
Abstract
The innate host response to influenza virus infection plays a critical role in determining the subsequent course of infection and the clinical outcome of disease. The host has a diverse array of detection and effector mechanisms that are able to recognize and initiate effective antiviral responses. In opposition, the virus utilizes a number of distinct mechanisms to evade host detection and effector activity in order to remain “stealthy” throughout its replication cycle. In this review, we describe these host and viral mechanisms, including the major pattern recognition receptor families (the TLRs, NLRs, and RLRs) in the host and the specific viral proteins such as NS1 that are key players in this interaction. Additionally, we explore nonreductive mechanisms of viral immune evasion and propose areas important for future inquiry.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Influenza virus is a pleomorphic enveloped virus of approximately 100 nm in diameter, containing a single-stranded negative sense segmented genome. The genus Influenzavirus belongs to the family Orthomyxoviridae and consists of three serotypes: Influenza A, B, and C. Influenza A viruses show considerable sequence variation in their surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA). Based on phylogenetic and serological analyses, Influenza A virus (IAV) is known to contain 17 HA and 10 NA subtypes (Shaw and Palese 2007). Only H1, H2, H3 and N1 and N2 subtypes have been known to cause sustained human-human infection and transmission. Occasionally, animal viruses such as H5N1, H9N2, and H7N9 infected humans as well and are capable of causing severe disease; additional, subclinical infections likely occur with other strains but are not detected due to the absence of symptoms (Wang et al. 2009; Uyeki et al. 2012; Imai et al. 2012; Herfst et al. 2012; Gao et al. 2013; Song et al. 2014). The influenza B virus (IBV) is structurally similar to IAV but only one subtype of HA has been found. IAV and IBV each contain eight segments in their genomes, whereas Influenza C virus (ICV) consists of only seven segments. Influenza C virus (ICV) has a surface glycoprotein known as the hemagglutinin-esterase-fusion (HEF) protein that mediates both binding and fusion and possesses an additional receptor destroying enzymatic activity (analogous to the NA of IAV and IBV) that destroys the HEF-9-O-acetylated sialic acid by enzymatic removal of the acetyl group. Thus, the ICV do not have a separate protein with neuraminidase activity (Luo 2012).
The eight segments of influenza A virus encodes 13 proteins (Palese and Shaw 2007). Of these 8 segments, segment 1, 4, 5, 6 are monocistronic and encode for PB2, HA, NP, and NA proteins, respectively. The other segments (2, 3, 7, and 8) in addition to their primary transcripts and proteins, also generate additional proteins by alternative splicing or frame shifts. Segment 2 that encodes the polymerase subunit PB1 also expresses two other proteins in some strains by ribosomal frame shift due to leaky ribosomal scanning: PB1-F2 and PB1-N40 (Chen et al. 2001; Wise et al. 2009). PB1-F2 is 87 amino acids in length and has been described as a virulence factor causing cell death. However, the contribution of PB1-F2 to virulence is still not clear since the protein is normally found to be expressed in avian strains and eliminated by truncation when adapted to mammalian hosts (Zell et al. 2007). Notably, the swine strains of IAV lack expression of PB1-F2 (Chen et al. 2001; Zell et al. 2007; McAuley et al. 2010). In addition, the effect of PB1-F2 on cell death was found to be cell (Chen et al. 2001; Yamada et al. 2004) and strain specific (McAuley et al. 2010). For some strains, an additional virulence factor, PB1-N40, was recently identified (Wise et al. 2009), although the function of this protein is not well-known. In vitro, though not essential, it has been reported to support virus replication (Wise et al. 2009). The third segment of IAV primarily expresses the viral protein PA, and recently has been reported to express an alternate product termed PA-X (Jagger et al. 2012). Additionally, the mRNA from segments 7 and 8 of IAV that encode for M1 and NS1, respectively, also give rise to alternatively spliced mRNA that express M2 and NS2, also known as NEP (Palese and Shaw 2007).
2 Influenza Virus Infection of Host Cell
The primary target cells for influenza virus infection are the respiratory epithelial cells of the upper respiratory (e.g., nasal airways and trachea) and the lower respiratory (small airways and lungs) tracts (Sanders et al. 2011). The prevailing view is that infection by the virus is achieved by HA-binding to these cells via sialic acid moieties on the cell surface, which triggers internalization of the virus via endocytosis. Early studies on influenza virus entry using electron microscopy and radioisotope-labeled viruses suggested that phagocytosis (perhaps indistinguishable from receptor-mediated endocytosis at that time) is a major route for virus entry (Patterson et al. 1979; Matlin et al. 1981). The endocytic pathway involves both clathrin-mediated and nonclathrin-, noncaveolin-mediated endocytosis (Sieczkarski and Whittaker 2002; Rust et al. 2004). An alternative pathway, macropinocytosis, has been described for entry of large filamentous and spherical influenza virions (de Vries et al. 2011; Rossman et al. 2012). Though it is speculated that internalization of viruses through pinocytic vesicles will converge with the endocytic pathway at some point, the exact route and kinetics of merging is not known (de Vries et al. 2011). Apart from endocytosis and pinocytosis, reports have described influenza virus infection of cells by attachment to the plasma membrane followed by direct release of the viral contents into the cytoplasm (Hoyle and Finter 1957; Morgan and Rose 1968; White et al. 1981). This pathway is similar to a mechanism used by many other viruses, e.g., SFV (White et al. 1980), HIV (27, 28), CMV (Compton et al. 1992), RSV (Srinivasakumar et al. 1991), and HSV (Sarmiento et al. 1979). It was shown that, in mildly acidic pH conditions, influenza virus can fuse to the plasma membrane of MDCK cells (Matlin et al. 1981). Another study observed direct fusion by electron microscopy in infected chorioallantoic membrane (an ex vivo, polarized, epithelial structure), suggesting that viral entry pathways may be dependent on the type and differentiation of the target cells (Morgan and Rose 1968). Indeed, work with well-differentiated human airway epithelial cell cultures found that broad-spectrum neuraminidase treatment did not affect influenza virus entry, though it did alter the entry of hPIV3, another sialic acid receptor-using virus (Kogure et al. 2006; Thompson et al. 2006). Further, cells lacking sialylated N-glycans can be infected with influenza virus (Stray et al. 2000; Thompson et al. 2006; Nicholls et al. 2007; Oshansky et al. 2011; de Vries et al. 2012) suggesting an alternative, nonsialic acid receptor-dependent route exists.
However, in most cell types and experimental systems that have been studied, the influenza virus HA initiates host cell infection by binding to sialic acid receptors. The HA subtypes of the well-adapted human IAV have been described to bind to α (2, 6)-sialic acid linkages preferentially. In contrast, the avian-adapted HA subtypes preferentially bind to α (2, 3)-sialic acid linkages. In polarized cells, influenza virus infection through the endocytic route results in internalization of the virus into early endosomes in an actin-dependent manner (Sun and Whittaker 2007) followed by trafficking through the endocytic network in a multistep process (Lakadamyali et al. 2003, 2006; Sieczkarski and Whittaker 2003). Release of viral nucleic acids from the endosome following binding and endocytosis of the virus is a key step in initiating productive infection and requires trafficking of virus containing endosomes. During this trafficking to the late endosomal stage, the interior pH of the endosome drops, which is sensed by the tetrameric viral M2 protein causing the M2 ion channel to assume an open configuration (Pinto et al. 1992). The H+ ions enter into the virion through this opening causing destabilization of the M1 protein from the ribonucleic protein (RNP) complex of the virus. Lowering of pH further induces conformational changes of the HA2 subunit resulting in exposure of the hydrophobic fusion peptide motif of the HA. When the fusion peptide comes in close apposition of the endosomal membrane, the peptide is inserted into the membrane (White and Wilson 1987; Xu and Wilson 2011; Fontana et al. 2012). Several HA molecules come together to form a cluster known as the “fusogenic unit” and the individual HA unit undergoes further conformational changes resulting in fusion of the viral envelope and capsid with the endosomal membrane and formation of the pore (Hamilton et al. 2012). The pH of activation of HA varies among IAV strains, thus playing an important role in determining the fitness of the virus in the infection process.
Following the pH-dependent fusion and formation of the pore between the virion and endosomal membranes, the viral RNPs are released into the cytoplasm (frequently described as uncoating). The RNPs are then transported to the nucleus where transcription of the viral genome occurs using the viral RNA-dependent RNA polymerase (RdRp), generating 5′ cap structures with short nucleotides derived from host cell mRNA. This is achieved by the binding of the PB2 subunit of the viral RdRp to the 5′ cap of host mRNAs and subsequent cleaving by the PA subunit approximately 10–15 nt downstream of the cap structure. The elongation of the mRNA from the viral genome is carried out by the PB1 subunit using the 5′cap + nucleotides as primers, which also adds a polyadenylated [poly(A)] tail by means of a stuttering mechanism on a sequence of uridine residues near the 5′ end of the negative-strand genome. The viral mRNAs are transported back to the cytoplasm for translation. In the nucleus, the genome of the virus is also replicated through an intermediate RNA template (cRNA) step by the RdRp. This intermediate lacks the poly (A) tail or any 5′ modification. In the nucleus, newly synthesized genomes are encapsidated with NP, RdRP subunits, PB2, PB1, and PA to form RNPs and are transported to the cytoplasm by binding with M1 and NEP. Other viral components such as HA, NA, and M2 are transported to the plasma membrane via the Golgi network and assemble with RNPs to form mature virus particles that are released from the cell via budding (Palese and Shaw 2007)
3 Host Cell Response to Influenza Virus
Upon infection of respiratory epithelial cells with influenza virus, the host rapidly initiates an innate immune response . The first responder to the virus infection is the infected cell itself, which plays a major role in coordinating the ensuing innate immune response. Viral induction of the innate immune response can be divided into early (initiation and amplification) and late responses (local systemic response) that cause the recruitment of innate cells to the site of infection. Sensors on the cell surface, endosomes, and cytosol have been found to detect ligands such as incoming virus particles or their unique molecular signatures. These sensors activate downstream signaling and induce two types of responses: an antiviral response and a proinflammatory response. Subsequently, concurrent viral replication generates more of these ligands (within the first few hours) which amplify these responses in infected (in the first replication cycle of the virus) and neighboring cells (following release of progeny virus). Production of a variety of cytokines and chemokines in the initiation phase of the early response locally attracts other cellular mediators of the innate response including neutrophils, macrophages, monocytes, and NK cells. In this review, we discuss the early innate immune response to IAV and the viral strategies and characteristics that evade this host cell response.
As mentioned above, the host cell possesses an array of innate immune sensors to detect components of invading pathogens that are nonself. Alternatively, there are also sensors that recognize host cell-derived components under certain pathologic conditions such as the cellular stress response often associated with infection, metabolic disorders, and cancer. These sensors are collectively termed as pattern recognition receptors (PRRs ). The PRRs recognize unique molecular signatures known as pathogen-associated molecular patterns (PAMPs), which are nonself. The self-derived molecular patterns arising following pathogen infection and/or cellular stress responses, such as reactive oxygen species (ROS), ATP, changes in ion flux, and denatured cytosolic and nuclear contents, including DNA from necrotic cells, are broadly termed danger-associated molecular patterns (DAMPs). Thus, PRRs, by sensing DAMPs, can also play a role in tissue homeostasis (Marshak-Rothstein 2006). To date, known PRRs have been classified into five major groups: Toll-like receptors (TLR s), retinoic acid-inducible gene-I (RIG-I) like receptors (RLRs), nucleotide oligomerization and binding domain (NOD) like receptors (NLR s), C-type lectin receptors (CLR s) and the Pyrin-HIN (PYHIN) domain containing family that includes AIM2. It has been shown that at least three (TLRs, RLRs and NLRs) out of the above five PRR families play a role in innate immunity to influenza virus infection (Lupfer and Kanneganti 2013). We will discuss them in detail below.
There is considerable variation in the expression pattern of the PRRs in tissues and at the cellular level. In general, the TLRs are both intracellular (compartmentalized and cytosolic) and on the cell surface, whereas the RLRs and NLRs are exclusively cytosolic. Consequently, these PRRs demonstrate differential recognition for various PAMPs and DAMPs.
3.1 Toll-Like Receptors
Among the PRRs, the Toll receptor in Drosophila was first shown to be important for protection against fungal infection (Lemaitre et al. 1996). Subsequently, the mammalian homologue of the Toll receptors, the TLRs, were described for humans and mice (Kawai and Akira 2011). Thus far, there have been 10 and 12 TLRs described for humans and mice, respectively. Based on sequence comparisons of several vertebrate genomes, the TLRs are classified into six major families based on PAMP recognition (Roach et al. 2005). They are: TLR1 (TLR 1, TLR2, TLR6 and TLR10 (only in human)), TLR3 (TLR3), TLR4 (TLR4), TLR5 (TLR5), TLR7 (TLR7, TLR8 (human-only) and TLR9) and TLR11 (mouse only, including TLR11, TLR12 and TLR13). Each family recognizes a general class of PAMPs (Roach et al. 2005). The TLRs reported to play a role in antiviral immunity include TLR3, TLR7, TLR8, and TLR9. TLR expression varies across different cell types. There have been differing reports on the location of TLR3 on epithelial cells, perhaps resulting from the differentiation status of the cells in cultures across different experiments. Using primary cell cultures, it was shown that TLR3 expression is limited to the cell surface in human bronchial epithelial cell lines (BEAS-2B) (Hewson et al. 2005) and fibroblasts (Matsumoto et al. 2002). Expression was upregulated upon rhinovirus infection and active virus replication (Hewson et al. 2005). In contrast, Guilott et al., showed intracellular expression of TLR3 in uninfected BEAS-2B cells and the human lung carcinoma cell line A549 (Guillot et al. 2005). Expression of TLRs 1 to 6 has been reported in polarized mouse uterine epithelial cells (Soboll et al. 2006). Similarly, cervical epithelial cells of female mice express mRNA for TLR3, TLR9, and TLR7, but had only a weak signal for TLR8 (Andersen et al. 2006). Macrophages, fibroblasts, and some epithelial cell lines express TLR3 both on the cell surface and in the early endosome (Cario and Podolsky 2000). Recently, using an immunofluorescence technique on an in vitro polarized model of human alveolar epithelial cells (AEC) and frozen human tracheal tissues, which are important cells for influenza virus infection, Ioannidis et al. reported that TLR1 and TLR3 are expressed on both apical and basolateral surfaces of the hAEC (Ioannidis et al. 2013). In contrast, TLR2 and TLR6 were predominantly expressed basolaterally. TLR4, TLR5, TLR7, TLR9, and TLR10 were expressed weakly on the apical side or the luminal surface of the epithelial cells. It was also reported that TLR3, TLR7, and TLR9, which have been reported to be expressed only on the endosomal compartment of the DC (Muzio et al. 2000; Matsumoto et al. 2003; Funami et al. 2004), were found to be present in both intracellular and apical surfaces mostly restricted to the cilial border of human tracheal epithelium (Ioannidis et al. 2013). No TLR8 expression was found in respiratory epithelial cells in vivo and in vitro (Ioannidis et al. 2013).
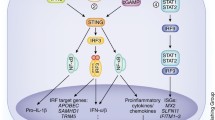
The TLRs are classified as type I trans-membrane proteins with extracellular leucine-rich repeats that recognize cognate ligands, a trans-membrane domain, and a cytoplasmic domain consisting of a conserved signaling Toll/interleukin-1 receptor (TIR) domain. It has been reported that endosomal acidification is required for the activation of TLRs such as TLR3 (Ioannidis et al. 2013) and TLR9 (Macfarlane and Manzel 1998). Stimulation of TLRs with PAMPs can lead to downstream signaling in two main pathways: the MyD88 signaling pathway (proinflammatory) and the TRIF signaling pathway (antiviral). All TLRs except TLR3 activate NFκB and the IRF3/7 response in a MyD88-dependent manner for signaling. TLR3 exclusively uses the TRIF-dependent pathway, whereas TLR4 uses both the TRIF and MyD88 pathways (Takeuchi and Akira 2007).
The prominent cytokines of the antiviral response are the type I and type III interferons (IFN). In humans, the type I IFNs comprises IFN-α, IFN-β, IFN-ω, IFN-ε, and IFN-κ (Samuel 2001). There are 13 different subtypes of IFN-α and only one IFN-β characterized in human (Díaz and Testa 1996). The type III IFNs consist of IFN-λ1, λ2, and λ3 (also known as IL29, IL28A and IL28B, respectively) (Kotenko et al. 2003; Sheppard et al. 2003) and have been described to play important roles in epithelial antiviral responses (Durbin et al. 2013).
Since IAV infects through the endocytic route, one might expect that TLR3, TLR7, and TLR8 should be major sensors for IFN induction by viral infection. Early studies showed that TLR7 plays a major role in sensing influenza viral RNA (Diebold et al. 2004; Lund et al. 2004). pDC derived from mice deficient in TLR7 showed marked decreases in IFNα production in tissue culture supernatants after infection (Diebold et al. 2004; Lund et al. 2004). Since uncoating of the viral RNA from the endocytosed virion into the cytoplasm occurs in a carefully orchestrated maturation process, the precise mechanisms by which the IAV RNA exposure to TLR7 occurs in the endosome is currently not known. It was suggested that proteosomal degradation of some virions in the endosomes may lead to exposure of the viral RNA to TLR7 (Diebold et al. 2004). It also can be speculated that following viral replication de novo generated viral RNA is released from dead or dying cells or by phagocytosis of dead cells allowing the viral RNA to enter the endocytic route, resulting in TLR7 mediated recognition and signaling. Alternatively, differential entry such as pinocytosis or isolated endocytic vesicles containing virions that failed to achieve the optimal pH of activation (see discussion below) may lead to lysosomal degradation, thus exposing the viral RNA to TLR7.
The role for TLR3 in recognition of IAV was shown to be more proinflammatory than antiviral. It was found that after TLR3 stimulation the BEAS-B2 cells produced more of the proinflammatory cytokines IL6 and IL8 and less IFN-β (Le Goffic et al. 2007). Interestingly, TLR3-/- mice were shown to have a survival advantage after infection with IAV, despite higher viral loads and lower viral clearance (Le Goffic et al. 2006) indicating that perhaps these proinflammatory cytokines are not essential for effective viral clearance. A similar phenomenon was seen in TLR4 knockout mice, which had better survival than the TLR4 sufficient animals after IAV infection (Imai et al. 2008; Nhu et al. 2010). Though a specific ligand derived from IAV is not known for TLR4, it was shown that endogenous oxidized phospholipids (Imai et al. 2008) and proteins such as S100A9 (Tsai et al. 2014) produced in response to the acute lung injury in IAV infection can act as DAMPs and stimulate the TLR4-TRIF-NF-κB signaling pathway. The authors of these two reports went on to demonstrate that enhanced acute lung injury was alleviated by the elimination of IL6 in H5N1 and IAV PR8 infected mice, though contributions by cytokines other than IL6 cannot be ruled out.
A role for TLR10 has recently been described in influenza infection (Lee et al. 2014). The authors reported that H1N1 and H5N1 infection of primary human macrophages and a human monocyte cell line induced type I and III IFNs and proinflammatory cytokines IL8 and IL6 in a TLR10-dependent manner. H5N1 virus infection induced robust TLR10 expression, indicating that the severe inflammation seen in influenza infection can be amplified by positive feedback through TLR10. TLR10 induction required active replication of the virus; however, the viral component(s) that induced the TLR10-dependent inflammatory response have not been identified. Although TLR10-knockdown reduced the expression of IFNs and cytokines, it did not have any effect on viral replication (Lee et al. 2014), suggesting that TLR10 signaling may play a role in shaping local innate effectors such as monocytes and macrophages and influencing the adaptive immune response, rather than modulating the antiviral response within the epithelial cell. While these reports highlight a negative role in clinical outcomes for TLR3 and TLR4 in mice and TLR10 in in vitro cultures, associated with heightened inflammation, it is possible that at least one PRR is necessary for effective control of the virus through initiation of an appropriate inflammatory response leading to functional wound healing. These situations might represent a special case of virulent viruses, as is the case for both pathogenic H5N1 and to a lesser extent PR8, where stimulation of these TLRs results in an inappropriate inflammatory response and pathogenesis.
Finally, the nonclassical PRR PAR2 (proteinase-activated receptor) is expressed on epithelial cells of the respiratory and gastrointestinal tract and works synergistically with TLR4 to suppress TLR3—mediated IRF3 activation and induction of ISGs. Mice deficient in PAR2 or TLR4 were protected from influenza–associated disease, potentially as a result of derepression of the TLR3-IRF3 signaling pathway (Nhu et al. 2010).
3.2 RIG-I-Like Receptors (RLRs)
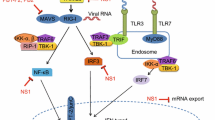
The RIG-I-like receptors or the RLRs consist of three members identified in humans and mice: the Retinoic acid-inducible gene-I (RIG-I, also known as DDX58), melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2, also known as DHX58). RIG-I and MDA5 consist of two N-terminal caspase recruitment domains (CARD), a central DExD/H box helicase with RNA binding and ATPase functions, and a C-terminal domain consisting of a regulatory domain (absent in MDA-5). In contrast, the LGP-2 does not have the CARD domain but contains a DExD/H box helicase and a regulatory domain similar to that of RIG-I, which is absent in MDA-5. The regulatory domain keeps the molecule in a closed conformation, inhibiting downstream signaling prior to activation. Upon ligand interaction, RIG-I and LGP are thought to undergo a conformational change leading to activation of antiviral signaling (Saito et al. 2007). The RLRs have been reported to be expressed in most tissue types and are maintained typically at low levels (Loo and Gale 2011). However, following infection or interferon exposure, the expression of the RLRs was shown to be greatly increased (Kang et al. 2004; Yoneyama et al. 2004; Tatsuta et al. 2012). Increased expression following ectopic overexpression (Yoneyama et al. 2005) or infection in the absence of IFN signaling (Yount et al. 2007) suggests that this low intrinsic basal expression pattern in part regulates unnecessary and potentially detrimental RLR activation in the homeostatic condition.
RIG-I and MDA-5 are key sensors of infection mediated by RNA viruses. They share structural and functional similarities but detect mostly nonoverlapping groups of viruses. Over the last few years, RIG-I has been shown to play a role in triggering innate immune responses to infection by negative-strand viruses such as paramyxoviruses, orthomyxoviruses, rhabdoviruses such as vesicular stomatitis virus, Ebola virus, and EBER RNA, carried by the Epstein-Barr virus (Kato et al. 2006; Cárdenas et al. 2006; Samanta et al. 2006; Yoneyama and Fujita 2007). In contrast, MDA-5 has been implicated in sensing positive strand RNA viruses such as picornaviruses (Kato et al. 2006; Gitlin et al. 2006). Certain flaviviruses induce innate immune responses via RIG-I and MDA5 (Sumpter et al. 2005; Chang et al. 2006; Fredericksen and Gale 2006; Kato et al. 2006; Loo et al. 2008). The ligands for both RIG-I and MDA-5 have been identified primarily through infection with a variety of viruses or transfection with viral or synthetic RNA such as Poly (I)(C) (Sumpter et al. 2005; Kato et al. 2006; Saito et al. 2007; Liu et al. 2007). Using RIG-I-deficient animals, Kato et al. showed that RIG-I plays an important role in initiating antiviral responses in fibroblasts and conventional DC but not plasmacytoid DC (Kato et al. 2005). Conversely, TLRs drive the initiation of antiviral responses in pDC with no contribution from RIG-I (Kato et al. 2005). Initially it was suggested that RIG-I and MDA5 were involved in sensing dsRNA generated from viral infection (Kang et al. 2002; Yoneyama et al. 2004; Andrejeva et al. 2004; Yoneyama et al. 2005; Rothenfusser et al. 2005). It was further shown that while RIG-I may be involved in recognition of short dsRNAs of less than 1 kb, MDA-5 is responsible for sensing long dsRNA (>1 kb) (Kato et al. 2008). Subsequently, it was reported that the 5′ triphosphate-linked ssRNA characteristic of viral RNA products can be recognized by RIG-I (Hornung et al. 2006; Pichlmair et al. 2006). Later, it was found that RIG-I responds to short dsRNAs with a triphosphorylated 5′ terminus and poly-U/A-rich sequences, while MDA5 activation is more dependent on complex dsRNA structures (Hornung et al. 2006; Kato et al. 2008; Saito and Gale 2008; Schlee et al. 2009; Schmidt et al. 2009; Pichlmair et al. 2009; Binder et al. 2011). A role for RIG-I in the detection of RNA products from DNA virus infections has also been described. Cellular RNA polymerase III can convert double-stranded DNA poly(dA-dT) to a short dsRNA species with a 5′-triphosphate that triggers the RIG-I pathway in human and mouse cells (Chiu et al. 2009).
In contrast, LGP2 which lacks signaling CARDs, binds to both dsRNA (Pippig et al. 2009) and single-stranded RNA with 5′-triphosphates via its C-terminal regulatory domain with greater affinity than RIG-I and MDA5 (Takahasi et al. 2009). There have been contrasting reports on the role of LGP2 in viral RNA sensing and signaling by RLR. While it was suggested that LGP2 inhibits RIG-I signaling and activity both in vitro (Yoneyama et al. 2005) and in vivo (Rothenfusser et al. 2005), augmentation of MDA5 signaling by LGP2 was also shown (Venkataraman et al. 2007; Satoh et al. 2010). LGP2 functions in cDC but not in pDC (Satoh et al. 2010) and is dispensable for the recognition of synthesized dsRNA and 5′ triphosphate RNA (Saito et al. 2007).
While many of the above studies involved chemically synthesized or virus-derived RNA, the exact molecular signature of the RNA (analogous to the specific epitope in adaptive immunity) is poorly studied. Saito et al mapped these unique signatures to two regions of the HCV genome: nucleotides 2406–3256 and 8872–9616, by measuring IFN-β promoter activity (Saito et al. 2008). Similarly, in IAV, single stranded viral RNA with a 5′ triphosphate derived from in vitro RNP reconstitution assays were shown to be the ligand for RIG-I, with the strength of IFNβ induction similar for all IAV gene segments (Rehwinkel et al. 2010). Surprisingly, in the absence of mRNA transcription, IFNβ induction was enhanced, suggesting the vRNA and cRNA (which do not have 5′ triphosphate), but not the mRNA, are potent inducers of IFN via RIG-I pathway (Rehwinkel et al. 2010) indicating molecular signatures in the termini may be playing a role. Indeed, recently a short region (27 nt) of 5′ UTR and a longer region of 3′ UTR of the IAV NS gene segment was shown to induce RIG-I dependent IFN-β induction (Davis et al. 2012). Further, a U/A rich region within the 3′ UTR was shown to be a potent inducer in a 5′ triphosphate-independent manner (Davis et al. 2012).
Interestingly, it was shown that infection of mice with a virus (PR8) stock containing high defective interfering (DI) particle content had increased protection compared to the group infected with low DI content (Tapia et al. 2013). The DI influenza particles are incomplete virus particles with shorter genome segments but identical termini (Tapia et al. 2013). In the case of influenza, the DI genome is mostly enriched for PB2, PB1, PA (Saira et al. 2013), and M gene (Noble and Dimmock 1995). Tapia and colleagues showed Sendai virus DI particles generated in cells during the course of a normal infection induced stronger antiviral responses than the cells with full length virus genomes (Tapia et al. 2013). Though a direct measurement of the antiviral cytokines produced between the high and low DI particle-containing IAV infections was not shown in those studies, it is tempting to assume that the 3′ and 5′ termini of the IAV DI may be a better stimulant of RIG-I and thus cause greater IFN production. More recently, direct recognition of IAV RNPs by RIG-I has been reported (Weber et al. 2013) suggesting that our knowledge of the nature of the ligands that activate these sensors is incomplete.
3.3 Nod-Like Receptors (NLRs)
The NLR family consists of a large number (20) of intracellular sensors with a conserved nucleotide oligomerization domain (NOD) (Lupfer and Kanneganti 2013). At the N-terminus, the NLRs contain a protein interacting caspase activation and recruitment (CARD) or pyrin domain (PYD) that drive recruitment and binding to the protein caspase-1 directly, or through an intermediate interaction with the adaptor protein, apoptosis-associated speck-like protein containing a CARD (ASC) . At the C-terminus, NLRs contain a variable leucine-rich repeat (LRR) involved in PAMP sensing (Strowig et al. 2012). The NLRs have been shown to be expressed in a variety of cells and are also induced after influenza infection (Ichinohe et al. 2009; Allen et al. 2009).
Of the known NLRs, stimulation of NLRP1, NLRP3, and NLRC4 with PAMPs and DAMPs causes activation of the caspase-1 containing inflammasome (Thomas et al. 2009; Malireddi et al. 2010; Masters et al. 2012). The activated caspase-1 cleaves inactive pro-IL1β and pro-IL18 to their active form (Black et al. 1989) which then signals through target cell receptors to induce an array of proinflammatory cytokines, including, KC, MIP1α, IL6, TNFα, and IFN-γ (Dinarello et al. 1998; Allen et al. 2009; Thomas et al. 2009). Alternatively, caspase-1 activation can lead to a cell death known as pyroptosis (Bergsbaken et al. 2009). Other NLRs such as NLRP12 (Zaki et al. 2011) and NLRP6 (Anand et al. 2012), have been implicated in down-modulating canonical NF-κB signaling, thus acting as anti-inflammatory stimuli.
The ligands that induce NLR signaling are thus far not well-defined. Initially, it was suggested that dsRNA but not ssRNA could induce activation of the NLRP3 inflammasome in a TLR3-independent manner (Kanneganti et al. 2006; Rajan et al. 2010). Infection of NLRP3-deficient macrophages with the IAV PR8 strain did not activate caspase-1, while wild type macrophages had robust activation (Kanneganti et al. 2006). However, it is known that influenza virus does not generate dsRNA during replication (Weber et al. 2006). Indeed, intraperitoneal injection of NLRP3-deficient mice with influenza viral RNA derived from stock virus resulted in reduced IL1β and Il18 production compared to wild type animals (Allen et al. 2009; Thomas et al. 2009). Additionally, our own studies and related work observed decreased survival to influenza virus infection in NLRP3-deficient mice (Allen et al. 2009; Thomas et al. 2009). Another report using similar mice found defective adaptive immune responses to IAV, but failed to see any difference in mortality (Ichinohe et al. 2009). This work used a different infection dose than the other studies, which may account for the difference in mortality. The absence of NLRP3 also caused reduced cellular infiltration to the lungs and an improper wound healing response, but did not compromise type IFN induction (Thomas et al. 2009).
While direct recognition of the RNA ligands by NLRs is still under investigation, other viral components have been reported to induce antiviral responses. The IAV M2 protein ion channel activates the inflammasome in an NLRP3-dependent manner (Ichinohe et al. 2010). Other chemical stimuli such as uric acid crystals (Martinon et al. 2006), silica, and asbestos, (Dostert et al. 2008; Hornung et al. 2008), changes in intracellular ion flux (Franchi et al. 2007; Pétrilli et al. 2007), abnormal reactive oxygen species (ROS) (Dostert et al. 2008; Allen et al. 2009), and abnormal protein aggregates such as amyloid-β (Halle et al. 2008) have been suggested to activate NLRs as second messengers. Endosomal destabilization has also been implicated in the activation of NLRP3 inflammasome (Hornung and Latz 2010; Tschopp 2011). The absence of a known ligand and the diverse nature of the ligands that have found to stimulate NLRs have led to a two signal model for inflammasome activation (Meylan et al. 2006; Ogura et al. 2006). In this model, initial upstream signals such as TLR, RLR, or the cellular stress response (ATP/ROS/endosomal destabilization) induces transcription and translation of proIL1β, proIL18, and NLRP3 as a part of the proinflammatory program, which is followed by a second signal triggering NLRP3 to activate caspase-1 and produce active IL1β and IL18.
Apart from these indirect roles in inflammasome activation, recent reports have suggested a direct interaction of the PRRs in the assembly of the inflammasome that does not require NLRs. Poeck et al. showed that after infection with the negative-strand virus VSV, RIG-I, rather than initiating its canonical sensing of ssRNA and signaling through MAVS, instead interacts with CARD9–Bcl-10-ASC causing inflammasome assembly and caspase-1 activation in an NLRP3-independent manner (Poeck et al. 2010). Similar observations were also found by Pothichet et al., who reported an overlapping TLR3/RIG-I/NLRP3 pathway in inflammasome activation by IAV in normal human bronchial epithelial cells (Pothlichet et al. 2013). The authors suggest that RIG-I occupies the most upstream position in this pathway. When it senses viral ssRNA, it induces the MAVS-IFN pathway. In addition, as shown by Poeck et al., they also found RIG-I interactions with ASC and caspase-1. They went on to suggest that IFN-driven upregulation of TLR3 and NLRP3 sets off a positive feedback loop of inflammasome activation (Pothlichet et al. 2013). This provided a direct example of a two signal model of NLR activation as suggested before.
NLRC1 (NOD1) and NLRC2 (NOD2) have been known to signal through the adapter protein RIPK2 (also known as RICK or RIP2) and induce NFκB and MAPK signaling. A role for NOD2 as a viral PRR has been shown. Transfection of viral genomes derived from RSV, IAV and PIV into A549 cells showed IRF3 activation in a NOD2-dependent manner (Sabbah et al. 2009). Another indirect role for NOD2 in inhibition of viral replication has been described (Dugan et al. 2009). NOD2 increases the enzymatic activity of OAS2 which in turn enhances the activity of RNAse L to degrade viral RNA (Malathi et al. 2007; Dugan et al. 2009). Finally, Lupfer et al. showed that IAV infection results in RIPK2-induced mitophagy in infected cells. This process is not NFkB or MAPK dependent (Lupfer et al. 2013).
4 Evasion Strategy by Influenza Virus
Due to their clinical importance, significant work has been conducted to understand how influenza viruses can evade effective antiviral responses. The NS1 protein has received most of the attention, along with contributions from some other viral determinants. However, other “nonreductive” features of the viral life cycle also appear to contribute to a stealthy phenotype but cannot be easily ascribed to an isolate feature of the viral genome. We discuss both types of strategies below.
4.1 Viral Determinant of Innate Immune Evasion
The eighth segment of influenza virus encodes the NS gene, consisting of two separate products: NS1 and NEP (NS2). NS1 plays a major role in regulating the host cell response (García-Sastre 2011). It has been implicated in the inhibition of dsRNA sensors such as TLR3 by sequestering dsRNA intermediates (Talon et al. 2000). Though a known dsRNA ligand from IAV infection has not been detected (Weber et al. 2006), it is possible that NS1 may be targeting downstream pathways and RNA sensors, including RIG-I (Guo et al. 2007; Rehwinkel et al. 2010) to inhibit antiviral responses. NS1 has been shown to interact directly with RIG-I (Mibayashi et al. 2007) and indirectly via binding to ligand RNA (Pichlmair et al. 2006; Rehwinkel et al. 2010). However, NS1 inhibition of the ubiquitin ligase tripartite-motif containing protein 25 (TRIM25) has been observed, preventing the ubiquitination of RIG-I which limits its signal transduction activity (Gack et al. 2009). This suggests there are multiple, possibly overlapping, pathways for NS1 to inhibit RIG-I activation. Further downstream, NS1 has also been found to inhibit activation and nuclear translocation of the transcription factors IRF3 (Talon et al. 2000), NF-κB (Wang et al. 2000) and ATF-2/c-Jun (Ludwig et al. 2002). In vivo infections of mice and pigs with IAV NS1 mutants have demonstrated reduced pathogenesis (Garcia-Sastre et al. 1998; Talon et al. 2000; Donelan et al. 2003; Falcon et al. 2005; Solórzano et al. 2005).
Limiting host cell protein synthesis as a viral evasion strategy has been described for multiple viruses (157), reducing the antiviral response and channeling the host cell protein synthesis machinery for replication of the virus. IAV has been observed to utilize this strategy via the viral protein PA-X, which is translated from the PA gene segment in an alternative reading frame (Desmet et al. 2013). Similarly, NS1 has been implicated in the alteration of host cell mRNA by binding to cleavage and polyadenylation specificity factor (CPSF30) (Nemeroff et al. 1998; Noah et al. 2003). Other polymerase proteins have been implicated in suppression of host cell protein synthesis. The well-described “cap snatching mechanism” involving PB2 and PA for viral mRNA transcription (Plotch et al. 1981; Dias et al. 2009) has been suggested to reduce the levels of capped host mRNAs, attenuating host cell gene expression including the antiviral response (Nakhaei et al. 2009).
The phosphatidylinositol-3-kinases (PI3K) are a family of enzymes involved in cellular functions such as cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking (Datta et al. 1999). IAV NS1 has been shown to induce PI3K activation by direct binding of the p85 subunit (Hale et al. 2006). Later in the infection, NS1 induced activation of PI3K and its downstream effector AKT/Protein kinase B prevents premature apoptosis and favors viral replication.
Recombinant IAV with a deletion of NS1 induces a stronger caspase-1 activity and higher IL1-β and IL18 levels in primary human macrophages (Stasakova et al. 2005). This result indicates a role for NS1 in antagonizing the NLR pathway. Although the precise mechanism of such antagonism is not known, it is possible that NS1, by virtue of its ability to perturb PKR and RIG-I pathways, can eliminate the priming signal for subsequent NLRP3 inflammasome activation.
Apart from NS1 and PA-X, the other protein that has been well studied for a role in innate immune evasion is PB1-F2. PB1-F2 is a small protein of 87aa encoded by an alternate reading frame within the PB1 gene and originally described to be proapoptotic (Chen et al. 2001). Later, it was shown that PB1-F2 mediates cell death via interaction with the mitochondrial proteins ANT3 and VDAC1 (Zamarin et al. 2005). Murine infection with IAV deficient in PB1-F2 showed reduced pathogenesis in mice (Zamarin et al. 2006). It was further shown that the N66S amino acid residue may contribute to the increased virulence associated with H5N1 and the 1918 pandemic H1N1 (Conenello and Palese 2007; McAuley et al. 2007). However, the recent pandemic 2009 H1N1 strain lacked a functional PB1-F2, yet showed increased virulence over prior seasonal strains. Engineering PB1-F2 into the 2009 strain had a minimal effect on virulence in animal models (Hai et al. 2010; Pena et al. 2012) indicating that virulence can represent a strain-specific combination of multiple determinants of host and viral factors. Additionally, PB1-F2 has been described to decrease mitochondrial membrane potential by binding to MAVS and inhibiting induction of IFN (Varga et al. 2012).
4.2 Nonreductive Determinants of Viral Fitness
Apart from specific viral proteins and domains that play a role in the evasion of the innate immune response, other features of the virus such as its spherical or elongated shape, its replication fitness, which represents a compound result of many specific features, its secondary RNA structure may also play a role in the ability of the virus to be stealthy and establish infection without detection by host cell recognition machinery.
Role of HA in Entry
The HA in different virus subtypes and strains can vary with regard to the optimum pH of activation (Mittal et al. 2003). For example, the HA molecule of H2 subtype undergoes a slow pH-dependent conformational change compared to the H3-HA molecule (Puri et al. 1990; Leikina et al. 2002). Similarly, the stability of the pH dependent conformational change has also been reported to be influenced by the density of the surface HA molecules (Markovic et al. 2001; Leikina et al. 2002). Experimental work with H5N1 viruses and mutants in the A/chicken/Vietnam/C58/04 (H5N1) background found that the optimal pH of activation is a key contributor to the observed increased pathogenesis. Using mutants that differ only, or primarily, in their optimum pH of activation, Zarket et al. showed that a higher pH of activation is highly pathogenic in avian species whereas a lower pH of activation favors pathogenesis in mice (Zaraket et al. 2013b). In another study, the same group showed that a mutant IAV (VN/1203-K58I) whose pH of activation is 5.5 compared to the wild type strain’s pH of 6.0 grew to similar titers as the wild type virus in MDCK and NHBE cells, but not in A549 cells (which have a lower endosomal pH than either of those cell types), resulting in reduced growth (Zaraket et al. 2013a). Interestingly, they found that an increase in acid stability resulted in increased virus growth in the upper respiratory tract. However, the mutation was not sufficient to confer transmission in ferrets, indicating that additional cell type specific features of endosomal pH activation may be playing a role in this in vivo model. Thus, it is clear that the HA sequence determine the stability of the HA with regard to pH, and by extension the genetic and environmental stability of the virus and its replication potential (Reed et al. 2010).
Role of HA in Exit
The HA also plays a role in the exit of the virus from the cell by budding. HA proteins in infected cells are synthesized as polypeptides (HA0) which lack membrane fusion activity (Skehel and Wiley 2000). To acquire this functionality, the HA0 must be cleaved by host cell proteases at the linker sequence between the HA1 and HA2 subunits, which are then reorganized to the mature HA molecule that is expressed on the surface of the virion (Garten and Klenk 1999). Thus, the expression pattern of host cell proteases has been suggested to play a critical role in the tissue restriction of IAV replication. Although the array of proteases that can mediate cleavage of HA0 is only beginning to be detailed, the established role of specific proteases in tissue tropism and pathogenesis provides an illustrative example. It has been reported that the HA in low pathogenic avian influenza viruses (LPAIV) can only be cleaved by certain trypsin like proteases expressed exclusively in the respiratory tract (of terrestrial birds) or the gastrointestinal tract (of water fowl and other terrestrial birds), resulting in the restriction of viral replication in those tissue compartments (Bertram et al. 2010). In contrast, the highly pathogenic avian influenza virus (HPAIV) contains a polybasic cleave site in its HA that is readily cleaved by a ubiquitously expressing protease, furin, in the trans-golgi network, resulting in systemic infection and severe disease (Bosch et al. 1981; Webster and Rott 1987; Perdue et al. 1997). However, it appears that the cleavability of the HA is not the only determinant of pathogenesis as HPAIV and known pandemic strains lacking furin multibasic cleavage sites have caused severe disease in human populations (Bertram et al. 2010).
RNA Accessibility
One may hypothesize that influenza virus infections utilizing distinct methods of entry (endocytosis, macropinocytosis, or penetration of RNP by plasma membrane fusion) may result in very different host responses, considering the localization of innate immune receptor distribution. Especially in the physiological setting with a low multiplicity of infection, such differential entry into the target cells will result in a variation in the kinetics of accessibility of the viral RNA to the host cytoplasmic sensors. It has been reported that IAV entering by endocytosis localizes to early endosome within 10 min and to the late endosome by 40 min after infection in HeLa cells (Sieczkarski and Whittaker 2003). However, the entry kinetics appear to vary by cell type. Viral fusion with the endosome has been detected as early as 10 min in MDCK and CHO cells (Yoshimura et al. 1982; Yoshimura and Ohnishi 1984; Lakadamyali et al. 2003). It is interesting to note that influenza infection of HeLa cells is found to be inefficient (Lohmeyer et al. 1979; Gujuluva et al. 1994). In contrast, we might speculate that virus internalized by macropinocytosis may take an even longer time (compared to endocytosis in HeLa cells) to reach the optimum low pH for virus uncoating. Another consideration is that the endocytic vesicle, regardless of the method of internalization, may fail to achieve fusion with the virus particle as a result of stochastic processes, resulting in diversion of the virion into the lysosomal pathway and autophagic destruction. It has been suggested that autophagy can present viral nucleic acid to TLRs in endolysomes (Kawai and Akira 2009). Thus, viruses with increased HA acid stability may enter endolysomes and the autophagy pathway, resulting in induction of IFN via TLR7/RIG I pathway. On the other hand, viruses that fuse at the plasma membrane and release RNPs may be more likely to encounter the RIG-I pathway (Weber et al. 2013) and initiate the antiviral response more quickly. Of note, differentiated human bronchial epithelial cells derived from asthmatic patients mounted an enhanced activation of the inflammasome and innate immune signaling after IAV infection (Bauer et al. 2012). It is reported that the pH of the bronchial tree mucous in asthmatic patients is around 5.3 (Ryley and Brogan 1968) as opposed to the normal range of 6.9–9.0 in the trachea (Karnad et al. 1990). Although apical pH data from these cultures was not reported, it has been suggested that cultures from cystic fibrosis patients also exhibit a more acidic pH than those from healthy individuals (Coakley et al. 2003). In summary, under acidic pH conditions, IAV can fuse directly to the plasma membrane (Matlin et al. 1981) and release RNPs to the cytosol directly (Morgan and Rose 1968). Thus, we can hypothesize that those viral genomes may be recognized faster than viruses entering through the endocytic route, resulting in faster initiation of the innate immune response. However, very little research has been done on the role of such differential entry affecting the proinflammatory and antiviral responses, and further investigation is warranted.
Genomic Features
Another aspect of RNA accessibility in determining the stealthy phenotype of influenza virus is the viral genome itself. Detection by both RIG-I and MDA-5 of flaviviruses suggests that these viruses may have genomic features that trigger both sensors (Loo et al. 2008). Similarly, within a given virus, distinct strains may have more “innate immunogenic” features by virtue of different sequences. Limited studies attempting to define the exact features of the RNA ligand stimulating immune responses from HCV (Saito et al. 2008) and IAV (Davis et al. 2012) indicated that certain regions of the genome may be more immunogenic than others. While studies from Davis et al., suggested that the 3′ and 5′ UTR of the IAV containing U/A tracts are potent innate immune inducers, little is known about the rest of the viral genome. It can be assumed that certain subtypes and strains may have more of these immunogenic features (potentially defined by secondary structures and sequence composition) than others.
Another factor that may contribute to viral stealthiness is the presence of defective interfering (DI) particle, which, until recently, has been shown to be a phenomenon of in vitro viral propagation (Von Magnus 1954; Nayak 1980). Recently, the presence of DI particles in human samples infected with pandemic H1N1 has been described (Saira et al. 2013). Another group has also described the presence of “semi infectious” particles in vitro as well as in vivo propagated virus inoculums (Brooke et al. 2013). Using immunofluorescence staining for four different viral proteins, the authors proposed that 90 % of infectious inocula failed to establish productive infection by lacking expression of one or more viral proteins (Brooke et al. 2013). This was independent of cell type in the presence of an intact IFN system, though presence of partial RNA similar to DI particles in this “semi-infectious” population could not be ruled out (Brooke et al. 2013). We can imagine that the presence or absence of DI particles or “semi-infectious” particles in inocula may play an important role in determining in the outcome of infection and the efficiency of transmission.
5 Conclusion and Future Direction
In sum, multiple compound features of viral entry, trafficking and replication can combine to allow the virus to evade host recognition to greater or lesser extents. Given the exponential growth of the virus with each round of replication, even a slight advantage in early replication kinetics can result in a significant alteration in viral titers and clinical and pathological outcomes (Askovich et al. 2013). Dissecting these “whole virus” features presents a new challenge for influenza research, which made great strides in the last decade using reverse genetics technology. Systems biology and other high-throughput approaches will be necessary to provide insights into these areas of influenza virus biology.
References
Allen IC, Scull MA, Moore CB et al (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30:556–565. doi:10.1016/j.immuni.2009.02.005
Anand PK, Malireddi RKS, Lukens JR et al (2012) NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488:389–393. doi:10.1038/nature11250
Andersen JM, Al-Khairy D, Ingalls RR (2006) Innate immunity at the mucosal surface: role of toll-like receptor 3 and toll-like receptor 9 in cervical epithelial cell responses to microbial pathogens. Biol Reprod 74:824–831. doi:10.1095/biolreprod.105.048629
Andrejeva J, Childs KS, Young DF et al (2004) The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-β promoter. Proc Natl Acad Sci U S A 101:17264–17269. doi:10.1073/pnas.0407639101
Askovich PS, Sanders CJ, Rosenberger CM et al (2013) Differential host response, rather than early viral replication efficiency, correlates with pathogenicity caused by influenza viruses. PloS One 8:e74863. doi:10.1371/journal.pone.0074863
Bauer RN, Brighton LE, Mueller L et al (2012) Influenza enhances caspase-1 in bronchial epithelial cells from asthmatic volunteers and is associated with pathogenesis. J Allergy Clin Immunol 130:958–967 e14. doi:10.1016/j.jaci.2012.07.013
Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7:99–109. doi:10.1038/nrmicro2070
Bertram S, Glowacka I, Steffen I et al (2010) Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev Med Virol 20:298–310. doi:10.1002/rmv.657
Binder M, Eberle F, Seitz S et al (2011) Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J Biol Chem 286:27278–27287. doi:10.1074/jbc.M111.256974
Black RA, Kronheim SR, Sleath PR (1989) Activation of interleukin-1 beta by a co-induced protease. FEBS Lett 247:386–390
Bosch FX, Garten W, Klenk HD, Rott R (1981) Proteolytic cleavage of influenza virus hemagglutinins: primary structure of the connecting peptide between HA1 and HA2 determines proteolytic cleavability and pathogenicity of Avian influenza viruses. Virology 113:725–735
Brooke CB, Ince WL, Wrammert J et al (2013) Most influenza a virions fail to express at least one essential viral protein. J Virol 87:3155–3162. doi:10.1128/JVI.02284-12
Cárdenas WB, Loo Y-M, Gale M Jr et al (2006) Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80:5168–5178. doi:10.1128/JVI.02199-05
Cario E, Podolsky DK (2000) Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 68:7010–7017
Chang T-H, Liao C-L, Lin Y-L (2006) Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect Inst Pasteur 8:157–171. doi:10.1016/j.micinf.2005.06.014
Chen W, Calvo PA, Malide D et al. (2001) A novel influenza A virus mitochondrial protein that induces cell death. Nat Med 7:1306–1312. http://dx.doi.org/10.1038/nm1201-1306
Chiu Y-H, Macmillan JB, Chen ZJ (2009) RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–591. doi:10.1016/j.cell.2009.06.015
Coakley RD, Grubb BR, Paradiso AM et al (2003) Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc Natl Acad Sci U S A 100:16083–16088. doi:10.1073/pnas.2634339100
Compton T, Nepomuceno RR, Nowlin DM (1992) Human cytomegalovirus penetrates host cells by pH-independent fusion at the cell surface. Virology 191:387–395
Conenello GM, Palese P (2007) Influenza A virus PB1-F2: a small protein with a big punch. Cell Host Microbe 2:207–209. http://dx.doi.org/10.1016/j.chom.2007.09.010
Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13:2905–2927
Davis WG, Bowzard JB, Sharma SD et al (2012) The 3’ untranslated regions of influenza genomic sequences are 5’PPP-independent ligands for RIG-I. PloS One 7:e32661. doi:10.1371/journal.pone.0032661
Desmet EA, Bussey KA, Stone R, Takimoto T (2013) Identification of the N-terminal domain of the influenza virus PA responsible for the suppression of host protein synthesis. J Virol 87:3108–3118. doi:10.1128/JVI.02826-12
Dias A, Bouvier D, Crépin T et al (2009) The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918. doi:10.1038/nature07745
Díaz MO, Testa D (1996) Type I interferon genes and proteins. Biotherapy Dordr Neth 8:157–162
Diebold SS, Kaisho T, Hemmi H et al (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–1531. doi:10.1126/science.1093616
Dinarello CA, Novick D, Puren AJ et al (1998) Overview of interleukin-18: more than an interferon-gamma inducing factor. J Leukoc Biol 63:658–664
Donelan NR, Basler CF, Garcia-Sastre A (2003) A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J Virol 77:13257–13266
Dostert C, Pétrilli V, Van Bruggen R et al (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320:674–677. doi:10.1126/science.1156995
Dugan JW, Albor A, David L et al (2009) Nucleotide oligomerization domain-2 interacts with 2’-5’-oligoadenylate synthetase type 2 and enhances RNase-L function in THP-1 cells. Mol Immunol 47:560–566. doi:10.1016/j.molimm.2009.09.025
Durbin RK, Kotenko SV, Durbin JE (2013) Interferon induction and function at the mucosal surface. Immunol Rev 255:25–39. doi:10.1111/imr.12101
Falcon AM, Fernandez-Sesma A, Nakaya Y, et al. (2005) Attenuation and immunogenicity in mice of temperature-sensitive influenza viruses expressing truncated NS1 proteins. J Gen Virol 86:2817–2821. http://dx.doi.org/10.1099/vir.0.80991-0
Fontana J, Cardone G, Heymann JB et al (2012) Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J Virol 86:2919–2929. doi:10.1128/JVI.06698-11
Franchi L, Kanneganti T-D, Dubyak GR, Núñez G (2007) Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem 282:18810–18818. doi:10.1074/jbc.M610762200
Fredericksen BL, Gale M Jr (2006) West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J Virol 80:2913–2923. doi:10.1128/JVI.80.6.2913-2923.2006
Funami K, Matsumoto M, Oshiumi H et al (2004) The cytoplasmic “linker region” in Toll-like receptor 3 controls receptor localization and signaling. Int Immunol 16:1143–1154. doi:10.1093/intimm/dxh115
Gack MU, Albrecht RA, Urano T et al (2009) Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5:439–449. doi:10.1016/j.chom.2009.04.006
Gao H-N, Lu H-Z, Cao B et al (2013) Clinical findings in 111 cases of influenza A (H7N9) virus infection. N Engl J Med 368:2277–2285. doi:10.1056/NEJMoa1305584
García-Sastre A (2011) Induction and evasion of type I interferon responses by influenza viruses. Virus Res 162:12–18. doi:10.1016/j.virusres.2011.10.017
Garcia-Sastre A, Egorov A, Matassov D et al (1998) Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252:324–330
Garten W, Klenk HD (1999) Understanding influenza virus pathogenicity. Trends Microbiol 7:99–100
Gitlin L, Barchet W, Gilfillan S et al (2006) Essential role of mda-5 in type I IFN responses to polyriboinosinic: polyribocytidylic acid and encephalo myocarditis picornavirus. Proc Natl Acad Sci U S A 103:8459–8464. doi:10.1073/pnas.0603082103
Le Goffic R, Balloy V, Lagranderie M et al (2006) Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2:e53. doi:10.1371/journal.ppat.0020053
Le Goffic R, Pothlichet J, Vitour D et al (2007) Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol Baltim Md 1950 178:3368–3372
Guillot L, Le Goffic R, Bloch S et al (2005) Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem 280:5571–5580. doi:10.1074/jbc.M410592200
Gujuluva CN, Kundu A, Murti KG, Nayak DP (1994) Abortive replication of influenza virus A/WSN/33 in HeLa229 cells: defective viral entry and budding processes. Virology 204:491–505. doi:10.1006/viro.1994.1563
Guo Z, Chen L, Zeng H et al (2007) NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am J Respir Cell Mol Biol 36:263–269. doi:10.1165/rcmb.2006-0283RC
Hai R, Schmolke M, Varga ZT et al (2010) PB1-F2 Expression by the 2009 Pandemic H1N1 Influenza Virus Has Minimal Impact on Virulence in Animal Models. J Virol 84:4442–4450. doi:10.1128/JVI.02717-09
Hale BG, Jackson D, Chen Y-H et al (2006) Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc Natl Acad Sci U S A 103:14194–14199. doi:10.1073/pnas.0606109103
Halle A, Hornung V, Petzold GC et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865. doi:10.1038/ni.1636
Hamilton BS, Whittaker GR, Daniel S (2012) Influenza virus-mediated membrane fusion: determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses 4:1144–1168. doi:10.3390/v4071144
Herfst S, Schrauwen EJA, Linster M et al (2012) Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336:1534–1541. doi:10.1126/science.1213362
Hewson CA, Jardine A, Edwards MR et al (2005) Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J Virol 79:12273–12279. doi:10.1128/JVI.79.19.12273-12279.2005
Hornung V, Bauernfeind F, Halle A et al (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9:847–856. doi:10.1038/ni.1631
Hornung V, Ellegast J, Kim S et al (2006) 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997. doi:10.1126/science.1132505
Hornung V, Latz E (2010) Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol 40:620–623. doi:10.1002/eji.200940185
Hoyle L, Finter NB (1957) The use of influenza virus labelled with radio-sulphur in studies of the early stages of the interaction of virus with the host cell. J Hyg (Lond) 55:290–297
Ichinohe T, Lee HK, Ogura Y et al (2009) Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 206:79
Ichinohe T, Pang IK, Iwasaki A (2010) Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11:404–410. doi:10.1038/ni.1861
Imai M, Watanabe T, Hatta M et al (2012) Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486:420–428. doi:10.1038/nature10831
Imai Y, Kuba K, Neely GG et al (2008) Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133:235–249. doi:10.1016/j.cell.2008.02.043
Ioannidis I, Ye F, McNally B et al (2013) Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J Virol 87:3261–3270. doi:10.1128/JVI.01956-12
Jagger BW, Wise HM, Kash JC et al (2012) An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204. doi:10.1126/science.1222213
Kang D, Gopalkrishnan RV, Wu Q et al (2002) mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A 99:637–642. doi:10.1073/pnas.022637199
Kang D-C, Gopalkrishnan RV, Lin L et al (2004) Expression analysis and genomic characterization of human melanoma differentiation associated gene-5, mda-5: a novel type I interferon-responsive apoptosis-inducing gene. Oncogene 23:1789–1800. doi:10.1038/sj.onc.1207300
Kanneganti T-D, Body-Malapel M, Amer A et al (2006) Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem 281:36560–36568. doi:10.1074/jbc.M607594200
Karnad DR, Mhaisekar DG, Moralwar KV (1990) Respiratory mucus pH in tracheostomized intensive care unit patients: Effects of colonization and pneumonia. Crit Care Med 18:699–701
Kato H, Sato S, Yoneyama M et al (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19–28. doi:10.1016/j.immuni.2005.04.010
Kato H, Takeuchi O, Mikamo-Satoh E et al (2008) Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205:1601–1610. doi:10.1084/jem.20080091
Kato H, Takeuchi O, Sato S et al (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi:10.1038/nature04734
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650. doi:10.1016/j.immuni.2011.05.006
Kawai T, Akira S (2009) The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 21:317–337. doi:10.1093/intimm/dxp017
Kogure T, Suzuki T, Takahashi T et al (2006) Human trachea primary epithelial cells express both sialyl(alpha2-3)Gal receptor for human parainfluenza virus type 1 and avian influenza viruses, and sialyl(alpha2-6)Gal receptor for human influenza viruses. Glycoconj J 23:101–106. doi:10.1007/s10719-006-5442-z
Kotenko SV, Gallagher G, Baurin VV et al (2003) IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4:69–77. doi:10.1038/ni875
Lakadamyali M, Rust MJ, Babcock HP, Zhuang X (2003) Visualizing infection of individual influenza viruses. Proc Natl Acad Sci U A 100:9280–9285. http://dx.doi.org/10.1073/pnas.0832269100
Lakadamyali M, Rust MJ, Zhuang X (2006) Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 124:997–1009. doi:10.1016/j.cell.2005.12.038
Lee SMY, Kok K-H, Jaume M et al (2014) Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc Natl Acad Sci U S A 111:3793–3798. doi:10.1073/pnas.1324266111
Leikina E, Ramos C, Markovic I et al (2002) Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J 21:5701–5710
Lemaitre B, Nicolas E, Michaut L et al (1996) The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86:973–983
Liu P, Jamaluddin M, Li K et al (2007) Retinoic acid-inducible gene i mediates early antiviral response and toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J Virol 81:1401–1411. doi:10.1128/JVI.01740-06
Lohmeyer J, Talens LT, Klenk H-D (1979) Biosynthesis of the influenza virus envelope in abortive infection. J Gen Virol 42:73–88. doi:10.1099/0022-1317-42-1-73
Loo Y-M, Fornek J, Crochet N et al (2008) Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol 82:335–345. doi:10.1128/JVI.01080-07
Loo Y-M, Gale M Jr (2011) Immune signaling by RIG-I-like receptors. Immunity 34:680–692. doi:10.1016/j.immuni.2011.05.003
Ludwig S, Wang X, Ehrhardt C et al (2002) The influenza A virus NS1 protein inhibits activation of Jun N-terminal kinase and AP-1 transcription factors. J Virol 76:11166–11171
Lund JM, Alexopoulou L, Sato A et al (2004) Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A 101:5598–5603. doi:10.1073/pnas.0400937101
Luo M (2012) Influenza virus entry. In: Rossmann MG, Rao VB (eds) Viral Molecular Machines. Springer, Boston, pp 201–221
Lupfer C, Kanneganti T-D (2013) The expanding role of NLRs in antiviral immunity. Immunol Rev 255:13–24. doi:10.1111/imr.12089
Lupfer C, Thomas PG, Anand PK et al (2013) Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 14:480–488. doi:10.1038/ni.2563
Macfarlane DE, Manzel L (1998) Antagonism of immunostimulatory CpG-oligodeoxynucleotides by quinacrine, chloroquine, and structurally related compounds. J Immunol Baltim Md 1950 160:1122–1131
von Magnus P (1954) Incomplete forms of influenza virus. Adv Virus Res 2:59–79
Malathi K, Dong B, Gale M Jr, Silverman RH (2007) Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–819. doi:10.1038/nature06042
Malireddi RKS, Ippagunta S, Lamkanfi M, Kanneganti T-D (2010) Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J Immunol Baltim Md 185:3127–3130. doi:10.4049/jimmunol.1001512
Markovic I, Leikina E, Zhukovsky M et al (2001) Synchronized activation and refolding of influenza hemagglutinin in multimeric fusion machines. J Cell Biol 155:833–844. doi:10.1083/jcb.200103005
Marshak-Rothstein A (2006) Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 6:823–835. doi:10.1038/nri1957
Martinon F, Pétrilli V, Mayor A et al (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440:237–241. doi:10.1038/nature04516
Masters SL, Gerlic M, Metcalf D et al (2012) NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 37:1009–1023. doi:10.1016/j.immuni.2012.08.027
Matlin KS, Reggio H, Helenius A, Simons K (1981) Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol 91:601–613
Matsumoto M, Funami K, Tanabe M et al (2003) Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol Baltim Md 1950 171:3154–3162
Matsumoto M, Kikkawa S, Kohase M et al (2002) Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem Biophys Res Commun 293:1364–1369. doi:10.1016/S0006-291X(02)00380-7
McAuley JL, Hornung F, Boyd KL, et al. (2007) Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2:240–249. http://dx.doi.org/10.1016/j.chom.2007.09.001
McAuley JL, Zhang K, McCullers JA (2010) The effects of influenza A virus PB1-F2 protein on polymerase activity are strain specific and do not impact pathogenesis. J Virol 84:558–564. doi:10.1128/JVI.01785-09
Meylan E, Tschopp J, Karin M (2006) Intracellular pattern recognition receptors in the host response. Nature 442:39–44
Mibayashi M, Martinez-Sobrido L, Loo Y-M, et al. (2007) Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol 81:514–524. http://dx.doi.org/10.1128/JVI.01265-06
Mittal A, Leikina E, Chernomordik LV, Bentz J (2003) Kinetically differentiating influenza hemagglutinin fusion and hemifusion machines. Biophys J 85:1713–1724. doi:10.1016/S0006-3495(03)74601-3
Morgan C, Rose HM (1968) Structure and development of viruses as observed in the electron microscope. 8 Entry of influenza virus. J Virol 2:925–936
Muzio M, Bosisio D, Polentarutti N et al (2000) Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol Baltim Md 1950 164:5998–6004
Nakhaei P, Genin P, Civas A, Hiscott J (2009) RIG-I-like receptors: sensing and responding to RNA virus infection. Semin Immunol 21:215–222. doi:10.1016/j.smim.2009.05.001
Nayak DP (1980) Defective interfering influenza viruses. Annu Rev Microbiol 34:619–644. doi:10.1146/annurev.mi.34.100180.003155
Nemeroff ME, Barabino SM, Li Y et al (1998) Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′ end formation of cellular pre-mRNAs. Mol Cell 1:991–1000
Nhu QM, Shirey K, Teijaro JR et al (2010) Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol 3:29–39. doi:10.1038/mi.2009.120
Nicholls JM, Chan MCW, Chan WY et al (2007) Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat Med 13:147–149. doi:10.1038/nm1529
Noah DL, Twu KY, Krug RM (2003) Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3’ end processing of cellular pre-mRNAS. Virology 307:386–395
Noble S, Dimmock NJ (1995) Characterization of putative defective interfering (DI) A/WSN RNAs isolated from the lungs of mice protected from an otherwise lethal respiratory infection with influenza virus A/WSN (H1N1): a subset of the inoculum DI RNAs. Virology 210:9–19. doi:10.1006/viro.1995.1312
Ogura Y, Sutterwala FS, Flavell RA (2006) The inflammasome: first line of the immune response to cell stress. Cell 126:659–662. doi:10.1016/j.cell.2006.08.002
Oshansky CM, Pickens JA, Bradley KC et al (2011) Avian influenza viruses infect primary human bronchial epithelial cells unconstrained by sialic acid α2,3 residues. PloS One 6:e21183. doi:10.1371/journal.pone.0021183
Palese P, Shaw ML (2007) Orthomyxoviridae: the viruses and their replication, 5th ed. Lippincott Williams and Wilkins, Philadelphia
Patterson S, Oxford JS, Dourmashkin RR (1979) Studies on the mechanism of influenza virus entry into cells. J Gen Virol 43:223–229
Pena L, Vincent AL, Loving CL et al (2012) Restored PB1-F2 in the 2009 pandemic H1N1 influenza virus has minimal effects in swine. J Virol 86:5523–5532. doi:10.1128/JVI.00134-12
Perdue ML, García M, Senne D, Fraire M (1997) Virulence-associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res 49:173–186
Pétrilli V, Papin S, Dostert C et al (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14:1583–1589. doi:10.1038/sj.cdd.4402195
Pichlmair A, Schulz O, Tan CP et al (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001. doi:10.1126/science.1132998
Pichlmair A, Schulz O, Tan C-P et al (2009) Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol 83:10761–10769. doi:10.1128/JVI.00770-09
Pinto LH, Holsinger LJ, Lamb RA (1992) Influenza virus M2 protein has ion channel activity. Cell 69:517–528
Pippig DA, Hellmuth JC, Cui S et al (2009) The regulatory domain of the RIG-I family ATPase LGP2 senses double-stranded RNA. Nucleic Acids Res 37:2014–2025. doi:10.1093/nar/gkp059
Plotch SJ, Bouloy M, Ulmanen I, Krug RM (1981) A unique cap(m7G pppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23:847–858
Poeck H, Bscheider M, Gross O et al (2010) Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol 11:63–69. doi:10.1038/ni.1824
Pothlichet J, Meunier I, Davis BK et al (2013) Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog 9:e1003256. doi:10.1371/journal.ppat.1003256
Puri A, Booy FP, Doms RW et al (1990) Conformational changes and fusion activity of influenza virus hemagglutinin of the H2 and H3 subtypes: effects of acid pretreatment. J Virol 64:3824–3832
Rajan JV, Warren SE, Miao EA, Aderem A (2010) Activation of the NLRP3 inflammasome by intracellular poly I:C. FEBS Lett 584:4627–4632. doi:10.1016/j.febslet.2010.10.036
Reed ML, Bridges OA, Seiler P et al (2010) The pH of activation of the hemagglutinin protein regulates H5N1 influenza virus pathogenicity and transmissibility in ducks. J Virol 84:1527–1535. doi:10.1128/JVI.02069-09
Rehwinkel J, Tan CP, Goubau D et al (2010) RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397–408. doi:10.1016/j.cell.2010.01.020
Roach JC, Glusman G, Rowen L et al (2005) The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci U S A 102:9577–9582. doi:10.1073/pnas.0502272102
Rossman JS, Leser GP, Lamb RA (2012) Filamentous influenza virus enters cells via macropinocytosis. J Virol 86:10950–10960. doi:10.1128/JVI.05992-11
Rothenfusser S, Goutagny N, DiPerna G et al (2005) The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol Baltim Md 1950 175:5260–5268
Rust MJ, Lakadamyali M, Zhang F, Zhuang X (2004) Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol 11:567–573. http://dx.doi.org/10.1038/nsmb769
Ryley HC, Brogan TD (1968) Variation in the composition of sputum in chronic chest diseases. Br J Exp Pathol 49:625–633
Sabbah A, Chang TH, Harnack R et al (2009) Activation of innate immune antiviral responses by Nod2. Nat Immunol 10:1073–1080. doi:10.1038/ni.1782
Saira K, Lin X, DePasse JV et al (2013) Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J Virol 87:8064–8074. doi:10.1128/JVI.00240-13
Saito T, Gale M Jr (2008) Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. J Exp Med 205:1523–1527. doi:10.1084/jem.20081210
Saito T, Hirai R, Loo Y-M et al (2007) Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A 104:582–587. doi:10.1073/pnas.0606699104
Saito T, Owen DM, Jiang F et al (2008) Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523–527. doi:10.1038/nature07106
Samanta M, Iwakiri D, Kanda T et al (2006) EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J 25:4207–4214. doi:10.1038/sj.emboj.7601314
Samuel CE (2001) Antiviral actions of interferons. Clin Microbiol Rev 14:778–809. http://dx.doi.org/10.1128/CMR.14.4.778-809.2001
Sanders CJ, Doherty PC, Thomas PG (2011) Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res 343:13–21. doi:10.1007/s00441-010-1043-z
Sarmiento M, Haffey M, Spear PG (1979) Membrane proteins specified by herpes simplex viruses. III. Role of glycoprotein VP7(B2) in virion infectivity. J Virol 29:1149–1158
Satoh T, Kato H, Kumagai Y et al (2010) LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A 107:1512–1517. doi:10.1073/pnas.0912986107
Schlee M, Roth A, Hornung V et al (2009) Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31:25–34. doi:10.1016/j.immuni.2009.05.008
Schmidt A, Schwerd T, Hamm W et al (2009) 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci U S A 106:12067–12072. doi:10.1073/pnas.0900971106
Shaw M, Palese P (2007) Orthomyxoviridae: The Viruses and Their Replication, 6th ed. Lippincott Williams and Wilkins, Philadelphia
Sheppard P, Kindsvogel W, Xu W et al (2003) IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 4:63–68. doi:10.1038/ni873
Sieczkarski SB, Whittaker GR (2003) Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic Cph Den 4:333–343
Sieczkarski SB, Whittaker GR (2002) Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol 76:10455–10464. doi:10.1128/JVI.76.20.10455-10464.2002
Skehel JJ, Wiley DC (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569. doi:10.1146/annurev.biochem.69.1.531
Soboll G, Shen L, Wira CR (2006) Expression of Toll-like receptors (TLR) and responsiveness to TLR agonists by polarized mouse uterine epithelial cells in culture. Biol Reprod 75:131–139. doi:10.1095/biolreprod.106.050690
Solórzano A, Webby RJ, Lager KM et al (2005) Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J Virol 79:7535–7543. doi:10.1128/JVI.79.12.7535-7543.2005
Song R, Pang X, Yang P et al (2014) Surveillance of the first case of human avian influenza A (H7N9) virus in Beijing, China. Infection 42:127–133. doi:10.1007/s15010-013-0533-9
Srinivasakumar N, Ogra PL, Flanagan TD (1991) Characteristics of fusion of respiratory syncytial virus with HEp-2 cells as measured by R18 fluorescence dequenching assay. J Virol 65:4063–4069
Stasakova J, Ferko B, Kittel C et al (2005) Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J Gen Virol 86:185–195. doi:10.1099/vir.0.80422-0
Stray SJ, Cummings RD, Air GM (2000) Influenza virus infection of desialylated cells. Glycobiology 10:649–658
Strowig T, Henao-Mejia J, Elinav E, Flavell R (2012) Inflammasomes in health and disease. Nature 481:278–286. doi:10.1038/nature10759
Sumpter R Jr, Loo Y-M, Foy E et al (2005) Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 79:2689–2699. doi:10.1128/JVI.79.5.2689-2699.2005
Sun X, Whittaker GR (2007) Role of the actin cytoskeleton during influenza virus internalization into polarized epithelial cells. Cell Microbiol 9:1672–1682. doi:10.1111/j.1462-5822.2007.00900.x
Takahasi K, Kumeta H, Tsuduki N et al (2009) Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: identification of the RNA recognition loop in RIG-I-like receptors. J Biol Chem 284:17465–17474. doi:10.1074/jbc.M109.007179
Takeuchi O, Akira S (2007) Signaling pathways activated by microorganisms. Curr Opin Cell Biol 19:185–191. doi:10.1016/j.ceb.2007.02.006
Talon J, Horvath CM, Polley R et al (2000) Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 74:7989–7996
Tapia K, Kim W-K, Sun Y et al (2013) Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog 9:e1003703. doi:10.1371/journal.ppat.1003703
Tatsuta T, Imaizumi T, Shimoyama T et al (2012) Expression of melanoma differentiation associated gene 5 is increased in human gastric mucosa infected with Helicobacter pylori. J Clin Pathol 65:839–843. doi:10.1136/jclinpath-2011-200590
Thomas PG, Dash P, Aldridge JR Jr et al (2009) The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30:566–575. doi:10.1016/j.immuni.2009.02.006
Thompson CI, Barclay WS, Zambon MC, Pickles RJ (2006) Infection of human airway epithelium by human and avian strains of influenza a virus. J Virol 80:8060–8068. doi:10.1128/JVI.00384-06
Tsai S-Y, Segovia JA, Chang T-H et al (2014) DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog 10:e1003848. doi:10.1371/journal.ppat.1003848
Tschopp J (2011) Mitochondria: Sovereign of inflammation? Eur J Immunol 41:1196–1202. doi:10.1002/eji.201141436
Uyeki TM, Nguyen DC, Rowe T et al (2012) Seroprevalence of antibodies to avian influenza A (H5) and A (H9) viruses among market poultry workers, Hanoi, Vietnam, 2001. PloS One 7:e43948. doi:10.1371/journal.pone.0043948
Varga ZT, Grant A, Manicassamy B, Palese P (2012) Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol 86:8359–8366. doi:10.1128/JVI.01122-12
Venkataraman T, Valdes M, Elsby R et al (2007) Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol Baltim Md 1950 178:6444–6455
De Vries E, Tscherne DM, Wienholts MJ et al (2011) Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog 7:e1001329. doi:10.1371/journal.ppat.1001329
De Vries E, de Vries RP, Wienholts MJ et al (2012) Influenza A virus entry into cells lacking sialylated N-glycans. Proc Natl Acad Sci U S A 109:7457–7462. doi:10.1073/pnas.1200987109
Wang M, Fu C-X, Zheng B-J (2009) Antibodies against H5 and H9 avian influenza among poultry workers in China. N Engl J Med 360:2583–2584. doi:10.1056/NEJMc0900358
Wang X, Li M, Zheng H et al (2000) Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol 74:11566–11573
Weber F, Wagner V, Rasmussen SB et al (2006) Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80:5059–5064. doi:10.1128/JVI.80.10.5059-5064.2006
Weber M, Gawanbacht A, Habjan M et al (2013) Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 13:336–346. doi:10.1016/j.chom.2013.01.012
Webster RG, Rott R (1987) Influenza virus A pathogenicity: the pivotal role of hemagglutinin. Cell 50:665–666
White J, Kartenbeck J, Helenius A (1980) Fusion of Semliki forest virus with the plasma membrane can be induced by low pH. J Cell Biol 87:264–272
White J, Matlin K, Helenius A (1981) Cell fusion by Semliki Forest, influenza, and vesicular stomatitis viruses. J Cell Biol 89:674–679
White JM, Wilson IA (1987) Anti-peptide antibodies detect steps in a protein conformational change: low-pH activation of the influenza virus hemagglutinin. J Cell Biol 105:2887–2896
Wise HM, Foeglein A, Sun J et al (2009) A complicated message: Identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J Virol 83:8021–8031. doi:10.1128/JVI.00826-09
Xu R, Wilson IA (2011) Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J Virol 85:5172–5182. doi:10.1128/JVI.02430-10
Yamada H, Chounan R, Higashi Y et al (2004) Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett 578:331–336. doi:10.1016/j.febslet.2004.11.017
Yoneyama M, Fujita T (2007) Function of RIG-I-like receptors in antiviral innate immunity. J Biol Chem 282:15315–15318. doi:10.1074/jbc.R700007200
Yoneyama M, Kikuchi M, Matsumoto K et al (2005) Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol Baltim Md 1950 175:2851–2858
Yoneyama M, Kikuchi M, Natsukawa T et al (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. doi:10.1038/ni1087
Yoshimura A, Kuroda K, Kawasaki K et al (1982) Infectious cell entry mechanism of influenza virus. J Virol 43:284
Yoshimura A, Ohnishi S (1984) Uncoating of influenza virus in endosomes. J Virol 51:497–504
Yount JS, Moran TM, López CB (2007) Cytokine-independent upregulation of MDA5 in viral infection. J Virol 81:7316–7319. doi:10.1128/JVI.00545-07
Zaki MH, Vogel P, Malireddi RKS et al (2011) The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20:649–660. doi:10.1016/j.ccr.2011.10.022
Zamarin D, Garcia-Sastre A, Xiao X, et al. (2005) Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog 1:e4. http://dx.doi.org/10.1371/journal.ppat.0010004
Zamarin D, Ortigoza MB, Palese P (2006) Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol 80:7976–7983. http://dx.doi.org/10.1128/JVI.00415-06
Zaraket H, Bridges OA, Duan S et al (2013a) Increased acid stability of the hemagglutinin protein enhances H5N1 influenza virus growth in the upper respiratory tract but is insufficient for transmission in ferrets. J Virol 87:9911–9922. doi:10.1128/JVI.01175-13
Zaraket H, Bridges OA, Russell CJ (2013b) The pH of activation of the hemagglutinin protein regulates H5N1 influenza virus replication and pathogenesis in mice. J Virol 87:4826–4834. doi:10.1128/JVI.03110-12
Zell R, Krumbholz A, Eitner A et al (2007) Prevalence of PB1-F2 of influenza A viruses. J Gen Virol 88:536–546. doi:10.1099/vir.0.82378-0
Acknowledgment
The authors were supported by NIH/NIAID Contracts HHSN266200700005C (St. Jude Center of Excellence for Influenza Research and Surveillance) and HHSN272200800058C (Systems Influenza).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Dash, P., Thomas, P.G. (2014). Host Detection and the Stealthy Phenotype in Influenza Virus Infection. In: Oldstone, M., Compans, R. (eds) Influenza Pathogenesis and Control - Volume II. Current Topics in Microbiology and Immunology, vol 386. Springer, Cham. https://doi.org/10.1007/82_2014_412
Download citation
DOI: https://doi.org/10.1007/82_2014_412
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-11157-5
Online ISBN: 978-3-319-11158-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)