
Abstract
Proinflammatory cytokines perturb brain development and neurotransmission and are implicated in various psychiatric diseases, such as schizophrenia and depression. These cytokines often induce the production of reactive oxygen species (ROS) and regulate not only cell survival and proliferation but also inflammatory process and neurotransmission. Under physiological conditions, ROS are moderately produced in mitochondria but are rapidly scavenged by reducing agents in cells. However, brain injury, ischemia, infection, or seizure-like neural activities induce inflammatory cytokines and trigger the production of excessive amounts of ROS, leading to abnormal brain functions and psychiatric symptoms. Protein phosphatases, which are involved in the basal silencing of cytokine receptor activation, are the major targets of ROS. Consistent with this, several ROS scavengers, such as polyphenols and unsaturated fatty acids, attenuate both cytokine signaling and psychiatric abnormalities. In this review, we list the inducers, producers, targets, and scavengers of ROS in the brain and discuss the interaction between ROS and cytokine signaling implicated in schizophrenia and its animal models. In particular, we present an animal model of schizophrenia established by perinatal exposure to epidermal growth factor and illustrate the pathological role of ROS and antipsychotic actions of ROS scavengers, such as emodin and edaravone.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Oxidative Stress Is Implicated in the Pathogenesis of Schizophrenia
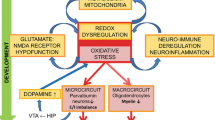
Oxygen radicals are molecules essential for energy production, metabolic processes, and germicide in our body; however, they are also toxic to cellular components, such as membrane lipids and DNA (Brewer et al. 2015). Therefore, our tissues and cells contain a large variety of reducing agents (i.e., scavengers) as well as reactive oxygen species (ROS)-degrading enzymes (Chan 2001; Table 1). This is especially true in the brain, which produces high energy accompanying ROS generation and is enriched with these antioxidants to protect the tissue from oxidative stress. Following brain injury, ischemia, infection, or seizures, NADPH oxidase (NOX) is activated in microglia and GABAergic neurons and produces large amounts of ROS in the brain (Kaur et al. 2015). When ROS is not scavenged immediately, they act not only on cell components (membrane lipids and DNA) but also on the molecules that carry ROS (redox) sensors, such as the protein tyrosine phosphatase PTP1B, nuclear factor kappa B (NF-κB), potassium channels, hypoxia-inducible factors, or N-methyl-d-aspartic acid (NMDA) receptor. For example, ROS acts on free cysteine residues of the NMDA receptor, altering its channel conformation to decrease NMDA-mediated cation movements, presumably attenuating neuronal cell death (Sanchez et al. 2000; Dukoff et al. 2014). This anti-ROS process functions as a self-defense system in the brain; however, when ROS generation is prolonged, the reduction in NMDA receptor channel activity can perturb normal neurotransmission and brain function. Chronic suppression of NMDA receptor activity is known to result in abnormal cognition and behaviors relevant to schizophrenia (Mohn et al. 1999; Takahashi et al. 2006).
Several reports on ROS generation show that contents of reducing agents are decreased and oxidizing agents are increased in patients and animal models with schizophrenia (Yao and Keshavan 2011; Bokkon and Antal 2011). For example, the products of reactions between ROS and membrane lipids (i.e., peroxidized lipids) are increased in patients in the acute phase of schizophrenia (Mahadik and Scheffer 1996; Ben Othmen et al. 2008). ROS also reacts with the guanosine residue of DNA in nuclei, converting it to 8-hydroxy-2-deoxyguanosine and leading to DNA fragmentation. The brain contents of 8-hydroxy-2-deoxyguanosine and fragmented DNA are elevated in patients with schizophrenia (Nishioka and Arnold 2004; Buttner et al. 2007). Blood levels of pentosidine, an advanced glycation end product, are also elevated as a result of sugar oxidation and are higher in patients with schizophrenia (Arai et al. 2010, 2014).
Conversely, the total amount of ROS scavengers (TAR, the total antioxidative response) is reduced in patients with schizophrenia (Ustundag et al. 2006). Blood concentrations of vitamins C and E are decreased in patients with schizophrenia (Suboticanec et al. 1990; McCreadie et al. 1995). In addition to these small molecules, the levels of enzymes that produce or degrade ROS are also altered. The activities of superoxide dismutase (SOD) and catalase are downregulated in the erythrocytes of patients with schizophrenia (Ben Othmen et al. 2008). Glutamate cysteine ligase is also decreased in patients with schizophrenia (Gysin et al. 2007). These findings suggest that the magnitude of ROS generation appears to be upregulated in patients with schizophrenia.
The pathologic interactions between ROS and psychosis have been investigated in animal models for drug-induced psychosis to monitor ROS generation following exposure to psychostimulants such as amphetamine, phencyclidine, ketamine, or the NMDA receptor blocker dizocilpine (MK-801). Behrens et al. (2007) showed that a ketamine-induced abnormality in GABAergic neurotransmission and behavior involves ROS generation by NOX. Zuo et al. (2007) also observed increases in hydroxyl radicals in the brain following MK-801 and ketamine challenges. It is established that psychostimulants upregulate ROS production in the brain and contribute to psychostimulant-driven behavioral impairments.
However, caution is necessary when relating these findings to schizophrenia. One problem is the disease specificity of ROS pathology (Cobb and Cole 2015). The pathologic implications of ROS are not limited to schizophrenia. ROS is also implicated in Alzheimer’s disease and Parkinson’s disease (Lovell and Markesbery 2007; Nakabeppu et al. 2007). Neural activity itself involves higher energy metabolism and results in higher ROS production, as evident in patients with epilepsy (Puttachary et al. 2015). Medication for schizophrenia also enhances the production of ROS; the metabolism of antipsychotic drugs necessitates cytochrome P450 recruitment and also promotes dopamine release and monoamine oxidase activation, all of which drive ROS generation (Martins et al. 2008; Reinke et al. 2004; Table 1). In this context, the pathologic and pharmacologic roles of ROS generation in patients with schizophrenia are controversial and remain to be characterized further (Naviaux 2012).
2 Pharmacologic Actions of Antioxidants in Patients with and Animal Models for Schizophrenia
In contrast to arguments supporting the pathologic role of ROS in patients with schizophrenia, many reports more consistently describe the antipsychotic actions of reducing agents. A variety of reducing agents and radical scavengers, including ω-3 fatty acids, N-acetyl cysteine, minocycline, and vitamins C and E, have been administered to patients, and their therapeutic effects evaluated. Based on this theory, Sivrioglu et al. (2007) supplemented the food of patients with schizophrenia with ω-3 fatty acids and reported a reduction in antipsychotic side effects and an improvement in psychotic symptoms. Farokhnia et al. (2013) administered a more potent antioxidant, N-acetyl cysteine, to patients with schizophrenia and observed its effectiveness on negative symptoms. In combination with atypical antipsychotics, minocycline was administered to patients in the acute phase, resulting in an improvement in cognition and negative symptoms (Miyaoka et al. 2007; Levkovitz et al. 2010).
The antipsychotic effects of reducing agents have also been tested in several animal models. Both MK-801 and methamphetamine induce hyperlocomotion and social interaction deficits and cause neurodegeneration in various parts of the brain (Ozyurt et al. 2007). The administration of vitamin E into MK-801-challenged rats decreased cell death in the cingulated cortex (Zhang et al. 2006; Willis and Ray 2007), whereas the administration of the radical scavenger edaravone inhibited cell death in striatal neurons (Kawasaki et al. 2006). In addition to cell death, the behavioral deficits induced by phencyclidine are also ameliorated by the scavenger of a SOD mimetic (Wang et al. 2003). Neonatal hippocampal lesions result in behavioral impairments at the postpubertal stage and serve as an animal model of schizophrenia (Lipska et al. 1993). The antipsychotic effects of N-acetyl cysteine have been verified in this animal model (Cabungcal et al. 2014). Despite discrepancies in the arguments supporting an etiologic or pathologic role of ROS in schizophrenia, almost all antioxidative agents exhibit beneficial effects on cognitive abnormalities relevant to schizophrenia, although the cellular and molecular mechanisms underlying these pharmacologic phenomena remain unclear.
3 Strong Interactions Between ROS and Cytokine Signaling
ROS signaling is known to link to cytokine production and cytokine receptor signaling (Landskron et al. 2014). Proinflammatory cytokines and growth factors activate NOX and produce ROS. Brain neurons and microglia release ROS in response to cytokines. Conversely, generated ROS stimulate cytokine production by activating the redox sensor NF-κB (Lian and Zheng 2009) or accelerate cytokine receptor signaling by inhibiting the redox sensor phosphatase. Notably, ROS alone induces the phosphorylation and activation of cytokine receptors in the absence of cytokine ligands such as epidermal growth factor (EGF). In this review, we discuss the interaction between ROS and EGF signaling most extensively demonstrated in the field of carcinogenesis (Chiarugi et al. 2003; Goldkorn et al. 2005).
EGF signaling is highly implicated and serves as a drug target in tumor biology, because this cytokine potently produces ROS, which are known to be DNA mutagens. An EGF challenge to cancer cells and cultured neurons elevates intracellular ROS concentrations (Bae et al. 1997; Cha et al. 2000). Table 2 shows the potency of the cytokines that induce ROS production in cultured neocortical neurons. EGF oxidizes the ROS indicator dihydroethidium and changes the indicator to a fluorescent form. This EGF-triggered increase in ROS levels is blocked by the co-application of antioxidants (apocynin, Trolox; Fig. 1). These results indicate that EGF indeed induces ROS production in the brain.


The effects of EGF and antioxidants on the levels of ROS in neocortical cultures. a Cultured cortical neurons (DIV7) were stimulated with or without 20 ng/ml EGF. ROS production was assessed by mitotracker, which sensed mitochondrial membrane potential. Scale bar 50 µm. b Cultured cortical neurons were treated with antioxidants (0.5 mM apocynin, 100 µM Trolox) in the absence or presence of 20 ng/ml EGF. c Cultured cortical neurons were stimulated with 20 ng/ml EGF, 20 ng/ml neuregulin-1, or 1 µM ketamine. The MitoTracker fluorescence intensity was measured in 10 cells per well using Image J (National Institutes of Health, Bethesda, MD, USA; arbitrary units, A.U.). n = 8, *P < 0.05, EGF epidermal growth factor
In such inflammatory conditions, the main source of ROS is NOX, which is highly potentiated by EGF signals (Fan et al. 2005a, b; Chen et al. 2008). De Yulia et al. (2005) proposed that hydrogen peroxide is produced by the EGF receptor (EGFR, also known as ErbB1) itself. Conversely, ROS converts the thiol group of a cysteine residue to sulfuric acid in the core region of the tyrosine phosphatase PTP1B, inactivating this enzyme (Yip et al. 2010). Compared with many receptor-type protein tyrosine kinases, the EGFR undergoes a significant amount of basal autophosphorylation in the absence of its ligand and requires phosphatases for its silencing in a basal condition. Thus, the ROS-driven inactivation of PTP1B alone increases phosphorylation of the EGFR, resulting in the activation of EGF signaling without ligands (Lee et al. 1998; De Wit et al. 2001). Thus, hydrogen peroxide-oxidized low-density lipoprotein can also phosphorylate EGFR in the absence of EGF and evoke EGFR signaling (Suc et al. 1998). If acting on peripheral cells, hydrogen peroxide may promote cell proliferation and induce carcinogenesis. In this context, the EGFR signaling cascade appears to be one of the most crucial ROS targets.
Figure 2 summarizes the production of and interaction between EGF and ROS. The ligand EGF binds to EGFR and activates calcium signaling, in turn activating the ROS generators NOX, cyclooxygenase, and cytochromes in mitochondria. The ROS produced acts on phosphatases such as PTP1B and enhances and prolongs the EGF signal transduction. The enhanced phosphorylation of EGFR also hinders its internalization (Ravid et al. 2002). Additionally, ROS acts on the redox site of the transcription factor NF-κB and accelerates NF-κB-mediated gene expression, leading to the synthesis of mRNA for EGF and other cytokines. When ROS reacts with ion channels, neurotransmission in the brain is perturbed unless ROS are trapped by various scavenger molecules (glutathione, thioredoxin, SOD, catalase, etc.).


Schematic diagram of the interaction between EGF and ROS. EGF and its homologues (transforming growth factor alpha, amphiregulin, heparin-binding EGF-like growth factor, betacellulin, epiregulin, neuregulins 1–6) as well as poxvirus virokines (SMDF, VGF, etc.) directly and indirectly interact with the EGFR (also known as ErbB1) to evoke calcium signaling. This activates NOX, cyclooxgenase (COX), and mitochondrial cytochrome c, which produce ROS. In turn, ROS act on and inhibit protein tyrosine phosphatases (SHP2, the protein tyrosine phosphatase PTP1B, etc.) and promote the phosphorylation and activation of EGFR, leading to the production of other cytokines and the further activation of NOX, COX, and mitochondrial cytochromes. EGF epidermal growth factor; EGFR EGF receptor; NOX NADPH oxidase; ROS reactive oxygen species
As shown in Table 2, the EGF homologue neuregulin-1 (NRG-1) triggers ROS production in the brain. In contrast to EGF, NRG-1 binds to the ErbB3 or ErbB4 receptor tyrosine kinases, but NRG-1 signal transduction also involves ROS-mediated autoregulation. However, compared with EGF, NRG-1-driven ROS production is more persistent (Goldsmit et al. 2001).
4 Establishment of Animal Models of Schizophrenia by Exposure to EGF and NRG-1
Maternal viral infection and obstetric complication are implicated as environmental risk factors for schizophrenia (Nawa and Takei 2006; Iwakura and Nawa 2013). EGF is known to be highly enriched in human amniotic fluids and contributes to obstetric complication (Varner et al. 1996). EGF derivatives are known to be encoded in viral genome of poxviruses and might be supplied to human fetus and neonates via their infection (Tzahar et al. 1998). Genetic linkage studies and neuropathologic investigations also suggest an association between schizophrenia and EGF and its derivative NRG-1 (Futamura et al. 2002; Anttila et al. 2004; Stefansson et al. 2004; Groenestege et al. 2007). On the premise of the neurodevelopmental hypothesis for this illness, therefore, we analyzed animal models of schizophrenia established by perinatal exposure to EGF and its derivatives (Futamura et al. 2003; Tohmi et al. 2004; Watanabe et al. 2004; Tsuda et al. 2008; Kato et al. 2011). Treatment of neonatal rats and mice with EGF, NRG-1, and their homologues and paralogues (i.e., transforming growth factor alpha, epiregulin) all showed the behavioral deficits relevant to schizophrenia at the postpubertal stage (Fig. 3). Injected factors penetrate the immature blood–brain barrier and bind to brain neurons carrying EGFR or ErbB receptors, such as GABAergic and dopaminergic neurons (Abe et al. 2009; Namba et al. 2009). In addition to dopaminergic neurons, GABAergic neurons and glial cells also respond to these cytokines, but their responses gradually diminish after the cessation of cytokine treatment (Nagano et al. 2007; Abe et al. 2011). The most prominent and persistent influences of cytokine treatment are those on dopaminergic neurons, which cause the animals to display cognitive and behavioral impairments (Sotoyama et al. 2011, 2013). These behavioral impairments include abnormalities in sensorimotor gating (prepulse inhibition [PPI]), social interaction, exploratory movement, latent inhibition of learning, and sensitivity to psychostimulants (methamphetamine and/or MK-801), although the directions and magnitudes of these deficits are significantly altered by the genetic background of the animal species used (Mizuno et al. 2004; Tohmi et al. 2005). This observation agrees with the current theory that the onset of schizophrenia involves both genetic and environmental factors.


Behavioral impairments in the animal model of schizophrenia established by perinatal exposure to EGF. EGF epidermal growth factor. Various behavioral abnormalities emerge at the post-pubertal stage
Although many animal models of schizophrenia have been established, our animal models have the following three characteristic features (Nawa et al. 2014):
-
The emergence of behavioral abnormalities occurs not before or during puberty, but around the postpubertal stage. The EGF-exposure-generated monkey model of schizophrenia shows striking behavioral deficits from 5 years of age (Sakai et al. 2014).
-
EGF- and NRG-1-exposure-generated models have no apparent deficits in learning. In the eight-arm radial maze test, context fear-learning paradigm, and active avoidance test, these models are indistinguishable from controls (Futamura et al. 2003; Kato et al. 2011).
-
Atypical antipsychotics, such as risperidone and clozapine, are effective for the above behavioral deficits, whereas the pharmacologic action of typical antipsychotics (i.e., haloperidol) is limited (Futamura et al. 2003, Sotoyama et al. 2013).
We found similar behavioral abnormalities in transgenic mice in which EGF or NRG-1 was overexpressed from their transgenes (Kato et al. 2010; Eda et al. 2013). However, these approaches to cytokine administration obscure their cellular target(s). The behavioral abnormalities may therefore be ascribed not only to the dysfunction of midbrain dopaminergic neurons but also to that of other peripheral organs. To limit the target organ of the cytokines, we conducted intracerebroventricular administration of EGF into adult rats and verified the reproducibility of the behavioral impairments achieved by perinatal pre-exposure to EGF (Mizuno et al. 2007). This result suggests that peripherally administered EGF targets brain neurons in the neonatal EGF model for schizophrenia.
Subsequent analyses of these models revealed that NRG-1 mainly acts on dopaminergic neurons in the ventral tegmental area of the midbrain, whereas EGF interacts with those in the substantia nigra to promote dopamine synthesis and terminal arborization (Abe et al. 2009; Iwakura et al. 2011a, b). Neonatal mice treated with NRG-1 exhibit a hyperdopaminergic state in the prelimbic cortex (i.e., the medial prefrontal cortex) as adults (Kato et al. 2011). Similarly, neonatal rats treated with EGF display a hyperdopaminergic state in the globus pallidus (Sotoyama et al. 2011). Dopaminergic innervation and release in the globus pallidus are significantly elevated in this model. However, their PPI deficits and abnormal dopamine release are simultaneously normalized by the local administration of an antipsychotic drug to the globus pallidus (Sotoyama et al. 2011, 2013). The globus pallidus is enriched with dopamine D2 receptors and is the major target of antipsychotic drugs in the indirect pathway of the basal ganglia.
5 Pharmacologic Actions of ROS Scavengers in the EGF-Exposure-Generated Model
As mentioned earlier, EGF is a potent ROS producer, and knockdown of protein phosphatases by ROS in turn enhances phosphorylation-based signaling cascades emanating from EGFR. In agreement, Finch et al. (2006) found that catalase degrades hydrogen peroxide and reduces EGF signaling. To assess the direct interaction between ROS production and EGF signaling, we administered the three ROS scavengers (Trolox, edaravone, and 2,2,6,6-tetramethyl-4-piperidinol-N-oxyl [TEMPOL]) during EGF treatment in neonatal rats. The effects of these scavengers on the PPI deficits of EGF-treated rats were evaluated at the adult stage (Fig. 4). Co-administration of TEMPOL and edaravone with EGF resulted in attenuation of the PPI deficits of the EGF-exposure-generated model. This result suggests that ROS production during neonatal EGF treatment is required to generate the model.


Competition by antioxidants with neonatal EGF treatment. Epidermal growth factor (EGF; 0.875 µg/g, administered subcutaneously) or saline was administered daily on postnatal days 2–11 together with antioxidants (3 mg/kg TEMPOL, 10 mg/kg edaravone, and 10 mg/kg Trolox). Interference with the effects of EGF on prepulse inhibition scores was monitored at the adult stage. n = 5 in each group, *P < 0.05 and **P < 0.01, EGF epidermal growth factor
We also examined the medication effects of ROS scavengers (emodin, Trolox, edaravone) on the behavioral impairments of the EGF- pretreatment model at the adult stage (Fig. 5). Emodin is a polyphenol ROS scavenger known as 3-methyl-1,6,8-trihydroxyanthraquinone and attenuates EGFR signaling (Mizuno et al. 2008). Edaravone is otherwise known as 3-methyl-1-phenyl-2-pyrazolin-5-one and is prescribed to patients who have suffered a stroke to mitigate the expansion of cerebral ischemic injuries. This drug is verified to reduce EGFR signaling in cancer cells (Suzuki et al. 2005). Subchronic treatment with emodin at the adult stage suppressed the acoustic startle response and abolished the PPI deficits of the EGF-pretreatment model (Mizuno et al. 2008). The medication effects of emodin on social interaction impairments were undetectable. However, we observed an antipsychotic-like activity of emodin in the methamphetamine model (Mizuno et al. 2010). In contrast to emodin, intraperitoneal injection of edaravone improved social interaction scores and decreased an abnormally high acoustic startle response of the EGF-pretreatment model, although it had no effect on their PPI deficits (Fig. 5). Both the results verified the pharmaceutical effectiveness of ROS scavengers on behavioral impairments relevant to schizophrenia. However, the antipsychotic-like profiles of the individual scavengers differed significantly. Presumably, the preference for the target ROS species differs between those ROS scavengers and results in the diversity between their pharmacologic profiles (Suzuki et al. 2005).


Antipsychotic effects of edaravone on the EGF model relevant to schizophrenia. Epidermal growth factor (0.875 µg/g) or saline was administered subcutaneously daily on postnatal days 2–11. Adult rats were treated with or without the radical scavenger edaravone (5 mg/kg/day for 7 days, administered intraperitoneally). n = 6–11 in each group, *P < 0.05, **P < 0.01, and ***P < 0.001. EGF epidermal growth factor
Although the therapeutic mechanism of ROS scavengers remains undetermined, we believe the EGFR signaling cascade to be one of their molecular targets, as suggested by Suzuki et al. (2005). We postulate that an elevation in EGFR signaling (e.g., ErbB1, ErbB2) is sustained until the adult stage even when EGF treatment is completed during the neonatal stage (Futamura et al. 2002). The hypothesis stems from the fact that the administration of specific EGFR blockers also ameliorated the behavioral deficits of the EGF-exposure-generated model (Mizuno et al. 2008, 2013). In light of the strong interaction between EGF signaling and ROS production, further study is warranted into which plays the crucial role in the pathogenesis of both the EGF-exposure-generated model and patients with schizophrenia.
6 Conclusion
There are tight interactions between ROS and cytokine signaling. In particular, such interactions are evident for the EGF signal cascade in which basal EGFR autophosphorylation is not negligible. ROS itself inactivates its phosphatases and provokes EGF receptor signaling without any ligands. Conversely, EGFR phosphorylation efficiently activates various ROS-generating enzymes such as NOX and COX to produce excess amounts of ROS. The cytokine–ROS system appears to be recruited in the pathologic conditions of brain injury, infection, and seizures to protect the organ with their given neurotrophic actions as well as to promote the regeneration processes from the insults. When the activation of the cytokine–ROS system is prolonged, the unfavorable side effects emerge in dopaminergic and glutamatergic development/neurotransmission, leading to abnormal brain functions. Our latest results from the EGF model indicate that oxygen radical scavengers are beneficial for the medication of their cognitive deficits. However, the pharmacological profiles differ significantly among scavengers. We hope that safe and novel antipsychotic drugs will be developed from any of oxygen radical scavengers.
References
Abe Y, Namba H, Zheng Y, Nawa H (2009) In situ hybridization reveals developmental regulation of ErbB1-4 mRNA expression in mouse midbrain: implication of ErbB receptors for dopaminergic neurons. Neuroscience 161(1):95–110
Abe Y, Namba H, Kato T, Iwakura Y, Nawa H (2011) Neuregulin-1 signals from the periphery regulate AMPA receptor sensitivity and expression in GABAergic interneurons in developing neocortex. J Neurosci 31(15):5699–5709
Anttila S, Illi A, Kampman O, Mattila KM, Lehtimäki T, Leinonen E (2004) Association of EGF polymorphism with schizophrenia in Finnish men. NeuroReport 15(7):1215–1218
Arai M, Yuzawa H, Nohara I, Ohnishi T, Obata N, Iwayama Y, Haga S, Toyota T, Ujike H, Arai M, Ichikawa T, Nishida A, Tanaka Y, Furukawa A, Aikawa Y, Kuroda O, Niizato K, Izawa R, Nakamura K, Mori N, Matsuzawa D, Hashimoto K, Iyo M, Sora I, Matsushita M, Okazaki Y, Yoshikawa T, Miyata T, Itokawa M (2010) Enhanced carbonyl stress in a subpopulation of schizophrenia. Arch Gen Psychiatry 67(6):589–597
Arai M, Miyashita M, Kobori A, Toriumi K, Horiuchi Y, Itokawa M (2014) Carbonyl stress and schizophrenia. Psychiatry Clin Neurosci 68(9):655–665
Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG (1997) Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem 272(1):217–221
Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL (2007) Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318(5856):1645–1647
Ben Othmen L, Mechri A, Fendri C, Bost M, Chazot G, Gaha L, Kerkeni A (2008) Altered antioxidant defense system in clinically stable patients with schizophrenia and their unaffected siblings. Prog Neuropsychopharmacol Biol Psychiatry 32(1):155–159
Bokkon I, Antal I (2011) Schizophrenia: redox regulation and volume neurotransmission. Curr Neuropharmacol 9:289–300
Brewer TF, Garcia FJ, Onak CS, Carroll KS, Chang CJ (2015) Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu Rev Biochem 84:765–790
Buttner N, Bhattacharyya S, Walsh J, Benes FM (2007) DNA fragmentation is increased in non-GABAergic neurons in bipolar disorder but not in schizophrenia. Schizophr Res 93(1–3):33–41
Cabungcal JH, Counotte DS, Lewis EM, Tejeda HA, Piantadosi P, Pollock C, Calhoon GG, Sullivan EM, Presgraves E, Kil J, Hong LE, Cuenod M, Do KQ, O’Donnell P (2014) Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron 83(5):1073–1084
Cha YK, Kim YH, Ahn YH, Koh JY (2000) Epidermal growth factor induces oxidative neuronal injury in cortical culture. J Neurochem 75(1):298–303
Chan PH (2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21(1):2–14
Chen W, Shang WH, Adachi Y, Hirose K, Ferrari DM, Kamata T (2008) A possible biochemical link between NADPH oxidase (Nox) 1 redox-signalling and ERp72. Biochem J 416(1):55–63
Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G (2003) Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol 161(5):933–944
Cobb CA, Cole MP (2015) Oxidative and nitrative stress in neurodegeneration. Neurobiol Dis. doi:10.1016/j.nbd.2015.04.020
De Wit R, Makkinje M, Boonstra J, Verkleij AJ, Post JA (2001) Hydrogen peroxide reversibly inhibits epidermal growth factor (EGF) receptor internalization and coincident ubiquitination of the EGF receptor and Eps15. FASEB J 15(2):306–308
De Yulia GJ, Jr Cárcamo JM, Bórquez-Ojeda O, Shelton CC, Golde DW (2005) Hydrogen peroxide generated extracellularly by receptor-ligand interaction facilitates cell signaling. Proc Natl Acad Sci USA 102(14):5044–5049
Dukoff DJ, Hogg DW, Hawrysh PJ, Buck LT (2014) Scavenging ROS dramatically increase NMDA receptor whole-cell currents in painted turtle cortical neurons. J Exp Biol 217(Pt 18):3346–3355
Eda T, Mizuno M, Araki K, Iwakura Y, Namba H, Sotoyama H, Kakita A, Takahashi H, Satoh H, Chan SY, Nawa H (2013) Neurobehavioral deficits of epidermal growth factor-overexpressing transgenic mice: impact on dopamine metabolism. Neurosci Lett 547:21–25
Fan CY, Katsuyama M, Yabe-Nishimura C (2005a) PKCdelta mediates up-regulation of NOX1, a catalytic subunit of NADPH oxidase, via transactivation of the EGF receptor: possible involvement of PKCdelta in vascular hypertrophy. Biochem J 390(Pt 3):761–767
Fan C, Katsuyama M, Nishinaka T, Yabe-Nishimura C (2005b) Transactivation of the EGF receptor and a PI3 kinase-ATF-1 pathway is involved in the upregulation of NOX1, a catalytic subunit of NADPH oxidase. FEBS Lett 579(5):1301–1305
Farokhnia M, Azarkolah A, Adinehfar F, Khodaie-Ardakani MR, Hosseini SM, Yekehtaz H, Tabrizi M, Rezaei F, Salehi B, Sadeghi SM, Moghadam M, Gharibi F, Mirshafiee O, Akhondzadeh S (2013) N-acetylcysteine as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia: a randomized, double-blind, placebo-controlled study. Clin Neuropharmacol 36(6):185–192
Finch JS, Tome ME, Kwei KA, Bowden GT (2006) Catalase reverses tumorigenicity in a malignant cell line by an epidermal growth factor receptor pathway. Free Radic Biol Med 40(5):863–875
Futamura T, Toyooka K, Iritani S, Niizato K, Nakamura R, Tsuchiya K, Someya T, Kakita A, Takahashi H, Nawa H (2002) Abnormal expression of epidermal growth factor and its receptor in the forebrain and serum of schizophrenic patients. Mol Psychiatry 7(7):673–682
Futamura T, Kakita A, Tohmi M, Sotoyama H, Takahashi H, Nawa H (2003) Neonatal perturbation of neurotrophic signaling results in abnormal sensorimotor gating and social interaction in adults: implication for epidermal growth factor in cognitive development. Mol Psychiatry 8(1):19–29
Goldkorn T, Ravid T, Khan EM (2005) Life and death decisions: ceramide generation and EGF receptor trafficking are modulated by oxidative stress. Antioxid Redox Signal 7(1–2):119–128
Goldsmit Y, Erlich S, Pinkas-Kramarski R (2001) Neuregulin induces sustained reactive oxygen species generation to mediate neuronal differentiation. Cell Mol Neurobiol 21(6):753–769
Groenestege WM, Thébault S, van der Wijst J, van den Berg D, Janssen R, Tejpar S, van den Heuvel LP, van Cutsem E, Hoenderop JG, Knoers NV, Bindels RJ (2007) Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest 117(8):2260–2267
Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P, Deppen P, Preisig M, Ruiz V, Steullet P, Tosic M, Werge T, Cuénod M, Do KQ (2007) Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc Natl Acad Sci USA 104(42):16621–16626
Iwakura Y, Zheng Y, Sibilia M, Abe Y, Piao YS, Yokomaku D, Wang R, Ishizuka Y, Takei N, Nawa H (2011a) Qualitative and quantitative re-evaluation of epidermal growth factor-ErbB1 action on developing midbrain dopaminergic neurons in vivo and in vitro: target-derived neurotrophic signaling (Part 1). J Neurochem 118(1):45–56
Iwakura Y, Wang R, Abe Y, Piao YS, Shishido Y, Higashiyama S, Takei N, Nawa H (2011b) Dopamine-dependent ectodomain shedding and release of epidermal growth factor in developing striatum: target-derived neurotrophic signaling (Part 2). J Neurochem 118(1):57–68
Iwakura Y, Nawa H (2013) ErbB1-4-dependent EGF/neuregulin signals and their cross talk in the central nervous system: pathological implications in schizophrenia and Parkinson’s diseasse. Front Cell Neurosci 7:4
Kato T, Abe Y, Sotoyama H, Kakita A, Kominami R, Hirokawa S, Ozaki M, Takahashi H, Nawa H (2011) Transient exposure of neonatal mice to neuregulin-1 results in hyperdopaminergic states in adulthood: implication in neurodevelopmental hypothesis for schizophrenia. Mol Psychiatry 16(3):307–320
Kato T, Kasai A, Mizuno M, Fengyi L, Shintani N, Maeda S, Yokoyama M, Ozaki M, Nawa H (2010) Phenotypic characterization of transgenic mice overexpressing neuregulin-1. PLoS ONE 5(12):e14185
Kaur U, Banerjee P, Bir A, Sinha M, Biswas A, Chakrabarti S (2015) Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: the NF-κB connection. Curr Top Med Chem 15(5):446–457
Kawasaki T, Ishihara K, Ago Y, Nakamura S, Itoh S, Baba A, Matsuda T (2006) Protective effect of the radical scavenger edaravone against methamphetamine-induced dopaminergic neurotoxicity in mouse striatum. Eur J Pharmacol 542(1–3):92–99
Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA (2014) Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014:149185
Lee SR, Kwon KS, Kim SR, Rhee SG (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem 273(25):15366–15372
Levkovitz Y, Mendlovich S, Riwkes S, Braw Y, Levkovitch-Verbin H, Gal G, Fennig S, Treves I, Kron S (2010) A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry 71(2):138–149
Lian M, Zheng X (2009) HSCARG regulates NF-kappaB activation by promoting the ubiquitination of RelA or COMMD1. J Biol Chem 284(27):17998–18006
Lipska BK, Jaskiw GE, Weinberger DR (1993) Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology 9(1):67–75
Lovell MA, Markesbery WR (2007) Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res 35(22):7497–7504
Mahadik SP, Scheffer RE (1996) Oxidative injury and potential use of antioxidants in schizophrenia. Prostaglandins Leukot Essent Fatty Acids 55(1–2):45–54
Martins MR, Petronilho FC, Gomes KM, Dal-Pizzol F, Streck EL, Quevedo J (2008) Antipsychotic-induced oxidative stress in rat brain. Neurotox Res 13:63–69
McCreadie RG, MacDonald E, Wiles D, Campbell G, Paterson JR (1995) The Nithsdale Schizophrenia Surveys. XIV: plasma lipid peroxide and serum vitamin E levels in patients with and without tardive dyskinesia, and in normal subjects. Br J Psychiatry 167:610–617
Miyaoka T, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J (2007) Possible antipsychotic effects of minocycline in patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 31(1):304–307
Mizuno M, Malta RS Jr, Nagano T, Nawa H (2004) Conditioned place preference and locomotor sensitization after repeated administration of cocaine or methamphetamine in rats treated with epidermal growth factor during the neonatal period. Ann NY Acad Sci 1025:612–618
Mizuno M, Sotoyama H, Narita E, Kawamura H, Namba H, Zheng Y, Eda T, Nawa H (2007) A cyclooxygenase-2 inhibitor ameliorates behavioral impairments induced by striatal administration of epidermal growth factor. J Neurosci 27(38):10116–10127
Mizuno M, Kawamura H, Takei N, Nawa H (2008) The anthraquinone derivative Emodin ameliorates neurobehavioral deficits of a rodent model for schizophrenia. J Neural Transm 115(3):521–530
Mizuno M, Kawamura H, Ishizuka Y, Sotoyama H, Nawa H (2010) The anthraquinone derivative emodin attenuates methamphetamine-induced hyperlocomotion and startle response in rats. Pharmacol Biochem Behav 97(2):392–398
Mizuno M, Sotoyama H, Namba H, Shibuya M, Eda T, Wang R, Okubo T, Nagata K, Iwakura Y, Nawa H (2013) ErbB inhibitors ameliorate behavioral impairments of an animal model for schizophrenia: implication of their dopamine-modulatory actions. Transl Psychiatry 30(3):e252
Mohn AR, Gainetdinov RR, Caron MG, Koller BH (1999) Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98(4):427–436
Nagano T, Namba H, Abe Y, Aoki H, Takei N, Nawa H (2007) In vivo administration of epidermal growth factor and its homologue attenuates developmental maturation of functional excitatory synapses in cortical GABAergic neurons. Eur J Neurosci 25(2):380–390
Namba H, Zheng Y, Abe Y, Nawa H (2009) Epidermal growth factor administered in the periphery influences excitatory synaptic inputs onto midbrain dopaminergic neurons in postnatal mice. Neuroscience 158(4):1731–1741
Naviaux RK (2012) Oxidative shielding or oxidative stress? J Pharmacol Exp Ther 342(3):608–618
Nawa H, Takei N (2006) Recent progress in animal modeling of immune inflammatory processes in schizophrenia: implication of specific cytokines. Neurosci Res 56(1):2–13
Nawa H, Sotoyama H, Iwakura Y, Takei N, Namba H (2014) Neuropathologic implication of peripheral neuregulin-1 and EGF signals in dopaminergic dysfunction and behavioral deficits relevant to schizophrenia: their target cells and time window. Biomed Res Int. 2014:697935
Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K (2007) Oxidative damage in nucleic acids and Parkinson’s disease. J Neurosci Res 85(5):919–934
Nishioka N, Arnold SE (2004) Evidence for oxidative DNA damage in the hippocampus of elderly patients with chronic schizophrenia. Am J Geriatr Psychiatry 12(2):167–175
Ozyurt B, Ozyurt H, Akpolat N, Erdogan H, Sarsilmaz M (2007) Oxidative stress in prefrontal cortex of rat exposed to MK-801 and protective effects of CAPE. Prog Neuropsychopharmacol Biol Psychiatry 31(4):832–838
Puttachary S, Sharma S, Stark S, Thippeswamy T (2015) Seizure-induced oxidative stress in temporal lobe epilepsy. Biomed Res Int 2015:745613
Ravid T, Sweeney C, Gee P, Carraway KL 3rd, Goldkorn T (2002) Epidermal growth factor receptor activation under oxidative stress fails to promote c-Cbl mediated down-regulation. J Biol Chem 277(34):31214–31219
Reinke A, Martins MR, Lima MS, Moreira JC, Dal-Pizzol F, Quevedo J (2004) Haloperidol and clozapine, but not olanzapine, induces oxidative stress in rat brain. Neurosci Lett 372(1–2):157–160
Sanchez RM, Wang C, Gardner G, Orlando L, Tauck DL, Rosenberg PA, Aizenman E, Jensen FE (2000) Novel role for the NMDA receptor redox modulatory site in the pathophysiology of seizures. J Neurosci 20(6):2409–2417
Sakai M, Kashiwahara M, Kakita A, Nawa H (2014) An attempt of non-human primate modeling of schizophrenia with neonatal challenges of epidermal growth factor. J Addict Res Ther 5:170
Sivrioglu EY, Kirli S, Sipahioglu D, Gursoy B, Sarandöl E (2007) The impact of omega-3 fatty acids, vitamins E and C supplementation on treatment outcome and side effects in schizophrenia patients treated with haloperidol: an open-label pilot study. Prog Neuropsychopharmacol Biol Psychiatry 31(7):1493–1499
Sotoyama H, Namba H, Chiken S, Nambu A, Nawa H (2013) Exposure to the cytokine EGF leads to abnormal hyperactivity of pallidal GABA neurons: implications for schizophrenia and its modeling. J Neurochem 126(4):518–528
Sotoyama H, Zheng Y, Iwakura Y, Mizuno M, Aizawa M, Shcherbakova K, Wang R, Namba H, Nawa H (2011) Pallidal hyperdopaminergic innervation underlying D2 receptor-dependent behavioral deficits in the schizophrenia animal model established by EGF. PLoS ONE 6(10):e25831
Stefansson H, Steinthorsdottir V, Thorgeirsson TE, Gulcher JR (2004) Stefansson K (2004) Neuregulin 1 and schizophrenia. Ann Med 36(1):62–71
Suboticanec K, olnegovic-Smalc V, Korbar M, Mestrovic B, Buzina R (1990) Vitamin C status in chronic schizophrenia. Biol Psychiatry 28:959–966
Suc I, Meilhac O, Lajoie-Mazenc I, Vandaele J, Jürgens G, Salvayre R, Nègre-Salvayre A (1998) Activation of EGF receptor by oxidized LDL. FASEB J 12(9):665–671
Suzuki R, Gopalrao RK, Maeda H, Rao P, Yamamoto M, Xing Y, Mizobuchi S, Sasaguri S (2005) MCI-186 inhibits tumor growth through suppression of EGFR phosphorylation and cell cycle arrest. Anticancer Res 25(2A):1131–1138
Takahashi M, Kakita A, Futamura T, Watanabe Y, Mizuno M, Sakimura K, Castren E, Nabeshima T, Someya T, Nawa H (2006) Sustained brain-derived neurotrophic factor up-regulation and sensorimotor gating abnormality induced by postnatal exposure to phencyclidine: comparison with adult treatment. J Neurochem 99(3):770–780
Tohmi M, Tsuda N, Mizuno M, Takei N, Frankland PW, Nawa H (2005) Distinct influences of neonatal epidermal growth factor challenge on adult neurobehavioral traits in four mouse strains. Behav Genet 35(5):615–629
Tohmi M, Tsuda N, Watanabe Y, Kakita A, Nawa H (2004) Perinatal inflammatory cytokine challenge results in distinct neurobehavioral alterations in rats: implication in psychiatric disorders of developmental origin. Neurosci Res 50(1):67–75
Tsuda N, Mizuno M, Yamanaka T, Komurasaki T, Yoshimoto M, Nawa H (2008) Common behavioral influences of the ErbB1 ligands transforming growth factor alpha and epiregulin administered to mouse neonates. Brain Dev 30(8):533–543
Tzahar E, Moyer JD, Waterman H, Barbacci EG, Bao J, Levkowitz G, Shelly M, Strano S, Pinkas-Kramarski R, Pierce JH, Andrews GC, Yarden Y (1998) Pathogenic poxviruses reveal viral strategies to exploit the ErbB signaling network. EMBO J 17(20):5948–5963
Ustundag B, Atmaca M, Kirtas O, Selek S, Metin K, Tezcan E (2006) Total antioxidant response in patients with schizophrenia. Psychiatry Clin Neurosci 60(4):458–464
Varner MW, Dildy GA, Hunter C, Dudley DJ, Clark SL, Mitchell MD (1996) Amniotic fluid epidermal growth factor levels in normal and abnormal pregnancies. J Soc Gynecol Investig. 3:17–19
Wang C, McInnis J, West JB, Bao J, Anastasio N, Guidry JA, Ye Y, Salvemini D, Johnson KM (2003) Blockade of phencyclidine-induced cortical apoptosis and deficits in prepulse inhibition by M40403, a superoxide dismutase mimetic. J Pharmacol Exp Ther 304(1):266–271
Watanabe Y, Hashimoto S, Kakita A, Takahashi H, Ko J, Mizuno M, Someya T, Patterson PH, Nawa H (2004) Neonatal impact of leukemia inhibitory factor on neurobehavioral development in rats. Neurosci Res 48(3):345–353
Willis CL, Ray DE (2007) Antioxidants attenuate MK-801-induced cortical neurotoxicity in the rat. Neurotoxicology 28(1):161–167
Yao JK, Keshavan MS (2011) Antioxidants, redox signaling, and pathophysiology in schizophrenia: an integrative view. Antioxid Redox Signal 15(7):2011–2035
Yip SC, Saha S, Chernoff J (2010) PTP1B: a double agent in metabolism and oncogenesis. Trends Biochem Sci 35(8):442–449
Zhang L, Kitaichi K, Fujimoto Y, Nakayama H, Shimizu E, Iyo M, Hashimoto K (2006) Protective effects of minocycline on behavioral changes and neurotoxicity in mice after administration of methamphetamine. Prog Neuropsychopharmacol Biol Psychiatry 30(8):1381–1393
Zuo DY, Wu YL, Yao WX, Cao Y, Wu CF, Tanaka M (2007) Effect of MK-801 and ketamine on hydroxyl radical generation in the posterior cingulate and retrosplenial cortex of free-moving mice, as determined by in vivo microdialysis. Pharmacol Biochem Behav 86(1):1–7
Acknowledgments
We are grateful to Tomoyuki Sugano and Akira Yarimizu for their technical assistances. The authors’ original work was partly supported by Grants-in-Aid for Scientific Research on Innovative Areas and for Challenging Exploratory Research (No. 24116010, No. 21659273) and a grant for Promotion of Niigata University Research Projects. Human recombinant EGF was kindly provided by Higeta Shoyu Co. Ltd. Except this gift, all the authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Nagano, T., Mizuno, M., Morita, K., Nawa, H. (2015). Pathological Implications of Oxidative Stress in Patients and Animal Models with Schizophrenia: The Role of Epidermal Growth Factor Receptor Signaling. In: Kostrzewa, R.M., Archer, T. (eds) Neurotoxin Modeling of Brain Disorders—Life-long Outcomes in Behavioral Teratology. Current Topics in Behavioral Neurosciences, vol 29. Springer, Cham. https://doi.org/10.1007/7854_2015_399
Download citation
DOI: https://doi.org/10.1007/7854_2015_399
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-34134-7
Online ISBN: 978-3-319-34136-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)