
Abstract
The reconstruction of partial bacterial divisomes inside artificial containers forms part of the bottom-up synthetic biology approach aiming at completing and exploiting our knowledge on bacterial proliferation. The proto-ring, formed in Escherichia coli by FtsZ, FtsA and ZipA, is the molecular complex that initiates divisome assembly. For its elementary reconstruction, very simple soluble structures, as nanodiscs, can be used. They are complemented by the use of more organized containers as maxicells, in which these nucleoid-free cells that retain many macromolecular synthetic pathways allow to study proteins produced from plasmid-carried genes. Between these extremes, artificial structures of an intermediate complexity, as lipid bilayers and vesicles, can be used to test some functions of the proto-ring. We describe several specific procedures for the production of nanodiscs, lipid monolayers, giant vesicles and maxicells to be used as containers. Procedures to purify ZipA, one of the proto-ring elements, in vitro and also to produce it inside maxicells are provided. Examples of the techniques, as protein labelling and immunostaining, used for the subsequent detection and analysis of the reconstructed assemblies are included. A short note on how to grow bacterial cultures suitable for specific purposes completes the collection of techniques that we find valuable to study division in bacteria and to reproduce it outside the cell.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Keywords:
- Bilayers
- Division
- Escherichia coli
- Maxicells
- Nanodiscs
- Proto-ring
- Streptococcus pneumoniae
- Vesicles
- ZipA
1 Introduction
Although our knowledge on bacterial division has progressed considerably due to the use of novel technologies, including genetic analysis and microscopy, a full biochemical and structural description of the process is still hampered by technical difficulties [1, 2]. Among them, the lack of a complete toolbox of purified divisome components from a coherent biological system needs to be solved to allow a full analytical description as a prerequisite for the application of meaningful synthetic biology approaches. Published results often rely on elements obtained from a diversity of biological sources and are even analysed using conditions that hardly reproduce the environment, as temperature, ligands, crowding, etc., found inside the living bacteria [3, 4].
We compile here procedures that we have used to reconstruct partial divisome assemblies that perform limited functions inside artificial containers. From their analysis in Escherichia coli, we have obtained relevant information on topics such as the strength of the interaction operating in the anchoring of the FtsZ ring to the membrane by ZipA (using nanodiscs [5]) and the ability of the Min proteins to precisely localize the FtsZ ring at midcell in the total absence of the nucleoid occlusion system (using maxicells [6]). An intermediate system, between soluble nanodiscs and maxicells that retain a large degree of structural complexity, is the use of vesicles. Using pore-forming proteins, such as hemolysins, we have reconstructed a contractile system contained within permeable vesicles in which the polymerization of FtsZ, driven by the addition of GTP, pulls from the ZipA molecules attached to the membrane and shrinks the vesicle [7]. The assembly of the Streptococcus pneumoniae FtsA polymers onto a lipid bilayer has been instrumental to evidence the function of its carboxyl terminus as an intramolecular switch to regulate polymerization and membrane association [8].
2 Molecular Assemblies
2.1 Nanodiscs
Nanodiscs allowed the specific integration of the purified membrane protein ZipA into a soluble membrane environment [5]. Once incorporated into the nanodisc, ZipA was probed for protein–protein interaction with the cell division protein FtsZ in solution. The procedure avoids aggregation artefacts encountered in whole membrane vesicle systems. The process starts by mixing the membrane scaffold protein (MSP) [9, 10] with purified ZipA in the presence of detergent-solubilized phospholipids. After removal of the detergent, the product is a 10 nm disc-like particle formed by two MSP molecules encircling a lipid bilayer cylinder carrying the ZipA protein inserted into the lipids (Fig. 1). The protocols in this section include the heterologous overproduction and purification of the MSP from a cell-free extract, the overproduction and purification of E. coli ZipA and the assembly of either empty nanodiscs or those in which the E. coli ZipA is incorporated.


Integration of ZipA in nanodiscs. Sedimentation coefficient c(s) distribution of Nd-ZipA bound to FtsZ polymers in the presence of GMPCPP (dashed green line). The red line represents the c(s) distribution of Nd-ZipA in the presence of FtsZ-GMPCPP polymers and a peptide containing the C terminal end of FtsZ used as a competitor of the interaction. The inset shows a cartoon model of ZipA integrated in a nanodisc. Modified from [5] and [11]
Materials
-
Plasmid pMSP1D1 [9] encoding the smallest variant of the original MSP proteins under the control of T7 promoter for overproduction of the protein in E. coli strains carrying the DE3 cassette producing bacteriophage T7 RNA polymerase (see Table 1 and [12–14]).
Table 1 Commonly used promoter systems -
LB (Luria-Bertani) broth and LB agar plates [15]
-
20% (w/v) glucose. G8270 Sigma
-
CaCl2-competent E. coli strain BL21(DE3) [15]
-
100 mg/ml ampicillin stock solution [15]
-
1M IPTG (Isopropyl β-d-1-thiogalactopyranoside). I1658 Sigma
-
Storage buffer: 50 mM Tris–HCl (pH 8), 20% (w/v) sucrose
-
TB buffer: 50 mM Tris–HCl (pH 8), 2 mM DTT (dithiothreitol), 0.1 mM PMSF (phenylmethanesulfonyl fluoride) and 1 mM DNase I (deoxyribonuclease I)
-
5 ml IMAC (immobilized metal ion affinity chromatography) Column, i.e. HiTrap Chelating, HiTrap IMAC or HisTrap, all from GE Healthcare
-
MSP IMAC equilibration buffer: 50 mM Tris–HCl (pH 8), 100 mM NaCl and 10% (v/v) glycerol
-
MSP wash buffer: 50 mM Tris–HCl (pH 8), 100 mM NaCl, 10% (v/v) glycerol and 50 mM imidazole
-
MSP elution buffer: 50 mM Tris–HCl (pH 8), 100 mM NaCl, 10% (v/v) glycerol and 600 mM imidazole
-
Protein concentrator, i.e. Amicon (Merck Millipore) or Vivaspin (Sartorius AG)
-
Liquid nitrogen or dry ice/ethanol
-
Plasmid pET-15ZIP [16] encoding the E. coli ZipA protein under the control of T7 promoter for overproduction of ZipA in E. coli strains carrying the DE3 cassette producing bacteriophage T7 RNA polymerase
-
TNG buffer: 50 mM Tris–HCl (pH 8), 100 mM NaCl, 10% glycerol
-
RC DC Protein Assay Kit. Bio-Rad
-
10% (v/v) Triton X-100. T8787, Sigma
-
ZipA IMAC buffer: 50 mM Tris–HCl (pH 8), 100 mM NaCl, 0.05%, Triton X-100, 10% glycerol
-
ZipA wash buffer: 50 mM Tris–HCl, pH 8, 100 mM NaCl, 10% glycerol, 0.05%, Triton-X100, 50 mM imidazole
-
ZipA elution buffer: 50 mM Tris–HCl, pH 8, 100 mM NaCl, 10% glycerol, 0.05%, Triton-X100, 600 mM imidazole
-
E. coli total lipid extract (100500) or E. coli polar lipid extract (100600) from Avanti Polar Lipids, Inc.
-
Bio-Beads SM2 Adsorbents. 152–3920. Bio-Rad
-
DDM (n-dodecyl-β-d-maltopyranoside) 324355. Calbiochem
-
Sodium cholate. C9282. Sigma
-
Nd-buffer: 20 mM Tris–HCl, pH 8, 0.1 M NaCl, 0.5 mM EDTA
-
Superdex 200 HR10/30 gel filtration column. GE Healthcare
Methods
2.1.1 Heterologous Overproduction of the Membrane Scaffold Protein, MSP
-
1.
Transform 1 μl of plasmid pMSP1D1 to 100 μl CaCl2-competent E. coli BL21(DE3) cells and plate transformed cells on selective LB broth agar plates supplemented with 0.2% glucose and 100 μg/ml of ampicillin and incubate overnight at 37°C.
-
2.
Pick one colony and transfer it with a sterile toothpick to 10 ml of LB supplemented with 0.2% glucose and 100 μg/ml of ampicillin and incubate for 8 h in a shaker at 37°C. Next transfer the 10 ml culture to 200 ml of fresh LB supplemented with 0.2% (v/v) glucose and 100 μg/ml of ampicillin and incubate overnight in a shaker at 37°C.
-
3.
Dilute the overnight culture 1:50 in 1 l of fresh LB supplemented with 0.2% glucose and 100 μg/ml of ampicillin and incubate at 37°C in an orbital shaker until it reaches an OD600 of 0.5–0.6.
-
4.
Add IPTG to the culture to a final concentration of 0.5 mM to induce the production of MSP1D1, and incubate it for 2 h at 37°C in an orbital shaker.
-
5.
To harvest cultivated cells, centrifuge for 15 min at 7,000×g.
-
6.
Discard the supernatant and resuspend the bacterial pellet in 30 ml storage buffer. If the cells are not processed immediately, flash-freeze the pellet by immersion in liquid nitrogen or dry ice/ethanol bath and store them at −80°C.
2.1.2 Preparation of Cell-Free Extract
-
1.
Gently thaw the frozen pellet on ice and add one volume of TB buffer.
-
2.
Break the cells using a French press (3 cycles at 8,000 psi) or by sonic disruption (20 on/off cycles of 30 s with a 50% amplitude, refrigerate between the cycles).
-
3.
Centrifuge the disrupted cells (15 min at 7,000g at 4°C) to discard the cell debris and use the supernatant for the following steps.
-
4.
Centrifuge the supernatant for 90 min at 180,000×g at 4°C to separate membranes from the soluble extract and discard the pellet.
-
5.
This cell-free extract contains high amounts of MSP.
2.1.3 Purification of the Membrane Scaffold Protein, MSP
-
1.
Equilibrate an IMAC column with MSP IMAC equilibration buffer.
-
2.
Load the cell-free extract containing the overproduced soluble MSP protein on the column.
-
3.
Wash non-specific contaminants bound to the column with ten column volumes of MSP wash buffer.
-
4.
Elute the MSP protein with MSP elution buffer.
-
5.
Run a sample from each eluted fraction on SDS-PAGE to detect the fractions that contain the desired protein [17].
-
6.
Pool the fractions containing the MSP protein and dialyse them overnight against MSP IMAC equilibration buffer.
-
7.
Concentrate the dialysed protein with a protein concentrator to obtain a sample containing approximately 10 mg/ml and flash-freeze it in liquid nitrogen or dry ice/ethanol.
-
8.
Store at −80°C until further use.
2.1.4 Overproduction and Purification of E. coli ZipA
-
1.
Transform 1 μl of plasmid pET-15ZIP into 100 μl CaCl2-competent E. coli BL21(DE3) cells and plate transformed cells on selective LB agar plates supplemented with 0.2% (w/v) glucose and 100 μg/ml of ampicillin and incubate at 37°C overnight.
-
2.
Follow steps 2–6 described in Sect. 2.1.1.
-
3.
Follow steps 1–4 described in Sect. 2.1.2, but discard the supernatant instead of the membrane pellet.
-
4.
Resuspend the membrane pellet in 1 ml TNG buffer and determine total protein concentration using the RC DC Protein Assay Kit according to manufacturer’s instructions.
-
5.
Dilute the membrane fraction with TNG down to a total protein concentration of 1 mg/ml.
-
6.
Add Triton X-100 (1% (v/v) final concentration) to solubilize membrane proteins and incubate at 4°C for 1 h on a rocking platform.
-
7.
Centrifuge the supernatant at 4°C and 180,000 g for 90 min to separate non-solubilized material from the solubilized membrane proteins including ZipA.
-
8.
Dilute this soluble fraction with ZipA IMAC buffer to obtain a final concentration of 0.05% Triton-X 100.
-
9.
Load the solubilized protein extract on a ZipA IMAC buffer-equilibrated IMAC column.
-
10.
Discard contaminants using ten column volumes of ZipA wash buffer.
-
11.
Elute the ZipA with ZipA elution buffer.
-
12.
Run a sample from each eluted fraction on SDS-PAGE to detect the fractions that contain the desired protein [17].
-
13.
Pool fractions containing the ZipA protein and dialyse against TNG buffer.
-
14.
Concentrate ZipA to 10 mg/ml with a protein concentrator and flash-freeze aliquots with liquid nitrogen or dry ice/ethanol.
-
15.
Store at −80°C until further use.
2.1.5 Nanodisc Production and Integration of ZipA into Nanodiscs
-
1.
Mix MSP1D1 with lipids (seeNote 1) at a 1:60 molar ratio to obtain empty nanodiscs. Alternatively use a mixture of ZipA, MSP1D1 and lipids at a 1:4:100 molar ratio to produce nanodiscs containing the ZipA protein inserted. Keep the concentration of lipids at 9 mM and of cholate at least at 18 mM.
-
2.
Incubate the specific mixture at room temperature for 1 h.
-
3.
Add ~50 ml of the Bio-Bead suspension (seeNote 2) to each and incubate at 4°C overnight on a rocking table.
-
4.
Allow the Bio-Beads to separate by gravity and recover the supernatant (Note 3).
-
5.
Centrifuge the nanodisc containing supernatant at 4°C at 80,000g for 30 min to remove aggregates and precipitates.
-
6.
Recover the soluble material and load it on a Superdex 200 HR10/30 column equilibrated with Nd-buffer.
-
7.
Collect eluted fractions of ZipA-containing nanodiscs, empty nanodiscs or detergent-solubilized ZipA.
-
8.
Run a sample from each eluted fraction on native PAGE [18] to detect the fractions that contain the desired protein.
-
9.
Estimate the concentration of the ZipA-containing nanodiscs and the empty nanodiscs. For the calculations use a molar extinction coefficient at 280 nm (ε 280) of 57,930 or 42,000 M−1 cm−1 respectively [5].
-
10.
Pool the ZipA-containing nanodisc fractions and freeze in liquid nitrogen or dry ice/ethanol and store at −80°C (seeNote 4).
Notes
-
1.
Dissolve E. coli total lipid extract or E. coli polar lipid to a final concentration of 1 mM in chloroform and distribute it in samples in screw cap microcentrifuge tubes. Evaporate the solvent under a gentle stream of nitrogen and dry it further overnight in a vacuum desiccator. Next, hydrate the lipid film in Nd-buffer to a final concentration of 5 nM. Resuspension and rehydration of the of the dry lipid film is achieved by incubating the tube in a water bath at 37°C and 3–4 alternated cycles of vortexing (30 s) and sonication using a bath sonicator. The suspension will turn slightly opaque when completely dissolved. Addition of 0.5% (v/v) DDM (final concentration) will clear the suspension.
-
2.
-
1.
Place approximately 10–15 ml (dry volume) of Bio-Beads in a 50 ml falcon tube and wash them with 50 ml of 100% methanol.
-
2.
Sediment the Bio-Beads by gravity and discard the liquid phase.
-
3.
Wash the methanol-washed Bio-Beads with 50 ml of 95% ethanol.
-
4.
Sediment the Bio-Beads by gravity and discard the liquid phase.
-
5.
Wash the ethanol-washed Bio-Beads twice with 50 ml of H2O (Milli-Q).
-
6.
Sediment the Bio-Beads by gravity and discard the liquid phase.
-
7.
Wash the washed Bio-Beads twice with 50 ml of Nd-buffer.
-
8.
Sediment the Bio-Beads by gravity and discard the liquid phase.
-
9.
Resuspend washed Bio-Beads in approximately 10 ml Nd-buffer and store at 4°C until further use.
-
1.
-
3.
The supernatant is almost detergent-free and contains nanodiscs.
-
4.
Before use, gently thaw stored nanodiscs and centrifuge them at 4°C at 80,000×g for 30 min to remove possible aggregates and precipitates. Load a small portion of the sample on Nd-buffer-equilibrated Superdex 200 HR10/30 gel filtration column to evaluate the integrity of the sample.
2.2 Lipid Monolayers
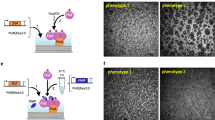
The assay of divisome proteins bound to lipid monolayers is based on the orientation of the phospholipids, whose hydrophobic tails face towards the air and the hydrophilic heads point to the aqueous buffer. Membrane-associated proteins are dispersed in the aqueous phase, and they become concentrated as a two-dimensional array when incorporated into the lipid monolayer by their interaction with lipids. The interaction of a hydrophobic electron microscope grid with the lipid tails allows removing the monolayer from the buffer. Then, proteins on the monolayer can be negatively stained and visualized under an electron microscope.
This lipid monolayer assay has been used to study by electron microscopy the arrangement of streptococcal FtsA molecules anchored to lipids [8]. FtsA binds to the membrane by an amphipathic helix located at the end of its C terminus [19]. Using this technique a role of the FtsA C terminus as a switch that temporarily disturbs the protein polymerization in the cytoplasm and facilitates it when anchored to lipids was revealed.
Materials
-
E. coli PG: l-α-phosphatidylglycerol (E. coli) (sodium salt). 841188P. Avanti Polar Lipids, Inc. (seeNote 6).
-
E. coli CA: cardiolipin (E. coli) (sodium salt). 841199P Avanti Polar Lipids, Inc. (seeNote 6).
-
Chloroform. 102445. Merck.
-
Teflon block, custom made with wells allowing injection through the side (Fig. 2).
Fig. 2 
Schematic representation of the custom-made Teflon block used to coat electron microscope grids with lipid monolayers. Upper view (a) and a magnified section (b) showing a detail of one of the wells (1) and its side injection channel (2)
-
Filtered polymerization buffer, optimized composition to maintain the stability of the protein of interest (seeNote 7).
-
400-mesh carbon-coated copper electron microscope grid.
-
Buffered ATP (seeNote 8). A2383. Sigma.
-
2% (w/v) uranyl acetate solution. 22400. EMS
-
Hamilton syringe and needle.


Methods
2.2.1 Lipid Monolayer Assay
-
1.
Prepare a fresh lipid mixture at the concentration of 0.1 μg/μl (use 2 μl per sample).
-
2.
Place the Teflon block in a wet chamber (a Petri dish covered with a water-soaked filter paper) and fill the wells with 100 μl of the polymerization buffer.
-
3.
Gently pour 2 μl of the lipid chloroform solution on top of the buffer using a Hamilton syringe.
-
4.
Incubate the Teflon block at room temperature inside the wet chamber for 60 min. This will allow the chloroform to evaporate resulting in the formation of a lipid monolayer on the buffer surface (seeNote 11).
-
5.
Place the electron microscope grid horizontally on top of each monolayer. The carbon side should be facing the buffer.
-
6.
Carefully inject 2 μM of the purified protein FtsA [8] into the buffer through the side injection channel.
-
7.
Gently pipette back and forth to distribute the protein homogenously in the buffer (seeNote 11).
-
8.
Incubate at room temperature for 40 min.
-
9.
Start FtsA polymerization by adding buffered ATP to the final concentration of 4 mM and mix gently as in step 7.
-
10.
Incubate at room temperature for 20 min.
-
11.
Replace 50 μl of the droplet with fresh buffer to decrease the amount of soluble protein remaining unadsorbed in the droplet. Repeat a total of ten times.
-
12.
Remove the grid with tweezers (seeNotes 12 and 13).
-
13.
Blot the grid by gently touching a piece of filter paper.
-
14.
Stain the grid with 15 μl of 2% uranyl acetate for 30 s and blot the grid as described in step 13.
-
15.
Check the presence of FtsA polymers under a transmission electron microscope.
Notes
-
5.
All reagents used in the assay should be of the highest purity. Buffers should be filtered prior to use.
-
6.
The lipid composition should be defined depending on the bacteria used as a protein source. Phosphatidylglycerol and cardiolipin mixed at a 1:1 ratio provide a lipid composition mimicking the streptococcal membrane [20, 21].
-
7.
The best conditions for S. pneumoniae FtsA are 5 μM FtsA in FtsA polymerization buffer (20 mM Tris–HCl, pH 7.5, 50 mM NaCl, 5 mM MgCl2, 10% glycerol).
-
8.
Buffered ATP: to avoid a decrease in pH, prepare a stock solution of 100 mM of nucleotide in 1 M Tris–HCl (pH 7.5).
-
9.
To remove undissolved crystals, the uranyl acetate solution should be centrifuged before use (12,000 g, 30 s).
-
10.
Trace lipid contaminations on the Teflon block (see step 2) or Hamilton syringes can result in misleading negative controls. To avoid them, the Teflon block should be washed with hot water and ethanol and finally soaked overnight in a chloroform/methanol mixture (1:1). To remove any protein or lipid residues, the Hamilton syringes should be washed with chloroform (at least three cycles by pumping back and forth) and methanol (once).
-
11.
Pure chloroform can be used as a control to exclude the non-specific binding of membrane proteins to the hydrophobic grid. Chloroform should always be evaporated inside a fume hood or in a desiccator to avoid inhalation.
-
12.
To avoid disturbing the lipid monolayer on the surface, handle the Teflon block and inject the protein solution with outmost care.
-
13.
The grid can be easily removed from the block by injecting 30 μl of buffer to the side injection well. Stained grids can be stored at room temperature and are stable for several years.
3 Confined Environments
3.1 Giant Unilamellar Vesicles
Giant unilamellar vesicles (GUVs) provide a powerful tool to study biological processes associated to membranes, e.g. the assembly and subsequent constriction of the bacterial division ring [7, 8, 22]. GUVs serve as biochemical reaction chambers providing a membrane-confined environment, intermediate between the cell and the lipid monolayer. In addition, they can be easily observed using optical microscopy.
Vesicles consist of an aqueous interior surrounded by a self-closed lipid matrix arranged into single or multilamellar bilayers, depending on the chemical nature and architecture of the lipids. The term “giant vesicles” refers to their diameter, as these vesicles are 1–100 μm in size, in the range of most cells, differing from the large or small unilamellar vesicles used as drug delivery systems that have a mean diameter of 120–140 or 20–50 nm, respectively. The spherical or nonspherical shape of GUVs depends on the lipid structure, the experimental conditions such as temperature or pH, the presence of intercalating molecules like surfactants or the differences in the osmotic pressure between the vesicle lumen and the external solution [23–25].
Different methods to obtain vesicles have been extensively documented [24, 26]. This section provides an experimental procedure to obtain the maximum benefit of permeable GUVs made of egg phosphatidylcholine (eggPC), with a high encapsulation efficiency of biological macromolecules. With this approach we have shown the constriction and shrinkage of permeable GUVs containing E. coli FtsZ in the lumen and ZipA inserted into the inner surface of the membrane through DGS-NTA-Ni lipids into the inner surface of their membrane [7]. Dynamic polymers of FtsZ produce the initial constriction force, and ZipA anchors them to the cytoplasmic membrane to exert the pulling force that finally collapses the vesicle (Fig. 3). We have also recently demonstrated that upon the external addition of ATP, FtsA from S. pneumoniae polymerizes inside GUVs and binds to the vesicle membrane leading to its deformation [8]. The protocol provided describes protein labelling, vesicle preparation, permeabilization and visualization under a confocal microscope (Fig. 3).


Vesicle shrinkage and collapse induced upon interaction of membrane-bound ZipA with FtsZ polymers. Equatorial cross-sectional merged images of permeable vesicles containing 5 M sZipA-Alexa Fluor 647 (red) attached to the membrane through DOGS-NTA lipids and 12 M Alexa Fluor 488-FtsZ (green) in the absence (top row) or presence of the CTZ-MUT peptide inhibitor of FtsZ-ZipA interaction (bottom row). Frames were taken at the times (seconds) indicated after external addition of GTP and Mg2. Modified from [7]
Materials
-
Purified E. coli FtsZ [7]
-
Purified E. coli sZipA, a soluble version of ZipA lacking the membrane-anchoring domain [7]
-
Labelling buffer: 50 mM HEPES/HCl, pH 8.0, 100 mM KCl and 5 mM MgCl2
-
Alexa-488 carboxylic acid succinimidyl ester dye. A20000. Life Technologies
-
Alexa-647 succinimidyl ester dye. A20006. Life Technologies
-
1 M Tris–HCl (pH 8.0)
-
HiTrap Desalting column. 29-0486-84, GE Healthcare
-
Chloroform. 102445, Merck
-
Acetone. 100014. Merck
-
Compressed nitrogen gas
-
18:1 DGS-NTA-Ni: 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] nickel salt. Powder, 790404P, Avanti Polar Lipids, Inc.
-
Egg PC: l-α-phosphatidylcholine (Egg, Chicken). Powder, 840051P, Avanti Polar lipids, Inc.
-
Egg Liss Rhod PE: l-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (Ammonium salt) (Egg-transphosphatidylated, Chicken). Powder, 810146P, Avanti Polar Lipids, Inc.
-
Mineral oil. M5904, Sigma
-
I-buffer: 100 mM KCl, 100 mM sucrose, 50 mM HEPES (pH 7.5), 0.1 mM GDP and 1 mM EDTA
-
O-buffer: 100 mM KCl, 100 mM glucose, 50 mM HEPES (pH 7.5)
-
100 mM glucose
-
Ficoll 70. 17031050. GE Healthcare
-
DiIC18: 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine. D-282 Life Technologies
-
α-Hemolysin from Staphylococcus aureus. H9395, Sigma-Aldrich. Stock solution prepared in O-buffer at 150 μM
-
14:0 PC (DMPC): 1,2-dimyristoyl-sn-glycero-3-phosphocholine. Powder, 850345P. Avanti Polar Lipids, Inc.
-
8-well visualization chambers. 155411, LabTech
Methods
3.1.1 Protein Labelling
To facilitate visualization of proteins encapsulated in GUVs, they are differentially labelled with fluorescent dyes. No difference between the activities of labelled and unlabelled proteins used in the referred publications has been detected.
-
1.
Covalently label polymerized E. coli FtsZ (99 μM) in the amine groups with Alexa 488 dye (seeNote 14). E. coli ZipA (10 μM) can be labelled with Alexa 647. Mix a tenfold molar excess of each dye to each protein preparation in labelling buffer. Incubate on ice for 30 min with shaking.
-
2.
Stop the reactions with 10% (v/v) 1 M Tris–HCl and incubate at room temperature for 15 min with shaking. Labelling efficiency was 0.9 ± 0.2 mol of fluorophore per mol of protein.
-
3.
Eliminate free dye from labelled proteins using a gel filtration column (seeNote 15).
-
4.
Divide labelled proteins in convenient samples, freeze in liquid nitrogen and store at −80°C.
3.1.2 Vesicle Preparation
GUVs are prepared by reverse emulsion. This method achieves a high encapsulation efficiency and protein entrapment control without loss of activity, when compared to other vesicle preparation methods [7, 24].
-
1.
Rinse the glass tubes to be used with acetone three times and once with chloroform.
-
2.
Mix the phospholipid stock solutions as needed, e.g. DOGS-NTA-Ni (ranging from 2 to 10 mol%) with eggPC.
-
3.
Dry chloroform from stock lipid solutions under a gentle stream of nitrogen to obtain films.
-
4.
Dissolve phospholipid mixture in 5 ml mineral oil at a final concentration of 0.5 mg/ml (seeNotes 16 and 17).
-
5.
Place the oil–lipid suspension in a water bath at room temperature and sonicate (30 W output) for 30 min.
-
6.
Heat the suspension to 50°C for 3 h and then cool it down to room temperature. The oil–lipid mixture can be stored at 4°C for up to 1 week. Sonicate suspension for 10 min before using it.
-
7.
Mix labelled and unlabelled proteins at a 9:1 molar ratio in I-buffer (seeNote 18).
-
8.
Add proteins dissolved in 10–25 μl of I-buffer to 500 μl of the lipid–oil mixture (step 6). Pipette gently back and forth for approximately 1 min with a 1 ml pipette until the emulsion looks homogeneously cloudy to disperse the mixture into droplets.
-
9.
Place 500 μl of the oil–lipid mixture (step 6) on top of an equal volume of O-buffer to assemble a lipid monolayer at the interface (seeNote 19).
-
10.
Gently pour 500 μl of the emulsion from step 7 on top of the tube containing the oil–lipid mixture and the O-buffer to produce three different phases within the same tube.
-
11.
Spin this tube on a tabletop centrifuge for first 10 min at 100×g and then for 10 min at 350×g to allow emulsion droplets of all sizes to pass through the lipid monolayer. Wash vesicles between runs with 100 mM glucose to discard oil contaminants that would appear as droplets under the microscope.
-
12.
Stain lipids by incorporating DiIC18 at a final concentration of 11 mM during the washing steps when needed. Alternatively Egg Liss Rhod PE (0.01% final concentration) can be added in step 2 to the initial lipid mixture.
-
13.
Store vesicles in O-buffer.
3.1.3 Vesicle Permeabilization
Permeabilization can be achieved using two different pore-forming systems that allow with similar efficiency the passage of small molecules across the membrane (seeNote 20):
-
Addition of the pore-forming protein α-hemolysin (7 μM) to the O-buffer after vesicle formation (step 11)
-
Incorporation of 10% of the acyl zwitterionic lipid 14:0 PC (DMPC) to the lipid mixture (step 2)
3.1.4 Vesicle Analysis
The behaviour of proteins inside GUVs can be tracked using an inverted confocal microscope equipped, in this case, with Ar and He–Ne ion laser lines to excite the specific Alexa fluorophores used.
-
1.
Place 500 μl samples of GUVs on 8-well visualization chambers.
-
2.
Collect images using a 63x oil immersion objective (N.A. = 1,4–0,6/Oil HCX PL APO, Lbd.Bl.).
Notes
-
14.
To avoid any interference of the dye during intra-vesicle protein assembly, the fluorescent labelling of E. coli FtsZ has to be performed under conditions that promote protein polymerization [27].
-
15.
-
1.
Equilibrate the column with three volumes of ddH2O and five volumes of the corresponding protein buffer.
-
2.
Pass the protein solution through the column and collect 250 μl samples.
-
3.
Measure the absorbance at 280 nm and at 488 for FtsZ or 647 nm for ZipA to calculate the labelling ratio for each protein.
-
4.
Wash the column according to manufacturer’s instructions. Store the column until further use at 4°C.
-
1.
-
16.
To avoid spoilage of the mineral oil due to hydration, the bottle must be opened in a chamber under a stream of nitrogen.
-
17.
The lipid mixture should approximate the composition of the lipids present in the cytoplasmic membrane of the microorganism originating the macromolecules to be studied.
-
18.
Ficoll (50 g/l) is added during vesicle formation together with the proteins when their lumen needs to be crowded. As reported [7] the FtsZ protein (12 μM) was diluted in I-buffer with different Ficoll concentrations (ranging from 0 to 150 g/l). When used, ZipA (from 2 to 10 μM) was diluted in I-buffer with 50 g/l of Ficoll.
-
19.
Changes in the I-buffer composition should be used to adapt it to the optimal biochemical activities of the proteins to be encapsulated. Note that O-buffer must maintain I-buffer osmolarity.
-
20.
Heptamerization of the α-hemolysin in the membrane forms 1.4 nm diameter pores allowing the entrance of molecules up to 3 kDa into the vesicle [28].
The use of DMPC with a melting temperature (Tm: 23°C) near the room temperature allows the appearance of heterogeneities in the vesicle membrane [29].
3.2 Maxicells
Maxicells [6] are nucleoid-free bacterial cells used as a whole cell in vitro system to probe protein–protein interactions without the need of prior protein purification. Nucleoid degradation in maxicells is achieved by the accumulation of DNA damage, introduced by ultraviolet light (UV) irradiation [30] or by the addition of small DNA binding peptides as, e.g. microcin B17 [31], in recA minus strains [32, 33] that are unable to repair the accumulated damage; these treatments activate DNA degradation. Plasmids contained in maxicells are able to escape to the UV-irradiation damage due to their smaller size and higher copy number [30] and allow the specific expression of plasmid-encoded genes (Fig. 4).


Procedure to obtain maxicells. The procedure involves the irradiation of an exponentially growing culture and incubation with D-cycloserine as illustrated in the left-hand side column. A cartoon showing the appearance of cells present at each stage of the procedure is shown at the right. Discontinuous lines indicate damaged nucleoids (middle row) or lysed cell walls (bottom row)
Materials
-
E. coli CSR603 strain (F−thr-1 leuB6 proA2 phr-1 recA1 argE3 thi-1 uvrA6 ara-14 lacY1 galK2 xyl-5 mtl-1 gyrA98 (nalA98) rpsL31 tsx-33 lambda−supE44) deficient in the DNA repair system for UV-inducible damage
-
LB broth [15]
-
UV germicidal lamp. TUV TL D 15 W SLV, Philips
-
d-cycloserine: (R)-4-Amino-3-isoxazolidone, 4-Amino-3-isoxazolidinone, C6880 Sigma
-
1% (w/v) agarose in water to coat microscope slides
-
DAPI: 4′, 6-diamidino-2-phenylindole, D-8417, Sigma. Stock solution in water at 1 mg/ml
Methods
3.2.1 Preparation of E. coli Maxicells
E. coli CSR603 maxicells were prepared with modifications [30].
-
1.
Inoculate E. coli CSR603 strain in LB broth and incubate at 37°C overnight in a shaker (seeNote 21).
-
2.
Next day, inoculate 20 ml of fresh LB 1:100 with the overnight grown cell culture and incubate at 37°C in a shaker until the cell culture reaches an OD600 of 0.4.
-
3.
Transfer the culture to 4°C for 15 min to slow down bacterial metabolism.
-
4.
Transfer 10 ml of the bacterial suspension to an open Petri dish and irradiate with UV while shaking in complete darkness (seeNote 22).
-
5.
Transfer 10 ml of UV-irradiated cell suspension to a flask wrapped with aluminium foil (to prevent activation of residual photo-repair) and incubate at 37°C for 3 h in a shaker in complete darkness.
-
6.
At 3 h after UV irradiation, add d-cycloserine at a final concentration of 200 μg/ml (seeNotes 23 and 24).
-
7.
Incubate cells at 37°C for additional 16 h in a shaker in complete darkness.
-
8.
To image the DNA content within the maxicells by phase contrast and fluorescence microscopy, mix to 1 μg/ml of DAPI in 5 μl of cells and spot the mixture on agarose-coated microscope slide.
-
9.
Observe cells on a microscope equipped with a 100× oil immersion lens, phase-contrast optics and band-pass filter of 360–370 nm to observe DAPI fluorescence.
Notes
-
21.
LB broth should be supplemented with the suitable antibiotic if the strain carries a plasmid with a selective antibiotic marker.
-
22.
To ensure the maximal brightness and a good reproducibility of the cell mortality, the UV-lamp should be switched on at least 15 min before use. UV irradiation of the cell suspension should be carried out while shaking to ensure a homogenous irradiation of all cells, and flasks should be kept in complete darkness to avoid the photo damage repair during or after UV irradiation. The UV dose at 254 nm is 105.98 J/m2. In our case this corresponds to a minimum of 30 s of irradiation at 30 cm distance. For simplicity and better reproducibility, we set the UV-irradiation time at 1 min. The UV dose should be measured with a UV light meter or determined empirically for every germicidal lamp. A mortality of 1 × 106 cells per ml corresponds to the required UV dose [33]. These parameters should be previously calibrated for each individual lamp and the lamp recalibrated after prolonged use as the irradiation energy diminishes after usage.
-
23.
d-cycloserine should be freshly prepared before each use. This antibiotic inhibits the condensation of the pentapeptide peptidoglycan precursors during peptidoglycan synthesis and therefore causes the lysis of any surviving cells that escaped UV irradiation and did not degrade their nucleoid [34].
-
24.
Measurement of the OD600 during the preparation of maxicells can be used as a quality marker to evaluate the efficacy of the procedure. The OD600 value of an irradiated culture doubles at 3 h after UV irradiation. This value should not change in the maxicell preparation; if OD600 increases significantly after addition of d-cycloserine, the culture should be discarded.
3.3 Bacterial Cells
Considering bacterial cells as living reaction chambers or biovessels, their physiology, protein–DNA or protein–protein interactions can be studied in vivo and in situ. Experimental methods used to analyse protein–protein or protein–DNA interactions include two-hybrid or one-hybrid systems, respectively, extensively described by Dammeyer and Schobert [35]. The Bimolecular Fluorescence Complementation (BiFC) assay is based on the association of two structural complementary nonfluorescent fragments of a fluorescent protein. When brought close enough by two interacting proteins, the two nonfluorescent moieties can reconstruct a fluorescent structure [36, 37]. The interactions of divisome proteins have also been studied using a polar recruitment system mediated by S. pneumonia Div IVA, a protein that gets incorporated into the E. coli poles [38]. Useful assays to define the assembly sequence of the divisome components take advantage of the FtsA* property to bypass the need for ZipA in the sequence [39, 40].
These studies rely on attaining a precise control of the expression levels of the proteins being analysed. This can be achieved through the control of plasmid copy number, based on the type of replicon carried by the vectors used, or the fine-tuning of inducible promoters. The gene of interest can thus be cloned downstream of a regulated promoter in a plasmid (the method most often used [7]), or less frequently the regulated promoter of choice is introduced in the genome upstream of the gene [41]. Many expression systems have been developed allowing to overproduce to different extents the protein of interest and to control the overproduction through the addition at defined concentrations of catabolites to the growth media. Examples of well-characterized and regulated promoters commonly used are described in Table 1.
Materials
-
E. coli MG1655 strain (F−λ−ilvG−rfb-50 rph-1) K-12 sequenced and reference strain [49] or MC1061 strain (F−Δ(ara-leu)7697 [araD139]B/r Δ(codB-lacI)3 galK16 galE15 λ−e14−mcrA0 relA1 rpsL150(strR) spoT1 mcrB1 hsdR2(r−m+)) [50, 51].
-
pASV003: pBAD24 [47] derivative plasmid carrying a His-tagged version of the zipA gene under the control of the arabinose promoter [7].
-
20% (w/v) glucose, filter-sterilized; keep at room temperature.
-
20% (w/v) arabinose, filter-sterilized; keep at room temperature. Do not use arabinose solutions older than 2 weeks for in vivo experiments.
-
Formaldehyde solution, min. 37%. Merck.
-
0.85% (w/v) NaCl. Keep at room temperature, but filter before each use to discard crystals.
-
Propidium iodide: 1.0 mg/ml solution in water. P3566, Life Technologies.
-
DiOC2(3): 3,3′-diethyloxacarbocyanine iodide. 3 mM solution in DMSO. D-14730, Life Technologies.
-
ISOTON® II Diluent. 8448011, Beckman Coulter, Inc.
-
Isopropanol.
-
1% (w/v) agarose in water to coat microscope slides.
-
Fixation solution: acetic acid: methanol (1:4).
-
Cover slip circles (15 mm diam.). CB00150RA1, Menzel-Gläser.
-
1× PBS (phosphate buffer saline), filter-sterilized.
-
GTE: 50 mM glucose, 10 mM EDTA, 20 mM Tris–HCl (pH 7.5).
-
75% ethanol.
-
Poly-l-lysine solution. P8920, Sigma.
-
Lysozyme 100 μg/ml in water. Keep aliquots frozen.
-
Anti-polyhistidine monoclonal antibody clone His-1 (peroxidase conjugate), A7058 Sigma.
-
Secondary antibodies (Life Technologies) Alexa 488 anti-mouse (A-11029) or Alexa 594 anti-rabbit (A-11037).
-
Blocking solution: 2% (w/v) BSA in 1× PBS. Keep aliquots frozen.
-
VECTASHIELD mounting medium, Vector Laboratories.
-
DAPI: 4′, 6-diamidino-2-phenylindole, D-8417, Sigma. Stock solution in water at 1 mg/ml.
-
Uncoloured nail polish (any brand).
Methods
3.3.1 Overproduction of ZipA for In Vivo Studies
A protocol to study the cellular effects of a controlled overproduction of ZipA is provided below:
-
1.
Transform a wild-type E. coli strain (MG1655, MC1061) with pASV003. Select transformants in LB broth containing 100 μg/μl ampicillin and supplemented with 0.2% glucose to repress the expression of the cloned zipA.
-
2.
Grow cells at 37°C in LB supplemented with 0.2% glucose and 100 μg μl−1 ampicillin overnight in a shaking water bath with aeration.
-
3.
Dilute the culture 1:100 into fresh prewarmed medium supplemented as above.
-
4.
Measure optical density at 600 nm (OD600) periodically (seeNote 25).
-
5.
After six mass doublings filter the cells through a pre-wetted 0.2 μm pore diameter filter to exclude glucose from the medium. While they are still in the filter, wash the cells twice with prewarmed LB without supplements (seeNote 26).
-
6.
Transfer the filter containing the cells to a prewarmed medium, containing 0.2% arabinose and no antibiotic. Grow for 150 min to overproduce His-ZipA. Withdraw samples at 30 min intervals. Maintained cell density within an OD600 range of 0.2–0.3 units by serial dilutions.
-
7.
To measure cell parameters (number, size, length) the samples are fixed with formaldehyde (25 μl per 1 ml of culture). Keep samples refrigerated until use.
-
1.
Count cell number and measure cell volume with a multichannel analyser Beckman Coulter Multisizer (Beckman), equipped with a 30 μm diameter orifice.
-
2.
Spread cells (5 μl) on an agarose-coated microscope slide. Cells are observed and photographed using a microscope with a 100× oil immersion lens with a charge-coupled device camera. Analyse cell length or number with the image analysis ObjectJ plug-in from the software Image J version 2.21 [52].
-
1.
-
8.
To analyse membrane integrity or potential with flow cytometry, cells are not fixed but used directly.
-
1.
Mix one volume of cells with the same volume of 0.85% NaCl. Incubate on ice for 5 min. Spin briefly in a tabletop centrifuge.
-
2.
Discard the supernatant and resuspend the cells in the same starting volume of 0.85% NaCl containing 1 μg/ml propidium iodide (to study membrane integrity) or 3 μM DiOC2(3) (to study membrane potential) (seeNote 27).
-
3.
Mix the pelleted cells (200 μl) with ISOTON (1 ml) and record the fluorescence signal of at least 105 cells for each time point (seeNote 28).
-
1.
3.3.2 Immunostaining of Bacterial Cells
Immunolocalize His-ZipA and additional division proteins with fluorescence microscopy using a combination of several antibodies [7, 53]:
-
1.
Incubate 1 volume of cells with 1 volume fixing solution.
-
2.
Leave mixture at room temperature for 10 min and then incubate on ice for 50 min.
-
3.
Centrifuge cells at 4°C at 4,200×g 5 min.
-
4.
Wash cells three times by adding 500 μl of 1× PBS to the pellet and repeat centrifugation.
-
5.
Resuspend pellet in GTE and store at 4°C (up to 2 months at 4°C).
-
6.
Place cover slip circles in a Petri dish on a shaker to wash them with ddH2O for 15 min.
-
7.
Discard the water, add 75% ethanol and wash for 15 min on a shaker.
-
8.
Remove ethanol and let the cover slip circles dry in between sheets of blotting paper.
-
9.
Add 70–80 μl of 10% (v/v) poly-l-lysine solution in water on each cover slip circle to cover the entire surface.
-
10.
Incubate for 15 min and gently remove poly-l-lysine solution with a pipette.
-
11.
Allow the fixed cells to warm to room temperature and perform all subsequent steps at room temperature, unless otherwise stated.
-
12.
Pipette 30 μl of fixed cells onto poly-l-Lysine-coated circular cover slips.
-
13.
Add 2.4 μl of 100 μg/μl lysozyme to the fixed cells and incubate at room temperature for 2 min (seeNote 29).
-
14.
Remove the liquid from the cover slip with a pipette and replace twice 60 μl of 1× PBS.
-
15.
Add 60 μl blocking solution and incubate 30 min without shaking.
-
16.
Dilute the primary antibody in blocking solution (1:3,000):
-
1.
Anti-polyhistidine monoclonal antibody His-1 is used to specifically detect the overproduced His-tagged protein [7].
-
2.
Endogenous ZipA and overproduced His-ZipA are simultaneously detected using the anti-ZipA MVC1 polyclonal antibody [54].
-
3.
FtsZ or FtsA are detected using the MCV2 or MVC3 polyclonal antibodies, respectively [53].
-
1.
-
17.
Add 30 μl of the diluted primary antibody to each lens and incubate at 4°C overnight.
-
18.
Wash the cover slips containing the stained cells ten times with 60 μl of 1× PBS.
-
19.
Dilute the secondary antibody in blocking solution (1:500).
-
20.
Add 30 μl of the diluted secondary antibody to each cover slip and incubate in the dark at room temperature for 90 min.
-
21.
Wash the cover slip circles 10 times with 60 μl of 1× PBS.
-
22.
Add 3 μl VECTASHIELD previously mixed with 0.5 μl of 25 μg/μl DAPI to each cover slip.
-
23.
Turn cover slip circles and place them on microscope slides. Seal the slips with nail polish to protect the samples from drying during observation. Slides can be stored in the dark at 4°C for 1 month.
-
24.
Cells are observed and photographed using a fluorescence microscope with a 100× immersion oil lens coupled to a charge-coupled device camera.
-
25.
Process images with Adobe Photoshop, ImageJ (http://imagej.nih.gov/ij/docs/) or a similar software.
Notes
-
25.
Maintain optical density between 0.2 and 0.3 OD600 units by serial dilutions with prewarmed medium to attain exponential balanced growth (seeNote 30). Under uninduced conditions, transformed cells at 37°C should grow as wild-type cells with 20–30 min duplication times.
-
26.
To calculate the volume of culture to be filtered, consider that once transferred to the arabinose-containing medium at time zero, the culture needs to be at an OD600 between 0.2 and 0.3 units. Use therefore a dilution factor of at least 1:3. As a rule, filter at least 10 ml of culture and place the filter into a flask containing 30 ml of fresh medium.
-
27.
Propidium iodide and DiOC2(3) are fluorescent molecules that are used to study bacterial membrane physiology. Propidium iodide is a red fluorescent dye that intercalates in dsDNA and is used to detect membrane integrity as it can only pass through fully permeable, i.e. unhealthy cytoplasmic membranes. The DiOC2(3) is a positively charged lipophilic carbocyanine dye that produces green fluorescence in all bacteria. When the membrane proton gradient is intact, the uptake of DiOC2(3) increases and the fluorescence shifts to red by autoassociation of the molecules of the dye inside the cells. Once the membrane potential is disrupted, the ratio of red to green fluorescence might drop to nearly zero. Before measuring fluorescence by flow cytometry, the concentration of dye and the staining time should be adjusted to suit each bacterial species analysed.
-
28.
To calibrate the flow cytometer parameters, a sample of healthy control cells should be fixed with an equal volume of isopropanol and then stained with each dye. Isopropanol will damage membrane integrity and potential, thus giving the maximum fluorescence intensity for cells stained with propidium iodides or the minimum red to green or ratio for DiOC2(3).
-
29.
Never prolong the treatment with lysozyme beyond 2 min. Lysozyme is used to produce pores in the bacterial wall large enough to allow the entry of the antibodies, but longer times may fully destroy the wall and lyse the cell rendering the sample useless for observation.
-
30.
From cells to population. The art of growing bacterial cultures.
A clear distinction between the cell cycle and the population cycle has to be made. The cell cycle comprises the events that take place in a single cell from birth to division. The population cycle is defined by the growth phases occurring since a bacterial population is transferred to a fresh medium until the cell density of the culture is too high to allow further growth. Among other events, the cell cycle includes birth, DNA replication, nucleoid segregation and septation leading to division. During its course one newly born cell grows until its mass is doubled and finally divides producing two daughters that are statistically equal between themselves and also equal to the initial newly born cell. In the population cycle the freshly inoculated culture shows a lag phase in which the old resting cells restart growth and once their mass has doubled, they accomplish the first division event. After this, growth allows the duplication of mass and cell numbers in an exponential fashion until the medium becomes so crowded that limitations in the aeration and accumulation of metabolic off-products prevent further growth. Division is then arrested, causing the population to enter into stationary phase. In a culture of E. coli cells growing in rich medium (as Nutrient Broth), stationary phase occurs at cell densities above 1–2 × 109 cells per ml (http://bionumbers.hms.harvard.edu//bionumber.aspx?id=100984&ver=1).
The molecular genetics description of the E. coli cell cycle has been derived from experimental evidence obtained using cells growing exponentially under balanced growth conditions. In these cells, their measurable parameters, including all their macromolecular contents, increase along time at an equal rate keeping the mean values of cell mass, length and volume constant. These balanced growth conditions are only maintained at cell densities lying within a narrow range. In practical terms values of approximately 5 × 107 cells per ml are the most adequate to obtain physiologically meaningful measurements. At much lower densities, the carbon dioxide tension has been described to be too low to sustain the maximum growth rate allowed by the carbon source [55]. On the contrary, at higher cell densities, over 108 cells per ml, even if mass increase exponentially the increase in protein or RNA contents is not balanced (MV unpublished observation). These observations highlight the convenience to monitor growth using cell counts as the preferred measurement. In particular particle increases provide a more meaningful measurement when determining the physiological state of the cells and the cell cycle parameters. Particles are measured using a particle counter as a Coulter Counter instrument fitted with a 30 μm measuring orifice.
More conveniently, optical density measurements are often used instead of particle counts when the cultures are used as a source for protein purification. It should be noted, nevertheless, that the correlation between optical density and particles is variable depending on the mass growth rate, cells growing faster are larger than those growing at slower rates. For example, E. coli B/r grown in M9 medium containing succinate as a carbon source (supporting 0.6 particle doublings per hour) has a mean mass of 1.56 × 10−9 OD540 units per cell. On the other hand, when grown in M9 containing glucose supplemented with casamino acids and tryptophan (2.2 particle doublings per hour), the mean mass is 3.44 × 10−9 OD540 units per cell (MV unpublished).
An exponentially growing culture is usually started with a 100-fold, or better 200-fold, dilution of an overnight grown culture. Successive fourfold dilutions with prewarmed medium are made when the cell density approaches 5 × 107 cells per ml, and they are repeated for at least six or preferably ten doublings to ensure that growth is both exponential and balanced. A shorter procedure can be implemented by preparing overnight cultures using serial dilutions and starting the experiment using the culture in which cell density is closer to 5 × 107 cells per ml. This procedure offers the advantage of avoiding the delay caused by the lag phase found when diluting saturated overnight cultures.
References
Natale P, Pazos M, Vicente M (2013) The Escherichia coli divisome: born to divide. Environ Microbiol 15:3169–3182
Rico AI, Krupka M, Vicente M (2013) In the beginning, Escherichia coli assembled the proto-ring: an initial phase of division. J Biol Chem 288:20830–20836
Osawa M, Erickson HP (2013) Liposome division by a simple bacterial division machinery. Proc Natl Acad Sci U S A 110:11000–11004
Szwedziak P, Wang Q, Bharat TA, Tsim M, Löwe J (2014) Architecture of the ring formed by the tubulin homologue FtsZ in bacterial cell division. Elife 3:e04601
Hernández-Rocamora VM, Reija B, García C et al (2012) Dynamic interaction of the Escherichia coli cell division ZipA and FtsZ proteins evidenced in nanodiscs. J Biol Chem 287:30097–30104
Pazos M, Natale P, Vicente M (2013) A specific role for the ZipA protein in cell division: stabilization of the FtsZ protein. J Biol Chem 288:3219–3226
Cabré EJ, Sánchez-Gorostiaga A, Carrara P et al (2013) Bacterial division proteins FtsZ and ZipA induce vesicle shrinkage and cell membrane invagination. J Biol Chem 288:26625–26634
Krupka M, Cabré EJ, Jiménez M, Rivas G, Rico AI, Vicente M (2014) Role of the FtsA C terminus as a switch for polymerization and membrane association. MBio 5:e02221
Bayburt TH, Sligar SG (2003) Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci 12:2476–2481
Denisov IG, Grinkova YV, Lazarides AA, Sligar SG (2004) Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc 126:3477–3487
Hernández-Rocamora VM, García-Montañes C, Rivas G, Llorca O (2012) Reconstitution of the Escherichia coli cell division ZipA-FtsZ complexes in nanodiscs as revealed by electron microscopy. J Struct Biol 180:531–538
Studier FW, Moffatt BA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130
Dubendorff JW, Studier FW (1991) Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J Mol Biol 219:45–59
Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW (1990) Use of T7 RNA-polymerase to direct expression of cloned genes. Methods Enzymol 185:60–89
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbour Laboratory Press, Cold Spring Harbour, NY
RayChaudhuri D (1999) ZipA is a MAP-Tau homolog and is essential for structural integrity of the cytokinetic FtsZ ring during bacterial cell division. EMBO J 18:2372–2383
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Schagger H, Cramer WA, von Jagow G (1994) Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem 217:220–230
Pichoff S, Lutkenhaus J (2005) Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol Microbiol 55:1722–1734
Epand RF, Savage PB, Epand RM (2007) Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim Biophys Acta 1768:2500–2509
Trombe MC, Laneelle MA, Laneelle G (1979) Lipid composition of aminopterin-resistant and sensitive strains of Streptococcus pneumoniae. Effect of aminopterin inhibition. Biochim Biophys Acta 574:290–300
Osawa M, Anderson DE, Erickson HP (2008) Reconstitution of contractile FtsZ rings in liposomes. Science 320:792–794
Tanaka T, Sano R, Yamashita Y, Yamazaki M (2004) Shape changes and vesicle fission of giant unilamellar vesicles of liquid-ordered phase membrane induced by lysophosphatidylcholine. Langmuir 20:9526–9534
Walde P, Cosentino K, Engel H, Stano P (2010) Giant vesicles: preparations and applications. Chembiochem 11:848–865
Chang H-Y, Sheng Y-J, Tsao H-K (2014) Structural and mechanical characteristics of polymersomes. Soft Matter 10:6373–6381
Jiménez M, Martos A, Cabré EJ, Raso A, Rivas G (2013) Giant vesicles: a powerful tool to reconstruct bacterial division assemblies in cell-like compartments. Environ Microbiol 15:3158–3168
González JM, Jiménez M, Vélez M et al (2003) Essential cell division protein FtsZ assembles into one monomer-thick ribbons under conditions resembling the crowded intracellular environment. J Biol Chem 278:37664–37671
Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE (1996) Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274:1859–1866
Cisse I, Okumus B, Joo C, Ha T (2007) Fueling protein DNA interactions inside porous nanocontainers. Proc Natl Acad Sci U S A 104:12646–12650
Sancar A, Hack AM, Rupp WD (1979) Simple method for identification of plasmid-coded proteins. J Bacteriol 137:692–693
Mayo O, Hernández-Chico C, Moreno F (1988) Microcin B17, a novel tool for preparation of maxicells: identification of polypeptides encoded by the IncFII minireplicon pMccB17. J Bacteriol 170:2414–2417
Skarstad K, Boye E (1993) Degradation of individual chromosomes in recA mutants of Escherichia coli. J Bacteriol 175:5505–5509
Kannan PR, Dharmalingam K (1987) Restriction alleviation and enhancement of mutagenesis of the bacteriophage T4 chromosome in recBCsbcA strains of Escherichia coli. Mol Gen Genet 209:413–418
Lovering AL, Safadi SS, Strynadka NC (2012) Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem 81:451–478
Dammeyer T, Schobert M (2010) Interactomics. In: Handbook of hydrocarbon and lipid microbiology. Springer, Berlin Heidelberg, pp 4407–4428
Pazos M, Natale P, Margolin W, Vicente M (2013) Interactions among the early Escherichia coli divisome proteins revealed by bimolecular fluorescence complementation. Environ Microbiol 15:3282–3291
Hu C-D, Kerppola TK (2003) Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol 21:539–545
Corbin B, Geissler B, Sadasivam M, Margolin W (2004) Z-ring-independent interaction between a subdomain of FtsA and late septation proteins as revealed by a polar recruitment assay. J Bacteriol 186:7736–7744
Goehring N, Gueiros-Filho F, Beckwith J (2005) Premature targeting of a cell division protein to midcell allows dissection of divisome assembly in Escherichia coli. Genes Dev 19:127–137
Goehring N, Gonzalez M, Beckwith J (2006) Premature targeting of cell division proteins to midcell reveals hierarchies of protein interactions involved in divisome assembly. Mol Microbiol 61:33–45
Garrido T, Sánchez M, Palacios P, Aldea M, Vicente M (1993) Transcription of ftsZ oscillates during the cell cycle of Escherichia coli. EMBO J 12:3957–3965
Baneyx F (1999) Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol 10:411–421
de Boer HA, Comstock LJ, Vasser M (1983) The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci U S A 80:21–25
Bass SH, Yansura DG (2000) Application of the E. coli trp promoter. Mol Biotechnol 16:253–260
Villaverde A, Benito A, Viaplana E, Cubarsi R (1993) Fine regulation of cI857-controlled gene expression in continuous culture of recombinant Escherichia coli by temperature. Appl Environ Microbiol 59:3485–3487
Winstanley C, Morgan JA, Pickup RW, Jones JG, Saunders JR (1989) Differential regulation of lambda pL and pR promoters by a cI repressor in a broad-host-range thermoregulated plasmid marker system. Appl Environ Microbiol 55:771–777
Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130
Lee N, Francklyn C, Hamilton EP (1987) Arabinose-induced binding of AraC protein to araI2 activates the araBAD operon promoter. Proc Natl Acad Sci 84:8814–8818
Blattner FR, Plunkett G 3rd, Bloch CA et al (1997) The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462
Casadaban MJ, Cohen SN (1980) Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138:179–207
Durfee T, Nelson R, Baldwin S et al (2008) The complete genome sequence of Escherichia coli DH10B: insights into the biology of a laboratory workhorse. J Bacteriol 190:2597–2606
Vischer NOE, Huls PG, Woldringh CL (1994) Object-image: an interactive image analysis program using structured point collection. Binary 6:160–166
Rico AI, García-Ovalle M, Palacios P, Casanova M, Vicente M (2010) Role of Escherichia coli FtsN protein in the assembly and stability of the cell division ring. Mol Microbiol 76:760–771
Rueda S, Vicente M, Mingorance J (2003) Concentration and assembly of the division ring proteins FtsZ, FtsA, and ZipA during the Escherichia coli cell cycle. J Bacteriol 185:3344–3351
Alexeeva S, Hellingwerf KJ, Teixeira de Mattos MJ (2002) Quantitative assessment of oxygen availability: perceived aerobiosis and its effect on flux distribution in the respiratory chain of Escherichia coli. J Bacteriol 184:1402–1406
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this protocol
Cite this protocol
Sánchez-Gorostiaga, A., Rico, A.I., Natale, P., Krupka, M., Vicente, M. (2015). Marrying Single Molecules to Single Cells: Protocols for the Study of the Bacterial Proto-ring Components Essential for Division. In: McGenity, T.J., Timmis, K.N., Nogales, B. (eds) Hydrocarbon and Lipid Microbiology Protocols. Springer Protocols Handbooks. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8623_2015_79
Download citation
DOI: https://doi.org/10.1007/8623_2015_79
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-49129-4
Online ISBN: 978-3-662-49131-7
eBook Packages: Springer Protocols