
Abstract
Disorders of intracellular cobalamin (vitamin B12) metabolism result from deficient synthesis of the coenzymes derived from vitamin B12: adenosylcobalamin and methylcobalamin. Disturbances of cobalamin-cofactor synthesis result in elevated levels of homocysteine and/or methylmalonic acid. Nine defects of intracellular cobalamin metabolism have been defined. The most common of these disorders is cblC (combined methylmalonic aciduria and homocystinuria). The cblD disorder is rare with fewer than twenty cases reported in the literature. Some cblD patients have combined methylmalonic aciduria and homocystinuria (referred to as “cblD original,” “cblD-combined,” or herein “cblD-MMA/HC”); some have isolated homocystinuria (referred to as “cblD-variant 1” or herein “cblD-HC”); and others have isolated methylmalonic aciduria (called “cblD-variant 2” or herein “cblD-MMA”). Only six cases of cblD-HC have been defined thus far. We report the 7th case of cblD-HC. The clinical manifestations, biochemical profile, genetic mutation, and plausible ancestry are discussed.
Competing interests: None declared
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Mean Corpuscular Volume
- Megaloblastic Anemia
- Methylmalonic Acid
- Serum Homocysteine
- Global Developmental Delay
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Disorders of intracellular cobalamin (vitamin B12) metabolism result from deficient synthesis of two coenzymes derived from vitamin B12, namely, adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl). AdoCbl is required for activity of the mitochondrial enzyme methylmalonyl-CoA mutase, which catalyzes the conversion of methylmalonyl-CoA (generated during catabolism of branched-chain amino acids, odd-chain fatty acids, and cholesterol) to succinyl-CoA which enters the Krebs cycle. MeCbl is needed for activity of the cytoplasmic enzyme methionine synthase, which catalyzes the methylation of homocysteine to form methionine (Watkins and Rosenblatt 2011).
Inborn errors of cobalamin-cofactor synthesis are a group of rare disorders affecting multiple steps between the lysosomal release of cobalamin and the subsequent generation of mitochondrial AdoCbl and cytosolic MeCbl. Disturbances of cobalamin-cofactor synthesis result in elevated levels of homocysteine and/or methylmalonic acid, depending on which step is affected in the pathway. To date, nine distinct defects have been identified using in vitro somatic complementation and molecular genetic analysis. The disorders have been designated cblA, cblB, cblC, cblD, cblE, cblF, cblG, cblJ, and mut (Watkins and Rosenblatt 2011; Coelho et al. 2012; Yu et al. 2013). The clinical manifestations can be highly variable even within a single complementation group and can span the prenatal period through adolescence and adulthood. The clinical phenotype is believed to be influenced by the severity and location in the pathway of the defect. Clinical manifestations include intrauterine growth retardation, microcephaly, congenital heart disease, feeding difficulty, megaloblastic anemia/cytopenia, global developmental delay, hypotonia, seizures, neuropsychiatric symptoms, and thromboembolic complications. The prototype and best understood is cblC, which is also the most common of these disorders. The cblC disorder causes combined methylmalonic aciduria and homocystinuria.
The cblD disorder has been among the rarest of the disorders of intracellular cobalamin metabolism. Some cblD patients have combined methylmalonic aciduria and homocystinuria (referred to as “cblD original,” “cblD-combined,” or herein “cblD-MMA/HC”); some have isolated homocystinuria (referred to as “cblD-variant 1” or herein “cblD-HC”); and others have isolated methylmalonic aciduria (called “cblD-variant 2” or herein “cblD-MMA”) (Suormala et al. 2004). Goodman et al. (1970) described the first cblD disorder cases in a sibship in 1970 with combined methylmalonic aciduria and homocystinuria. No additional patients with the cblD disorder were reported until 2004 when Suormala et al. (2004) described two patients with cblD-HC and one patient with cblD-MMA using complementation analysis. To date, eighteen cblD patients have been reported; including five patients with the classic form of the disorder, six with cblD-HC, and seven with cblD-MMA (Coelho et al. 2008; Miousse et al. 2009; Parini et al. 2013; Stucki et al. 2011). Mutations at the MMADHC gene have been shown to underlie the cblD disorder in all of these patients. This manuscript describes the seventh patient to be reported with the cblD-HC disorder. The clinical presentation, biochemical profile, and genetic mutation that underlie the cblD-HC disease in this patient are also discussed.
Case Presentation
Clinical Presentation
The patient is a girl of East Indian descent who came to our attention at the age of 5 years. The parents are first cousins and in good health. Her only sibling, an 11-year-old sister, was also in good health. The patient was born at term in India following a pregnancy that was significant for hypertension and hyperemesis. The birth weight was 3.25 kg. Other measurements at birth were not available. Her neonatal course was unremarkable.
Beyond the neonatal period, the patient was generally well with the exception of hypotonia and microcephaly (according to the parents). Reportedly, by 18 months of age, she was deemed to have global developmental delay. She did not achieve good head control until 18 months of age. She sat independently by 2 years. She walked by 2 1/2–3 years. By 5 years of age, the time of immigration of the family to Canada, she was still unable to pick up finger foods with a pincer grasp. She also had profound delay in her expressive and receptive speech and language skills (at 15-month level). Additionally, she displayed multiple repetitive/stereotypical mannerisms. She was noted to have significant deficits in her social and emotional reciprocity. She lacked ability to spontaneously seek shared enjoyment (joint attention). She could not maintain eye-to-eye gaze. She was unable to maintain any peer relationships as she preferred solitary play. She had profound difficulty with transitioning as she would throw a temper tantrum when her routine was broken. Hence, she was deemed to be on the autistic disorder spectrum.
Biochemical and Radiological Parameters
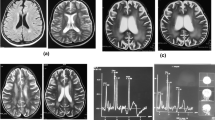
While in India (at 18 months of age), her serum homocysteine (total) was elevated at 71 μmol/L (upper limit of reference range 10 μmol/L). Methionine was decreased at 1 μmol/L (reference range 11–16 μmol/L). Urine organic acids were not available. Hematological parameters were also not available. A brain MRI at 15 months of age revealed frontotemporal and corpus callosum atrophy. Hypomyelination in peripheral white matter of bilateral cerebral hemispheres was also noted. The electroencephalogram was normal. She was diagnosed with an inborn error of cobalamin metabolism. She was commenced on vitamin B12 injections (the exact type, dose, and frequency were not known to us). Following 3 months of treatment, her serum homocysteine dropped to 32 μmol/L.
At the time of referral to our center (at 5-years of age), serum homocysteine was elevated at 45 μmol/L, methionine was elevated at 33 μmol/L, and methylmalonic acid was within the reference range. The acylcarnitine profile was unremarkable. There was no evidence of megaloblastic anemia or cytopenia. Her white blood cells (WBC) were 7.9 × 109/L (normal 4–10 × 109/L), hemoglobin (Hgb) was 121 g/L (normal 120–160 g/L), mean corpuscular volume (MCV) was 81.4 fL (normal 77–96 fL), hematocrit (Hct) was 0.357 (normal 0.36–0.48), and platelets (Plt) were 309 × 109/L (normal 150–400 × 109/L). The brain MRI was normal. She was commenced on daily hydroxycobalamin 1 mg intramuscular injections, betaine, and folic acid. Homocysteine dropped to 18 μmol/L. Hydroxycobalamin injections were, however, discontinued after 3 months due to significant administration difficulties encountered by the parents due to her difficult behavior. She remained on betaine and folic acid. While off treatment, her homocysteine levels have varied between 25 and 40 μmol/L, and her hematological profile remained essentially unchanged (WBC 12 × 109/L, Hgb 121 g/L, MCV 81.1 fL, Hct 0.37, and Plt 384 × 109/L). Recently, hydroxocobalamin injections were restarted weekly at a dose of 5 mg. There have been progressive gains in her development with the support of multidisciplinary input. Currently, at 10 years of age, she is at grades 1–2 level with vocabulary consisting of approximately 100 words and using 2–3 word sentences. Her behavior has improved significantly with increasing attention and fewer temper tantrums.
Studies of Cultured Patient Fibroblasts
Skin fibroblasts were obtained from the patient. Ability of her fibroblasts to incorporate label from [1-14C] propionate and 5-[14C]methyltetrahydrofolate into trichloroacetic acid-precipitable cellular macromolecules (measures of function of methylmalonyl-CoA mutase and methionine synthase, respectively) was performed as described previously (Miousse et al. 2009). The patient had decreased methyltetrahydrofolate incorporation (54 pmol/mg protein/18 h; reference 225 ± 165 pmol/mg protein/18 h), indicating decreased function of methionine synthase. Function of methylmalonyl-CoA mutase was within the reference range.
Synthesis of the two cobalamin coenzyme derivatives, AdoCbl and MeCbl, was assessed by incubation of her fibroblasts in medium containing [57Co] cyanocobalamin bound to transcobalamin followed by extraction and separation of cobalamin derivatives by high-performance liquid chromatography (Miousse et al. 2009). The proportion of intracellular cobalamin present as MeCbl was 3.5% (reference 58.0 ± 6.7). AdoCbl levels were within the reference range. Somatic cell complementation classified this patient as cblD.
Molecular Diagnosis
Genomic DNA was isolated from patient fibroblasts with the QIAamp DNA minikit (Qiagen) and exons and flanking intronic sequence of the MMADHC gene amplified using previously described primers (Coelho et al. 2008). Sequencing of PCR amplicons identified a homozygous missense mutation: c.746A>G (p.Tyr249Cys).
Discussion
The clinical manifestations of disorders of intracellular cobalamin metabolism can be highly variable even within a single complementation group. The cblC disorder (combined methylmalonic acidemia and homocystinuria due to mutations in MMACHC) is the most common inherited disorder of cobalamin metabolism, with over 550 patients reported. The cblD disorder has been rare with only eighteen cases reported in the literature (a single sibship identified before 2004). This disorder includes patients with the classic presentation of combined methylmalonic acidemia and homocystinuria (cblD-MMA/HC), isolated homocystinuria (cblD-HC), and isolated methylmalonic acidemia (cblD-MMA).
Mutations in the MMADHC (methylmalonic aciduria, cblD type, and homocystinuria) gene on chromosome 2q23.2 have been reported in all identified cblD patients (Coelho et al. 2008; Miousse et al. 2009; Parini et al. 2013; Stucki et al. 2011). The MMADHC protein has sequence homology with a bacterial ATP-binding cassette transporter and contains a mitochondrial targeting sequence (Coelho et al. 2008). It does not appear to bind cobalamin. The MMADHC protein is present both in the cytoplasm, where it binds the cobalamin chaperone protein MMACHC (mutated in the cblC inborn error) and in the mitochondria (Deme et al. 2012; Gherasim et al. 2013; Mah et al. 2013). It is not understood how the protein directs cobalamin to different cellular compartments. The identification of mutations in cblD patients has allowed investigation of the relationship between the genotypes and the three biochemical phenotypes of this disorder. Coelho et al. (2008) examined the mutations in seven unrelated patients with cblD disorder. The group reported that mutations found in the patients with cblD-MMA were located toward the N-terminal part of the protein and consisted of a nonsense mutation, a duplication, and a frame-shift deletion. Mutations found in the patients with cblD-HC were located toward the C-terminal part of the protein and consisted of missense mutations. Mutations found in the patients with the combined phenotype were located toward the C-terminal and consisted of a nonsense mutation, a splice-site deletion, and a frame-shift duplication. This correlation between the location of the mutations and the biochemical phenotype was further confirmed by a recent study by Jusufi et al. using mutant constructs of the MMADHC protein (Jusufi et al. 2014). It was postulated that the N-terminal domain, which contains a mitochondrial targeting sequence, was required for the synthesis of AdoCbl, and the C-terminal domain was required for MeCbl synthesis. Mutations predicted to result in a nonfunctional protein resulted in combined homocystinuria and methylmalonic aciduria (Coelho et al. 2008). Subsequently identified MMADHC mutations have been consistent with this hypothesis (Miousse et al. 2009; Parini et al. 2013).
Although mutations in six cblD-HC patients have been reported, clinical findings have been reported in only three cases (Suormala et al. 2004; Miousse et al. 2009). The first clinical description of a patient with cblD-HC was published by Suormala et al. (2004), a boy of Irish descent who was diagnosed at the age of six. He was the product of an uneventful pregnancy and labor/delivery to related parents and suspected “high-grade consanguinity.” At 6 years of age, he presented with global developmental delay, learning disability, spastic ataxia, and deterioration of gait. Brain MRI analysis revealed cerebral and cerebellar atrophy. Visual evoked potentials were delayed. The mean corpuscular volume (MCV) was elevated at 94 fL (normal range 70–87 fL). Hemoglobin (Hgb) was normal at 11.9 g/dL (reference range not given for the authors’ laboratory). Homocysteine was elevated at 9 μM (normal not detectable) and methionine was low at 14 μM (normal 15–40 μM). Methylmalonic acid was not detectable in the urine. Treatment consisted of hydroxocobalamin 1 mg intramuscular daily and then weekly injections. After 1 week on therapy, the patient was reported to be more alert with improved muscle tone. Six months later, he was able to walk and became more verbal.
The second patient with cblD-HC was also described by Suormala et al. (2004), a boy of Italian heritage who was diagnosed at the age of 3 months. He was the product of an uneventful pregnancy and labor/delivery to unrelated parents. At 3 months of age, he presented with severe hypotonia, nystagmus, dystonic movements, and seizures refractory to anticonvulsants. Brain MRI analysis revealed reduced myelination and a small cerebellar vermis. Megaloblastic anemia was found (MCV 105 fL and Hg 8.5 g/dL). Homocysteine was elevated at 128 μM and methionine was decreased at 4 μM, with undetectable methylmalonic acid in the urine. He was treated with hydroxocobalamin 1 mg intramuscular daily injections. Seizures disappeared within 10 days and the hematological status normalized. By 4 years of age, his gross and fine motor skills were reported as normal. Speech delay was still present. Brain MRI was normal.
The clinical description of the third patient with cblD-HC was reported by Miousse et al. (2009), a girl of Mexican background whose parents are first cousins. At 4 months of age, she was admitted to hospital with respiratory syncytial virus pneumonia and was noted to have developmental delay (inability to roll, inconsistent visual tracking, and limited responsiveness to voice), macrocephaly, and hair loss. A computed tomography scan revealed nonobstructive hydrocephalus. Homocysteine was 52.2 μmol/L (normal 0–14 μmol/L), methionine was not detected, and plasma methylmalonic acid level was normal. MCV was normal at 97.5 fL (normal 74–108 fL). The patient was started on a regimen of hydroxocobalamin (details not provided), folate, and betaine. Although the biochemical parameters improved, the clinical status did not. The patient developed a nearly obstructive vena cava clot. Medical support was then withdrawn due to progressive hydrocephalus and prolonged ventilatory support.
All six patients with cblD-HC had missense mutations affecting the C-terminal domain of MMADHC: two patients homozygous for c.737A>G (p.D246G), two patients homozygous for c.746A>G (p.Y249C), one patient homozygous for c.776T>C (p.L259P), and one patient heterozygous for c.746A>G and c.545C>A (T182N). Here, we report the seventh case of cblD-HC in a girl of East Indian origin homozygous for the c.746A>G nucleotide change. As reported previously, the 746A>G mutation occurred in a region of the MMADHC gene that is highly conserved among species and is sufficient to cause deficient synthesis of methylcobalamin (Coelho et al. 2008). Interestingly, although our patient was born in India to East Indian consanguineous parents, her last name is of Portuguese origin. Given that the c.746A>G mutation was previously reported in a European (Italian) boy, it is plausible that there is a shared ancestry for the mutated allele in these two patients.
With the recognition of the cblD-HC and cblD-MMA types of cblD disease and with the advances in the understanding of the molecular basis of cblD and the causative role of the MMADHC gene, the clinical, biochemical, and molecular diagnosis of cblD disease has improved considerably. Although cblD can be diagnosed early in life, through expanded newborn screening, cases can be missed as screening programs do not screen for low methionine levels seen in the classic and cblD-HC form. Given that patients generally respond well to treatment with vitamin B12, clinicians must be aware of this inborn error of cobalamin metabolism.
References
Coelho D, Suormala T, Stucki M et al (2008) Gene identification for the cblD defect of vitamin B12 metabolism. N Engl J Med 358:1454–1464
Coelho D, Kim JC, Miousse IR et al (2012) Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat Genet 44:1152–1155
Deme JC, Miousse IR, Plesa M et al (2012) Structural features of recombinant MMADHC isoforms and their interactions with MMACHC, proteins of mammalian vitamin B12 metabolism. Mol Genet Metab 107:352–362
Gherasim C, Hannibal L, Rajagopalan D, Jacobsen DW, Banerjee R (2013) The C-terminal domain of cblD interacts with cblC and influences intracellular cobalamin partitioning. Biochimie 95:1023–1032
Goodman SI, Moe PG (1970) Homocystinuria with methylmalonic aciduria: two cases in a sibship. Biochem Med 4:500–515
Jusufi J, Suormala T, Burda P, Fowler B, Froese DS, Baumgartner MR (2014) Characterization of functional domains of the cblD (MMADHC) gene product. JIMD Rep, doi:10.1007/s10545-014-9709-4.
Mah W, Deme JC, Watkins D et al (2013) Subcellular localization of MMACHC and MMADHC, two human proteins central to intracellular vitamin B12 metabolism. Mol Genet Metab 108:112–118
Miousse IR, Watkins D, Coelho D et al (2009) Clinical and molecular heterogeneity in patients with the cblD inborn error of cobalamin metabolism. J Pediatr 154:551–556
Parini R, Furlan F, Brambilla A et al (2013) Severe neonatal metabolic decompensation in methylmalonic acidemia caused by cblD defect. JIMD Rep 11:133–137
Stucki M, Coelho D, Suormala T, Burda P, Fowler B, Baumgartner MR (2011) Molecular mechanisms leading to three different phenotypes in the cblD defect of intracellular cobalamin metabolism. Human Mol Genet 21:1410–1418
Suormala T, Baumgartner MR, Coelho D et al (2004) The cblD defect causes either isolated or combined deficiency of methylcobalamin and adenosylcobalamin synthesis. J Biol Chem 279:42742–42749
Watkins D, Rosenblatt DS (2011) Inborn errors of cobalamin absorption and metabolism. Am J Med Genet C (Sem Med Genet) 157:33–44
Yu HC, Sloan JL, Scharer G et al (2013) An X-linked cobalamin disorder caused by mutations in a transcription coregulator HCFC1. Am J Hum Genet 93:506–514
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Matthias Baumgartner
Compliance with Ethics Guidelines
Compliance with Ethics Guidelines
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible.
Committee on human experimentation (institutional and national) and with the Helsinki
Declaration of 1975, as revised in 2000. Informed consent was obtained from the parents of the patient for being included in the study. It can be available upon request.
Proof that informed consent was obtained can be available upon request.
Additional informed consent was obtained from the parents of the patient for whom identifying information is included in this article.
Conflict of Interest
Celia Atkinson, Isabelle R. Miousse, David Watkins, David S. Rosenblatt, and Julian AJ Raiman declare that they have no conflict of interest.
Details of Contributions of Authors
Dr. Celia Atkinson: Wrote the manuscript and participated in clinical care and clinical diagnosis of the patient.
Dr. Isabelle R. Miousse: Revised the manuscript and participated in the biochemical and molecular diagnosis of the patient.
Dr. David Watkins: Revised the manuscript and participated in the biochemical and molecular diagnosis of the patient.
Dr. David S. Rosenblatt: Revised the manuscript and participated in the biochemical and molecular diagnosis of the patient.
Dr. Julian AJ Raiman: Revised the manuscript and participated in the clinical care and clinical diagnosis of the patient. GUARANTOR.
Rights and permissions
Copyright information
© 2014 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Atkinson, C., Miousse, I.R., Watkins, D., Rosenblatt, D.S., Raiman, J.A.J. (2014). Clinical, Biochemical, and Molecular Presentation in a Patient with the cblD-Homocystinuria Inborn Error of Cobalamin Metabolism. In: Zschocke, J., Gibson, K., Brown, G., Morava, E., Peters, V. (eds) JIMD Reports, Volume 17. JIMD Reports, vol 17. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2014_340
Download citation
DOI: https://doi.org/10.1007/8904_2014_340
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44577-8
Online ISBN: 978-3-662-44578-5
eBook Packages: MedicineMedicine (R0)