
Abstract
An increased number of clinicopathological studies on autoimmune pancreatitis, cholangitis, and sialoadenitis have led to the recognition of immunoglobulin G4-related disease (IgG4-RD) as a novel disorder, characterized by elevated levels of serum IgG4 and infiltration of IgG4-expressing plasma cells in the affected organs. Although the immunological background associated with the development of IgG4-RD remains poorly understood, recent studies have suggested involvement of the innate immune response in its pathogenesis. Peripheral blood innate immune cells, such as plasmacytoid dendritic cells and monocytes isolated from patients with IgG4-RD, promote IgG4 production by B cells. Activation of the innate immune response by microbe- and/or damage-associated molecular patterns stimulates production of type I interferon and B cell-activating factor by innate immune cells and results in IgG4 production by B cells. Elucidation of the innate immune response associated with IgG4-RD may help identify a new therapeutic target for this immune disorder.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
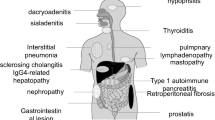
Immunoglobulin G4-related disease (IgG4-RD) is a chronic multiorgan fibroinflammatory disorder (Stone et al. 2012). It was first described by Japanese physicians and researchers who found elevated levels of serum IgG4 in most patients with autoimmune pancreatitis (AIP) (Hamano et al. 2001; Kamisawa and Okamoto 2006; Masaki et al. 2009; Okazaki et al. 2008). Physicians’ awareness and recognition of IgG4-RD have expanded substantially, enabling gastroenterologists and clinical immunologists to establish specific diagnostic criteria. These include the presence of elevated serum IgG4, as well as the massive infiltration of IgG4-expressing plasmacytes in the affected organs (Stone et al. 2012). Various single-organ diseases, such as AIP, sialoadenitis, and retroperitoneal fibrosis, are now considered as organ-specific manifestations of systemic IgG4-RD, because they share an enhanced IgG4 response. Thus, our knowledge of the clinical and epidemiological features of IgG4-RD is expanding rapidly, and IgG4-RD is attracting increasing attention from gastroenterologists and clinical immunologists.
Although the clinical manifestations of IgG4-RD have been established in several organs, our understanding of its immune pathogenesis remains limited. Given the elevated IgG4 serum levels and abundant accumulation of IgG4-expressing plasmacytes in tissues, it is likely that adaptive immunity, rather than innate immunity, plays a pathogenic role in disease development. In fact, the remarkable efficacy of B cell depletion therapy (Carruthers et al. 2015; Khosroshahi et al. 2010) provides strong evidence that adaptive immune cells, such as B and T cells, are involved in the pathogenesis of IgG4-RD. In line with this, recent studies have suggested the involvement of abnormal adaptive immune responses, such as excessive T helper type 2 (TH2), regulatory T cells (Tregs), and plasmablasts (Mattoo et al. 2014b; Miyoshi et al. 2008; Satoguina et al. 2008; Wallace et al. 2015; Zen et al. 2007). However, these studies have not elucidated the abnormal immunological environment that leads to the development of IgG4-RD, and a number of unresolved questions have been raised. These include the identification of antigens recognized by elevated IgG4 levels and the pathogenicity of IgG4 itself. In addition, recent investigations have highlighted the importance of innate immunity in preceding and/or augmenting IgG4 responses driven by adaptive immunity (Akitake et al. 2010; Arai et al. 2015; Fukui et al. 2015; Watanabe et al. 2012, 2013). Thus, the cross talk between adaptive and innate immune responses is associated with the immune pathogenesis of IgG4-RD. In this review, we discuss the innate immune responses associated with IgG4-RD.
2 IgG4-Related Disease (IgG4-RD) and Microbe-Associated Molecular Patterns (MAMPs) and Damage-Associated Molecular Patterns (DAMPs)
2.1 MAMPs and DAMPs
Danger signals are initially recognized by the innate immune system, which then promotes antigen-specific adaptive immune responses. Innate immunity exerts its host–defense functions immediately upon recognition of microbe-associated molecular patterns (MAMPs) (Akira and Takeda 2004; Chen et al. 2009; Strober et al. 2006) and damage-associated molecular patterns (DAMPs) (Kono and Rock 2008), through germline-encoded pathogen recognition receptors (PRRs). In contrast, adaptive immunity is responsible for delayed effector functions, since it relies on the selection and clonal expansion of antigen-specific B cells and T cells via gene rearrangement. At present, PRRs are categorized into four types: Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), RIG-I-like receptors, and C-type lectin-like receptors (Brubaker et al. 2015). These PRRs are mainly expressed on the cell surface or the cytosolic regions of innate immune cells such as antigen-presenting cells and epithelial cells. Upon microbial infection, MAMPs activate PRRs to eradicate or control microorganisms, in concert with adaptive immune responses against microbe-specific antigens. Under sterile inflammatory conditions, endogenous non-microbial danger molecules released from dying and necrotic cells activate PRRs and have been implicated in the development of autoimmune diseases (Kono and Rock 2008). Recent studies have revealed a possible involvement of MAMPs and DAMPs in the immune pathogenesis of IgG4-RD.
2.2 IgG4-RD and MAMPs
It is generally accepted that excessive immune reactions toward the intestinal microflora cause inflammatory bowel disease (IBD) (Bouma and Strober 2003). Indeed, significant numbers of patients with Crohn’s disease present mutations in caspase recruitment domain 15 (Hugot et al. 2001; Ogura et al. 2001), which encodes NOD2, a sensor for small peptides derived from the bacterial peptidoglycan (Strober et al. 2014). NOD2 polymorphisms associated with Crohn’s disease are thought to alter the immune response to gut bacteria and, consequently, the composition of the intestinal microflora (Strober et al. 2014). Thus, studies on the immune pathogenesis of Crohn’s disease in the presence of NOD2 mutations have revealed an excessive immune response to MAMPs derived from the intestinal microflora.
Given the lack of definitive genome-wide association or microbiome studies on IgG4-RD, it remains to be seen whether scenario described above applies to this disorder. Some studies suggest a possible involvement of MAMPs in the development of IgG4-RD. IBD is categorized in two subsets, Crohn’s disease and ulcerative colitis. The intestinal mucosa of IBD is characterized by enhanced production of TH1 cytokines, such as interleukin (IL)-12 and interferon (IFN)-γ, in Crohn’s disease, and TH2 cytokines, such as IL-5 and IL-13, in ulcerative colitis (Bouma and Strober 2003). Extensive immunohistochemical analysis of IgG4-positive plasma cells in the colonic mucosa of IBD patients revealed a significantly higher number of such cells in patients with ulcerative colitis than in those with Crohn’s disease (Virk et al. 2014). In addition, in ulcerative colitis, colonic infiltration of IgG4-positive plasma cells was accompanied by severe inflammation (Kuwata et al. 2014). These clinicopathological studies suggest that accumulation and infiltration of IgG4-expressing plasma cells in the colonic mucosa of patients with ulcerative colitis are probably caused by an excessive immune reaction against the intestinal microflora. Supporting this idea, Akitake et al. (2010) reported a case of IgG4-RD presenting abundant infiltration of IgG4-expressing plasmacytes in the ileum and colon without any symptoms of IBD. Furthermore, peripheral blood mononuclear cells isolated from this patient produced large amounts of TH2 cytokines upon stimulation with TLR ligands (Akitake et al. 2010). In line with this case report, the NOD2 ligand, muramyl dipeptide (MDP), has been shown to strongly induce IgG4 production in healthy individuals (Watanabe et al. 2012), which suggests that MAMPs derived from the intestinal microflora may trigger TH2-driven pathogenic responses in IgG4-RD. Repeated administration of heat-killed Escherichia coli to C57BL/6 mice induces activation of the innate immune system and the development of chronic fibroinflammatory disorder of the pancreas, akin to human AIP (Haruta et al. 2010; Yanagisawa et al. 2014). Taken together, both human and animal studies provide evidence that MAMPs derived from the intestinal microflora might trigger IgG4-RD through activation of PRRs.
One major question arising from the above hypothesis is whether a significant population of patients with IgG4-RD exhibits clinical symptoms of IBD. To this end, a case–control study revealed that four of 71 patients with AIP had concurrent IBD (Ravi et al. 2009). The simultaneous occurrence of IBD is generally observed in patients with type 2 non-IgG4-associated AIP rather than in patients with type 1 IgG4-associated AIP (Hart et al. 2015). Moreover, Ueki et al. (2015) reported an extremely low incidence of AIP in patients with IBD (Ueki et al. 2015). Thus, previous case–control studies do not support the hypothesis that MAMPs derived from the intestinal microflora are involved in the development of IgG4-RD. Given that intestinal tissue injury is absent even in the presence of massive infiltration of IgG4-expressing plasma cells (Akitake et al. 2010), the underlying immune reactions do not play a pathogenic role in the gut. In this regard, several mechanisms, including Tregs-mediated immune suppression, might protect the intestinal mucosa from tissue injury (Strober et al. 2007). Thus, one possible explanation for the low incidence of IBD in IgG4-RD patients is that pathogenic immune responses leading to colonic injury are suppressed by activation of Tregs. In contrast, these responses might cause tissue injury in sterile organs, such as the pancreas and salivary glands, due to the absence of regulatory mechanisms. Future studies addressing global immune reactions in the gut mucosa of patients with AIP will confirm this hypothesis.
2.3 IgG4-RD and DAMPs
DAMPs released from dying or necrotic cells are now recognized as potent activators of the innate immune system (Kono and Rock 2008). The role of DAMPs is highlighted in acute pancreatitis (Hoque et al. 2011, 2012). Double-stranded DNA (dsDNA) released from dying acinar cells upon experimental induction of acute pancreatitis activates TLR9 and NLR family, pyrin domain-containing 3 (NLRP3). These receptors are expressed in antigen-presenting cells and stimulate production of proinflammatory cytokines, such as IL-1β and IL-18 (Hoque et al. 2011, 2012). High-mobility group box chromosomal protein 1 (HMGB1) is a well-studied DAMP capable of inducing sterile inflammation by activating TLR4 (Zong et al. 2013). Yasuda et al. (2006) reported a marked increase in serum levels of HMGB1 in patients with acute pancreatitis. These studies support the idea that DAMPs released from necrotic pancreatic tissues induce the development of acute pancreatitis through activation of the innate immune system. Similarly, several reports suggest the involvement of DAMPs, such as dsDNA, monosodium urate crystals (MSU), and asbestos in the pathogenesis of IgG4-RD. Serum levels of dsDNA are higher in patients with IgG4-RD than in those with chronic pancreatitis or healthy individuals (Arai et al. 2015). Antigen-presenting cells and neutrophils isolated from patients with IgG4-RD induce IgG4 production by B cells upon MSU stimulation (Arai et al. 2015). Furthermore, Toyoshima et al. (2010) reported a case of IgG4-related lung disease in a worker exposed to asbestos, one of the prototypical DAMPs to activate the NLRP3 inflammasome (Toyoshima et al. 2010). It should nevertheless be noted that our knowledge of DAMPs in IgG4-RD is very limited and definitive proof is still lacking at present.
3 IgG4-RD and T Helper Type 2 Cytokines
Lesions in patients with IgG4-RD are characterized by enhanced expression of TH2 cytokines, such as IL-4, IL-10, and IL-13, rather than TH1 cytokines, such as IFN-γ and IL-12 (Della-Torre et al. 2015; Moriyama et al. 2014; Tanaka et al. 2012). In vitro studies have shown that stimulation of peripheral blood mononuclear cells with IL-4 and/or IL-10 increases IgG4 production in healthy subjects (Jeannin et al. 1998; Punnonen et al. 1993). The involvement of TH2 cytokines partly explains the allergic symptoms and elevated serum IgE levels commonly detected in IgG4-RD patients. It should be noted, however, that such classical TH2 responses might play a pathogenic role only in a defined subpopulation of IgG4-RD. Expansion of cells expressing IL-4, IL-5, or IL-13 and their lineage transcription factor GATA binding protein 3 (GATA3) (Crotty 2014) has been detected only in IgG4-RD patients with atopic symptoms and not in those without these symptoms (Mattoo et al. 2014a). Co-localization analyses on cells expressing both GATA3 and IL-4, IL-5, or IL-13 have not been performed in the lesions of IgG4-RD patients. Therefore, it is possible that IL-4 and IL-13 may be derived from other types of cells, such as T follicular helper (Tfh) cells (Akiyama et al. 2015; Maehara et al. 2012; Moriyama et al. 2014) and mast cells (Takeuchi et al. 2014, 2015) rather than conventional TH2 cells.
Tfh is a newly defined T helper subset that induces the development of germinal centers and the generation of high-affinity antibodies by B cells (Crotty 2014). Tfh cells express the master transcription factor B cell lymphoma 6 (Bcl6) and the chemokine receptor CXCR5 and produce large amounts of IL-21 (Crotty 2014). Maehara et al. (2012) reported increased IL-21 and BCL6 expression in the ectopic germinal centers of salivary glands from a patient with IgG4-RD (Maehara et al. 2012). Furthermore, flow cytometric analysis by Akiyama et al. (2015) found elevated numbers of CXCR5+ CD45RA− CD4+ CXCR3− CCR6− Tfh2 cells in the peripheral blood of IgG4-RD patients. Interestingly, the number of Tfh2 cells correlated with IL-4 and IgG4 serum levels, suggesting that Tfh cells may enhance the production of IgG4 in concert with conventional TH2 cells.
Another important feature of the T cell subset in IgG4-RD is infiltration of Tregs in the lesions (Miyoshi et al. 2008; Zen et al. 2007). Tregs express the master transcription factor, forkhead box protein p3 (Foxp3), and produce immunosuppressive cytokines, such as IL-10 and tumor growth factor (TGF)-β1 (Morikawa and Sakaguchi 2014), as observed in the liver of IgG4-RD patients (Zen et al. 2007). In vitro studies have shown that Tregs induce IgG4 production by B cells through IL-10 and TGF-β1 (Satoguina et al. 2008). Thus, it is likely that Tregs-derived IL-10 and TGF-β1 are associated with enhanced IgG4 production. One major question concerns the mechanism underlying the occurrence of chronic inflammation even in the presence of Tregs in IgG4-RD. In this regard, it should be noted that no studies have addressed the function of Tregs isolated from IgG4-RD patients by using conventional suppression assays. This leaves the possibility that Tregs accumulating in the lesions of IgG4-RD patients cannot fulfill their suppressive activity due to impaired immune regulatory functions, thus leading to chronic fibroinflammatory disorders. Alternatively, activation of Tregs in IgG4-RD may reflect some sort of counter-regulatory response to strong and persistent inflammation.
As suggested above, adaptive immune responses associated with IgG4-RD are mediated by a variety of T cell subsets, including classical TH2 cells, Tfh cells, and Tregs. Even if the elevated cytokine expression observed in IgG4-RD is confirmed to be derived from these T cell subsets, further studies are required to elucidate the cellular and molecular mechanisms accounting for pathological adaptive immune responses.
4 T Cell-Independent IgG4 Production (Fig. 1)
Innate immune responses mediated by MAMPs and DAMPs may be important for the onset and maintenance of abnormal immunological environments leading to IgG4-RD. Watanabe et al. (2012) have addressed the role of TLR- or NLR-mediated signaling pathways in the production of IgG4, and some key findings are summarized herein (Watanabe et al. 2012, 2013). First, MDP was found to be a potent inducer of IgG4 production (Watanabe et al. 2012). Second, MDP activation of NOD2 in monocytes induced the production of B cell-activating factor (BAFF), thereby enhancing IgG4 production through inhibition of B cell apoptosis. These results were obtained from peripheral blood mononuclear cells (PBMCs) of healthy subjects, suggesting the importance of NOD2 activation for IgG4 production in innate immune cells. Third, PBMCs isolated from IgG4-RD patients produced large amounts of IgG4 and BAFF upon stimulation with TLR and NLR ligands. Finally, B cells from healthy controls produced large quantities of IgG4 upon stimulation with TLR and NLR ligands via a T cell-independent manner only when co-cultured with monocytes isolated from patients with IgG4-RD. These results suggest that activation of the innate immune system by TLR and NLR ligands may be a critical step for the increased production of IgG4 and that IgG4 production can be induced without any help by T cells (Watanabe et al. 2012). As for the molecular mechanism accounting for BAFF production, involvement of NF-κB activation has been suggested. Inhibition of NF-κB signaling pathways reduces BAFF production by monocytes upon stimulation with MDP (Watanabe et al. 2012). Moreover, the DNA sequence of the BAFF promoter has several functional binding motifs for the NF-κB subunit (He et al. 2003).

Innate immune responses associated with IgG4-RD. Damage-associated molecular patterns (DAMPs) and microbe-associated molecular patterns (MAMPs) activate innate immune receptors, such as Toll-like receptors (TLRs) and NOD-like receptors (NLRs) expressed in plasmacytoid dendritic cells (pDCs), monocytes, and basophils. Activation of pDCs by neutrophil extracellular traps (NETs) leads to robust production of IFN-α and BAFF, whereas activation of TLRs and NLRs leads to robust production of BAFF. IgG4 production by B cells is enhanced upon co-culture with pDCs, monocytes, and basophils in a T cell-independent manner. Additionally, regulatory T (Treg), T helper type 2 (Th2), and T follicular helper (Tfh) cells stimulate IgG4 production by B cells through IL-4, IL-10, IL-13, and IL-21
Although basophils have been considered effector cells for TH2 and IgE responses, recent studies have highlighted their role in initiating allergic responses (Paul and Zhu 2010; Sokol and Medzhitov 2010). It seems likely that activation of basophils is involved in the immune pathogenesis of IgG4-RD, which is often characterized by elevated serum IgE levels. Indeed, Watanabe et al. (2013) showed that activation of TLRs in basophils stimulated IgG4 production by B cells in a T cell-independent manner (Watanabe et al. 2013). Basophils were found to release large quantities of BAFF upon stimulation with TLR ligands, thereby triggering IgG4 production. More importantly, as with monocytes (Watanabe et al. 2012), it was shown that B cells from healthy controls produced considerable amounts of IgG4 upon stimulation with TLR and NLR ligands only when co-cultured with basophils from IgG4-RD patients. These two studies suggest that activation of TLRs and/or NLRs in monocytes or basophils induces IgG4 production by B cells in a T cell-independent but BAFF-dependent manner. Moreover, monocytes or basophils from IgG4-RD patients produce large quantities of BAFF upon exposure to MAMPs via NF-κB signaling pathways.
Consistent with the above in vitro studies (Watanabe et al. 2012, 2013), BAFF serum levels are significantly higher in IgG4-RD patients than in healthy controls or in patients with chronic pancreatitis or pancreatic cancer (Kiyama et al. 2012; Yamanishi et al. 2011). Interestingly, BAFF-mediated signaling pathways seem to be operating in the inflamed pancreas, as confirmed by expression of BAFF and BAFF receptors in cells infiltrating the pancreas of IgG4-related AIP (Yamanishi et al. 2011). In addition, IgD+ B cells stimulated with BAFF and IL-4 induce Ig class-switch DNA recombination (CSR), giving rise to IgG1, IgG2, IgG3, and IgG4 (Litinskiy et al. 2002). Taken together, these reports have identified BAFF and TH2 cytokines derived from antigen-presenting cells and T cells, respectively, to be responsible for the generation of pathogenic immune responses in IgG4-RD. It should be noted, however, that enhanced IgG4 responses in IgG4-RD cannot be explained by BAFF-mediated pathways alone, since BAFF is a survival and activation factor for B cells rather than a specific inducer for IgG4 CSR (Mackay and Schneider 2009; Mackay et al. 2007).
The above studies on IgG4 and BAFF production in response to MAMPs support the idea that commensal flora-mediated innate immunity is involved in the immunopathogenesis of IgG4-RD. In this scenario, tissue injury initiated by an autoimmune process leads to impaired gut barrier function, followed by entry of intestinal microflora into the splanchnic vascular bed. MAMPs derived from intestinal microflora activate tissue-residing antigen-presenting cells and B cells, thus stimulating production of BAFF and IgG4. This idea is consistent with the observation that IgG4-expressing plasma cells are sometimes seen in the intestinal mucosa of patients with IgG4-RD (Akitake et al. 2010; Deheragoda et al. 2007).
5 IgG4-RD and Type I IFN (Fig. 1)
Type I IFNs (IFN-α and IFN-β) are indispensable components of host defenses against viral and bacterial infections (Akira and Takeda 2004). Although rapid induction of type I IFN production is particularly beneficial against viral infections, persistent and excessive release of these pluripotent cytokines causes autoimmune diseases. Detailed and extensive studies using clinical samples from lupus patients have established that excessive production of type I IFN and activation of downstream signaling pathways play a central role in the immune pathogenesis of the disease (Crow 2014a, b; Huang et al. 2015). This was confirmed by enhanced expression of type I IFN-related genes in peripheral blood mononuclear cells from lupus patients (Baechler et al. 2003; Bennett et al. 2003). Since they produce large quantities of IFN-α (Gilliet et al. 2008), plasmacytoid dendritic cells (pDCs) are considered the main promoters of type I IFN signaling pathways in lupus (Crow 2014a, b; Huang et al. 2015). This is particularly true, if they are exposed to immune complexes containing nucleic acids (Crow 2014a, b). Thus, the innate immune response mediated by pDC-derived IFN-α plays a predominant role in the initiation and progression of lupus. This notion is supported by recent genome-wide association studies, which have identified type I IFN-related genes as susceptible loci for lupus (Deng and Tsao 2010).
A recent study by Arai et al. (2015) provides evidence that activation of pDCs and IFN-α production are prominent features of human IgG4-RD and an experimental model of AIP (Arai et al. 2015). MRL/MpJ mice treated with polyinosinic–polycytidylic acid, poly (I:C), exhibit several histological features of chronic autoimmune pancreatitis, such as massive destruction of acinar cell architecture, infiltration of immune cells, and fibrosis (Nishio et al. 2011; Schwaiger et al. 2014). Although human IgG4-related AIP and MRL/MpJ mice treated with poly (I:C) share important clinical findings, common abnormal immune responses have been poorly described. Cytokine and chemokine arrays of pancreatic lysates from MRL/MpJ mice treated with poly (I:C) revealed that type I IFN-related chemokines, such as chemokine (C-X-C motif) ligand (CXCL) 9, 10, and 11, as wells as prototypic inflammatory cytokines, such as IL-6 and tumor necrosis factor-α, increased upon induction of experimental AIP (Arai et al. 2015). Indeed, administration of poly (I:C) led to a marked increase of IFN-α and IFN-β in the serum of MRL/MpJ mice and accumulation of pDCs in the pancreas. Since both depletion of pDCs and blockade of type I IFN signaling pathways prevent pancreatic inflammation, Arai et al. (2015) propose a pivotal role for pDC-mediated IFN-α signaling pathways in experimental AIP (Arai et al. 2015). In line with the above results, serum levels of IFN-α are significantly higher in patients with IgG4-associated AIP than in healthy controls, or in patients with chronic pancreatitis (Arai et al. 2015). Consistent with a report showing that BAFF expression is directly induced by type I IFN via interferon regulatory factors 1 and 2 (Sjöstrand et al. 2016), elevated IFN-α serum levels are accompanied by BAFF levels. Moreover, infiltration of pDCs producing both IFN-α and BAFF is observed in the pancreas of patients with IgG4-associated AIP, but not in those with chronic pancreatitis (Arai et al. 2015). Regarding activators of pDCs, two recent studies show that neutrophil extracellular traps (NETs) containing self-DNA and neutrophil-derived proteins are potent inducers of IFN-α production by pDCs in patients with lupus (Garcia-Romo et al. 2011; Lande et al. 2011). In the case of IgG4-RD, NETs may also be involved in increased IFN-α release by pDCs. Arai et al. (2015) report that B cells isolated from healthy individuals produce large quantities of IgG4 when co-cultured with NET-stimulated pDCs isolated from patients with IgG4-RD. Such pDC-mediated IgG4 production by B cells is markedly suppressed by the abrogation of type I IFN signaling pathways, which suggests an important role by pDC-derived IFN-α. Taken together, these results strongly indicate that pDC activation, followed by IFN-α release, is one of the pathogenic immune responses associated with IgG4-RD.
The study described above provides a new insight into the pathogenesis of IgG4-RD, which, like lupus, is characterized by pDC activation and IFN-α production. In addition, autoimmune complexes released by NETs may function as potent activators of pDCs in both immune disorders. Nevertheless, it should be noted that clinicopathological features of IgG4-RD and lupus are completely different, with augmented IgG4 production and storiform fibrosis being observed only in IgG4-RD. Therefore, it is clear that the immunopathogenesis of IgG4-RD cannot be explained solely by pDC activation and ensuing IFN-α production. Future studies aiming to identify immune responses other than the pDC-mediated IFN-α signaling pathways will elucidate the immunopathogenesis of IgG4-RD and help distinguish it from lupus at a cellular and a molecular level.
6 Conclusions
IgG4-RD is a newly established disease first proposed by Japanese gastroenterologists and rheumatologists. Although IgG4-RD is characterized by an adaptive response by B cells, recent studies suggest possible involvement of the innate immune system. Expression of IFN-α and BAFF produced by innate immune cells is enhanced in the pancreas of IgG4-RD patients and experimental AIP. Furthermore, pDCs and monocytes isolated from IgG4-RD patients induce a marked increase in IgG4 production by B cells through IFN-α and BAFF, respectively. Although many questions remain to be addressed, these insights into the role of innate immune responses in IgG4-RD pathogenesis support the idea that patients with IgG4-RD can be treated with inhibitors of innate immune cytokines, such as IFN-α and BAFF.
References
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4:499–511
Akitake R, Watanabe T, Zaima C, Uza N, Ida H, Tada S, Nishida N, Chiba T (2010) Possible involvement of T helper type 2 responses to Toll-like receptor ligands in IgG4-related sclerosing disease. Gut 59:542–545
Akiyama M, Suzuki K, Yamaoka K, Yasuoka H, Takeshita M, Kaneko Y, Kondo H, Kassai Y, Miyazaki T, Morita R et al (2015) Number of circulating follicular helper 2 T cells correlates with IgG4 and Interleukin-4 levels and plasmablast numbers in IgG4-related disease. Arthritis Rheumatol 67:2476–2481
Arai Y, Yamashita K, Kuriyama K, Shiokawa M, Kodama Y, Sakurai T, Mizugishi K, Uchida K, Kadowaki N, Takaori-Kondo A et al (2015) Plasmacytoid dendritic cell activation and IFN-alpha production are prominent features of murine autoimmune pancreatitis and human IgG4-related autoimmune pancreatitis. J Immunol 195:3033–3044
Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V et al (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA 100:2610–2615
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V (2003) Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197:711–723
Bouma G, Strober W (2003) The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol 3:521–533
Brubaker SW, Bonham KS, Zanoni I, Kagan JC (2015) Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 33:257–290
Carruthers MN, Topazian MD, Khosroshahi A, Witzig TE, Wallace ZS, Hart PA, Deshpande V, Smyrk TC, Chari S, Stone JH (2015) Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis 74:1171–1177
Chen G, Shaw MH, Kim YG, Nunez G (2009) NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol 4:365–398
Crotty S (2014) T follicular helper cell differentiation, function, and roles in disease. Immunity 41:529–542
Crow MK (2014a) Advances in understanding the role of type I interferons in systemic lupus erythematosus. Curr Opin Rheumatol 26:467–474
Crow MK (2014b) Type I interferon in the pathogenesis of lupus. J Immunol 192:5459–5468
Deheragoda MG, Church NI, Rodriguez-Justo M, Munson P, Sandanayake N, Seward EW, Miller K, Novelli M, Hatfield AR, Pereira SP, Webster GJ (2007) The use of immunoglobulin G4 immunostaining in diagnosing pancreatic and extrapancreatic involvement in autoimmune pancreatitis. Clin Gastroenterol Hepatol 5:1229–1234
Della-Torre E, Lanzillotta M, Doglioni C (2015) Immunology of IgG4-related disease. Clin Exp Immunol 181:191–206
Deng Y, Tsao BP (2010) Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol 6:683–692
Fukui Y, Uchida K, Sakaguchi Y, Fukui T, Nishio A, Shikata N, Sakaida N, Uemura Y, Satoi S, Okazaki K (2015) Possible involvement of Toll-like receptor 7 in the development of type 1 autoimmune pancreatitis. J Gastroenterol 50:435–444
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL et al (2011) Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 3:73ra20
Gilliet M, Cao W, Liu YJ (2008) Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 8:594–606
Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, Fukushima M, Nikaido T, Nakayama K, Usuda N, Kiyosawa K (2001) High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 344:732–738
Hart PA, Zen Y, Chari ST (2015) Recent advances in autoimmune pancreatitis. Gastroenterology 149:39–51
Haruta I, Yanagisawa N, Kawamura S, Furukawa T, Shimizu K, Kato H, Kobayashi M, Shiratori K, Yagi J (2010) A mouse model of autoimmune pancreatitis with salivary gland involvement triggered by innate immunity via persistent exposure to avirulent bacteria. Lab Invest 90:1757–1769
He B, Raab-Traub N, Casali P, Cerutti A (2003) EBV-encoded latent membrane protein 1 cooperates with BAFF/BLyS and APRIL to induce T cell-independent Ig heavy chain class switching. J Immunol 171:5215–5224
Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, Mehal W (2011) TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 141:358–369
Hoque R, Malik AF, Gorelick F, Mehal WZ (2012) Sterile inflammatory response in acute pancreatitis. Pancreas 41:353–357
Huang X, Dorta-Estremera S, Yao Y, Shen N, Cao W (2015) Predominant role of plasmocytoid dendritic cells in stimulating systemic autoimmunity. Front Immunol 6:526
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M et al (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411:599–603
Jeannin P, Lecoanet S, Delneste Y, Gauchat JF, Bonnefoy JY (1998) IgE versus IgG4 production can be differentially regulated by IL-10. J Immunol 160:3555–3561
Kamisawa T, Okamoto A (2006) Autoimmune pancreatitis: proposal of IgG4-related sclerosing disease. J Gastroenterol 41:613–625
Khosroshahi A, Bloch DB, Deshpande V, Stone JH (2010) Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum 62:1755–1762
Kiyama K, Kawabata D, Hosono Y, Kitagori K, Yukawa N, Yoshifuji H, Omura K, Fujii T, Mimori T (2012) Serum BAFF and APRIL levels in patients with IgG4-related disease and their clinical significance. Arthritis Res Ther 14:R86
Kono H, Rock KL (2008) How dying cells alert the immune system to danger. Nat Rev Immunol 8:279–289
Kuwata G, Kamisawa T, Koizumi K, Tabata T, Hara S, Kuruma S, Fujiwara T, Chiba K, Egashira H, Fujiwara J et al (2014) Ulcerative colitis and immunoglobulin G4. Gut Liver 8:29–34
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V et al (2011). Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 3:73ra19
Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, Cerutti A (2002) DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol 3:822–829
Mackay F, Schneider P (2009) Cracking the BAFF code. Nat Rev Immunol 9:491–502
Mackay F, Silveira PA, Brink R (2007) B cells and the BAFF/APRIL axis: fast-forward on autoimmunity and signaling. Curr Opin Immunol 19:327–336
Maehara T, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Tanaka A, Shinozaki S, Kubo Y, Nakamura S (2012) Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related dacryoadenitis and sialoadenitis, so-called Mikulicz’s disease. Ann Rheum Dis 71:2011–2019
Masaki Y, Dong L, Kurose N, Kitagawa K, Morikawa Y, Yamamoto M, Takahashi H, Shinomura Y, Imai K, Saeki T et al (2009) Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis 68:1310–1315
Mattoo H, Della-Torre E, Mahajan VS, Stone JH, Pillai S (2014a) Circulating Th2 memory cells in IgG4-related disease are restricted to a defined subset of subjects with atopy. Allergy 69:399–402
Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, Deshpande V, Stone JH, Pillai S (2014b) De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol 134:679–687
Miyoshi H, Uchida K, Taniguchi T, Yazumi S, Matsushita M, Takaoka M, Okazaki K (2008) Circulating naïve and CD4+ CD25high regulatory T cells in patients with autoimmune pancreatitis. Pancreas 36:133–140
Morikawa H, Sakaguchi S (2014) Genetic and epigenetic basis of Treg cell development and function: from a FoxP3-centered view to an epigenome-defined view of natural Treg cells. Immunol Rev 259:192–205
Moriyama M, Tanaka A, Maehara T, Furukawa S, Nakashima H, Nakamura S (2014) T helper subsets in Sjögren’s syndrome and IgG4-related dacryoadenitis and sialoadenitis: a critical review. J Autoimmun 51:81–88
Nishio A, Asada M, Uchida K, Fukui T, Chiba T, Okazaki K (2011) The role of innate immunity in the pathogenesis of experimental autoimmune pancreatitis in mice. Pancreas 40:95–102
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH et al (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411:603–606
Okazaki K, Uchida K, Fukui T (2008) Recent advances in autoimmune pancreatitis: concept, diagnosis, and pathogenesis. J Gastroenterol 43:409–418
Paul WE, Zhu J (2010) How are TH2-type immune responses initiated and amplified? Nat Rev Immunol 10:225–235
Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G, de Waal Malefyt R, de Vries JE (1993) Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci USA 90:3730–3734
Ravi K, Chari ST, Vege SS, Sandborn WJ, Smyrk TC, Loftus EV Jr (2009) Inflammatory bowel disease in the setting of autoimmune pancreatitis. Inflamm Bowel Dis 15:1326–1330
Satoguina JS, Adjobimey T, Arndts K, Hoch J, Oldenburg J, Layland LE, Hoerauf A (2008) Tr1 and naturally occurring regulatory T cells induce IgG4 in B cells through GITR/GITR-L interaction, IL-10 and TGF-β. Eur J Immunol 38:3101–3113
Schwaiger T, van den Brandt C, Fitzner B, Zaatreh S, Kraatz F, Dummer A, Nizze H, Evert M, Broker BM, Brunner-Weinzierl MC et al (2014) Autoimmune pancreatitis in MRL/Mp mice is a T cell-mediated disease responsive to cyclosporine A and rapamycin treatment. Gut 63:494–505
Sjöstrand M, Johansson A, Aqrawi L, Olsson T, Wahren-Herlenius M, Espinosa A (2016) The expression of BAFF is controlled by IRF transcription factors. J Immunol 196:91–96
Sokol CL, Medzhitov R (2010) Emerging functions of basophils in protective and allergic immune responses. Mucosal Immunol 3:129–137
Stone JH, Zen Y, Deshpande V (2012) IgG4-related disease. N Engl J Med 366:539–551
Strober W, Murray PJ, Kitani A, Watanabe T (2006) Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol 6:9–20
Strober W, Fuss I, Mannon P (2007) The fundamental basis of inflammatory bowel disease. J Clin Invest 117:514–521
Strober W, Asano N, Fuss I, Kitani A, Watanabe T (2014) Cellular and molecular mechanisms underlying NOD2 risk-associated polymorphisms in Crohn’s disease. Immunol Rev 260:249–260
Takeuchi M, Sato Y, Ohno K, Tanaka S, Takata K, Gion Y, Orita Y, Ito T, Tachibana T, Yoshino T (2014) T helper 2 and regulatory T-cell cytokine production by mast cells: a key factor in the pathogenesis of IgG4-related disease. Mod Pathol 27:1126–1136
Takeuchi M, Ohno K, Takata K, Gion Y, Tachibana T, Orita Y, Yoshino T, Sato Y (2015) Interleukin 13-positive mast cells are increased in immunoglobulin G4-related sialadenitis. Sci Rep 5:7696
Tanaka A, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Maehara T, Shinozaki S, Kubo Y, Nakamura S (2012) Th2 and regulatory immune reactions contribute to IgG4 production and the initiation of Mikulicz disease. Arthritis Rheum 64:254–263
Toyoshima M, Chida K, Kono M, Kaida Y, Nakamura Y, Suda T, Sugimura H (2010) IgG4-related lung disease in a worker occupationally exposed to asbestos. Intern Med 49:1175–1178
Ueki T, Kawamoto K, Otsuka Y, Minoda R, Maruo T, Matsumura K, Noma E, Mitsuyasu T, Otani K, Aomi Y et al (2015) Prevalence and clinicopathological features of autoimmune pancreatitis in Japanese patients with inflammatory bowel disease. Pancreas 44:434–440
Virk R, Shinagare S, Lauwers GY, Yajnik V, Stone JH, Deshpande V (2014) Tissue IgG4-positive plasma cells in inflammatory bowel disease: a study of 88 treatment-naive biopsies of inflammatory bowel disease. Mod Pathol 27:454–459
Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, Kulikova M, Deshpande V, Pillai S, Stone JH (2015) Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis 74:190–195
Watanabe T, Yamashita K, Fujikawa S, Sakurai T, Kudo M, Shiokawa M, Kodama Y, Uchida K, Okazaki K, Chiba T (2012) Activation of toll-like receptors and nucleotide-binding oligomerization domain-like receptors is involved in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis Rheum 64:914–924
Watanabe T, Yamashita K, Sakurai T, Kudo M, Shiokawa M, Uza N, Kodama Y, Uchida K, Okazaki K, Chiba T (2013) Toll-like receptor activation in basophils contributes to the development of IgG4-related disease. J Gastroenterol 48:247–253
Yamanishi H, Kumagi T, Yokota T, Azemoto N, Koizumi M, Kobayashi Y, Abe M, Murakami H, Hiasa Y, Matsuura B et al (2011) Clinical significance of B cell-activating factor in autoimmune pancreatitis. Pancreas 40:840–845
Yanagisawa N, Haruta I, Shimizu K, Furukawa T, Higuchi T, Shibata N, Shiratori K, Yagi J (2014) Identification of commensal flora-associated antigen as a pathogenetic factor of autoimmune pancreatitis. Pancreatology 14:100–106
Yasuda T, Ueda T, Takeyama Y, Shinzeki M, Sawa H, Nakajima T, Ajiki T, Fujino Y, Suzuki Y, Kudoda Y (2006) Significant increase of serum high-mobility group box chromosomal protein 1 levels in patients with severe acute pancreatitis. Pancreas 33:359–363
Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, Nakanuma Y (2007) Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology 45:1538–1546
Zong M, Bruton JD, Grundtman C, Yang H, Li JH, Alexanderson H, Palmblad K, Andersson U, Harris HE, Lundberg IE, Westerblad H (2013) TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann Rheum Dis 72:1390–1399
Acknowledgments
This work was supported by Grant-in-Aid for Scientific Research (25293172, 15K15370) from the Japan Society for the Promotion of Science; the Kato Memorial Trust for Nambyo Research; and the Health and Labor Sciences Research Grants for Research on Intractable Diseases from the Ministry of Health, Labor and Welfare, Japan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Watanabe, T., Yamashita, K., Kudo, M. (2016). IgG4-Related Disease and Innate Immunity. In: Okazaki, K. (eds) IgG4-Related Disease. Current Topics in Microbiology and Immunology, vol 401. Springer, Cham. https://doi.org/10.1007/82_2016_42
Download citation
DOI: https://doi.org/10.1007/82_2016_42
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52541-9
Online ISBN: 978-3-319-52542-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)