
Abstract
Mining microarray data to unearth interesting expression profile patterns for discovery of in silico biological knowledge is an emerging area of research in computational biology. A group of functionally related genes may have similar expression patterns under a set of conditions or at some time points. Biclustering is an important data mining tool that has been successfully used to analyze gene expression data for biologically significant cluster discovery. The purpose of this chapter is to introduce interesting patterns that may be observed in expression data and discuss the role of biclustering techniques in detecting interesting functional gene groups with similar expression patterns.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Keywords:
1 Introduction
With the rapid growth of DNA microarray technology, it is now possible to analyze expression patterns of many genes in a systematic and comprehensive manner at the genomic level [1]. The study of expression patterns of genes in different experimental conditions may enable one to understand the dynamic behavior of genes and pathways involved in biological processes. A gene expression level is a numerical value that measures how a particular gene is over-expressed or under-expressed in comparison with its activity in normal conditions. Analysis of expression patterns can be helpful in discovering groups of genes that participate in similar biological processes or functions. Various biotechnology laboratories and pharmaceutical companies involved in in silico drug design can identify molecular targets that may interact with the drugs. Microarray analysis can assist drug companies in choosing the most appropriate candidates for participation in clinical trials of new drugs [2]. Wide availability of diagnostic DNA microarrays has positively impacted cancer research compared to other recent technologies since they are relatively easy to make and use.
One major goal of analyzing expression data is to discover functionally similar genes. Co-regulation is a common phenomenon in gene expression. Expression patterns with similar tendency or behavior are normally termed as positively regulated and inverted behavior as negatively regulated [3]. Finding positively and negatively co-regulated gene clusters from gene expression data is a real need. A group of co-regulated genes may form gene clusters that can encode proteins, which interact amongst themselves and take part in common biological processes. Genes with similar (or inverted) expression profiles are very likely to be regulators of one another or be regulated by some other common parent gene [4, 5]. It has been observed that small sets of genes are co-regulated and co-expressed under certain conditions, their behavior being almost inactive for other conditions. Discovering groups of genes with similar or inverted expression profiles under a set of conditions leads to the concept of biclustering expression data. We discuss here various expression patterns identified in microarray data and how, based on these patterns, biological knowledge can be extracted in the form of biclusters.
1.1 Patterns in Gene Expression Data
With the help of microarray experiments one can simultaneously monitor the expression levels of genes at a genome scale. Data generated from microarray experiments, measuring relative expression levels of genes in a sample and in a controlled population can be represented in the form of a matrix or vector [6], often called gene expression matrix. Formally, it can be defined as follows.
Definition 1 (Gene Expression Data).
Let G = {G 1, G 2, …, G m } be a set of m genes and R = {T 1, T 2, …, T n } be the set of n conditions or time points at which the genes’ expression levels are recorded in a microarray dataset. The gene expression dataset X can be represented as an m × n matrix, X m×n where each entry x i, j in the matrix corresponds to the logarithm of the relative abundance of mRNA corresponding to a gene.
To gain better understanding of genes and their behavior inside the cell, various patterns can be derived by analyzing the change in expression levels of the genes. The notion of patterns in microarray data is introduced in [7] as below.
Definition 2 (Expression Pattern).
Given a gene G i , its expression values under a single condition or a series of varying conditions lie within a certain range. G i is a vector of real numbers within the range [a, b], denoted as G i @[a, b], and is called an item. The values in G i are limited inclusively between a and b.
A set containing one single item is called a pattern. A set of several items, which come from different genes is also called a pattern. So, a pattern looks like:
where i t ≠ i s , 1 ≤ t, s ≤ k, if k > 1.
Example data (Table 1) from Homo sapiens microarray dataset, GDS825, taken from NCBIFootnote 1 and their respective profile plots are shown in Fig. 1.

Profile plot of Homo sapiens expression data
From a biological point of view, patterns play an important role in discovering functions of genes, disease targets, or gene interactions [8]. A number of different patterns have been identified in biologically significant gene groups.
1.1.1 Shifting and Scaling Patterns
In shifting patterns [7], the gene profiles show similar trends, but distance-wise, they may not be close to each other (see Fig. 2).

Expression profile plot shows Shifting and Scaling patterns
In terms of expression values, the gene patterns are separated by more or less constant vertical distances among them. Formally, shifting patterns can be defined as follows
Definition 3 (Shifting Pattern).
Given two gene expression profiles G i = {E i1, E i2, …, E ik } and G j = {E j1, E j2, …, E jk } with k expression values, a profile is called a shifting pattern with respect to another profile, if expression value E ip can be related to E jp with a constant additive factor π ij for the p = 1… k. For genes G i and G j , the fact can be represented as follows.
Similarly, scaling patterns in gene expression roughly have multiplicative distance among the patterns. Scaling pattern can be defined as follows.
Definition 4 (Scaling Pattern).
Given two gene expression profiles G i = {E i1, E i2, …, E ik } and G j = {E j1, E j2, …, E jk } with k expression values, a profile is called a scaling pattern with respect to another profile, if expression value E ip can be related to E jp with constant multiplicative factor ζ ij for the p = 1… k. For genes G i and G j , the fact can be represented as follows.
As shown in Fig. 2, values of G 2 are roughly three times larger than those of G 3, and values of G 1 are roughly three times larger than those of G 2. In nature, it may so happen that due to different environmental stimuli or conditions, the pattern G 3 responds to these conditions similarly, although G 1 is more responsive or more sensitive to the stimuli than the other two.
1.1.2 Coherent Patterns
A group of genes showing similar pattern tendency across different conditions is called coherent. Such a group shows predominantly one kind of co-expression in the expression profiles of all member genes. Co-expressed genes are likely to be involved in the same cellular processes. In practice, co-expressed genes may belong to the same or similar functional categories indicating co-regulated families [5]. Coherent gene expression patterns may characterize important cellular processes and may provide a foundation for understanding regulation mechanisms in the cells [9]. The patterns shown in Fig. 2 are examples of coherent patterns.
1.1.3 Co-regulated Patterns
Often, coherent patterns are divided into two categories, namely, positively regulated patterns and negatively regulated or inverted patterns. Sometimes, a group of genes that are positively or negatively regulated are also called co-regulated genes. In Fig. 1, human genes, GLANT5 and IDH3B, show similar patterns or positively regulated patterns. On the other hand, IDH3B and GLANT5 show inverted or negative patterns with APOE. Biologically all three genes are very significant. As suggested Gene Ontology, the three genes are involved in regulation of plasma lipoprotein particle levels and triglyceride-rich lipoprotein particle remodeling. Pronounced inverted or negative patterns can be observed in Fig. 3, taken from NCBI Rat dataset GDS3702. Gene Ontology suggests that both are responsible for regulation of interferon-beta production. A group of genes may share a combination of both positive and negative co-regulation under a few conditions or at some time points.

Expression profile of RAT genes showing negative-regulation
Thus, gene expression data analysis involves pattern finding. Data mining is the study of techniques that extract patterns from large amounts of data. As a result, data mining provides the primary tools for gene expression data analysis. Biclustering is an important data mining tool for analyzing biologically significant gene groups. Below we present a brief discussion of biclustering techniques.
1.2 Biclustering of Co-regulated Genes
Clustering is a popular data analysis tool in genomic studies, particularly in the context of gene-expression microarrays [10–12]. Each microarray provides expression measurements for thousands of genes and clustering is a useful exploratory technique to analyze gene expression data since it groups similar genes together and allows biologists to identify groups of potentially meaningful genes, which have related functions or are co-regulated, which in turn helps find the relationships among them in the form of gene regulatory networks [5]. It has frequently been observed that subsets of genes are co-regulated and co-expressed under a subset of environmental conditions or time points [13]. Biclustering algorithms tackle the problem of finding a set of sub-matrices where each sub-matrix or bicluster meets a certain homogeneity criterion.
Given a gene expression dataset D N×M , where G = {G 1, G 2, … G N } is a set of N genes and R = {T 1, T 2, …, T M } is the set of M conditions or time points, biclusters can be defined as follows.
Definition 5 (Biclusters).
Biclusters are a set of sub-matrices of the matrix D = (N, M) with dimensions I 1 × J 1, … I k × J k such that I i ⊆ N, J i ⊆ M ∀i{1, …, k}, where each sub-matrix (bicluster) meets a given homogeneity criterion.
Madeira and Oliveira [14] identify four different categories of biclusters based on homogeneity criterion, namely:
-
1.
Constant biclusters,
-
2.
Biclusters with constant values on either columns or rows,
-
3.
Biclusters with coherent values, and
-
4.
Biclusters with coherent evolutions.
A comprehensive survey of different biclustering techniques for gene expression data clustering can be found in [15, 16]. In gene expression analysis, patterns play a more important role than expression values [17]. As a result, the value based homogeneity criterion mentioned above may not be suitable for grouping biologically significant genes.
2 Materials
Technological improvements in high-throughput DNA microarray technology is instrumental in the tremendous growth of publicly available gene expression data. This growing amount of expression data requires concurrent development of adequate bioinformatics tools for comprehensive analysis of the data for extracting biological knowledge. A number of online and offline tools are available for biclustering of gene expression data. We mention here a few of the leading, freely available biclustering packages (Table 2).
2.1 Data Sources
A plethora of real expression data produced by different biotechnology labs are freely available online. In this chapter, we use some datasets from Table 3 for experimentation and demonstration.
2.2 Evaluating Quality of Biclusters
From the point of view of biological data analysis, a cluster is biologically significant if it can produce functionally enriched groups of genes. A majority of the literature on biclustering evaluates and reports results based on functional enrichment of the clusters against Gene Ontology (GO). To determine the statistical significance of the association of a particular GO term with a group of genes in a cluster, various online tools from the GO ProjectFootnote 2 are available. In Table 4, we report some freely available tools.
These tools use the hypergeometric distribution to calculate the p-value or q-value, which evaluates whether the clusters have significant enrichment in one or more function groups. The p-value is computed as follows:
The p-value gives the probability of seeing at least k genes out of the total n genes in a cluster annotated with a particular GO termf, given the total number of genes in the whole genome g and the number of genes in the whole genome that are annotated with that GO term f. It is important to note that p-value measures whether a cluster is enriched with genes from a particular category to a greater extent than what would be expected by chance. If the majority of genes in a cluster appears in one category, the p-value of the category is small. That is, the closer the p-value to zero, the more the probability that the particular GO term is associated with the group of genes. The Q-value is the minimal False Discovery Rate (FDR) at which this gene appears significant. Q-values are estimated using the Benjamini Hochberg procedure [25].
3 Methods
This approach to clustering was originally introduced by Hartigan [26] and later applied by Cheng and Church [27] to expression data to capture the coherence of a subset of genes under a subset of conditions. Several techniques have been proposed to find quality biclusters from expression data. In Cheng and Church’s approach, the degree of coherence is measured using the concept of mean squared residue (MSR) and the algorithm greedily inserts/removes rows and columns to arrive at a certain number of biclusters, achieving some predefined residue score. The lower the score, the stronger the coherence exhibited by the biclusters, and better is the quality of the biclusters. Following Cheng and Church, a number of biclustering techniques have been proposed [27–37] to determine quality biclusters.
A greedy iterative search [27, 28] based approach finds a local optimal solution with an expectation to finally obtain a globally good solution. A divide and conquer [26] approach divides the whole problem into sub-problems and solves them recursively. Finally, it combines all the solutions to solve the original problem. In exhaustive biclustering [35], the best biclusters are identified using exhaustive enumeration of all possible biclusters extant in the data, in exponential time. A detailed categorization of heuristic approaches is available in [29]. A number of techniques based on metaheuristics, such as evolutionary and multi-objective evolutionary framework, have also been explored [30] to generate and iteratively refine an optimal set of biclusters. All of them use MSR as the merit function.
An MSR based technique is effective in finding optimized maximal biclusters. From a biological point of view, the interest resides in finding biclusters with subsets of genes showing similar behaviors, not similar values. Interesting and relevant patterns from a biological point of view, such as shifting and scaling patterns, may not be detected using this measure as it considers only expression values, not the patterns or tendencies of gene expression profile. It is important to discover this type of patterns because frequently the genes can present similar behavior although their expression levels vary in range or magnitude. Aguilar-Ruiz [31] proves that MSR is not a good measure to discover patterns in data when the variance among gene values is high, that is, when the genes present scaling and shifting patterns. To detect biologically relevant biclusters with scaling and shifting patterns, a scatter search based approach has been proposed [32]. This method uses a fitness function based on linear correlation among genes and an improvement method to select just the positively correlated genes.
Often, it has been observed that genes share local rather than global similarity in their gene expression profiles and only under a few conditions or time points [13]. Thus, correlation based technique may not be effective when computing pair-wise similarity among gene expression profiles. Other than that, various pattern-based approaches have also been proposed [33, 34, 38, 39] for discovery of biclusters where expression levels of genes rise and fall at a subset of conditions or time points.
Recently, it has been observed that [3] co-regulated genes also share negative patterns or inverted behaviors, which existing pattern-based approaches are unable to detect. CoBi [24] (Coregulated Biclustering) captures biclusters among both positively and negatively regulated genes as co-regulated genes. It considers both up- and down-regulation trends and similarity in degrees of fluctuation under consecutive conditions for expression profiles of two genes as a measure of similarity between the genes. It uses a new BiClust tree for generating biclusters in polynomial time that needs a single pass over the dataset.
3.1 Performance Comparison
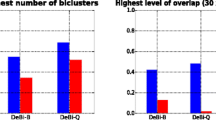
We compare the performance of a few biclustering methods taking into account functional enrichment of the biclusters. We consider four biclustering techniques: Bimax [40], Cheng and Church (CC) [27], OPSM [41], and CoBi [24]. For the purpose of comparison, we set the parameter values of other algorithms as recommended in the original papers. The functional enrichment of each bicluster is measured using Q-values associated with GO categories. For each bicluster, we calculate average of the percentage of the number of genes from the bicluster with a given function against all genes in the genome with the function. Figure 4 shows the average of functional enrichments of each bicluster obtained by different biclustering algorithms from four different datasets [24].

Comparison on functionally enriched biclusters obtained by different biclustering techniques
From the graphs it is clearly evident that CoBi outperforms all three algorithms in obtaining functionally enriched biclusters. However, for the YeastCho dataset, the Cheng and Church (CC) approach performs better than other algorithms.
4 Notes
Biclustering is a promising and important data mining tool for analyzing gene expression data. A number of techniques are available for biclustering. Most are greedy in nature and often computationally expensive. Moreover, they ignore positive- and negative-regulation patterns when performing biclustering. As mentioned in [42], a bicluster is considered a quality bicluster when participating genes exhibit consistent trends and similar degrees of fluctuation under consecutive conditions. We consider both up- and down-regulation trends and similarity in degrees of fluctuations under consecutive conditions for expression profiles of two genes as a measure of similarity between the genes. Compared to other methods discussed above, the design of CoBi has been motivated by a desire to handle the outstanding issues mentioned above and as a result, it exhibits promising results.
Notes
References
Kurella M, Hsiao L, Yoshida T, Randall J, Chow G, Sarang S, Jensen R, Gullans S (2001) Dna microarray analysis of complex biologic processes. J Am Soc Nephrol 12:1072–1078
Kraljevic S, Stambrook PJ, Pavelic K (2004) Accelerating drug discovery. EMBO Rep 5:837–842
Yu H, Luscombe N, Qian J, Gerstein M (2003) Genomic analysis of gene expression relationships in transcriptional regulatory networks. Trends Genet 19:422–427
Gasch A, Eisen M et al (2002) Exploring the conditional coregulation of yeast gene expression through fuzzy k-means clustering. Genome Biol 3:1–22
Tavazoie S, Hughes J, Campbell M, Cho R, Church G et al (1999) Systematic determination of genetic network architecture. Nat Genet 22:281–285
Grant R (2004) Computational genomics: theory and application. Horizon Bioscience, Cambridge
Li J, Wong L (2001) Emerging patterns and gene expression data. Genome Inform Ser 12:3–13
Alberts B, Johnson A et al (2002) Studying gene expression and function. In: Molecular biology of the cell, 4th edn
Spellman P, Sherlock G, Zhang M, Iyer V, Anders K, Eisen M, Brown P, Botstein D, Futcher B (1998) Comprehensive identification of cell cycle-regulated genes of the yeast saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell 9:3273–3297
Ben-Dor A, Shamir R, Yakhini Z (1999) Clustering gene expression patterns. J Comput Biol 6:281–297
Chipman H, Hastie TJ, Tibshirani R (2003) Clustering microarray data. In: Statistical analysis of gene expression microarray data, vol 1. Chapman & Hall/CRC, Boca Raton, pp 159–200
Ahmed HA, Mahanta P, Bhattacharyya D, Kalita JK (2011) Gerc: tree based clustering for gene expression data. In: 2011 I.E. 11th international conference on bioinformatics and bioengineering (BIBE), IEEE, pp 299–302
Mitra S, Banka H (2006) Multi-objective evolutionary biclustering of gene expression data. Pattern Recogn 39:2464–2477
Madeira SC, Oliveira AL (2004) Biclustering algorithms for biological data analysis: a survey. IEEE/ACM Trans Comput Biol Bioinform 1:24–45
Kriegel HP, Kröger P, Zimek A (2009) Clustering high-dimensional data: a survey on subspace clustering, pattern-based clustering and correlation clustering. ACM Trans Knowl Discov Data (TKDD) 3:1
Mahanta P, Ahmed H, Bhattacharyya D, Kalita JK (2011) Triclustering in gene expression data analysis: a selected survey. In: 2011 2nd national conference on emerging trends and applications in computer science (NCETACS), IEEE pp 1–6
Roy S, Bhattacharyya DK, Kalita JK (2014) Reconstruction of gene co-expression network from microarray data using local expression patterns. BMC Bioinf 15:S10
Shamir R, Maron-Katz A, Tanay A, Linhart C, Steinfeld I, Sharan R, Shiloh Y, Elkon R (2005) Expander—an integrative program suite for microarray data analysis. BMC Bioinf 6:232
Barkow S, Bleuler S, Prelić A, Zimmermann P, Zitzler E (2006) Bicat: a biclustering analysis toolbox. Bioinformatics 22:1282–1283
Gonçalves JP, Madeira SC, Oliveira AL (2009) Biggests: integrated environment for biclustering analysis of time series gene expression data. BMC Res Notes 2:124
Cheng KO, Law NF, Siu WC, Lau T (2007) Bivisu: software tool for bicluster detection and visualization. Bioinformatics 23:2342–2344
Zhou F, Ma Q, Li G, Xu Y (2012) Qserver: a biclustering server for prediction and assessment of co-expressed gene clusters. PloS one 7:e32660
Leung E, Bushel PR (2006) Page: phase-shifted analysis of gene expression. Bioinformatics 22:367–368
Roy S, Bhattacharyya DK, Kalita JK (2013) Cobi: pattern based co-regulated biclustering of gene expression data. Pattern Recogn Lett 34:1669–1678
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodological) 57:289–300
Hartigan JA (1972) Direct clustering of a data matrix. J Am Stat Assoc 67:123–129
Cheng Y, Church G (2000) Biclustering of expression data. In: Proceedings of 8th international conference on intelligent systems for molecular biology, ICISMB’00, vol 8, pp 93–103
Yang J, Wang H, Wang W, Yu P (2003) Enhanced biclustering on expression data. In: Proceedings of the 3rd IEEE symposium on bioinformatics and bioengineering, 2003, pp 321–327
Madeira S, Oliveira A (2004) Biclustering algorithms for biological data analysis: a survey. IEEE/ACM Trans Comput Biol Bioinf 1:24–45
Banka H, Mitra S (2006) Evolutionary biclustering of gene expressions. Ubiquity 7:1–12
Aguilar-Ruiz J (2005) Shifting and scaling patterns from gene expression data. Bioinformatics 21:3840–3845
Nepomuceno J, Troncoso A, Aguilar-Ruiz J et al (2011) Biclustering of gene expression data by correlation-based scatter search. BioData Min 4:3
Pei J, Zhang X Cho M, Wang H, Yu P (2003) Maple: a fast algorithm for maximal pattern-based clustering. In: Proceedings of the 3rd IEEE international conference on data mining, 2003 (ICDM’03), IEEE, pp 259–266
Wang H, Chu F, Fan W, Yu P, Pei J (2004) A fast algorithm for subspace clustering by pattern similarity. In: Proceedings of the 16th international conference on scientific and statistical database management, 2004, IEEE, pp 51–60
Tanay A, Sharan R, Shamir R (2002) Discovering statistically significant biclusters in gene expression data. Bioinformatics 18:S136–S144
Roy S, Bhattacharyya DK, Kalita JK (2012) Deterministic approach for biclustering of co-regulated genes from gene expression data. In: Proceedings of the 16th international conference on KES12, FAIA, vol 243, pp 490–499
Eren K, Deveci M, Küçüktunç O, Çatalyürek ÜV (2013) A comparative analysis of biclustering algorithms for gene expression data. Brief Bioinform 14:279–292
Wang H, Wang W, Yang J, Yu P (2002) Clustering by pattern similarity in large data sets. In: Proceedings of the international conference on management of data. ACM SIGMOD’02, ACM, pp 394–405
Zhao Y, Yu J, Wang G, Chen L, Wang B, Yu G (2008) Maximal subspace coregulated gene clustering. IEEE Trans Knowl Data Eng 20:83–98
Prelić A, Bleuler S et al (2006) A systematic comparison and evaluation of biclustering methods for gene expression data. Bioinformatics 22:1122–1129
Ben-Dor A, Shamir R, Yakhini Z (1999) Clustering gene expression patterns. J Comput Biol 6:281–297
Ji L, Mock K, Tan K (2006) Quick hierarchical biclustering on microarray gene expression data. In: Proceedings of the 6th IEEE symposium on bioinformatics and bioengineering, 2006 (BIBE’06), IEEE, pp 110–120
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this protocol
Cite this protocol
Roy, S., Bhattacharyya, D.K., Kalita, J.K. (2015). Analysis of Gene Expression Patterns Using Biclustering. In: Guzzi, P. (eds) Microarray Data Analysis. Methods in Molecular Biology, vol 1375. Humana Press, New York, NY. https://doi.org/10.1007/7651_2015_280
Download citation
DOI: https://doi.org/10.1007/7651_2015_280
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3172-9
Online ISBN: 978-1-4939-3173-6
eBook Packages: Springer Protocols