
Abstract
Intrinsic factor deficiency (OMIM #261000, IFD) is a rare inherited disorder of vitamin B12 metabolism due to mutations in the gastric intrinsic factor (GIF) gene.
We report three individuals from an Old Order Mennonite community who presented with B12 deficiency. Two cases are siblings born to consanguineous parents and the third case is not known to be closely related. The older male sib presented at 4 years with gastrointestinal symptoms, listlessness, and pallor. He had pancytopenia with megaloblastic anemia. Serum B12 was 61 (198–615 pmol/L). Methylmalonic aciduria was present. C3 was elevated on acylcarnitine profile. Homocysteine was high at 16.7 (5.0–12.0 umol/L). His asymptomatic female sibling was also found to have B12 deficiency. Genetic testing for methylmalonic aciduria (MMAA), transcobalamin deficiency (TCN2), and Imerslund-Gräsbeck syndrome (AMN) showed no mutation in both siblings. The third patient, a 34-year-old woman, had presented in infancy with a diagnosis of pernicious anemia. Mutation analysis of GIF revealed compound heterozygosity for a c.79+1G>A substitution and a c.973delG deletion in all three individuals. Oral or parenteral vitamin B12 has led to complete recovery of clinical parameters and vitamin B12 levels. Newborn screening samples on the siblings revealed normal methylcitrate, C3, and C3/C2 ratios thus indicating no disruption of propionic or methylmalonic acid metabolism.
A high index of suspicion should be maintained if children present with megaloblastic anemia since GIF deficiency is a treatable disorder and newborn screening may not be able to detect this condition.
Competing interests: None declared
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Inherited disorders of vitamin B12 metabolism can be due to many etiologies such as Imerslund-Gräsbeck syndrome (IGS; OMIM #261100, CUBN or AMN mutations) (Grasbeck 2006; Ament et al. 2009), transcobalamin deficiency (TC; OMIM #275350, TCN2 mutation) (Prasad et al. 2008; Trakadis et al. 2013), methylmalonic aciduria (MMA; OMIM #251100, MMAA mutation) (Trakadis et al. 2013), and intrinsic factor deficiency (IFD; OMIM #261000, GIF mutation) (Tanner et al. 2005).
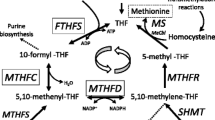
Gastric intrinsic factor (GIF) is a cofactor produced by the parietal cells of the stomach. GIF binds vitamin B12 in the duodenum and transports it to the terminal ileum (Kozyraki and Cases 2013). The GIF-B12 complex allows for endocytosis of B12 by the mucosal cells of the distal ileum via a cubam receptor composed of two proteins, cubilin and amnionless (Watkins and Rosenblatt 2011). Serum vitamin B12 is primarily transported by haptocorrin and transported into cells by transcobalamin (Trakadis et al. 2013) (see Fig. 1).

Enteral metabolism of B12 and GIF
Congenital IFD is a rare disorder of vitamin B12 metabolism presenting in infancy or early childhood. In congenital IFD, gastric acid secretion is normal and B12 deficiency results from a mutation in GIF leading to a low level or lack of GIF in gastric juices, abnormal susceptibility of GIF to pepsin degradation, or reduced affinity for ileal GIF-B12 receptor (Gordon et al. 2004; Chery et al. 2013). IFD differs from adulthood-acquired cobalamin deficiencies associated with atrophic gastritis in which normal GIF can be produced but is reduced in quantity due to decreased parietal cells or an autoimmune disorder with production of antibodies against GIF. The Schilling test can be used to differentiate between acquired and inherited causes of B12 deficiency; however this test is expensive, invasive, and rarely available in practice (Tanner et al. 2005).
Patients with GIF deficiency present with low serum cobalamin levels and megaloblastic anemia in comparison to TC and MMA deficiencies where B12 levels are usually normal (Trakadis et al. 2013). Other presentations include pancytopenia, splenomegaly, hepatomegaly, peripheral neuropathy, joint pain and swelling, anorexia, diarrhea, or infantile death (Gordon et al. 2004; Overgaard et al. 2010).
Material and Methods
Case Report
The proband (II-1 from Family 1) (see Fig. 2) presented at 4 years of age to the gastroenterology clinic with constipation and bloody stools. He is of Old Order Mennonite descent and his parents are consanguineous. Prenatal and birth history were unremarkable.

DNA molecular analysis of GIF identified heterozygous mutations for a c.79+1G>A substitution and a c.973delG deletion in Family 1 and Family 2. Closed symbols signify individuals with low serum vitamin B12
Prior to presentation he experienced gastrointestinal symptoms including loose stools, reduced appetite, vomiting, and mild jaundice as noted by his parents. He underwent dental surgery for caries and was noted to be pale and fatigued 2 weeks following this procedure. Over the next 6 months, he started having difficulty walking and getting up as well as paresthesias in his lower limbs. Initial bloodwork from the GI clinic disclosed the presence of pancytopenia and he was referred to the hematology-oncology clinic. Physical examination revealed a very pale child with a heart rate of 80 beats per minute without lymphadenopathy and organomegaly. He had no dysmorphic features. Cardiac examination was normal. Further hematological investigations revealed the diagnosis of megaloblastic anemia secondary to a very low B12 level (see Table 1). Nutritional deficiency and malabsorption causes were excluded. Infectious screen was negative. Barium swallow and 99mTc pertechnetate scintigraphy were negative. Urine organic acids showed elevated urine methylmalonic aciduria (MMA) which reinforced suspicions of a defect in cobalamin metabolism or transport. There was no proteinuria. No Schilling test was performed as it was unavailable.
The proband was started on oral B12 vitamin (methylcobalamin) supplementation 1,000 mcg/day. He had a rapid full recovery of his gastrointestinal and neurological symptoms and improvement of all hematological cell lineages as well as serum B12 levels. At 7 years of age he is thriving and succeeding at school. His weight is 29.7 kg (90th percentile), and height is 127 cm (97th percentile). He has a normal physical examination and no further urine MMA.
The sister of proband (II-2 from Family 1) was seen at 16 months of age on the basis of family history. She was clinically asymptomatic with a normal physical examination. Her initial hematologic parameters were normal. However, she had low serum B12 levels and elevated urine MMA. She was started on oral B12 supplementation with restoration of her serum B12 stores. A recent evaluation at 4 years of age shows a well child with a normal complete blood count, homocysteine level, and B12 serum levels as well as no urinary MMA.
A 24-year-old woman (II-4 from Family 2), also of Old Order Mennonite descent and with the same surname as the proband, had presented with low serum B12 and megaloblastic anemia in infancy. She had a bone marrow biopsy and has been on B12 injections since then. She had complete recovery of her clinical parameters and vitamin B12 levels. Three of her sibs had also been diagnosed with pernicious anemia and were on vitamin B12 therapy, but no further clinical details were available.
Results
Genetic Analysis
In Family 1, the proband had full sequencing of the MMAA, AMN, and TCN2 with no mutations identified. He then was evaluated for GIF mutations and was found to be compound heterozygous for a c.79+1G>A substitution and a c.973delG deletion. Subsequently, his sister (II-2) tested positive for the same GIF heterozygous mutations. In Family 2, II-4 had a negative test for AMN mutation. Molecular genetic testing for GIF deficiency was positive with the same heterozygous mutations as in Family 1 (See Fig. 2).
Biochemical Analysis
Urine organic acids of the proband showed urine MMA (quantitative values not available) on a qualitative analysis (see Table 1).
C3 was elevated on serum acylcarnitine profile. Serum homocysteine was high at 16.7 (5.0–12.0 umol/L). An initial serum IgA level was elevated at 2.40 g/L (0.20–1.00). Anti-tissue transglutaminase IgG and IgA antibodies were negative. There was no proteinuria on urine analysis. A serum creatine kinase level was normal at 89 U/L (40–280).
The sibling of the proband had urine MMA. Analysis of the newborn screening samples on the proband and his sibling revealed normal methylcitrate, C3, and C3/C2 ratios thus indicating no disruption of propionic/methylmalonic acid metabolism.
Hematological Analysis
See Table 1.
Discussion
The functional deficiency of cobalamin may affect multiple organ systems. Presentation can range from mild gastrointestinal symptoms to severe or fatal anemia (Sturm et al. 2013), pancytopenia, failure to thrive or weakness, leading to a delay in diagnosis or misdiagnosis (Gordon et al. 2004). Neurological symptoms of B12 deficiency may be subtle in presentation, making developmental delay a concern with untreated low serum cobalamin (Ament et al. 2009; Sturm et al. 2013).
When evaluating a patient presenting with low serum B12, many diagnostic investigations and specialists may be involved, making the process lengthy and costly (Carmel et al. 2003). Acquired causes of cobalamin deficiency such as autoimmune gastritis are rare in children. Therefore, in order to accelerate the diagnostic process, genetic causes should be considered in the initial evaluation of megaloblastic anemia in childhood including mutations in AMN, CUBN, GIF, TCN2, and MMAA genes (see Fig. 3) especially in context of consanguinity and Mennonite background. IGS, TC, and IFD are disease entities that can present with similar phenotypes (Overgaard et al. 2010). Lack of proteinuria and response to B12 supplementation can sometimes help to differentiate IFD from IGS in order to direct the sequence of genetic testing (Grasbeck and Tanner 2011; Sturm et al. 2013).

Diagnostic approach to megaloblastic anemia in childhood. Infectious and dietary causes are becoming less frequent in our society (Sturm et al. 2013). Imerslund-Grasbeck syndrome (IGS) with mutations in AMN and CUBN and IFD are disease entities that can present with similar phenotypes (Tanner et al. 2005; Ament et al. 2009). Proteinuria is sometimes used to differentiate IGS and IFD and direct the order of genetic testing but is not a specific finding (Grasbeck and Tanner 2011). *Multiple genes include MUT, MMAA, MMAB, MCEE and MMADHC
Dried blood spot C3 acylcarnitine is the newborn screening metabolite that is elevated in diseases of intracellular cobalamin metabolism, methylmalonic and propionic acidemias. TC deficiency can be detected on newborn screening (Prasad et al. 2012). C3 acylcarnitine was not elevated in newborn screening tests of GIF deficient patients II-1 and II-2 of Family 1. Presumably the infants are protected by hepatic stores of maternally originating cobalamin and not initially dependent on GIF-mediated cobalamin intestinal absorption.
Hewitt and Gordon et al. first identified GIF on chromosome 11 (Hewitt et al. 1991). IFD resulting from a mutation in GIF was first described in 2004 by Yassin et al. in a patient presenting with severe megaloblastic anemia (Yassin et al. 2004). Since then, multiple case reports have identified other GIF mutations resulting in B12 deficiency and megaloblastic anemia. The previous case reports have been summarized in Table 2.
The mode of inheritance of IFD was previously unclear (Gordon et al. 2004). With studies demonstrating homozygous and compound heterozygote mutations, the inheritance has been established as autosomal recessive for IFD (Yassin et al. 2004; Chery et al. 2013).
The Human Genome Mutational Database identifies 18 mutations in GIF (http://www.hgmd.org, retrieved from 2014/04/21). The three patients described here were compound heterozygous for c.79+1G>A and c.973 delG. The c.79+1G>A mutation (HGMD #CS051254) affects the intron 1 invariant donor splice site and has been described by Tanner et al. (2005). The c.973 delG is a novel mutation and results in a frame shift starting at codon 325 and produces a premature stop at codon 337 in the new reading frame.
All three patients are members of an Old Order Mennonite community in Southwestern Ontario, Canada, but interestingly all three are compound heterozygous for two GIF mutations. The phenotype associated with homozygosity for either of these two mutations has not yet been described. This report indicates that GIF deficiency should be considered as a cause of vitamin B12 deficiency in the Mennonite communities along with AMN deficiency as described by Strauss and Puffenberger in the Pennsylvania Mennonite community (Strauss and Puffenberger 2009).
B12 deficiency caused by GIF deficiency is a treatable disorder responding to oral B12 supplementation as gastric mucosa is normal. When diagnosed early many or all symptoms can be avoided. A high index of suspicion for GIF deficiency should be maintained if children present with megaloblastic anemia since prognosis is good once diagnosed.
Abbreviations
- AMN :
-
Amnionless gene
- B12 :
-
Vitamin B12 (cobalamin)
- CUBN :
-
Cubilin gene
- GIF:
-
Gastric intrinsic factor
- GIF :
-
Gastric intrinsic factor gene
- IFD:
-
Intrinsic factor deficiency
- IGS:
-
Imerslund-Gräsbeck syndrome
- MCEE :
-
Methylmalonic CoA epimerase gene
- MMA:
-
Methylmalonic aciduria
- MMAA :
-
Methylmalonic acidemia CblA type gene
- MMAB :
-
Methylmalonic acidemia CblB type gene
- MMADHC :
-
Methylmalonic acidemia CblD type gene
- MUT :
-
Methylmalonic CoA mutase gene
- TC:
-
Transcobalamin deficiency
- TCN2 :
-
Transcobalamin II gene
References
Ament AE, Li Z, Sturm AC et al (2009) Juvenile cobalamin deficiency in individuals of African ancestry is caused by a founder mutation in the intrinsic factor gene GIF. Br J Haematol 144(4):622–624
Boina Abdallah A, Ogier de Baulny H, Kozyraki R et al (2012) How can cobalamin injections be spaced in long-term therapy for inborn errors of vitamin B(12) absorption? Mol Genet Metab 107(1–2):66–71
Carmel R, Green R, Rosenblatt DS, Watkins D (2003) Update on cobalamin, folate, and homocysteine. Hematology Am Soc Hematol Educ Program 62–81
Chery C, Hehn A, Mrabet N et al (2013) Gastric intrinsic factor deficiency with combined GIF heterozygous mutations and FUT2 secretor variant. Biochimie 95(5):995–1001
Garcia Jimenez MC, Baldellou Vazquez A, Calvo Martin MT, Perez-Lungmus G, Lopez Pison J (2008) Hereditary juvenile cobalamin deficiency due to mutations in GIF gene. Anales de Pediatria (Barcelona, Spain: 2003) 69(1):56–58
Gordon MM, Brada N, Remacha A et al (2004) A genetic polymorphism in the coding region of the gastric intrinsic factor gene (GIF) is associated with congenital intrinsic factor deficiency. Hum Mutat 23(1):85–91
Grasbeck R (2006) Imerslund-Grasbeck syndrome (selective vitamin B(12) malabsorption with proteinuria). Orphanet J Rare Dis 1:17
Grasbeck R, Tanner SM (2011) Juvenile selective vitamin B(1)(2) malabsorption: 50 years after its description-10 years of genetic testing. Pediatr Res 70(3):222–228
Hewitt JE, Gordon MM, Taggart RT, Mohandas TK, Alpers DH (1991) Human gastric intrinsic factor: characterization of cDNA and genomic clones and localization to human chromosome 11. Genomics 10(2):432–440
Kozyraki R, Cases O (2013) Vitamin B12 absorption: mammalian physiology and acquired and inherited disorders. Biochimie 95(5):1002–1007
Overgaard UM, Tanner SM, Birgens HS (2010) Vitamin B12 deficiency in a 15-year old boy due to mutations in the intrinsic factor gene, GIF. Br J Haematol 150(3):369–371
Prasad C, Rosenblatt DS, Corley K, Cairney AE, Rupar CA (2008) Transcobalamin (TC) deficiency–potential cause of bone marrow failure in childhood. J Inherit Metab Dis 31(Suppl 2):S287–S292
Prasad C, Cairney AE, Rosenblatt DS, Rupar CA (2012) Transcobalamin (TC) deficiency and newborn screening. J Inherit Metab Dis 35(4):727
Strauss KA, Puffenberger EG (2009) Genetics, medicine, and the plain people. Annu Rev Genomics Hum Genet 10:513–536
Sturm AC, Baack EC, Armstrong MB et al (2013) Hereditary intrinsic factor deficiency in chaldeans. JIMD Rep 7:13–18
Tanner SM, Li Z, Perko JD et al (2005) Hereditary juvenile cobalamin deficiency caused by mutations in the intrinsic factor gene. Proc Natl Acad Sci U S A 102(11):4130–4133
Tanner SM, Sturm AC, Baack EC, Liyanarachchi S, de la Chapelle A (2012) Inherited cobalamin malabsorption. Mutations in three genes reveal functional and ethnic patterns. Orphanet J Rare Dis 7:56
Trakadis YJ, Alfares A, Bodamer OA et al (2013) Update on transcobalamin deficiency: clinical presentation, treatment and outcome. J Inherit Metab Dis 37(3):461–473
Watkins D, Rosenblatt DS (2011) Inborn errors of cobalamin absorption and metabolism. Am J Med Genet C Semin Med Genet 157C(1):33–44
Yassin F, Rothenberg SP, Rao S, Gordon MM, Alpers DH, Quadros EV (2004) Identification of a 4-base deletion in the gene in inherited intrinsic factor deficiency. Blood 103(4):1515–1517
Acknowledgments
We thank the patients and their families to allow us to share their information and Roger Dewar for DNA sequence analyses. This paper was presented as a poster in the Garrod Association meeting, May 2013, and as a poster at the Pediatric Research Day at London Health Sciences Centre, May 2014. A travel grant was received from the Garrod Association.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Verena Peters
Appendices
References to Electronic Databases
GeneReviews®: http://www.ncbi.nlm.nih.gov/books/NBK1116/
Human Genome Mutational Database HGMD®: http://www.hgmd.org
Online Mendelian Inheritance in Man: http://omim.org
Pubmed: http://www.ncbi.nlm.nih.gov/pubmed
Synopsis
A case report and literature review of inherited GIF deficiency mutation, a severe but treatable disorder of B12 metabolism whose presentation can mimic many disorders.
Compliance with Ethics Guidelines
Conflict of Interest
Amaryllis Cloelia Ferrand, Victoria Mok Siu, Melanie P Napier, Osama Al-Dirbashi, Pranesh Chakraborty, Chitra Prasad, and C Anthony Rupar declare no conflicts of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5).
Declaration
The paper is being submitted as an original article.
This paper has not been previously submitted nor is under consideration for publication in any other journal.
Author Contribution
The paper’s submission for publication has been approved by all of the authors.
Amaryllis C. Ferrand: Conception, design and drafting. First Author.
Victoria M. Siu: Conception, design and critical revision.
C Anthony Rupar: Analysis and interpretation of data, critical revision.
Melanie P. Napier: Conception, design and critical revision.
Osama Y. Aldirbashi: Analysis and interpretation of data, critical revision.
Pranesh Chakraborty: Analysis and interpretation of data, critical revision.
Chitra Prasad: Conception, design and drafting. Guarantor.
Rights and permissions
Copyright information
© 2014 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Ferrand, A. et al. (2014). Biochemical and Hematologic Manifestations of Gastric Intrinsic Factor (GIF) Deficiency: A Treatable Cause of B12 Deficiency in the Old Order Mennonite Population of Southwestern Ontario. In: Zschocke, J., Baumgartner, M., Morava, E., Patterson, M., Rahman, S., Peters, V. (eds) JIMD Reports, Volume 18. JIMD Reports, vol 18. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2014_351
Download citation
DOI: https://doi.org/10.1007/8904_2014_351
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44862-5
Online ISBN: 978-3-662-44863-2
eBook Packages: MedicineMedicine (R0)