967) Enzymatic abnormalities in diseases of sphingolipid metabolism. Clin Chem 14:565–577
Echevarria L, Benistan K, Toussaint A et al (2016) X-chromosome inactivation in female patients with Fabry disease. Clin Genet 89:44–54. https://doi.org/10.1111/cge.12613
Elstein Y, Eisenberg V, Granovsky-Grisaru S et al (2004) Pregnancies in Gaucher disease: a 5-year study. Am J Obstet Gynecol 190:435–441. https://doi.org/10.1016/j.ajog.2003.08.006
Germain DP, Bruneval P, Tran TC et al (2010) Uneventful pregnancy outcome after enzyme replacement therapy with agalsidase beta in a heterozygous female with Fabry disease: a case report. Eur J Med Genet 53:111–112. https://doi.org/10.1016/j.ejmg.2009.12.004
Holmes A, Laney D (2015) A retrospective survey studying the impact of Fabry disease on pregnancy. JIMD Rep 21:57–63. https://doi.org/10.1007/8904_2014_384
Kalkum G, Macchiella D, Reinke J et al (2009) Enzyme replacement therapy with agalsidase alfa in pregnant women with Fabry disease. Eur J Obstet Gynecol Reprod Biol 144:92–93. https://doi.org/10.1016/j.ejogrb.2009.01.007
Kint J (1970) Fabry disease: alpha-galactosidase deficiency. Science 167:1268–1269
Kłos J, Kwaśniak-Butowska M, Sławek J (2017) Alglucosidase alfa therapy for Pompe disease in pregnancy – case report. J Neurol Sci 375:167–169. https://doi.org/10.1016/j.jns.2017.01.068
Macdermot KD, Holmes A, Miners AH (2001) Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 38:769–807
Parent E, Wax JR, Smith W et al (2010) Fabry disease complicating pregnancy. J Matern Fetal Neonatal Med 23:1253–1256. https://doi.org/10.3109/14767050903580391
Politei JM (2010) Treatment with agalsidase beta during pregnancy in Fabry disease. J Obstet Gynaecol Res 36:428–429. https://doi.org/10.1111/j.1447-0756.2009.01164.x
Stewart FJ, Bentley A, Burton BK et al (2016) Pregnancy in patients with mucopolysaccharidosis: a case series. Mol Genet Metab Rep 8:111–115. https://doi.org/10.1016/j.ymgmr.2016.08.002
Tasci ES, Bicik Z (2015) Safe and successful treatment with agalsidase beta during pregnancy in Fabry disease. Iran J Kidney Dis 9:406–408
Thurberg BL, Politei JM (2012) Histologic abnormalities of placental tissues in Fabry disease: a case report and review of the literature. Hum Pathol 43:610–614. https://doi.org/10.1016/j.humpath.2011.07.020
Tsutsumi O, Sato M, Sato K et al (1985) Early prenatal diagnosis of inborn error of metabolism: a case report of a fetus affected with Fabry’s disease. Asia Oceania J Obstet Gynaecol 11:39–45
Vedder AC, Strijland A, Weerman MAB et al (2006) Manifestations of Fabry disease in placental tissue. J Inherit Metab Dis 29:106–111. https://doi.org/10.1007/s10545-006-0196-0
Vest AR, Cho LS (2012) Hypertension in pregnancy. Cardiol Clin 30:407–423. https://doi.org/10.1016/j.ccl.2012.04.005
Vinge L, Lees GE, Nielsen R et al (2010) The effect of progressive glomerular disease on megalin-mediated endocytosis in the kidney. Nephrol Dial Transplant 25:2458–2467. https://doi.org/10.1093/ndt/gfq044
Wendt S, Whybra C, Kampmann C et al (2005) Successful pregnancy outcome in a patient with Fabry disease receiving enzyme replacement therapy with agalsidase alfa. J Inherit Metab Dis 28:787–788. https://doi.org/10.1007/s10545-005-0018-9
Whybra C, Wendrich K, Ries M et al (2001) Clinical manifestation in female Fabry. Contrib Nephrol 136:245–250
Wilcox WR, Hopkin RJ, Ortiz A et al (2008) Females with Fabry disease frequently have major organ involvement: lessons from the Fabry registry. Mol Genet Metab 93:112–128. https://doi.org/10.1016/j.ymgme.2007.09.013
Acknowledgements
The patient is thanked for her kind cooperation and her willingness to help improving knowledge and management within the field of FD. The skilled assistance from technician Casper Kok and research nurse Ira Hagen Petersen is gratefully acknowledged.
Genzyme is thanked for providing the antihuman α-galactosidase A antibody. Hanne Sidelmann and Inger B. Kristoffersen are thanked for the skilful technical assistance. Mette Madsen (Dept. of Biomedicine, Aarhus University) is thanked for providing tissue from the control placenta.
Ulla Feldt-Rasmussen’s research salary is sponsored by an unrestricted research grant for NovoNordisk Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Uma Ramaswami
Appendices
Contribution of Individual Authors
EIC, UFR and CVM were involved in the conception and design of the paper. Data collection, analysis and interpretation were performed by EIC, RN, HM and CVM. The manuscript was drafted by EIC, UFR and CVM, while all authors have performed critical revision for important intellectual content.
Take-Home Message
ERT was continued during pregnancy in a severely affected female with Fabry disease and complications during pregnancy, in keeping with nine cases in the literature, resulting in a live birth: why ERT during pregnancy in Fabry disease should be considered safe and recommendations, which currently advise against ERT, should be changed.
Conflicts of Interest
CVM has received an unrestricted research grant from Genzyme. UF-R has received speaker honoraria, unrestricted research grants and appeared in advisory boards of Genzyme, Shire and Amicus and ad hoc advice to Protalix and Freeline. Erik Christensen, Rikke Nielsen, Helle Mogensen and Åse Rasmussen declare that they have no conflict of interest.
Details of Ethics Approval and Patient Statement of Consent
All material was collected with informed patient consent and was approved by the Danish National Committee on Health Research Ethics (approval no. KF 20060063). Informed consent for publication of data has been given by the patient.
Rights and permissions
Copyright information
© 2018 Society for the Study of Inborn Errors of Metabolism (SSIEM)
About this chapter

Cite this chapter
Madsen, C.V., Christensen, E.I., Nielsen, R., Mogensen, H., Rasmussen, Å.K., Feldt-Rasmussen, U. (2018). Enzyme Replacement Therapy During Pregnancy in Fabry Patients. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 44. JIMD Reports, vol 44. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2018_129
Download citation
DOI: https://doi.org/10.1007/8904_2018_129
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-58616-7
Online ISBN: 978-3-662-58617-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)