
Abstract
Serine is a nonessential amino acid that plays a vital role in proper development and functioning of the central nervous system (CNS). Serine deficiency leads to microcephaly, intellectual disability, seizures, and psychomotor retardation in children and severe axonal neuropathy in adults. Serine deficiency syndrome is due to a deficiency of one of three enzymes in the endogenous serine biosynthesis pathway: phosphoglycerate dehydrogenase, phosphoserine transaminase, or, most rarely, phosphoserine phosphatase. Of critical importance to clinical care, serine deficiency syndrome is treatable. Herein, we describe the novel presentation of phosphoserine phosphatase deficiency in an adult. The patient had intrauterine growth restriction, lifelong intellectual disability, childhood onset epilepsy, and borderline microcephaly. In adulthood, she developed progressively severe lower extremity hypertonia, axonal neuropathy, and hand contractures. Neuropathy was complicated by non-healing wounds. Fasting plasma amino acids showed low serine and glycine. Molecular analysis revealed compound heterozygous mutations in phosphoserine phosphatase (PSPH). Treatment with oral serine resulted in improvement of plasma serine levels, decreased neuropathic pain, and subjective improvement in energy level. Although the first case of phosphoserine phosphatase deficiency was described nearly 20 years ago, only eight cases have been reported, all in children. This is the first report of phosphoserine phosphatase deficiency in an adult.
Competing interests: None declared
An erratum to this chapter is available at 10.1007/8904_2015_540
An erratum to this chapter can be found at http://dx.doi.org/10.1007/8904_2016_540
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Intellectual Disability
- Cerebral Spinal Fluid
- Psychomotor Retardation
- Plasma Amino Acid
- Inherited Metabolic Disorder
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Background
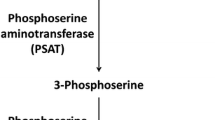
Serine is a nonessential amino acid that plays a critical role in the development and functioning of the central nervous system (CNS) (Furuya et al. 2000; Furuya 2008; de Koning 2006). Serine deficiency syndrome consists of a group of autosomal recessive, neurometabolic disorders due to a deficiency of one of three enzymes in the endogenous serine biosynthesis pathway: phosphoglycerate dehydrogenase (PHGDH, EC 1.1.1.95), phosphoserine transaminase (PSAT, EC 2.6.1.52), or phosphoserine phosphatase (PSPH, EC 3.1.3.3) (see Fig. 1) (Jaeken et al. 1996; Hart et al. 2007). Although the first case of phosphoserine phosphatase deficiency (OMIM #172480) was described nearly 20 years ago, only eight cases have been reported, all in children (Jaeken et al. 1997; Vincent et al. 2015). We describe a novel presentation of phosphoserine phosphatase deficiency in an adult with progressive myeloneuropathy and distal contractures of the upper extremities, expanding the currently understood phenotype. This patient had improvement of her neuropathic pain with serine supplementation.

Serine biosynthesis pathway
Case Report
The patient was born to a 31-year-old gravida 4, para 4 woman at 42 weeks gestation after an uncomplicated pregnancy. She weighed 4 pounds, 10 ounces at birth, 3 standard deviations (SD) below average, consistent with severe intrauterine growth restriction. The patient remained below the growth curve during childhood, though with reportedly proportionate growth. Microcephaly was never mentioned to the family and records are not available. She never had feeding difficulties. The patient met all developmental milestones until age 4 when she started performing poorly in preschool and was eventually diagnosed with absence seizures and intellectual disability. Diagnoses were confirmed with electroencephalogram (EEG) and formal neurologic testing. Absence seizures were difficult to treat, predominantly due to inconsistent medication administration, though the patient’s seizures have been well controlled on ethosuximide for the past 5 years. The patient graduated from high school and held manual labor jobs until severe neuropathy prevented her from working. Her demeanor is generally described as happy, though she can be intermittently extremely irritable. She was evaluated for acute paranoia at age 26, though this was later attributed to an adverse drug reaction. Currently, the patient lives independently with her boyfriend who also has mild intellectual disability. Her mother lives across the street, manages her finances, and is involved as a caretaker.
Throughout childhood, there was no concern for fine or gross motor abnormalities. At age 19, the patient began to complain of increasing lower extremity stiffness, starting in her Achilles tendons. Gross and fine motor skills subsequently deteriorated. Her gait became progressively abnormal, and she developed bilateral foot drop at age 27. Over the next ten years, the patient required multiple surgical debridements, hyperbaric oxygen treatments, casting and therapy for complications of neuropathy including non-healing ulcers, osteomyelitis, and finger tip amputations. Stiffness progressed to involve her upper extremities at age 32, and the patient began to note contractures of her hands and fingers (see Figs. 2 and 3).

Natural history timeline of one patient’s experience with phosphoserine phosphatase deficiency

Recent photographs (38 years old) demonstrate the patient’s borderline microcephaly, anterior receding hairline, and fingertip amputations. She does not have a wide mouth as has been mentioned in two previous reports on PSPH deficiency
Family history is negative for growth restriction, seizures, intellectual disability, microcephaly, recurrent pregnancy losses, and neuropathy. The patient has a 42-year-old unaffected full sister and two unaffected half-brothers. She is of Polish and Northern European ancestry; her parents are not consanguineous.
The neurology service at our tertiary academic medical center was consulted during an inpatient admission for osteomyelitis. On exam, she had borderline microcephaly (occipital-frontal circumference, 52 cm, −2 SD), short stature (height, 149 cm, −2.4 SD), and obesity (weight, 89 kg, BMI 40). She had a prominent widow’s peak with sparse anterior hair, though this was said to be a familial trait. Upper extremities showed an increased carrying angle. Ninety-degree contractures at the distal and proximal interphalangeal (DIP, PIP) joints could be actively extended to 180°. Her hands showed bilateral muscular atrophy of the thenar eminence and intrinsic muscles as well as two left DIP joint amputations. Skin exam showed diffuse, moderately severe eczema including on her face and a right foot ulcer. Her neurologic exam was notable for an immature affect. Speech was fluent and she was able to give a limited history. Cranial nerves were intact. Strength was fully intact in bilateral proximal upper extremities and hip flexors and severely reduced in finger abduction, finger flexion, and ankle dorsiflexion. Sensory exam was notable for severely decreased temperature and vibratory sensation in a length-dependent pattern to the midleg and mid-forearm bilaterally. Deep tendon reflexes were symmetric and diffusely brisk, with the exception of Achilles tendon reflexes, which were absent. She had mild spasticity in all four extremities. There was no dysmetria. Rapid alternative movements were limited by contractures and patient understanding. The patient was able to walk short distances. Gait was antalgic, with obvious foot drop and reduced flexion in the right knee.
Her exam was concerning for a myeloneuropathy, possibly due to an inherited metabolic disorder. Electrodiagnostic study demonstrated a chronic sensory and motor axonal polyneuropathy. Brain magnetic resonance images (MRI) performed at ages 27, 32, and 38 were all normal. Glucose, vitamin B12, folate, methylmalonic acid, copper, ceruloplasmin, and zinc were all within normal range. Plasma amino acids showed a markedly low serine (31 mcmol/L, reference range 60–170) and moderately low glycine (114 mcmol/L, reference range 130–400). Other amino acids were within normal range. The patient refused lumbar puncture. In conjunction with the biochemical genetics service, we ordered sequential sequencing of 3-phoshoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase (PSAT), and phosphoserine phosphatase (PSPH). There were no pathogenic mutations in PHGDH or PSAT. Molecular testing revealed two novel mutations in PSPH (NM_004577.3): c.131T>G (p.Val44Gly) and c.421G>A (p.Gly141Ser). Val44Gly was not found in dbSNP (http://www.ncbi.nlm.nih.gov/SNP), Exome Aggregate Consortium (ExAC, http://exac.broadinstitute.org), or NHLBI Exome Sequencing Project (ESP, http://evs.gs.washington.edu/EVS) databases. Gly141Ser was not found in dbSNP. It was reported with an allele frequency of 0.02% in ESP and 0.0025% in ExAC. In silico computational tools including Align GVGD, SIFT, MutationTaster, and Polyphen2 predict Gly141Ser and Val44Gly to be pathogenic variants. Both variants are evolutionarily conserved. Direct mutational analysis in the patient’s mother identified a heterozygous PSPH: c.131T>G (p.Val44Gly) nucleotide change. Based on our patient’s phenotype, biochemical, and molecular findings, we diagnosed her with phosphoserine phosphatase deficiency. Treatment with oral serine supplementation was initiated and titrated according to plasma serine levels. She currently takes 2.5 g orally, three times a day. Laboratory-measured plasma serine levels have improved; most recent serine and glycine plasma values are 96 and 222 mcmol/L, respectively. After four months of therapeutic treatment, the patient reported improved energy levels and increased sensation in her feet. Her foot ulcer has completely healed and not recurred. She has significantly reduced the dosage of pain medication necessary for neuropathic pain control.
Discussion
Phosphoserine phosphatase deficiency has been described in just two reports, all in children (Vincent et al. 2015; Jaeken et al. 1997; Veiga-da-Cunha et al. 2004). The first described patient was an infant with a separate, unrelated diagnosis of Williams–Beuren syndrome. Apart from features typical of Williams–Beuren syndrome, he had pre- and postnatal growth deficiency, psychomotor retardation, and congenital microcephaly. He was seizure free. Serine levels in the cerebral spinal fluid (CSF) and plasma were decreased, though plasma serine normalized after meals (CSF, 18 μmol/L, control range 27–57 μmol/L; plasma, 53–80 μmol/L, normal range 70–187). The patient was started on serine supplementation at one year of life. He experienced some catch-up head growth and developmental improvements; he was lost to follow-up after age two (Jaeken et al. 1996).
A second report described seven individuals, ages 5 to 19 years, from a multiplex consanguineous Pakistani family, all homozygous for PSPH c.103G>A (p.Ala35Thr) pathogenic mutation (Vincent et al. 2015). All individuals presented in infancy or early childhood with severe developmental delay. All had moderate to profound intellectual disability, seizures, and hypertonia. Most had microcephaly. Plasma amino acid analysis showed low serine and moderately low glycine in individuals with reported results (serine, 26–29 μmol/L, reference 75–175 μmol/L; glycine, 102–129 μmol/L; reference 148–324 μmol/L).
Recently, a molecular study of Neu-Laxova syndrome (OMIM #256520, #616038), a typically prenatally lethal condition characterized by profound neurodevelopmental abnormalities, found mutations in PHGDH, PSAT1, and PSPH. These findings suggest the most severe end of serine deficiency syndrome is incompatible with life (Acuna-Hidalgo et al. 2014). In contrast, our patient probably represents the mild end of the clinical spectrum.
PHGDH deficiency, the most common cause of serine deficiency syndrome, has been described in a single adult. PHGDH encodes the first enzymatic step in the serine biosynthesis pathway. Individuals with PHGDH deficiency typically present in infancy with severe neurodevelopmental abnormalities, including intractable seizures, severe intellectual disability, congenital microcephaly, and psychomotor retardation though more mild phenotypes have also been described (Jaeken et al. 1996; Tabatabaie et al. 2011). Méneret et al. describe a patient with congenital cataracts, mild psychomotor retardation, and slight cerebellar ataxia, who presented at age 31 with adult onset axonal sensorimotor polyneuropathy. He was started on serine supplementation and reported subjective improvement in his quality of life (Méneret et al. 2012).
Phosphoserine phosphatase (PSPH) encodes the third, final, and rate-limiting enzyme in the l-serine synthesis pathway, irreversibly hydrolyzing phosphoserine to l-serine and phosphate (Collet et al. 1997). PSPH is inhibited by l-serine, creating a pathway that is regulated by serine demand rather than supply (Collet et al. 1997). Thus, in contrast to other aminoacidopathies, there is no accumulation of a metabolite or precursor. Screening tests rely on detecting low serine concentration in CSF and/or fasting plasma (de Koning 2006). The serine level in the CSF is most clinically relevant, though invasive and not routinely available. While screening plasma amino acids is specific, sensitivity is affected by the fact that serine levels can normalize after meals (Jaeken et al. 1996). It behooves the physician with a high clinical suspicion for serine deficiency to collect the plasma samples during a fasting state and pursue CSF testing if inconclusive.
Identifying phosphoserine phosphatase deficiency in our adult patient expands the phenotype to include severe axonal neuropathy and contractures in addition to the previously described intellectual disability, epilepsy, microcephaly, and growth deficiency. This case suggests that inexpensive, basic metabolic screening can potentially identify this treatable syndrome. Fasting plasma amino acids should be considered in adult patients with atypical presentations of otherwise unexplained neuropathies and complex features, such as craniofacial deformities, concomitant myelopathy, epilepsy, and developmental delay.
References
Acuna-Hidalgo R, Schanze D, Kariminejad A et al (2014) Neu-Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am J Hum Genet 95(3):285–93
Collet JF, Gerin I, Rider MH, Veiga-da-Cunha M, Van Schaftingen E (1997) Human L-3-phosphoserine phosphatase: sequence, expression and evidence for a phosphoenzyme intermediate. FEBS Lett 408(3):281–4
de Koning TJ (2006) Treatment with amino acids in serine deficiency disorders. J Inherit Metab Dis 29(2-3):347–51
Furuya S (2008) An essential role for de novo biosynthesis of L-serine in CNS development. Asia Pac J Clin Nutr 17(Suppl 1):312–5
Furuya S, Tabata T, Mitoma J et al (2000) L-serine and glycine serve as major astroglia-derived trophic factors for cerebellar Purkinje neurons. Proc Natl Acad Sci U S A 97(21):11528–33
Hart CE, Race V, Achouri Y et al (2007) Phosphoserine aminotransferase deficiency: a novel disorder of the serine biosynthesis pathway. Am J Hum Genet 80:931–937
Jaeken J, Detheux M, Van Maldergem L et al (1996) 3-Phosphoglycerate dehydrogenase deficiency and 3-phosphoserine phosphatase deficiency: inborn errors of serine biosynthesis. J Inherit Metab Dis 19:223–226
Jaeken J, Detheux M, Fryns JP (1997) Phosphoserine phosphatase deficiency in a patient with Williams syndrome. J Med Genet 34(7):594–596
Méneret A, Wiame E, Marelli C, Lenglet T, Van Schaftingen E, Sedel F (2012) A serine synthesis defect presenting with a Charcot-Marie-Tooth-like polyneuropathy. Arch Neurol 69(7):908–11
Tabatabaie L, Klomp LWJ, Rubio-Gozalbo ME et al (2011) Expanding the clinical spectrum of 3-phosphoglycerate dehydrogenase deficiency. J Inherit Metab Dis 34(1):181–184
Veiga-da-Cunha M, Collet JF, Prieur B (2004) Mutations responsible for 3-phosphoserine phosphatase deficiency. Eur J Hum Genet 12(2):163–166
Vincent JB, Jamil T, Rafiq MA et al (2015) Phosphoserine phosphatase (PSPH) gene mutation in an intellectual disability family from Pakistan. Clin Genet 87(3):296–8
Acknowledgements
We would like to thank the patient and her family for their participation in this report and Dr. Angela Sun for her careful review of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Nicole Wolf, MD PhD
Appendices
Synopsis
This novel report of phosphoserine phosphatase deficiency in an adult expands our understanding of serine deficiency syndrome and describes an important and treatable condition for practitioners to consider when evaluating patients with unexplained metabolic neuropathy.
Compliance with Ethical Guidelines
Conflict of Interest
Heather M. Byers, Emily A. Malouf, Michael D. Weiss, Jie Feng, C. Ronald Scott, and Suman Jayadev declare that they have no conflicts of interest.
Robin L. Bennett is an author for John Wiley & Sons.
Informed Consent
This article does not contain any research studies with human or animal subjects performed by the any of the authors.
Additional informed consent for identifiable photography was obtained.
Details of the Contributions of Individual Authors
Dr. Heather Byers prepared the manuscript. Heather Byers, Robin Bennett, Emily Malouf, Michael Weiss, Jie Feng, C. Ronald Scott, and Suman Jayadev participated in the clinical care of this patient and critical review of the manuscript.
Rights and permissions
Copyright information
© 2015 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Byers, H.M. et al. (2015). Novel Report of Phosphoserine Phosphatase Deficiency in an Adult with Myeloneuropathy and Limb Contractures. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 30. JIMD Reports, vol 30. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2015_510
Download citation
DOI: https://doi.org/10.1007/8904_2015_510
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-53680-3
Online ISBN: 978-3-662-53681-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)