
Abstract
Over the last 60 years, poly-ADP-ribose polymerases (PARPs, 17 family members in humans) have emerged as important regulators of physiology and disease. Small-molecule inhibitors have been essential tools for unraveling PARP function, and recently the first PARP inhibitors have been approved for the treatment of various human cancers. However, inhibitors have only been developed for a few PARPs and in vitro profiling has revealed that many of these exhibit polypharmacology across the PARP family. In this review, we discuss the history, development, and current state of the field, highlighting the limitations and opportunities for PARP inhibitor development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Introduction
1.1 The PARP Family
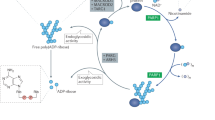
Poly-ADP-ribose polymerases (PARPs1-16; also known as ADP-ribosyltransferases or ARTDs) catalyze the transfer of ADP-ribose (ADPr) from nicotinamide adenine dinucleotide (NAD+) to their target. Most PARPs (PARP3, 4, 6–8, 10–12, 14–16) transfer a single unit of ADPr onto targets, a process known as mono-ADP-ribosylation (MARylation) (Fig. 1). Only four PARPs (PARP1, 2, 5a, 5b) have been shown to transfer multiple units of ADPr onto targets, a process known as poly-ADP-ribosylation (PARylation) (Fig. 1). In most cases, ADPr is transferred onto amino acids in proteins; however, recent studies demonstrate that PARP-mediated ADPr transfer can occur on DNA (Munnur and Ahel 2017; Belousova et al. 2018). Similar to phosphorylation and other more well-characterized posttranslational modifications (PTMs), both MARylation and PARylation are reversible suggesting that these PTMs are dynamic.

PARPs catalyze the transfer of ADPr from NAD+ to target proteins. The majority of PARPs transfer either a single unit of ADPr, a process known as MARylation, whereas only four PARP family members transfer multiple units of ADPr, a process known as PARylation
Though best known for their role in DNA damage repair, recent studies have revealed that PARPs play much wider roles in cells: from transcriptional regulation, miRNA processing, mRNA stability, and nuclear core complex biology to the unfolded protein response (Bock et al. 2015). Regarding the physiological role of PARPs, there is a wealth of evidence demonstrating the involvement of PARPs in the immune system and T cell regulation, which has been well reviewed (Rosado et al. 2013; Krishnakumar and Kraus 2010; Giansanti et al. 2010). For example, PARP1, 2, and 14 have been shown to mediate pro-inflammatory responses (Mehrotra et al. 2013; Zingarelli et al. 1998; Andreone et al. 2003; Bai and Virág 2012). A recent study demonstrates a role of PARP6 in neurodevelopment (Huang et al. 2016). Lastly, several PARPs are implicated in human diseases; for example, PARP14 mediates allergic responses in asthma (Mehrotra et al. 2013) and is a survival factor in multiple myeloma (Barbarulo et al. 2013) and hepatocellular carcinoma (Iansante et al. 2015).
1.2 Summary of Review
Despite the rapid expansion of the PARP field in recent years, most PARP family members that catalyze MARylation are poorly understood. This is due, in part, to the dearth of small-molecule inhibitors that selectively inhibit PARPs that catalyze MARylation. In contrast, there are numerous selective inhibitors for PARPs that catalyze PARylation and these have been useful not only as tools to reveal the functions of PARylation in the cell, but also validate PARPs as therapeutic targets. There are currently three FDA-approved PARP inhibitors (olaparib, rucaparib, and niraparib) that exhibit good selectivity for PARP1/2.
In this review, we discuss the history of PARP inhibitor development, structural features of PARP inhibitors, PARP inhibitor screening assays, and the current challenges and opportunities for PARP inhibitor development. For a general review of ADP-ribosylation and PARP biology, we direct the reader to the following references (Bock et al. 2015; Bai 2015; Vyas et al. 2014; Kraus 2015; Barkauskaite et al. 2015; Cohen and Chang 2018; Hottiger et al. 2010). To begin, we will describe structural features within the PARP catalytic domain.
2 Common and Distinct Features in the Catalytic Domain of PARPs
2.1 The NAD+ Binding Domain
While the PARP family is diverse in regard to their domain architecture, they all share a highly conserved catalytic domain known as the ADP-ribosyltransferase (ART) fold, which binds NAD+ in a conformation optimal for ADPr transfer. Nearly all PARP inhibitors target the ART fold and are competitive with NAD+. The key interaction between NAD+ and PARPs has been gleaned from crystal structures of related ART bacterial toxins (diphtheria, pertussis, cholera, and certain clostridial toxins) (Hottiger et al. 2010) bound to NAD+(Tsurumura et al. 2013) (Fig. 2a), and a recent structure of PARP1 bound to benzamide adenine dinucleotide (BAD) (Langelier et al. 2018), a non-hydrolyzable NAD+ analog (Fig. 2b). Two key interactions between NAD+ and PARPs are the interaction of the exocyclic amide of the nicotinamide moiety with the main chain of a conserved glycine (Gly863, human PARP1 numbering) and the side chain of a conserved serine (Ser 904, human PARP1 numbering) (Fig. 2c). These interactions are commonly exploited by PARP inhibitors, as will be illustrated below.

Structural features of the NAD+ binding site. a The bacterial toxin ExoA (PDB 2ZIT) bound to endogenous NAD+ shows how the H-Y-E catalytic triad, A-loop, and D-loop hold NAD+ in the binding site of a protein that catalyzes MARylation; b PARP1 in complex with a non-hydrolyzable NAD+ analog (PDB 6BHV) closely mirrors the structural interactions of endogenous NAD+ with ExoA; c Based on these crystal structures a simplified model of key interactions between NAD+ and the PARP catalytic domain reveals two important regions: the nicotinamide subsite (green) and the adenosine subsite (orange)
2.2 HYE Versus HYΦ PARPs
Active PARPs can be divided into two subfamilies based on the sequence of an active site triad motif in the ART fold (Fig. 2c)—the histidine–tyrosine–glutamate (HYE) PARPs (PARP1, 2, 3, 4, 5a and 5b) and the histidine–tyrosine–hydrophobic amino acid (HYΦ) PARPs (PARP 6–8, 10–12, 14–16) (Hottiger et al. 2010). In both the HYE and HYΦ, the conserved histidine forms a hydrogen bond with the 2-OH of the adenosine ribose and the conserved tyrosine π-stacks with the nicotinamide moiety (Fig. 2c). The glutamate in the HYE PARPs is necessary but not sufficient for the PARylation activity of several HYE PARPs (Marsischky et al. 1995; Rolli et al. 1997). HYΦ PARPs were recently shown to exclusively catalyze MARylation (Vyas et al. 2014).
2.3 Non-Conserved Loops
In addition to these conserved amino acids, there are two loop regions in the ART fold that are less well conserved yet thought to be critical for catalysis and NAD+ binding: the acceptor loop (A-loop) and the donor-loop (D-loop) (Fig. 2a–c). The A-loop varies widely in terms of length and amino acid composition across the PARP family and is thought to interact with PARP substrates (Fig. 2c). The D-loop is a variable region that interacts with the ADPr moiety of NAD+ (Fig. 2a–c). Based on mutagenesis studies of the related bacterial ARTs, the D-loop in PARPs is thought to be necessary for NAD+ binding and ADPr transfer (Pinto and Schüler 2015). The high variability of the A-loop and D-loop of PARPs could be exploited for the design of isoform-selective inhibitors.
3 Early Developments in the Field: Initial Focus on Small-Molecule Inhibitors of PARP1
3.1 The Founding PARP Inhibitors
Interest in PARP inhibitors emerged in the early 1980s following the discovery of PARP1, the most ubiquitous and abundant PARP family member (Fig. 3). The first-described PARP1 inhibitors focused on targeting the nicotinamide subsite and were simple biomimetics of nicotinamide such as 3-aminobenzamide (3-AB) (Purnell and Whish 1980) (Fig. 4), which inhibits PARP1 with a half-maximal inhibitory concentration (IC50) ~ 10 µM. These inhibitors played a crucial role in elucidating the role of PARP1 in DNA damage repair. Consistent with their role in DNA damage repair, these first-generation PARP inhibitors were shown to potentiate the cytotoxicity induced by DNA damaging agents in cancer cells (Purnell and Whish 1980; Nduka et al. 1980; Durrant and Boyle 1982). During the next two decades, significant advances were made in PARP inhibitor development, and by the mid-2000s inhibitors with nanomolar IC50 values against PARP1, and the closely related PARP2, were identified.

Timeline of PARP inhibitor development

Structures of common PARP inhibitors and their classifications
3.2 PARP1/2 Identified as Therapeutic Targets in Cancer
In 2005, two seminal papers demonstrated that potent PARP1/2 inhibitors induce synthetic lethality in BRCA-deficient cancers by blocking PARP1 mediated DNA repair pathways (Farmer et al. 2005; Bryant et al. 2005). These studies motivated the development of PARP1/2 inhibitors as adjuvants with DNA damaging reagents or as single agents in cancers that have defects in the DNA repair machinery. First disclosed in 2008, the potent PARP1/2 inhibitor olaparib (Fig. 4) effectively killed BRCA-deficient cancer cells at low nanomolar concentrations (Menear et al. 2008). In 2009, Fong et al. published the first Phase 1 trial of olaparib reporting its antitumor effects in BRCA1/2 mutated cancer (Fong et al. 2009), and in 2014 olaparib became the first FDA-approved PARP inhibitor. Since then off-label and approved uses of olaparib have expanded beyond its original approval as a monotherapy for BRCA-deficient ovarian cancers to include prostate cancer and germ line mutated metastatic breast cancer. Following the FDA approval of olaparib, two more potent PARP1/2 inhibitors have entered the clinic: rucaparib in 2016 and niraparib in 2017 (Fig. 4).
These FDA-approved PARP inhibitors have shifted the paradigm for cancer treatment. Because they exploit synthetic lethality they are relatively non-toxic to normal cells, thus avoiding many of the side effects of frontline chemotherapeutics. For a thorough review of PARP1 history, biology, and inhibitors see the following references (Kraus 2015; Ferraris 2010; Feng et al. 2015). For a recent review of the clinical applications of PARP1/2 inhibitors including olaparib, rucaparib, and niraparib, see the following references (Ohmoto and Yachida 2017; Mariappan et al. 2017).
4 Moving Beyond PARP1/2
4.1 Forward Chemical Genetic Screen Identifies a Small-Molecule Inhibitor of PARP5a/b
In contrast to the targeted approach used to identify potent and selective inhibitors of PARP1/2, the first inhibitor of the other PAR-generating PARPs, PARP5a/b (also known as tankyrase 1/2), was identified in a forward chemical genetic screen focused on identifying inhibitors of Wnt/β-catenin signaling (Huang et al. 2009). A small molecule, XAV939 (Fig. 4), was found to induce degradation of β-catenin by stabilizing axin, thereby blocking β-catenin-mediated transcription (Huang et al. 2009). Using an immobilized, active analog of XAV939, it was shown that the target of XAV939 is PARP5a/b (Huang et al. 2009). XAV939 potently inhibits the activity of PARP5a/b-mediated PARylation, and later structural studies demonstrate that XAV939 binds to the nicotinamide subsite in PARP5a (Fig. 5f) (Karlberg et al. 2010). Knockdown of PARP5a/b phenocopied the effects of XAV939 on axin stabilization, providing evidence that PARP5a/b are the targets of XAV939 (Huang et al. 2009). In this same study, the authors showed that a previously described axin-stabilizing compound, IWR-1-endo, also potently inhibited PARP5a/b-mediated PARylation, suggesting that this compound stabilizes axin by inhibiting PARP5a/b catalytic activity (Huang et al. 2009). Intriguingly, structural studies show IWR-1-endo binds exclusively to the adenosine subsite, which at the time, was the first example of this type if binding mode to a PARP (Narwal et al. 2012).

Different small molecules access different regions of the NAD+ binding pocket. a PARP1 in complex with niraparib (PDB 4R6E); b PARP1 in complex with rucaparib (PDB 4RV6); b PARP2 in complex with olaparib (PDB 4TVJ); d PARP5b in complex with EB-47 (PDB 4TK5); e PARP5b in complex with IWR-1-endo (PDB 3UA9); f PARP5b in complex with G007-LK; g PARP10 in complex with 3-aminobenzamide (PDB 3HKV); h PARP14 in complex with 3-aminobenzamide (PDB 3GOY); i PARP14 in complex with compound 4s (PDB 5NQE)
Since this study, several academic and industry efforts have led to the development of more potent and selective PARP5a/b inhibitors, including G007-LK (Fig. 4) (Menear et al. 2008), which targets the adenosine subsite of the NAD+ binding pocket (Fig. 5). This compound as well as structurally unrelated PARP5a/b inhibitors have been invaluable tools for uncovering the roles of PARP5a/b-mediated PARylation in cells and show promise as anticancer drugs (Vyas et al. 2014; Zhan et al. 2014; Kamal et al. 2014).
4.2 A Dearth of Selective and Potent Inhibitors for the HYΦ PARP Subfamily
Historically, PARP inhibitor design has focused on PARP1/2 and PARP5a/b with comparatively little attention given to the rest of the family, and in particular to the HYΦ PARPs. Recent years have seen a growing interest in the HYΦ PARP subfamily and a subsequent increase in HYΦ PARP inhibitor development. Of particular interest to many in drug discovery research has been PARP14 because of its role in several pathologies including asthma (Mehrotra et al. 2013), multiple myeloma (Barbarulo et al. 2013), and hepatocellular carcinoma (Iansante et al. 2015). Promising inhibitors of PARP14 have been published by Upton et al. (e.g. 4s, Fig. 4) (Upton et al. 2017) and Yoneyama-Hirozane et al. at Takeda Pharmaceuticals (Compound 1 and 2, Fig. 3) (Yoneyama-Hirozane et al. 2017). Upton et al. expanded on a previously identified small-molecule inhibitor of PARP14 (Andersson et al. 2012; Ekblad et al. 2015) based on 3-AB to develop potent but non-selective PARP14 inhibitors containing cis-maleic amide substituents emanating from the 3-amino group of the 3-AB scaffold (Upton et al. 2017). Though originally designed to interact with the D-loop and adenosine subsite, crystal structure analysis of 4s shows that the cis-maleic amide substituent accesses an induced pocket adjacent to the NAD+ binding site (further discussion below). Yoneyama-Hirozane et al. screened a small-molecule library to identify two compounds (Compound 1 and 2, Fig. 4) based on divergent scaffolds that showed potent PARP14 inhibition and no activity against PARP1 up to 25 µM (Yoneyama-Hirozane et al. 2017). Though untested against the majority of the PARP family, these scaffolds are promising starting points for further development of PARP14 inhibitors.
There have been some efforts to generate selective inhibitors for other HYΦ PARPs. For example, a mono-selective inhibitor of PARP10 was developed using a chemical genetic strategy (Fig. 4; Morgan et al. 2015). A screen of a small-molecule library from the National Cancer Institute led to the discovery of a PARP10 inhibitor (OUL35, Fig. 4) (Venkannagari et al. 2016). OUL35, an ether linked dimer of benzamide, appears to exhibit some selectivity for PARP10 when tested against several other PARP family members. Recently, a PARP11 inhibitor (ITK7, Fig. 4) with low nano-molar potency and greater than 200-fold selectivity over the entire PARP family was reported. This inhibitor was used to elucidate the connection between the catalytic activity of PARP11 its localization to the nuclear pore (Kirby et al. 2018).
A number of non-selective PARP inhibitors and promising scaffolds have been described, but broadly speaking these compounds lack the selectivity or potency necessary for use as probes for exploring the individual role of HYΦ PARPs in cells (Wahlberg et al. 2012; Thorsell et al. 2017).
5 Structural Studies of PARP Inhibitors: Insights into Inhibitor Potency and Selectivity
Structural studies of PARPs bound to various inhibitors have provided insight into inhibitor potency and selectivity and have been useful for structure-based design efforts. Here we discuss a few of these structures. The majority of structural studies have focused on HYE PARPs, such as PARP1 and PARP5a/b (Fig. 5).
5.1 Structural Studies of HYE PARP Inhibitors
The three FDA-approved inhibitors olaparib, rucaparib, and niraparib all share similar binding modes to PARP1/2 despite their divergent scaffolds (Figs. 4, 5a–c). A common and seemingly essential feature among these inhibitors is the interaction of an amide moiety (nicotinamide mimic) with the backbone glycine and side chain of an active site serine, which mimics the hydrogen bonding interaction observed with the nicotinamide of NAD+ (Figs. 2c, 5a–c). Additionally, these inhibitors contain various substituents that engage with, to varying degrees, the D-loop and adenosine subsite (Fig. 5a–c).
Another potent PARP inhibitor, EB47, which is designed to mimic the full NAD+ structure, occupies both the nicotinamide and adenosine subsites of PARP5b (Fig. 5d). Unsurprisingly, this molecule is not specific for PARP5b and has been shown to bind to several PARPs (Wahlberg et al. 2012). By contrast, two PARP5a/b inhibitors, IWR-1-endo and G007-LK, which do not contain nicotinamide mimics, exclusively occupy the adenosine subsite (Fig. 5e, f). PARP inhibitors that target the adenosine subsite but not the nicotinamide subsite are still fairly uncommon, and their efficacy against HYΦ PARPs has not been explored.
5.2 Structural Studies of HYΦ PARP Inhibitors
In recent years, greater attention has been given to the HYΦ PARPs, though inhibitors and crystal structures remain relatively rare by comparison to the better characterized HYE PARP family members. Some of the first reported crystal structures of HYΦ PARPs were PARP10 and PARP14 bound to 3-aminobenzamide (3-AB). As expected, 3-AB binds in the nicotinamide subsite in PARP10 and PARP14 and makes the same interactions with the conserved glycine backbone as the benzamide moiety of BAD bound to PARP1 (Fig. 5h, i). The PARP14 inhibitors mentioned above, which extend substituents from the 3-amino position of the 3-AB scaffold, occupy a unique, induced pocket in PARP14. The crystal structure of one of these compounds (4s) shows that 3-AB binds in the nicotinamide subsite as expected, but that the cis-maleic amide substituent emanating from 3-amino position reaches into a nascent pocket (near the A-loop) that likely results from a compound-induced movement of the D-loop (Fig. 5j). This finding reveals a new pocket in PARP14, and perhaps other HYΦ PARPs that could be targeted by other inhibitors.
5.3 Pharmacophore for PARP Inhibitors
From these structures, we can construct a model to summarize known inhibitor interactions with the NAD+ binding pocket, which can guide future PARP inhibitor development (Fig. 6). A crucial feature of many PARP inhibitors is mimicking the interaction between the nicotinamide moiety of NAD+ and the backbone glycine and side chain of an active site serine. Indeed, a wide range of scaffolds exploiting this interaction have been developed into successful PARP inhibitors. Targeting the nicotinamide site in concert with the adenosine subsite and D-loop appears optimal for potent PARP inhibition; however, many of these inhibitors are not selective (more on this below). Exclusively targeting the adenosine subsite, as has been shown for PARP5a/b, may result in more selective PARP inhibitors. Lastly, D-loop disrupting compounds (e.g., 4s) that can induce unique pockets outside the nicotinamide and adenosine subsites may turn out to be a generalizable approach for generating potent and selective PARP inhibitors.

Major structural interactions of known PARP inhibitors with the NAD+ binding pocket. Crystal structures of known PARP inhibitors demonstrate how various small molecules can exploit different features in the nicotinamide and adenosine subsites in the NAD+ binding pocket
6 Chemical and Biological Reagents for Measuring PARP Activity
6.1 NAD+ Analogs for In Vitro Analysis
As described above, PARPs use NAD+ as a substrate to mediate PARylation or MARylation. Historically, PARP activity was measured using radioactive NAD+ (e.g., adenylate phosphate-[32P] NAD+) (Surowy and Berger 1985) or using biotin-NAD+ (Zhang and Snyder 1992) (Fig. 7).Compared to [32P] NAD+, biotin-NAD+ can be used for identifying P/MARylated targets (Narendja and Sauermann 1994). Other NAD+ analogs include N-6-etheno-NAD+ (Barrio et al. 1972), a fluorescent NAD+ analog, and ADP-ribose-p-nitrophenoxy (Oei et al. 1999), an analog in which the nicotinamide is replace with a p-nitrophenol for use in colorimetric assays (Fig. 7). Additionally, various “clickable” NAD+ analogs have recently been developed, which contain an alkyne at various positions on the adenosine ring of NAD+ (Fig. 7) (Jiang et al. 2010; Wallrodt et al. 2016; Wang et al. 2014; Carter-O’Connell et al. 2014). These clickable NAD+ analogs can be coupled to a fluorescent-azide (visualize) or biotin-azide (visualize and identify) via the Huisgen 1,3-Dipolar Cycloaddition (“click reaction”).

Various NAD+ analogs and probes have been developed to monitor PARP activity. Salient modifications to NAD+ are highlighted in green
6.2 Detection Methods with Endogenous NAD+
In addition to using NAD+ analogs, several other strategies for detecting PARylation or MARylation have been described. For PARylation detection, the most commonly used reagents are antibodies that specifically recognize PARylated substrates (e.g. 10H) (Kawamitsu et al. 1984; Meyer and Hilz 1986; Küpper et al. 1996). Recently, protein-based reagents for detecting both PARylated and MARylated proteins have been described. These reagents consist of domains that recognize either ADPr (e.g., macro domain) or poly-ADPr (e.g., WWE domain) fused to Fc (Gibson et al. 2017). These reagents have been used in Western blot experiments as well as plate-based assays, as well as pull-down experiments. Lastly, an aminooxy-alkyne probe (AO-alkyne, Fig. 7) was described, which can readily detect proteins that are P/MARylated on acidic amino acids. AO-alkyne can also be used in cells for detecting cellular PARylation and MARylation (Morgan and Cohen 2015).
7 Assessing PARP Inhibitor Selectivity Across the PARP Family
7.1 Profiling Using Protein Stabilization Reveals Lack of Selectivity of Many PARP Inhibitors
Arguably the most important aspect of inhibitor development is assessing its target selectivity. Among other things, this is essential for understanding any cell-based or in vivo studies conducted with an inhibitor. Unfortunately, there are few studies that assess PARP inhibitor selectivity across multiple PARP family members, let alone the entire PARP family. One of the first examples of profiling inhibitor selectivity across multiple PARP family members was described in 2012. In this study, known and potential PARP inhibitors were screened against 13 PARPs using differential scanning fluorimetry (DSF), which assesses whether a compound can stabilize proteins (Wahlberg et al. 2012). One of the main findings of this study was that compounds previously described as selective PARP1/2 inhibitors, such as veliparib, rucaparib, and olaparib, stabilize several other HYE PARPs suggesting that these compounds may not be as selective as previously thought. In general, most of the known PARP inhibitors did not stabilize HYΦ PARPs, suggesting that they would not inhibit these PARPs. While this study provided the first insight into PARP inhibitor selectivity, DSF only assesses whether these PARP inhibitors can stabilize PARPs, which does not necessarily correlate with inhibition of catalytic activity.
7.2 Polypharmacology Among PARP Inhibitors
Recently, a high throughput 96-well-plate-based ADP-ribosylation assay using biotin-NAD+ was used for screening known PARP inhibitors against 11 PARP family members (Thorsell et al. 2017), most of which were the same PARPs used in the DSF study. In general, there was reasonable agreement between the ADP-ribosylation activity study and the DSF study, although the DSF study slightly overestimated PARP inhibitor selectivity. The selectivity profile of various PARP inhibitors is summarized in a heat map shown in Fig. 8 (Voronkov et al. 2013; Upton et al. 2017; Thorsell et al. 2017; Huang et al. 2009; Ishida et al. 2006; Papeo et al. 2014; Kirby et al. 2018). There are several important findings worth noting: 1. veliparib appears to be the most potent and selective PARP1/2 inhibitor, exhibiting greater than 100-fold selectivity for PARP1/2 versus other PARPs; 2. XAV939, which was previously described as selective inhibitor of PARP5a/b, potently inhibits PARP1 and PARP2, whereas IWR-1 is highly selective for PARP5a/b; 3. rucaparib and olaparib, while most selective for PARP1 and PARP2, inhibit several other PARP family members with sub-micromolar IC50 values.

Heat map showing the known IC50 values of PARP inhibitors shown in Fig. 3. The IC50 values used to generate this heat map derived from serval sources, as referenced in the text. In gray: values unknown
While a comprehensive assay for screening inhibitors across the entire PARP family is still needed to fully assess family-wide PARP inhibitor selectivity, these findings have several important implications for interpreting results from cell-based experiments using these compounds. For example, a PubMed search reveals that many papers describe studies using XAV939 in cell-based assays at concentrations that also inhibit PARP1 and PARP2, making it difficult to conclude that the effects of the compound were in fact due to PARP5a/b inhibition. For selective inhibition of PARP5a/b in cells the adenosine pocket binders IWR-1 or G007-LK are better options in our opinion.
Lastly, these findings have important implications for evaluating PARP inhibitors in a clinical setting for cancer treatment. The different selectivity profiles of the three FDA-approved PARP inhibitors (Fig. 8) could potentially contribute to efficacy and/or toxicity. Knowing the selectivity profiles will help assess the effectiveness of polypharmacology for certain cancers.
8 Conclusions and Future Directions
The growing interest in PARPs should catalyze the development of selective PARP inhibitors to use as tools for uncovering the role of PARPs in cells and as potential therapeutics. The multifarious PARP-inhibitor structures available should guide the design of the next generation of more potent and selective inhibitors, especially for the HYΦ PARP subfamily for which is there a dearth of inhibitors. Exploiting differences in the variable A-loop or D-loop or targeting regions outside the NAD+-binding site may provide strategies for the ultimate goal of generating potent and selective inhibitors for every PARP family member. Covalent inhibitors that target non-conserved nucleophilic amino acids are another potential strategy for the design of potent and selective inhibitors. With the exception of some early work (Watson et al. 1998), this inhibition strategy has not been pursued.
As new PARP inhibitors continue to be developed, it will be important to standardize in vitro PARP inhibitor screening assays. This is essential for comparing IC50 values, which depend on the concentration of NAD+ or NAD+ analog, obtained in different labs. Another consideration is the use of co-activators. PARP1, for example, requires single-stranded DNA for activation, whereas as PARP2 and PARP3 are optimally activated by 5’-phosphorylated double strand nicked DNA (Langelier et al. 2014). Most assays assessing PARP activity have focused on auto-P/MARylation; however, PARPs can also trans-P/MARylate targets in a cellular context (Carter-O’Connell et al. 2016; Gibson et al. 2016). It will be important to incorporate this into in vitro assays as auto-modification versus trans-modification may yield different inhibition profiles. In some cases, co-activators may be required for optimal trans-P/MARylation. For example, recently, it was shown that histone PARylation factor 1 (HPF1), which binds to PARP1, promotes PARP1 trans-PARylation of histones on serines (Gibbs-Seymour et al. 2016). Whether protein co-activators exist for other PARP family members is unclear, but as we learn more about PARP activation mechanisms these will need to be incorporated into in vitro PARP inhibitor screening assays.
Approaches to broadly assess PARP selectivity in a cellular context are desperately needed. Chemical proteomics approaches using resin bound PARP inhibitors is one potential strategy. This approach has the ability to identify potential non-PARP targets. Indeed, a recent study using resin bound olaparib, veliparib, rucaparib, and niraparib revealed that rucaparib and niraparib also target hexose-6-Phosphate Dehydrogenase (H6PD) and Deoxycytidine kinase (DCK), and inhibition of these targets may be clinically relevant (Knezevic et al. 2016). This chemical proteomics approach could also be used to profile PARP inhibitors in cell lysates, similar to the way Kinobeads have been used to profile kinase inhibitors (Golkowski et al. 2014); however, this approach requires a pan-PARP inhibitor with good potency, which unfortunately does not currently exist. Another approach could be to use activity-based protein profiling (ABPP), which exploits a conserved nucleophile in an enzyme active site for the development of a broad-spectrum probe for screening inhibitors across an enzyme family in cell lysates or cells (Cravatt et al. 2008). Such an approach for PARPs could be quite useful for profiling PARP inhibitors in a cellular context.
References
Andersson CD, Karlberg T, Ekblad T, Lindgren AEG, Thorsell A-G, Spjut S et al (2012) Discovery of ligands for ADP-ribosyltransferases via docking-based virtual screening. J Med Chem 55(17):7706–7718
Andreone TL, O’Connor M, Denenberg A, Hake PW, Zingarelli B (2003) Poly(ADP-Ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J Immunol 170(4):2113–2120. 15 Feb 2003. (American Association of Immunologists)
Bai P (2015) Biology of poly(ADP-Ribose) polymerases: the factotums of cell maintenance. Mol Cell 58(6):947–958
Bai P, Virág L (2012) Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett 586(21):3771–3777. 26 Sep 2012. (Wiley-Blackwell)
Barbarulo A, Iansante V, Chaidos A, Naresh K, Rahemtulla A, Franzoso G et al (2013) Poly(ADP-ribose) polymerase family member 14 (PARP14) is a novel effector of the JNK2-dependent pro-survival signal in multiple myeloma. Oncogene 32(36):4231–4242
Barkauskaite E, Jankevicius G, Ahel I (2015) Structures and mechanisms of enzymes employed in the synthesis and degradation of PARP-dependent protein ADP-ribosylation. Mol Cell 58(6):935–946. 18 Jun 2015. (Elsevier)
Barrio JR, Secrist JA, Leonard NJ (1972) A fluorescent analog of nicotinamide adenine dinucleotide. Proc Natl Acad Sci 69(8):2039–2042
Belousova EA, Ishchenko AA, Lavrik OI (2018) DNA is a new target of PARP3. Sci Rep. Nature Publishing Group 8(1):101. 8 Mar 2018
Bitler BG, Gynecologic ZW (2017) PARP inhibitors: clinical utility and possibilities of overcoming resistance. Gynecologiconcology
Bock FJ, Todorova TT, Chang P (2015) RNA Regulation by Poly(ADP-Ribose) Polymerases. Mol Cell 58(6):959–969
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E et al (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434(7035):913–917 Nature Publishing Group
Carter-O’Connell I, Jin H, Morgan RK, David LL, Cohen MS (2014) Engineering the substrate specificity of ADP-ribosyltransferases for identifying direct protein targets. J Am Chem Soc 136(14):5201–5204. 9 Apr 2014
Carter-O’Connell I, Jin H, Morgan RK, Zaja R, David LL, Ahel I et al Identifying Family-Member-Specific Targets of Mono-ARTDs by Using a Chemical Genetics Approach. Cell Rep 14(3):621–631
Chambon P, Weill JD, Doly J, Strosser MT, Mandel P (1966) On the formation of a novel adenylic compound by enzymatic extracts of liver nuclei. Biochem Biophys Res Commun 25(6):638–643
Chambon P, Weill JD, Mandel P (1963) Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun 11(1):39–43
Cohen MS, Chang P (2018) Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat Chem Biol 14(3):236–243 Nature Publishing Group
Cravatt BF, Wright AT, Kozarich JW (2008) Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem 77(1):383–414
Donawho CK, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD et al (2007) ABT-888, an orally active poly(ADP-Ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clini Cancer Res 13(9):2728–2737 American Association for Cancer Research
Durrant LG, Boyle JM (1982) Potentiation of cell killing by inhibitors of poly (ADP-ribose) polymerase in four rodent cell lines exposed to N-methyl-N-nitrosourea or UV light. Chem Biol Interact 38(3):325–338
Ekblad T, Lindgren AEG, Andersson CD, Caraballo R, Thorsell A-G, Karlberg T et al (2015) Towards small molecule inhibitors of mono-ADP-ribosyltransferases. Eur J Med Chem 95:546–551
Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434(7035):917–921 Nature Publishing Group
Feng FY, de Bono JS, Rubin MA, Knudsen KE (2015) Chromatin to clinic: the molecular rationale for PARP1 inhibitor function. Mol Cell 58(6):925–934
Ferraris DV (2010) Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J Med Chem 53(12):4561–4584
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M et al (2009) Inhibition of poly(ADP-Ribose) polymerase in tumors from BRCAMutation carriers. N Engl J Med 361(2):123–134
Giansanti V, Donà F, Tillhon M, Scovassi AI (2010) PARP inhibitors: new tools to protect from inflammation. Biochem Pharmacol 80(12):1869–1877
Gibbs-Seymour I, Fontana P, Rack JGM, Ahel I (2016) HPF1/C4orf27 Is a PARP-1-interacting protein that regulates PARP-1 ADP-ribosylation activity. Mol Cell 62(3):432–442
Gibson BA, Conrad LB, Huang D, Kraus WL (2017) Generation and characterization of recombinant antibody-like ADP-ribose binding proteins. Biochemistry 56(48):6305–6316
Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H et al (2016) Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 353(6294):45–50
Golkowski M, Brigham JL, Perera BGK, Romano GS, Maly DJ, Ong S-E (2014) Rapid profiling of protein kinase inhibitors by quantitative proteomics. Med Chem Commun 5(3):363–369 The Royal Society of Chemistry
Hottiger MO, Hassa PO, Lüscher B, Schüler H, Koch-Nolte F (2010a) Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci 35(4):208–219
Hottiger MO, Hassa PO, Lüscher B, Schüler H, Koch-Nolte F (2010) Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci 35(4):208–219. (Elsevier)
Huang JY, Wang K, Vermehren Schmaedick A, Adelman JP, Cohen MS (2016) PARP6 is a regulator of hippocampal dendritic morphogenesis. Sci Rep 6(1):208. 4 Jan 2016. (Nature Publishing Group)
Huang S-MA, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA et al (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461(7264):614–620. 16 Sept 2009. (Nature Publishing Group)
Huang S-MA, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA et al (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461(7264):614–620. 1 Oct 2009. (Nature Publishing Group)
Iansante V, Choy PM, Fung SW, Liu Y, Chai J-G, Dyson J et al (2015) PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat Commun 10(6):7882
Ishida J, Yamamoto H, Kido Y, Kamijo K, Murano K, Miyake H et al (2006) Discovery of potent and selective PARP-1 and PARP-2 inhibitors: SBDD analysis via a combination of X-ray structural study and homology modeling. Bioorg Med Chem 14(5):1378–1390
Jagtap PG, Southan GJ, Baloglu E, Ram S, Mabley JG, Marton A et al (2004) The discovery and synthesis of novel adenosine substituted 2,3-Dihydro-1H-isoindol-1-ones: potent inhibitors of poly(ADP-ribose) polymerase-1 (PARP-1). ChemInform 35(18):81 4 May 2004. (WILEY‐VCH Verlag)
Jiang H, Kim JH, Frizzell KM, Kraus WL, Lin H (2010) Clickable NAD Analogues for Labeling Substrate Proteins of Poly(ADP-ribose) Polymerases. J Am Chem Soc 132(27):9363–9372
Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C et al (2009) Discovery of 2-{4-[(3 S)-Piperidin-3-yl]phenyl}-2 H-indazole-7-carboxamide (MK-4827): A Novel Oral Poly(ADP-ribose)polymerase (PARP) Inhibitor Efficacious in BRCA-1 and -2 Mutant Tumors. J Med Chem 52(22):7170–7185
Kamal A, Riyaz S, Srivastava AK, Rahim A (2014) Tankyrase inhibitors as therapeutic targets for cancer. Curr Top Med Chem 14(17):1967–1976
Karlberg T, Markova N, Johansson I, Hammarström M, Schütz P, Weigelt J et al (2010) Structural basis for the interaction between tankyrase-2 and a potent Wnt-signaling inhibitor. J Med Chem 53(14):5352–5355
Kawamitsu H, Hoshino H, Okada H, Miwa M, Momoi H, Sugimura T (1984) Monoclonal antibodies to poly(adenosine diphosphate ribose) recognize different structures. Biochemistry 23(16):3771–3777
Kirby IT, Kojic A, Arnold MR, Thorsell A-G, Karlberg T, Vermehren Schmaedick A et al (2018) A potent and selective PARP11 inhibitor suggests coupling between cellular localization and catalytic activity. Cell Chem Biol
Knezevic CE, Wright G, Remsing Rix LL, Kim W, Kuenzi BM, Luo Y et al (2016) Proteome-wide profiling of clinical PARP inhibitors reveals compound-specific secondary targets. Cell Chem Biol 23(12):1490–1503
Kraus WL (2015) PARPs and ADP-Ribosylation: 50 Years … and Counting. Mol Cell 58(6):902–910
Krishnakumar R, Kraus WL (2010) The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 39(1):8–24
Küpper J-H, van Gool L, Müller M, Bürkle A (1996) Detection of poly(ADP-ribose) polymerase and its reaction product poly(ADP-ribose) by immunocytochemistry. Histochem J 28(5):391–395. (Kluwer Academic Publishers)
Langelier MF, Riccio AA, Pascal JM (2014) PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res 42(12):7762–75
Langelier M-F, Zandarashvili L, Aguiar PM, Black BE, Pascal JM (2018) NAD+ analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat Commun 9(1):844 Nature Publishing Group
Mabley JG, Jagtap P, Perretti M, Getting SJ, Salzman AL, Virág L et al (2001) Anti-inflammatory effects of a novel, potent inhibitor of poly (ADP-ribose) polymerase. Inflamm res 50(11):561–569. (Birkhäuser Verlag)
Mariappan L, Jiang XY, Jackson J, Drew Y (2017) Emerging treatment options for ovarian cancer: focus on rucaparib. IJWH 9:913–924 Dove Press
Marsischky GT, Wilson BA, Collier RJ (1995) Role of glutamic acid 988 of human poly-ADP-ribose polymerase in polymer formation. Evidence for active site similarities to the ADP-ribosylating toxins. J Biol Chem 270(7):3247–3254
Mehrotra P, Hollenbeck A, Riley JP, Li F, Patel RJ, Akhtar N et al (2013) Poly (ADP-ribose) polymerase 14 and its enzyme activity regulates TH2 differentiation and allergic airway disease. J Allergy Clin Immunol 131(2):521–531.e12. (Elsevier)
Menear KA, Adcock C, Boulter R, Cockcroft X-L, Copsey L, Cranston A et al (2008) 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem 51(20):6581–6591
Meyer T, Hilz H (1986) Production of anti-(ADP-ribose) antibodies with the aid of a dinucleotide-pyrophosphatase-resistant hapten and their application for the detection of mono(ADP-ribosyl)ated polypeptides. Eur J Biochem 155(1):157–165. (Blackwell Publishing Ltd)
Morgan RK, Carter-OConnell I, Cohen MS (2015) Selective inhibition of PARP10 using a chemical genetics strategy. Bioorg Med Chem Lett
Morgan RK, Cohen MS (2015) A clickable aminooxy probe for monitoring cellular ADP-ribosylation. ACS Chem Biol 10(8):1778–1784. 27 May 2015. (American Chemical Society)
Munnur D, Ahel I (2017) Reversible mono-ADP-ribosylation of DNA breaks. FEBS J. Wiley/Blackwell (10.1111); 2017 Nov 8;284(23):4002–16
Narendja FM, Sauermann G (1994) The use of biotinylated poly(ADP-ribose) for studies on poly(ADP-ribose)-protein interaction. Anal Biochem 220:415–419. (Vienna)
Narwal M, Venkannagari H, Lehtiö L (2012) Structural basis of selective inhibition of human tankyrases. J Med Chem 55(3):1360–1367
Nduka N, Skidmore CJ, Shall S (1980) The enhancement of cytotoxicity of N-Methyl-N-nitrosourea and of y-radiation by inhibitors of poly(ADP-ribose) Polymerase. Eur J Biochem 105(3):525–530. (Blackwell Publishing Ltd)
Oei SL, Griesenbeck J, Buchlow G, Jorcke D, Mayer-Kuckuk P, Wons T et al (1999) NAD+ analogs substituted in the purine base as substrates for poly(ADP-ribosyl) transferase. FEBS Lett 397(1):17–21
Ohmoto A, Yachida S (2017) Current status of poly(ADP-ribose) polymerase inhibitors and future directions. OTT 10:5195–5208 Dove Press
Papeo G, Avanzi N, Bettoni S, Leone A, Paolucci M, Perego R et al (2014) Insights into PARP inhibitors’ selectivity using fluorescence polarization and surface plasmon resonance binding assays. J Biomol Screen 19(8):1212–1219
Pinto AF, Schüler H (2015) Comparative structural analysis of the putative mono-ADP-ribosyltransferases of the ARTD/PARP family. Curr Top Microbiol Immunol 384 (Chapter 417):153–166. (Springer International Publishing, Cham)
Purnell MR, Whish WJ. Novel inhibitors of poly(ADP-ribose) synthetase. Biochem J 185(3):775–777. 1 Mar 1980. (Portland Press Limited)
Rolli V, O’Farrell M, Ménissier-de Murcia J, de Murcia G (1997) Random mutagenesis of the poly(ADP-ribose) polymerase catalytic domain reveals amino acids involved in polymer branching †. Biochemistry 36(40):12147–12154
Rosado MM, Bennici E, Novelli F, Pioli C (2013) Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology (10.1111); 139(4):428–437. 2 Jul 2013. (Wiley/Blackwell)
Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R et al (2013) BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res 19(18):5003–5015 American Association for Cancer Research
Surowy CS, Berger NA (1985) A, 3-aminobenzamide-resistant labeled protein in [32P]NAD+-labeled cells. Biochimica et Biophysica Acta (BBA)—Molecular. Cell Res 847(3):309–315
Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S et al (2007) A simple, sensitive, and generalizable plate assay for screening PARP inhibitors. In: Methods in molecular biology. American Association for Cancer Research, pp 945–56
Thorsell A-G, Ekblad T, Karlberg T, Löw M, Pinto AF, Trésaugues L et al (2017a) Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J Med Chem 60(4):1262–1271
Thorsell A-G, Ekblad T, Karlberg T, Löw M, Pinto AF, Trésaugues L et al (2017b) Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J Med Chem 60(4):1262–1271
Tsurumura T, Tsumori Y, Qiu H, Oda M, Sakurai J, Nagahama M et al (2013) Arginine ADP-ribosylation mechanism based on structural snapshots of iota-toxin and actin complex. Proc Natl Acad Sci 110(11):4267–4272. 12 Mar 2013
Upton K, Meyers M, Thorsell A-G, Karlberg T, Holechek J, Lease R et al (2017) Design and synthesis of potent inhibitors of the mono(ADP-ribosyl)transferase, PARP14. Bioorg Med Chem Lett 27(13):2907–2911
Venkannagari H, Verheugd P, Koivunen J, Haikarainen T, Obaji E, Ashok Y et al (2016) Small-molecule chemical probe rescues cells from mono-ADP-ribosyltransferase ARTD10/PARP10-induced apoptosis and sensitizes cancer cells to DNA damage. Cell Chem Biol 23(10):1251–1260. (Elsevier)
Voronkov A, Holsworth DD, Waaler J, Wilson SR, Ekblad B, Perdreau-Dahl H et al (2013) Structural basis and SAR for G007-LK, a lead stage 1,2,4-triazole based specific tankyrase 1/2 inhibitor. J Med Chem 56(7):3012–3023
Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT et al (2014) Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun 5:4426
Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell A-G et al (2012) Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol 30(3):283–288
Wallrodt S, Buntz A, Wang Y, Zumbusch A, Marx A (2016) Bioorthogonally functionalized NAD+ analogues for in-cell visualization of poly(ADP-ribose) formation. Angew Chem Int Ed 55(27):7660–7664
Wang Y, Rösner D, Grzywa M, Marx A (2014) Chain-terminating and clickable NAD+ analogues for labeling the target proteins of ADP-ribosyltransferases. Angew Chem Int Ed Engl 53(31):8159–8162. 28 July 2014. (WILEY-VCH Verlag)
Watson CY, Whish WJD, Threadgill MD (1998) Synthesis of 3-substituted benzamides and 5-substituted isoquinolin-1(2H)-ones and preliminary evaluation as inhibitors of poly(ADP-ribose)polymerase (PARP). Bioorg Med Chem 6(6):721–734
Yamada M, Miwa M, Sugimura T (1971) Studies on poly (adenosine diphosphate-ribose): X. Properties of a partially purified poly (adenosine diphosphate-ribose) polymerase. Arch Biochem Biophy 146(2):579–586
Yoneyama-Hirozane M, Matsumoto S-I, Toyoda Y, Saikatendu KS, Zama Y, Yonemori K et al (2017) Identification of PARP14 inhibitors using novel methods for detecting auto-ribosylation. Biochem Biophys Res Commun 486(3):626–631
Zhan P, Song Y, Itoh Y, Suzuki T, Liu X (2014) Recent advances in the structure-based rational design of TNKSIs. Mol BioSyst 10(11):2783–2799 The Royal Society of Chemistry
Zhang J, Snyder SH (1992) Nitricoxidestimulatesauto-ADP-ribosylationof glyceraldehyde-3-phosphatedehydrogenase. Proc Natl Acad Sci U S A 89:9382–9385
Zingarelli B, Salzman AL, Szabó C (1998) Genetic disruption of poly (ADP-Ribose) synthetase inhibits the expression of P-Selectin and intercellular adhesion molecule-1 in myocardial ischemia/reperfusion injury. Circ Res 13;83(1):85–94. 13 Jul 1998. (American Heart Association, Inc)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kirby, I.T., Cohen, M.S. (2018). Small-Molecule Inhibitors of PARPs: From Tools for Investigating ADP-Ribosylation to Therapeutics. In: Cravatt, B., Hsu, KL., Weerapana, E. (eds) Activity-Based Protein Profiling. Current Topics in Microbiology and Immunology, vol 420. Springer, Cham. https://doi.org/10.1007/82_2018_137
Download citation
DOI: https://doi.org/10.1007/82_2018_137
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-11142-7
Online ISBN: 978-3-030-11143-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)