
Abstract
Recently, mRNA-based therapeutics have been greatly boosted since the development of novel technologies of both mRNA synthesis and delivery system. Promising results were showed in both preclinical and clinical studies in the field of cancer vaccine, tumor immunotherapy, infectious disease prevention and protein replacement therapy. Recent advancements in clinical trials also encouraged scientists to attempt new applications of mRNA therapy such as gene editing and cell programming. These studies bring mRNA therapeutics closer to real-world application. Herein, we provide an overview of recent advances in mRNA-based therapeutics.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

1 Introduction
Great progress has been made in the development of nucleic acid-based therapeutic agents. Many drugs have already been approved for clinical applications by the regulatory bodies in the USA and in Europe. Gene therapy drugs Glybera (Watanabe et al. 2015), Strimvelis (Aiuti et al. 2017), Luxturna (Ginn et al. 2018) and Zolgensma (Rao et al. 2018), antisense oligonucleotide (ASO) drug Spinraza (Dolgin 2017), and small interfering ribonucleic acid (siRNA) drug Onpattro (Hoy 2018) are just a few examples of approved nucleic acid drugs. Messenger RNA (mRNA)-based therapeutics have a great potential in human applications. The mRNA molecule can be constructed to not only express conventional proteins for protein replacement therapy (Kormann et al. 2011) but also produce therapeutic antibodies (Kose et al. 2019). In addition, it can be tailored to encode chimeric proteins that are used for cell engineering and antigen proteins or peptides in vaccine development. Despite the great potential, clinical application of mRNA-based therapeutics has been lagged comparing to other types of nucleic acid drugs. One of the bottleneck issues has been difficulties in delivering mRNA molecules into the target organs and/or cells inside the body. Comparing to the ASOs and siRNA oligos, mRNA molecules are usually much bigger in size and are very sensitive to enzymatic degradation. Thus, technology platforms designed for ASO and siRNA cannot always be applied directly for mRNA delivery. Another technical challenge is synthesis of a large quantity of mRNA molecules for large-scale drug production. In this chapter, we will introduce recent progress in platform development and in vitro synthesis of mRNA molecules. In addition, we will review potential clinical applications of mRNA therapy, with primary focus on cancer immunotherapy and protein replacement therapy.
2 Delivery Platforms for mRNA Therapeutics
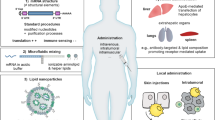
There has been a long history in the development of platforms for RNA transfection in vitro and RNA delivery in vivo. Verma and colleagues applied a synthetic cationic lipid, N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA), to prepare liposomes and applied it to transfect luciferase mRNA into NIH3T3 cells back in 1989 (Malone et al. 1989). Hoerr and colleagues used protamine-condensed mRNA and liposome-coated protamine/mRNA complex to enhance the stability of mRNA and uptake of mRNA into cells in vivo in 2000 (Hoerr et al. 2000). Multiple lipid-based platforms have been described since then. Among the variety of technology platforms, lipid nanoparticle (LNP) is one of the most widely used formulations for in vivo applications (Semple et al. 2010; Genc et al. 2011; Owen et al. 2018). Although initially designed for delivery of small molecule drugs, LNPs have been adapted to deliver siRNA oligos (Akinc et al. 2010), plasmid DNAs (Vijayanathan et al. 2002), and most recently mRNAs (Thess et al. 2015; Guimaraes et al. 2019). LNPs are usually less than 100 nm in diameter and are composed of structural phospholipids, cholesterol, and cationic lipids. DlinMC3DMA is one of the cationic lipids that have been successfully applied in mRNA delivery (Yanez Arteta et al. 2018). Many laboratories and companies have developed various forms of LNP-based platforms for mRNA delivery. Rosigkeit, S and colleagues developed a LPX delivery platform that was composed of DOTMA and dioleoyl phophotidylethanolamine (DOPE) and applied it to deliver mRNA and induce immune responses by targeting lung or spleen through the adjustable surface potential of nanoparticles (Rosigkeit et al. 2018). Kowalski, P. S. and colleagues applied liposome consisting of lipid and lipid-like polymer to deliver mRNA for protein replacement therapy (Kowalski et al. 2018). Patel, S. and colleagues applied LNP with novel lipid structures to increase protein expression efficiency (Patel et al. 2017; Hassett et al. 2019). Since most lipids can be dissolved in ethanol, the lipids can be mixed with nucleic acids in water in a microfluidic setting (Belliveau et al. 2012). This allows for controllable and standardized large-scale production. Indeed, precision nanosystems (www.precisionnanosysytems.com) have developed microfluidics-based instruments to prepare LNPs at different scales including one for GMP-grade drug manufacturing.
Other delivery platforms have also been optimized to achieve maximum delivery efficiency and protein expression. They include lipoplex (Koynova et al. 2007), cationic peptide (Yang et al. 2009), cationic polymer (Green et al. 2008), and micelle (Zheng et al. 2013). Persano, S. and colleagues applied a lipopolyplex (LPP) delivery platform which is composed of a polymer-condensed mRNA inner core surrounded by a lipid shell (Persano et al. 2017). Preliminary results suggest effective delivery of mRNA molecules to dendritic cells and potent protein expression.
Despite the tremendous advances in the field, there is still a big demand for new delivery platforms that are applicable for different routes of administration routes such as intravenous, intradermal, subcutaneous, intramuscular, and intratumor injections (Pardi et al. 2015). These platforms are expected to provide an optimal biodistribution pattern and to enable effective endosomal escape of mRNA molecules so as to improve protein expression. In addition, new platforms should also have an ideal biocompatibility.
3 In Vitro Synthesis of mRNA Molecules
Production of a large quantity of mRNA molecules is vital for the success of development of mRNA therapeutics. Although there are a few commercial entities that sell mRNA molecules, the list of mRNAs on their catalogs is not long. Thus, most laboratories and biopharmaceutic companies rely on their own facility or contract research organizations (CROs) to produce mRNAs, most through in vitro transcription (IVT). Although the technology has been improved dramatically in the last three decades, bacteriophage-derived RNA polymerases have served as key enzymes for in vitro mRNA synthesis throughout the time (Sarnow 1989). A single strand DNA that contains a 5′ untranslated region (UTR) including a T7 promoter, an open-reading frame of the gene-of-interest, and a 3′ polyA tail is used as the template in the IVT. An anti-reverse di-guanosine cap analog is included in the IVT to generate a 5′ cap (Warren et al. 2010). However, it was found that the synthetic mRNA molecules may cause undesirable immune responses (Linares-Fernández et al. 2020). High performance liquid chromatography (HPLC) has been applied to purify the IVT product so as to mitigate the side effects (Kariko et al. 2011). In addition, pseudouridine and other modified nucleosides have been incorporated into the synthesized mRNA molecules to suppress immune responses, to increase translational efficiency, and to enhance mRNA stability (Warren et al. 2010; Kariko et al. 2008).
4 Clinical Application of mRNA Therapeutics
4.1 mRNA in Cancer Immunotherapy
Cancer immunotherapy has gained much attention in recent years due to its huge success in patient care with therapeutic antibodies, T cell-based therapies, and cancer vaccines (Mellman et al. 2011). Although no approved mRNA drug is available for now, mRNA-based therapy has played a big role in the field of cancer immunotherapy (Foster et al. 2019; Diken et al. 2017). Owing to their versatile applicability, mRNA molecules have been used to generate tumor-associated antigens and neoantigens in cancer vaccine development (Tanyi et al. 2018), in therapeutic T cell manipulation (Beatty et al. 2014), in antibody production (Stadler et al. 2017), and in expression of therapeutic cytokines (Hewitt et al. 2019). Comparing to proteins and peptides, mRNA may offer unprecedented advantages, such as generating diversified tumor antigens from one single molecule as a result of alternative mRNA splicing or intron retention (Frankiw et al. 2019; Smart et al. 2018). In Sects. 4.2–4.4, we will describe mRNA-based cancer immunotherapeutics in detail. In the meantime, mRNA vaccines have also been successfully applied in the fight against infectious diseases, such as the Zika virus (Richner et al. 2017a, b). Details will be described in Sect. 4.5.
4.2 Cancer Vaccine
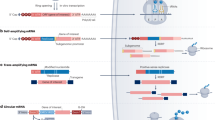
Applying mRNA encoding tumor antigens has unique advantages over the traditional protein or peptide-based vaccine strategies. mRNA molecules serve as self-adjuvants (Ziegler et al. 2017). There is no limitation on human leukocyte antigen (HLA)-type restrictions. Comparing to DNA-based therapies, mRNA vaccines do not integrate into the genome and therefore do not generate the risk of gene mutation. In addition, mRNA vaccines are adaptable to both dividing and non-dividing cells. Mechanistically, mRNAs encoding cancer antigens are delivered to the antigen-presenting cells (APCs) where they are translated in the cytoplasm. The newly synthesized protein is then processed into peptides, and the generated antigen peptides are presented by major histocompatibility complex (MHC) class I or MHC class II molecules which then activate T cells (Fiedler et al. 2016). Both MHC-TCR and B7-CD28 interactions are needed to generate antigen-specific T cells and to promote T cell proliferation. In order to develop a potent therapeutic cancer vaccine, it is essential to select the proper tumor antigen and adjuvant(s).
4.2.1 Tumor-Associated Antigen (TAA)-Based Cancer Vaccine
TAAs are antigens that are overexpressed in tumor cells. They are the primary choice for cancer vaccine development. In some of the pilot studies, scientists in CureVac demonstrated antigen generation from intradermally injected naked mRNA, cationic liposomal mRNA or protamine complex-encapsulated mRNA, and these mRNA vaccines induced both antigen-specific cytotoxic T lymphocytes (CTLs) and IgG antibodies (Hoerr et al. 2000). In addition, they found that T helper 2 (Th2)-type immune responses could be induced by intradermal vaccination of naked β-globin untranslated region (UTR)-stabilized mRNA, and that Th2-biased response could be shifted to a Th1-type response by co-delivering granulocyte–macrophage colony stimulating factor (GM-CSF) (Carralot et al. 2004). Early clinical trials demonstrated that intradermal injection of protamine-complexed mRNA or mRNA combined with GM-CSF was feasible, safe and effective, and treatment successfully induced antigen-specific T cell and antibody immune responses (Rittig et al. 2011; Weide et al. 2008). The strategy was further optimized by packaging two mRNA components in one formulation: a naked mRNA to encode the tumor antigen and a protamine-complexed mRNA to stimulate Toll-like receptor 7 signaling. The two-component mRNA vaccines with self-adjuvant property induce balanced adaptive immune responses and significantly better antitumor activity compared to single-component mRNA vaccine (Fotin-Mleczek et al. 2011). Apart from that, it is important to note that the strategy of using mRNA encoding a single antigen may not have enough immunogenicity to break central immune tolerance (Vansteenkiste et al. 2016). Therefore, strategies based on antigen cocktail were applied to maximize the immunogenicity of vaccines. Recent clinical results showed that CV9201, a RNA cancer vaccine encoding five non-small cell lung cancer antigens (NSCLC) (Sebastian et al. 2019), exhibited a well-tolerated safety profile and significantly improved immune responses against TAAs in patients who received a dosage of 1600 µg (NCT00923312). In another study, they combined BI1361849 (CV9202), a self-adjuvanted vaccine formulation consisting of protamine-complexed mRNA encoding six antigens (Papachristofilou et al. 2019), with local radiotherapy to reverse the immunosuppressive tumor microenvironment through induction of immunogenic tumor cell death and to enhance recruitment and stimulation of T cells in patients with stage IV NSCLC. Results from a phase Ib trial showed that treatment was well tolerated and induced antigen-specific response in 84% patients. In addition, 46.2% patients achieved stable disease after vaccination (NCT01915524). Current clinic trial is ongoing to evaluate the effect from CV9201 in combination with the checkpoint blockade antibody durvalumab (NCT03164772).
4.2.2 Neoantigens Antigen-Based Cancer Vaccine
Neoantigens are generated when mutations are introduced in cancer cells (Schumacher and Schreiber 2015). Compared with non-mutated self-antigens, neoantigens may contribute more significantly to tumor control, since T cells stimulated by neoantigens tend to avoid central immune tolerance (Gilboa 1999). In order to identify neoantigens, DNA samples from both tumor and normal tissues are sequenced, and results are used to predict binding affinity from mutant proteins to patient’s HLA alleles. Mutant antigen peptides are then ranked, and the information is applied to synthesize peptides or neoantigen-encoding mRNAs that will be used for vaccine preparation (Grabbe et al. 2016). DNA sequencing technology has been advanced so dramatically in recent years, and machine learning algorithms are being used to predict mutated peptides binding with HLA molecules (Linnemann et al. 2015; Abelin et al. 2017; Fritsch et al. 2014; Bulik-Sullivan et al. 2018). Applying mass spectrometry has also greatly advanced the field of tumor antigen identification (Creech et al. 2018). Consequently, personalized vaccines for cancer immunotherapy have been remarkably promoted. Preclinical studies from BioNTech AG revealed that one-third of mutated epitopes identified from B16F10 murine melanoma were immunogenic (Castle et al. 2012). Intravenous administration of neoantigen-encoding mRNA in lipoplex induced interferon-α secretion by plasmacytoid DCs and macrophages and promoted strong immune responses including maturation of DCs, and proliferation of antigen-specific effector and memory T cell. The immune responses subsequently mediated potent IFNα-dependent rejection of progressive tumors (Kranz et al. 2016). There have also been reports that showed an important role from CD4+ T cells in remodeling the tumor microenvironment after MHC class II-restricted epitopes administration (Sebastian et al. 2015). Proof-of-concept personalized cancer immunotherapy with mRNA cancer vaccine was first demonstrated in 2017 in patients with melanoma (Sahin et al. 2017). T cell immune responses to multiple neo-epitopes were induced in the patients who received treatment with mRNA cancer vaccines. Neoantigens-specific T cell responses were detected after vaccination in two patients with resected metastases, and one of them achieved a complete response after treatment with vaccine in combination with PD-1 inhibition therapy (Vallazza et al. 2015). Multiple clinical trials using mRNA-based cancer vaccines are currently being conducted in multiple cancer types, such as melanoma, colorectal cancer, glioblastoma, non-small-cell lung cancer, esophageal cancer, and bladder cancer (NCT03480152) (Li et al. 2014).
4.2.3 Dendritic Cell Vaccine
Since DCs are professional APCs, they can be loaded with tumor antigens to induce anti-cancer immune responses. mRNA in DCs can serve both as the source for antigen production and a potent adjuvant to stimulate TLR7/8 signaling (Laurent et al. 2012). First DC vaccine pulsed with mRNA was reported in 1996, and results showed that exposure of DCs to antigen-encoding mRNA or total mRNA extracted from tumor cells could induce significant T cell immune responses and inhibit growth of established tumors (Boczkowski et al. 1996). Many clinical trials using mRNA-based DC vaccines have been performed in cancer patients since then, and feasibility and safety of this treatment strategy have been well established (Gerold 2010; Daphné et al. 2015; Krug et al. 2014). In addition, efficacy from mRNA-transfected DCs can be further enhanced by combining cytokines and checkpoint blockade inhibitors in treatment (Mu et al. 2005; Kyte et al. 2006). It is important to point out that the cytokines and antibodies in combination treatment can also be produced by mRNA molecules inside the cells. DCs displayed a strong stimulatory potential after transfection with mRNAs encoding IL-12, IL-18 or other proinflammatory cytokines (Bontkes et al. 2007; Bontkes et al. 2008). Introduction of mRNA encoding the soluble extracellular part of PD-1 or PD-L1 resulted in elevated levels of CD80 (a DC maturation marker) and a group of cytokines and induced multifunctional T cells and cytokines secretion (Pen et al. 2014). Since DCs activation is mediated by several pathways, combination treatment of DCs to stimulate these pathways may achieve even better outcomes. TriMix-DC vaccine, a DC vaccine with mRNA molecules that encode TLR4, CD40L and CD70 (Pen et al. 2013), showed superior stimulatory capacity and suppressed the activity of regulator T cells (Treg), thus lifting CD8+ T cell activity.
4.3 Therapeutic mRNA Encoding Cytokines and Other Immune-Modulating Factors
mRNAs encoding therapeutic cytokines, checkpoint blockade antibodies, and immune agonists have the potential to convert an otherwise non-inflamed tumor (that lacks T cell infiltration, also known as immunologically “cold”) into an inflamed tumors (Galon and Bruni 2019). This group of reagents is under extensive investigation in both preclinical and clinical studies. Hewitt and colleagues designed a triple combination therapy for intratumor injection of mRNAs encoding IL-36 γ, IL-23, OX-40L to turn “cold” tumors into “hot” tumors. Animal studies showed that mRNA mainly expressed in tumor tissues and triple therapy successfully modified the tumor microenvironment. The reagents stimulated both the innate and adaptive immune system by stimulating production of cytokines including IL-6, IL-22, TNF-α, IFN-γ, and IL-1β, promoting proliferation and infiltration of immune cells (DCs, NK cells, CD4+/CD8+ T cells) in both tumor tissue and proximal lymph nodes without affecting Treg cells. As a result, treatment of MC-38 colorectal tumor-bearing mice with cytokine-encoding mRNA via intratumoral injection dramatically inhibited tumor growth (Hewitt et al. 2019). The result showed that intratumoral triplet mRNA therapy may avoid systemic toxicity and drive in vivo immune activation against tumor antigens and obtain a long-term therapeutic effect. Clinic trials have been initiated to evaluate potential toxicity from mRNA encoding IL-12, OX 40L monotherapy (NCT03323398) and mRNA encoding IL-36 γ, IL-23, OX-40L triple therapy (NCT03739931). Intratumoral delivery of mRNA has also been applied to produce other cytokines and chemokines including a fusion protein composed of interferon-β and the extracellular binding domain of the TGF-β receptor II (Van der Jeught et al. 2014). In addition, the strategy has been used to produce antibodies against cytokines IL-6 and TGF-β (Bialkowski et al. 2018). Furthermore, intratumoral delivery of mRNA has been applied to produce recombinant bacteriophage MS2 virus-like particles (VLPs) (Harper and Sardh 2014), mAbs targeting checkpoint molecules (PD-1, TIM-3, LAG-3), and necroptosis executioner mixed lineage kinase domain-like (MLKL) protein (Van Hoecke et al. 2018).
4.4 CAR-T Cells
Clinical trials showed that engineering T cells with chimeric antigen receptors (CARs) or T cell receptors (TCRs) have a significant therapeutic benefit on patients with relapsed or refractory hematological malignancies (Cummins and Gill 2018). The first CAR-T (Kymriah) product was approved in 2017 for treatment of patients with acute lymphoblastic leukemia. Transfection of T cells with mRNA to express CAR has the potential to temporally limit the targeting capacity of genetically modified T cells due to transient CAR expression and therefore to reduce the potential of sustained killing of normal cells that express the targeted TAAs such as mesothelin (Hung et al. 2018), EGFR (Caruso et al. 2016), CD19 (Caruso et al. 2016), CD20 (Panjwani et al. 2016), CD33 (Kenderian et al. 2015). Expression of CAR from introduced mRNA has been shown to transiently redirect T cell specificity to a desired TAA and mediate tumor regression in murine models of mesothelioma and leukemia (Yangbing et al. 2010; Barrett et al. 2013). A new approach of engineering T cells using IVT mRNA to transiently express CAR including both the CD3-ξ and 4-1BB for reducing off-target toxicity is under clinical evaluation. Clinical results indicated CAR-Ts generated from adoptively transferred mRNA to target mesothelin are feasible and safe (Beatty et al. 2014). The lifespan of CART-meso cells was short in the peripheral blood after intravenous injection, and these CAR-T cells effectively migrated to primary and metastatic tumor sites. Bai and colleagues reported an approach that used modified mRNA encoding telomerase reverse transcriptase to transfect CD19 CAR-T cells in order to improve their lifespan and proliferation (Bai et al. 2015). mRNA treatment instantly boosted telomerase activity in the new CD19 CAR-T cells, which promoted proliferation, delayed replicative senescence, and then, provided long-term antitumor activity in a mouse xenograft model of B-cell leukemia (Bai et al. 2015).
4.5 mRNA Vaccines in Infectious Diseases
mRNA vaccines have been applied for prevention of infectious diseases such as influenza viruses (Richner et al. 2017; Petsch et al. 2012), zika virus (Feldman et al. 2019), rabies virus(Schnee et al. 2016), and Dengue virus (Roth et al. 2019). In 2017, a research group published a study on mRNA vaccines to protect against Zika virus infection (Richner et al. 2017). In these vaccines, the mRNA molecules encoding Zika viral prM/M-E protein antigens contain a proprietary nucleoside modification to minimize indiscriminate activation of innate immunity, although detailed information on the modification was not provided. In addition, a donor methyl group S-adenosylmethionine was added to the methylated capped RNA to enhance translation efficiency. The mRNA molecules were then packaged into lipid nanoparticles before the vaccines were applied to treat mice. The researchers found that a modified mRNA vaccine could prevent Zika disease in animal models. In a follow-up study, the researchers demonstrated protection against Zika virus-induced congenital disease in mice (Richner et al. 2017). With the recent emergence of the corona virus, there is an international effort to develop both prophylactic vaccines and therapeutic vaccines (Steenhuysen and Kelland 2020). Moderna and CureVac announced their involvement in company news release, and Stemirna has applied the LPP platform in vaccine development. Exciting development is anticipated in the coming months. For a more detailed overview of this mRNA application, please refer to the review in the book chapter “Messenger RNA-based vaccines against infectious diseases” reviewed by Mohamad-Gabriel Alameh, Drew Weissman, and Norbert Pardi.
4.6 mRNA in Protein Replacement Therapy
Defective protein translation from DNA genetic information can give rise to various diseases, such as fabry disease, methylmalonic acidemia (MMA), acute intermittent porphyria (AIP), Hemophilia B, and cystic fibrosis (CF) (Kerem et al. 1989; Mehta et al. 2004; Lerner-Ellis et al. 2006; Koeberl et al. 1990). In addition, normal proteins may not be available in certain disease areas due to blood vessel damage, leading to further development of diseases such as heart failure and diabetes foot. Compared with conventional protein drugs for such indications, mRNA therapy provides an effective alternative, since a single mRNA molecule can be translated into a large quantity of protein molecules over the course of hours or days treatment time (Warner et al. 1963). Furthermore, the nascent protein will go through all the required post-translational modification procedures including phosphorylation, acetylation and glycosylation, a fully functional protein product is guaranteed (Helenius et al. 2013).
4.6.1 Local Injection-Based Protein Replacement Therapy
4.6.1.1 Myocardial Infarction
Despite advances in curative and preventive medicine, cardiovascular disease still remains one of the leading causes of morbidity and mortality worldwide (Savarese and Lund 2017). To date, a significant clinically feasible and/or verified targeting biologic strategy for treating congestive heart failure is still lacking. Vascular endothelial growth factor A (VEGF-A) has been previously reported to regulate new blood vessel formation, enhance endothelial proliferation from epicardial-derived progenitor cells, and with a pro-survival effect on vascular, endothelial, and cardiac cells (Ferrara et al. 2003; Lui et al. 2013). Same as mRNA molecules for cancer therapy, those used for protein replacement therapy need to overcome the same technical hurdles as immunogenicity, instability, and low expression efficiency, and nucleoside modifications and 5’ capping have been applied in mRNA production (Kariko et al. 2008; Mockey et al. 2006). The modified RNA synthesized by Zangi and colleagues showed high production efficiency and dose-dependent expression profile of VEGF-A in a murine myocardial infarction model (Zangi et al. 2013). Direct in vivo comparison of mRNA therapy and DNA therapy showed that the rapid, pulse-like expression profile of mRNA benefited growth of functional vessels, whereas the prolonged VEGF-A expression profile of DNA-induced toxicity due to redundant formation of leaky vessels (Zangi et al. 2013). An effect of expansion and directed differentiation of endogenous heart progenitor cells were shown from the intramyocardial injection of mRNA encoding VEGF-A in the murine myocardial infarction model. Furthermore, Leif and colleagues investigated potential therapeutic application of mRNA in ischemic heart disease in swine (Leif et al. 2018). The purified and optimized (optimization of nucleotide, UTR sequence, capping efficiency, and buffer solution) mRNA showed tissue specific and long-lasting expression of protein without triggering innate immune response. Moreover, the study showed that swine cardiac function was improved after a single intra-cardiac injection of VEGF mRNA one week after myocardial infarction through limiting cardiac infarct expansion and fibrosis, improving systolic function.
4.6.1.2 Diabetes Foot
Diabetic ischemic ulcer is an intractable and the most devastating diabetic complication. Similar to cardiovascular diseases, angiogenesis is a critical factor for diabetic wound healing (Varu et al. 2010; Harold and Marjana 2007). Application of VEGF-A and PDGF-β offers a promising approach for treatment of wound and ulceration (Shi et al. 2018). Sun and colleagues analyzed microvascular responses after mice were treated with mRNA therapy (mRNA encoding VEGF-A, AZD8601) or protein therapy (VEGF-A protein) (Sun et al. 2018). Intradermal injection of AZD8601 into mice resulted in dose-dependent vasodilation, upregulation of blood flow and formation of neo-vessel in injection site, results that were not observed in mice treated with the VEGF-A protein or phosphate buffer saline control. In addition, sequential dosing of AZD8601 in diabetic mice resulted in sustained vascularization and tissue oxygenation within wound area. A clinical trial aiming to assess safety and potential therapeutic effects of the mRNA encoding VEGF-A on treating type 2 diabetes mellitus (T2DM) was performed (NCT02935712) (Gan et al. 2019). It was revealed that intradermal VEGF-A mRNA injection was well tolerated and local functional VEGF-A protein was steadily expressed after administration, which led to transient skin blood flow enhancement in patients with T2DM.
4.6.2 Liver Diseases
Liver is the natural target for protein replacement therapy, since most nanoparticles tend to accumulate in this organ via intravenous injection (Fenton et al. 2016; Derosa et al. 2016). Fabry disease is one of these diseases that can be effectively treated with protein replacement therapy. This is a rare inherited disorder of glycosphigolipid metabolism caused by absence or markedly deficient activity in liver α-galactosidase A (α-Gal A), an enzyme that is normally produced by the liver. Patients suffer from progressive decline in renal and cardiac function and develop cardiomyopathy and end-stage renal disease (Hebert et al. 2013; Messalli et al. 2012). mRNA molecules encoding α-Gal A were packaged by two different research groups into LNPs (Zhu et al. 2019) or nanoparticles formulated with lipids and lipid-like materials (DeRosa et al. 2019). A single intravenous injection of α-Gal A mRNA caused not only dose-dependent protein expression and substrate reduction but also long-term (up to 6 weeks) substrate reductions in tissue and plasma in mice. In addition, the product proved to be safe after multiple administrations to non-human primates (Zhu et al. 2019). Hemophilia B is another liver disease that is caused by a deficiency of factor IX (FIX), a serine protease. FIX activation plays a major role in the signaling the coagulation cascade (Jiang et al. 2018). The disease is characterized as sustained, internal bleeding, and easy bruising. The prophylactic treatment is an intravenous application of purified FIX along with blood transfusion, but a heavy administrative burden with continuous treatment over a short period was needed to ensure adequate as-needed dosage given. Ramaswamy and colleagues applied lipid-enabled and unlocked nucleic acid modified RNA (LUNAR) to treat FIX-deficiency and demonstrated its feasibility in mouse model of FIX-deficiency (Ramaswamy et al. 2017). LUNAR is a unique LNP composed of four lipids including a proprietary lipid. Delivery of human FIX mRNA encapsulated in LUNAR resulted in rapid pulse of the FIX protein, and high protein concentration was maintained for 4–6 days. Therapeutic efficacy from mRNA therapy was comparable to recombinant human FIX protein therapy which is the current standard of care (Ramaswamy et al. 2017). An additional example of protein replaces therapy with mRNA for liver diseases include deficiency of methylmalonyl-CoA mutase (MUT), a vitamin B12-dependent mitochondrial enzyme that catalyzes the isomerization of methylmalonyl-CoA to succinyl-CoA (Chalmers and Lawson 1982; An et al. 2017).
4.6.3 Lung Diseases
Lung is another organ that offers easy access to drug nanoparticles. Non-invasive aerosol inhalation is a defined method for delivering drugs to the lung, and many technology platforms have been developed for this purpose. For an example, Patel and colleagues synthesized hyperbranched poly (beta amino esters) (hPBAEs) to prepare stable and concentrated mRNA polyplexes for inhalation (Patel et al. 2019). They achieved 24.6% transfection efficiency in lung epithelial cells after a single dose. Cystic fibrosis which is caused by mutations in the CFTR gene is the most widespread life-limiting autosomal-recessive disease in Caucasian (Cutting 2015). Robinson and colleagues applied LNP-packaged mRNA encoding cystic fibrosis transmembrane conductance regulator (CFTR) to treat cystic fibrosis (Robinson et al. 2018). Mutations in the CFTR gene cause abnormal flux of ions into and out of cells, leading to accumulation of thick airway mucosa and permanent tissue scarring and respiratory failure (Welsh 1990; Lyczak et al. 2002). Nasal application of LNP-CFTR mRNA in CFTR knockout mice recovered CFTR-mediated chloride secretion to conductive airway epithelia for at least 14 days and achieved comparable outcomes with currently approved drug ivacaftor (Robinson et al. 2018).
4.7 Genome Editing
Many genome editing tools have emerged in the past decade including zinc finger nucleases (ZFNs) (Miller et al. 2007), transcription-activator like effector nucleases (TALENs) (Wefers et al. 2013), and the clustered regularly interspersed palindromic repeats (CRISPR)/CRISPR-associated (Cas) enzyme system (Ran et al. 2013). However, off-target effects remain as a major concern. Since the nuclease activity is required for only a short period of time for the action, transient expression of mRNAs encoding ZFNs, LENs, and Cas9 provides a valid option to reduce off-target effects. Recently, Conway and colleagues showed that LNP-packaged mRNA encoding ZFNs to target the TTR and PCSK9 genes achieved over 90% knockout in gene expression in mice (Conway et al. 2019). Finn and colleagues applied LNP to load with Cas9 mRNA and sgRNA delivery system in order to edit the mouse transthyretin (Ttr) gene in the liver and achieved over 97% reduction of the protein level in serum, and the effect lasted for over 12 months after a single administration (Finn et al. 2018). Meanwhile, Miller and colleagues developed zwitterionic amino lipid (ZAL)-based delivery system to co-deliver Cas9 mRNA and sgRNAs and observed permanent DNA editing with 95% decrease in protein expression (Miller et al. 2017). These results suggest that mRNAs packaged in nanoparticles provide an excellent route for genome editing.
5 Conclusions
Application of novel therapies and medical technologies has revolutionized patient care. With the advance in mRNA synthesis and improvements in delivery platforms, mRNA-based therapies will play more and more important roles in the field. As with other types of therapeutic agents, the pharmaceutic industry has taken a very prominent role in advancing mRNA therapies. They have pioneered in mRNA modification in order to minimize innate immune responses. In the meantime, there is a constant need to understand the physical and biological barriers in mRNA delivery and to develop next-generation platforms so as to better overcome the barriers and achieve precision tissue- and cell-targeted delivery, beyond the liver and lung. In addition, approaches to enhance stability of mRNA molecules should continue to be explored. Furthermore, there is a need to standardize production of mRNA molecules to ensure high quality and efficacy of mRNA therapies. Finally, advances in machine learning and bioinformatic analysis will further facilitate sequence optimization of mRNA and medical application of therapeutic mRNA.
References
Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, Stevens J, Lane W, Zhang GL, Eisenhaure TM (2017) Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 46:315–326
Aiuti A, Roncarolo MG, Naldini L (2017) Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Mol Med 9:737–740
Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, Jayaraman M, Rajeev KG, Cantley WL, Dorkin JR et al (2010) Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther 18:1357–1364
An D, Schneller JL, Frassetto A, Liang S, Zhu X, Park JS, Theisen M, Hong SJ, Zhou J, Rajendran R (2017) Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep 21:3548–3558
Bai Y, Kan S, Zhou S, Wang Y, Xu J, Cooke JP, Wen J, Deng H (2015) Enhancement of the in vivo persistence and antitumor efficacy of CD19 chimeric antigen receptor T cells through the delivery of modified TERT mRNA. Cell Discovery 1:15040
Barrett DM, Xiaojun L, Shuguang J, June CH, Grupp SA, Yangbing Z (2013) Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther 24:717–727
Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Gabriela P, Anne C, Yangbing Z, Levine BL, Albelda SM (2014) Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2:112–120
Belliveau NM, Huft J, Lin PJ, Chen S, Leung AK, Leaver TJ, Wild AW, Lee JB, Taylor RJ, Tam YK et al (2012) Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA. Mol Ther Nucleic Acids 1:e37
Bialkowski L, Van der Jeught K, Bevers S, Tjok Joe P, Renmans D, Heirman C, Aerts JL, Thielemans K (2018) Immune checkpoint blockade combined with IL-6 and TGF-β inhibition improves the therapeutic outcome of m RNA-based immunotherapy. Int J Cancer 143:686–698
Boczkowski D, Nair SK, Snyder D, Gilboa E (1996) Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med 184:465–472
Bontkes HJ, Kramer D, Ruizendaal JJ, Kueter EWM, van Tendeloo VFI, Meijer CJLM, Hooijberg E (2007) Dendritic cells transfected with interleukin-12 and tumor-associated antigen messenger RNA induce high avidity cytotoxic T cells. Gene Therapy 14:366–375
Bontkes HJ, Duco K, Ruizendaal JJ, Meijer CJLM, Erik H (2008) Tumor associated antigen and interleukin-12 mRNA transfected dendritic cells enhance effector function of natural killer cells and antigen specific T-cells. Clin Immunol 127:375–384
Bulik-Sullivan B, Busby J, Palmer CD, Davis MJ, Murphy T, Clark A, Busby M, Duke F, Yang A, Young L et al (2018) Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat Biotechnol 37:55
Carralot J-P, Probst J, Hoerr I, Scheel B, Teufel R, Jung G, Rammensee H-G, Pascolo S (2004) Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell Mol Life Sci Cmls 61:2418–2424
Caruso HG, Torikai H, Zhang L, Maiti S, Dai J, Do KA, Singh H, Huls H, Lee DA, Champlin RE (2016) Redirecting T-cell specificity to EGFR using mRNA to self-limit expression of chimeric antigen receptor. J Immunother 39:205–217
Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C et al (2012) Exploiting the mutanome for tumor vaccination. Can Res 72:1081–1091
Chalmers RA, Lawson AM (1982) Disorders of propionate and methylmalonate metabolism
Conway A, Mendel M, Kim K, McGovern K, Boyko A, Zhang L, Miller JC, DeKelver RC, Paschon DE, Mui BL (2019) Non-viral delivery of zinc finger nuclease mRNA enables highly efficient in vivo genome editing of multiple therapeutic gene targets. Mol Ther 27:866–877
Creech AL, Ting YS, Goulding SP, Sauld JFK, Barthelme D, Rooney MS, Addona TA, Abelin JG (2018) The role of mass spectrometry and proteogenomics in the advancement of HLA epitope prediction. Proteomics 18:e1700259
Cummins KD, Gill S (2018) Anti-CD123 chimeric antigen receptor T-cells (CART): an evolving treatment strategy for hematological malignancies, and a potential ace-in-the-hole against antigen-negative relapse. Leuk Lymphoma 59:1539–1553
Cutting GR (2015) Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet 16:45–56
Daphné B, Carlo H, Aude B, Kris T, Karine B (2015) mRNA-based dendritic cell vaccines. Expert Rev Vaccines 14:161–176
Derosa F, Guild B, Karve S, Smith L, Love K, Dorkin JR, Kauffman KJ, Zhang J, Yahalom B, Anderson DG (2016) Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene Ther 23:699–707
DeRosa F, Smith L, Shen Y, Huang Y, Pan J, Xie H, Yahalom B, Heartlein MW (2019) Improved efficacy in a Fabry disease model using a systemic mRNA liver depot system as compared to enzyme replacement therapy. Mol Ther 27:878–889
Diken M, Kranz LM, Kreiter S, Sahin U (2017) mRNA: a versatile molecule for cancer vaccines. Curr Issues Mol Biol 22:113–128
Dolgin E (2017) Spinal muscular atrophy approval boosts antisense drugs. Nat Biotechnol 35:99–100
Feldman RA, Fuhr R, Smolenov I, Mick Ribeiro A, Panther L, Watson M, Senn JJ, Smith M, Almarsson O, Pujar HS et al (2019) mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37:3326–3334
Fenton OS, Kauffman KJ, Mcclellan RL, Appel EA, Dorkin JR, Tibbitt MW, Heartlein MW, Derosa F, Langer R, Anderson DG (2016) Bioinspired alkenyl amino alcohol ionizable lipid materials for highly potent in vivo mRNA delivery. Adv Mater 28:2939–2943
Ferrara N, Gerber HP, Lecouter J (2003) The biology of VEGF and its receptors. Nat Med 9:669–676
Fiedler K, Lazzaro S, Lutz J, Rauch S, Heidenreich R (2016) mRNA cancer vaccines. Recent Results Cancer Res 209:61–85
Finn JD, Smith AR, Patel MC, Shaw L, Youniss MR, Heteren JV, Dirstine T, Ciullo C, Lescarbeau R, Seitzer J (2018) A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent invivo genome editing. Cell Rep 22:2227
Foster JB, Barrett DM, Kariko K (2019) The emerging role of in vitro-transcribed mRNA in adoptive T cell immunotherapy. Mol Ther 27:747–756
Fotin-Mleczek M, Duchardt KM, Lorenz C, Pfeiffer R, Ojkić-Zrna S, Probst J, Kallen KJ (2011) Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J Immunother 34:1–15
Frankiw L, Baltimore D, Li G (2019) Alternative mRNA splicing in cancer immunotherapy. Nat Rev Immunol 19:675–687
Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ (2014) HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res 2:522–529
Galon J, Bruni D (2019) Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 18:197–218
Gan L-M, Lagerström-Fermér M, Carlsson LG, Arfvidsson C, Egnell A-C, Rudvik A, Kjaer M, Collén A, Thompson JD, Joyal J et al (2019) Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat Commun 10:871–871
Genc B, Novobrantseva TI, Nicole R, Tam YYC, Hafez IM, Wong MK, Tsukasa S, Ruda VM, June Q, Boris K (2011) Influence of cationic lipid composition on gene silencing properties of lipid nanoparticle formulations of siRNA in antigen-presenting cells. Mol Ther J Am Soc Gene Ther 19:2186–2200
Gerold S (2010) Dendritic cells in cancer immunotherapy. Annu Rev Immunol 40:2123–2130
Gilboa E (1999) The makings of a tumor rejection antigen. Immunity 11:263
Ginn SL, Amaya AK, Alexander IE, Edelstein M, Abedi MR (2018) Gene therapy clinical trials worldwide to 2017: an update. J Gene Med 20:e3015
Grabbe S, Haas H, Diken M, Kranz LM, Langguth P, Sahin U (2016) Translating nanoparticulate-personalized cancer vaccines into clinical applications: case study with RNA-lipoplexes for the treatment of melanoma. Nanomedicine 11:2723–2734
Green JJ, Robert L, Anderson DG (2008) A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc Chem Res 41:749–759
Guimaraes PPG, Zhang R, Spektor R, Tan M, Chung A, Billingsley MM, El-Mayta R, Riley RS, Wang L, Wilson JM et al (2019) Ionizable lipid nanoparticles encapsulating barcoded mRNA for accelerated in vivo delivery screening. J Control Release 316:404–417
Harold B, Marjana TC (2007) Cellular and molecular basis of wound healing in diabetes. J Clin Invest 117:1219–1222
Harper P, Sardh E (2014) Management of acute intermittent porphyria. Expert Opin Orphan Drugs 2:349–368
Hassett KJ, Benenato KE, Jacquinet E, Lee A, Woods A, Yuzhakov O, Himansu S, Deterling J, Geilich BM, Ketova T et al (2019) Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Molecular therapy–nucleic acids 15:1–11
Hebert A, Lacbawan L, Taber T, Goker-Alpan O (2013) Evaluation of long-term enzyme replacement therapy for children with Fabry disease. Mol Genet Metab 108:S47–S47
Helenius A, Tatu U, Marquardt T, Braakman I (2013) Protein folding in the endoplasmic reticulum. Cold Spring Harb Perspect Biol 5:a013201
Hewitt SL, Bai A, Bailey D, Ichikawa K, Zielinski J, Karp R, Apte A, Arnold K, Zacharek SJ, Iliou MS et al (2019) Durable anticancer immunity from intratumoral administration of IL-23, IL-36gamma, and OX40L mRNAs. Sci Transl Med 11:eaat9143
Hoerr I, Obst R, Rammensee HG, Jung G (2000) In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol 30:1–7
Hoy SM (2018) Patisiran: first global approval. Drugs 78:1625–1631
Hung C-F, Xu X, Li L, Ma Y, Jin Q, Viley A, Allen C, Natarajan P, Shivakumar R, Peshwa MV, Emens LA(2018) Development of anti-human mesothelin-targeted chimeric antigen receptor messenger RNA-transfected peripheral blood lymphocytes for ovarian cancer therapy. Human Gene Ther 29:614
Jiang L, Berraondo P, Jericó D, Guey LT, Sampedro A, Frassetto A, Benenato KE, Burke K, Santamaría E, Alegre M et al (2018) Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat Med 24:1899–1909
Kariko K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, Weissman D (2008) Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 16:1833–1840
Kariko K, Muramatsu H, Ludwig J, Weissman D (2011) Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res 39:e142
Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, Scholler J, Song D, Porter DL, Carroll M (2015) CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 29:1637
Kerem B, Rommens J, Buchanan J, Markiewicz D, Cox T, Chakravarti A, Buchwald M, Tsui L (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245:1073–1080
Koeberl DD, Bottema CD, Ketterling RP, Bridge PJ, Lillicrap DP, Sommer SS (1990) Mutations causing hemophilia B: direct estimate of the underlying rates of spontaneous germ-line transitions, transversions, and deletions in a human gene. Am J Hum Genet 47:202–217
Kormann MSD, Günther H, Aneja MK, Gabriela N, Flemmer AW, Susanne HJ, Marceline H, Mays LE, Marta I, Andrea S (2011) Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol 29:154–157
Kose N, Fox JM, Sapparapu G, Bombardi R, Tennekoon RN, de Silva AD, Elbashir SM, Theisen MA, Humphris-Narayanan E, Ciaramella G et al (2019) A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against Chikungunya infection. Sci Immunol 4, eaaw6647
Kowalski PS, Capasso PU, Huang Y, Rudra A, Langer R, Anderson DG (2018) Ionizable amino-polyesters synthesized via ring opening polymerization of tertiary amino-alcohols for tissue selective mRNA delivery. Adv Mater 1801151
Koynova R, Tarahovsky YS, Wang L, Macdonald RC (2007) Lipoplex formulation of superior efficacy exhibits high surface activity and fusogenicity, and readily releases DNA. Biochim Et Biophys Acta 1768:375–386
Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H (2016) Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534:396–401
Krug C, Wiesinger M, Abken H, Schuler-Thurner B, Schuler G, Dörrie J, Schaft N (2014) A GMP-compliant protocol to expand and transfect cancer patient T cells with mRNA encoding a tumor-specific chimeric antigen receptor. Cancer Immunol Immunother 63:999–1008
Kyte JA, Mu L, Aamdal S, Kvalheim G, Dueland S, Hauser M, Gullestad HP, Ryder T, Lislerud K, Hammerstad H (2006) Phase I/II trial of melanoma therapy with dendritic cells transfected with autologous tumor-mRNA. Cancer Gene Ther 13:905–918
Laurent B, Monica B, Weiquan L, Wels WS, Teresa M, Van Etten RA, Hans K (2012) Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leukemia Lymphoma 53:958–965
Leif C, Clarke JC, Christopher Y, Francine G, Tamsin A, Martin B, Ann-Charlotte E, Li-Ming G, Karin J, Edvin J (2018) Biocompatible, purified VEGF-A mRNA improves cardiac function after intracardiac injection 1 week post-myocardial infarction in swine. Mol Ther Methods Clin Develop 9:330–346
Lerner-Ellis JP, Tirone JC, Pawelek PD, Doré C, Atkinson JL, Watkins D, Morel CF, Fujiwara TM, Moras E, Hosack AR (2006) Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat Genet 38:93
Li J, Sun Y, Jia T, Zhang R, Zhang K, Wang L (2014) Messenger RNA vaccine based on recombinant MS2 virus-like particles against prostate cancer. Int J Cancer 134:1683–1694
Linares-Fernández S, Lacroix C, Exposito JY, Verrier B (2020) Tailoring mRNA vaccine to balance innate/adaptive immune response. Trends Mol Med 26:311–323
Linnemann C, Buuren MMV, Bies L, Verdegaal EME, Schotte R, Calis JJA, Behjati S, Velds A, Hilkmann H, Atmioui DE (2015) High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 21:81–85
Lui KO, Zangi L, Silva EA, Lei B, Sahara M, Li RA, Mooney DJ, Chien KR (2013) Driving vascular endothelial cell fate of human multipotent Isl1+ heart progenitors with VEGF modified mRNA. Cell Res 23:1172–1186
Lyczak JB, Cannon CL, Pier GB (2002) Lung infections associated with cystic fibrosis. Clin Microbiol Rev 15:194
Malone RW, Felgner PL, Verma IM (1989) Cationic liposome-mediated RNA transfection. Proc Natl Acad Sci U S A 86:6077–6081
Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, Linhart A, Sunder-Plassmann G, Ries M, Beck M (2004) Fabry disease defined: baseline clinical manifestations of 366 patients in the fabry outcome survey. Eur J Clin Invest 34:236–242
Mellman I, Coukos G, Dranoff G (2011) Cancer immunotherapy comes of age. Nature 480:480–489
Messalli G, Imbriaco M, Russo R, Iodice D, Spinelli L, Dellegrottaglie S, Cademartiri F, Salvatore M, Pisani A (2012) Role of cardiac MRI in evaluating patients with Anderson-Fabry disease: assessing cardiac effects of long-term enzyme replacement therapy. Radiol Med (Torino) 117:19–28
Miller JC, Holmes MC, Jianbin W, Guschin DY, Ya-Li L, Igor R, Beausejour CM, Waite AJ, Wang NS, Kim KA (2007) An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25:778–785
Miller JB, Zhang S, Kos P, Xiong H, Zhou K, Perelman SS, Zhu H, Siegwart DJ (2017) Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew Chem Int Ed 129:1059
Mockey M, Gonçalves C, Dupuy FP, Lemoine FM, Pichon C, Midoux P (2006) mRNA transfection of dendritic cells: synergistic effect of ARCA mRNA capping with Poly(A) chains in cis and in trans for a high protein expression level. Biochem Biophys Res Commun 340:1062–1068
Mu LJ, Kyte JA, Kvalheim G, Aamdal S, Dueland S, Hauser M, Hammerstad H, Waehre H, Raabe N, Gaudernack G (2005) Immunotherapy with allotumour mRNA-transfected dendritic cells in androgen-resistant prostate cancer patients. Br J Cancer 93:749–756
Owen F, Kevin K, Rebecca MC, James K, Manhao Z, Jason A, Luke R, Michael H, Frank DR, Anderson DG (2018) Customizable lipid nanoparticle materials for the delivery of siRNAs and mRNAs. Angew Chem 57:13582–13586
Panjwani MK, Smith JB, Schutsky K, Gnanandarajah J, O’Connor CM, Powell Dj Jr, Mason NJ (2016) Feasibility and safety of RNA-transfected CD20-specific chimeric antigen receptor T cells in dogs with spontaneous B cell lymphoma. Mol Ther 24:1602–1614
Papachristofilou A, Hipp MM, Klinkhardt U, Früh M, Sebastian M, Weiss C, Pless M, Cathomas R, Hilbe W, Pall G (2019) Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J Immunother Cancer
Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Ying KT, Madden TD, Hope MJ, Weissman D (2015) Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Controlled Release 217:345–351
Patel S, Ashwanikumar N, Robinson E, Duross A, Sun C, Murphy-Benenato KE, Mihai C, Almarsson Ö, Sahay G (2017) Boosting intracellular delivery of lipid nanoparticle-encapsulated mRNA. Nano Lett 17:5711
Patel AK, Kaczmarek JC, Bose S, Kauffman KJ, Mir F, Heartlein MW, DeRosa F, Langer R, Anderson DG (2019) Inhaled nanoformulated mRNA polyplexes for protein production in lung epithelium. Adv Mater 31:1805116
Pen JJ, Brenda DK, Maenhout SK, An MT, Nuffel Van, Carlo H, Jurgen C, David E, Aude B, Kris T, Karine B (2013) Modulation of regulatory T cell function by monocyte-derived dendritic cells matured through electroporation with mRNA encoding CD40 ligand, constitutively active TLR4, and CD70. J Immunol 191:1976–1983
Pen JJ, Keersmaecker BD, Heirman C, Corthals J, Liechtenstein T, Escors D, Thielemans K, Breckpot K (2014) Interference with PD-L1/PD-1 co-stimulation during antigen presentation enhances the multifunctionality of antigen-specific T cells. Gene Ther 21:262
Persano S, Guevara ML, Li Z, Mai J, Ferrari M, Pompa PP, Shen H (2017) Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 125:81–89
Petsch B, Schnee M, Vogel AB, Lange E, Hoffmann B, Voss D, Schlake T, Thess A, Kallen K, Stitz L (2012) Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol 30:1210–1216
Ramaswamy S, Tonnu N, Tachikawa K, Limphong P, Vega JB, Karmali PP, Chivukula P, Verma IM (2017) Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc Natl Acad Sci U S A 114:E1941
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308
Rao VK, Kapp D, Schroth M (2018) Gene therapy for spinal muscular atrophy: an emerging treatment option for a devastating disease. J Managed Care Specialty Pharm 24:S3–S16
Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC et al (2017a) Modified mRNA vaccines protect against Zika virus infection. Cell 168(1114–1125):e1110
Richner JM, Jagger BW, Shan C, Fontes CR, Dowd KA, Cao B, Himansu S, Caine EA, Nunes BTD, Medeiros DBA et al (2017b) Vaccine mediated protection against Zika virus-induced congenital disease. Cell 170(273–283):e212
Rittig SM, Haentschel M, Weimer KJ, Heine A, Muller MR, Brugger W, Horger MS, Maksimovic O, Stenzl A, Hoerr I (2011) Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol Ther 19:990–999
Robinson E, Macdonald KD, Kai S, Mckinney M, Patel S, Sun C, Sahay G (2018) Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol Ther 26:2034–2046
Rosigkeit S, Meng M, Grunwitz C, Gomes P, Bockamp E (2018) Monitoring translation activity of mRNA-loaded nanoparticles in mice. Mol Pharm 15:3909–3919
Roth C, Cantaert T, Colas C, Prot M, Casademont I, Levillayer L, Thalmensi J, Langlade-Demoyen P, Gerke C, Bahl K et al (2019) A modified mRNA vaccine targeting immunodominant NS epitopes protects against dengue virus infection in HLA class I transgenic mice. Front Immunol 10:1424
Sahin U, Derhovanessian E, Miller M, Kloke B, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B (2017) Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547:222–226
Sarnow P (1989) Role of 3′-end sequences in infectivity of poliovirus transcripts made in vitro. J Virol 63:467–470
Savarese G, Lund LH (2017) Global public health burden of heart failure. Cardiac Fail Rev 3:7
Schnee M, Vogel AB, Voss D, Petsch B, Baumhof P, Kramps T, Stitz L (2016) An mRNA vaccine encoding rabies virus glycoprotein induces protection against lethal infection in mice and correlates of protection in adult and newborn pigs. PLOS Negl Trop Dis 10
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348:69–74
Sebastian K, Mathias V, Niels VDR, Mustafa D, Martin LW, Jan D, Sebastian B, Barbara SR, Fulvia V, Castle JC (2015) Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520:692–696
Sebastian M, Schröder A, Scheel B, Hong HS, Muth A, Boehmer LV, Zippelius A, Mayer F, Reck M, Atanackovic D (2019) A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol Immunother
Semple SC, Akin A, Jianxin C, Sandhu AP, Mui BL, Cho CK, Sah DWY, Derrick S, Crosley EJ, Ed Y (2010) Rational design of cationic lipids for siRNA delivery. Nat Biotechnol 28:172–176
Shi R, Lian W, Han S, Cao C, Jin Y, Yuan Y, Zhao H, Li M (2018) Nanosphere-mediated co-delivery of VEGF-A and PDGF-B genes for accelerating diabetic foot ulcers healing in rats. Gene Ther 25:1
Smart AC, Margolis CA, Pimentel H, He MX, Miao D, Adeegbe D, Fugmann T, Wong KK, Van Allen EM (2018) Intron retention is a source of neoepitopes in cancer. Nat Biotechnol 36:1056–1058
Stadler CR, Bähr-Mahmud H, Celik L, Hebich B, Roth AS, Roth RP, Karikó K, Türeci Ö, Sahin U (2017) Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med 23:815–817
Steenhuysen J, Kelland K (2020) With Wuhan virus genetic code in hand, scientists begin work on a vaccine. Reuters
Sun N, Ning B, Hansson KM, Bruce AC, Seaman SA, Zhang C, Rikard M, DeRosa CA, Fraser CL, Wågberg M et al (2018) Modified VEGF-A mRNA induces sustained multifaceted microvascular response and accelerates diabetic wound healing. Sci Rep 8:17509–17509
Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R, Baumgartner P, Stevenson BJ, Iseli C, Dangaj D (2018) Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med 10:eaao5931
Thess A, Grund S, Mui BL, Hope MJ, Baumhof P, Fotin-Mleczek M, Schlake T (2015) Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol Ther 23:S55–S55
Vallazza B, Petri S, Poleganov MA, Eberle F, Kuhn AN, Sahin U (2015) Recombinant messenger RNA technology and its application in cancer immunotherapy, transcript replacement therapies, pluripotent stem cell induction, and beyond. Wiley Interdisc Rev RNA 6:471
Van der Jeught K, Joe PT, Bialkowski L, Heirman C, Daszkiewicz L, Liechtenstein T, Escors D, Thielemans K, Breckpot K (2014) Intratumoral administration of mRNA encoding a fusokine consisting of IFN-beta and the ectodomain of the TGF-beta receptor II potentiates antitumor immunity. Oncotarget 5:10100–10113
Van Hoecke L, Van Lint S, Roose K, Van Parys A, Vandenabeele P, Grooten J, Tavernier J, De Koker S, Saelens X (2018) Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat Commun 9:3417–3417
Vansteenkiste JF, Cho BC, Vanakesa T, Pas TD, Zielinski M, Kim MS, Jassem J, Yoshimura M, Dahabreh J, Nakayama H (2016) Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 17:822–835
Varu VN, Hogg ME, Kibbe MR (2010) Critical limb ischemia. Curr Treat Options Cardiovasc Med 51:230–241
Vijayanathan V, Thomas T, Thomas TJ (2002) DNA nanoparticles and development of DNA delivery vehicles for gene therapy. Biochemistry 41:14085–14094
Warner JR, Knopf PM, Rich A (1963) A multiple ribosomal structure in protein synthesis. Proc Natl Acad Sci USA 49:122–129
Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A et al (2010) Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7:618–630
Watanabe N, Yano K, Tsuyuki K, Okano T, Yamato M (2015) Re-examination of regulatory opinions in Europe: possible contribution for the approval of the first gene therapy product Glybera. Mol Ther Methods Clin Dev 2:14066
Wefers B, Ortiz O, Wurst W, Kühn R (2013) Generation of targeted mouse mutants by embryo microinjection of TALENs. Methods 8:2355–2379
Weide B, Carralot JP, Reese A, Scheel B, Eigentler TK, Hoerr I, Rammensee HG, Garbe C, Pascolo S (2008) Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J Immunother 1:180–188
Welsh MJ (1990) Abnormal regulation of ion channels in cystic fibrosis epithelia. Faseb J 4:2718–2725
Yanez Arteta M, Kjellman T, Bartesaghi S, Wallin S, Wu X, Kvist AJ, Dabkowska A, Szekely N, Radulescu A, Bergenholtz J et al (2018) Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc Natl Acad Sci U S A 115:E3351–E3360
Yang S, Coles DJ, Esposito A, Mitchell DJ, Toth I, Minchin RF (2009) Cellular uptake of self-assembled cationic peptide-DNA complexes: multifunctional role of the enhancer chloroquine. J Controlled Release 135:159–165
Yangbing Z, Edmund M, Carmine C, Paulos CM, Xiaojun L, Brennan AL, Anne C, Carroll RG, John S, Levine BL (2010) Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Can Res 70:9053–9061
Zangi L, Lui KO, von Gise A, Ma Q, Ebina W, Ptaszek LM, Spater D, Xu H, Tabebordbar M, Gorbatov R et al (2013) Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol 31:898–907
Zheng C, Zheng M, Gong P, Deng J, Yi H, Zhang P, Zhang Y, Liu P, Ma Y, Cai L (2013) Polypeptide cationic micelles mediated co-delivery of docetaxel and siRNA for synergistic tumor therapy. Biomaterials 34:3431–3438
Zhu X, Yin L, Theisen M, Zhuo J, Siddiqui S, Levy B, Presnyak V, Frassetto A, Milton J, Salerno T et al (2019) Systemic mRNA therapy for the treatment of Fabry disease: preclinical studies in wild-type mice, Fabry mouse model, and wild-type non-human primates. Am J Hum Genet 104:625–637
Ziegler A, Soldner C, Lienenklaus S, Spanier J, Trittel S, Riese P, Kramps T, Weiss S, Heidenreich R, Jasny E (2017) A new RNA-based adjuvant enhances virus-specific vaccine responses by locally triggering TLR- and RLH-dependent effects. J Immunol 198:1595
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Huang, L. et al. (2020). Advances in Development of mRNA-Based Therapeutics. In: Yu, D., Petsch, B. (eds) mRNA Vaccines. Current Topics in Microbiology and Immunology, vol 440. Springer, Cham. https://doi.org/10.1007/82_2020_222
Download citation
DOI: https://doi.org/10.1007/82_2020_222
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-18069-9
Online ISBN: 978-3-031-18070-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)