
Abstract
Since the advent of pluripotent stem cells, (embryonic and induced pluripotent stem cells), applications of such pluripotent stem cells are of prime importance. Indeed, scientists are involved in studying the basic biology of pluripotent stem cells, but equal impetus is there to direct the pluripotent stem cells into multiple lineages for cell therapy applications. Scientists across the globe have been successful, to a certain extent, in obtaining cells of definitive endoderm and also pancreatic β islets by differentiating human pluripotent stem cells. Pluripotent stem cell differentiation protocols aim at mimicking in vivo embryonic development. As in vivo embryonic development is a complex process and involves interplay of multiple cytokines, the differentiation protocols also involve a stepwise use of multiple cytokines. Indeed the novel markers for pancreas organogenesis serve as the roadmaps to develop new protocols for pancreatic differentiation from pluripotent stem cells. Earliest developed protocols for pancreas differentiation involved “Nestin selection pathway,” a pathway common for both neuronal and pancreatic differentiation lead to the generation of cells that were a combination of cells from neuronal lineage. Eventually with the discovery of hierarchy of β cell transcription factors like Pdx1, Pax4, and Nkx2.2, forced expression of such transcription factors proved successful in converting a pluripotent stem cell into a β cell. Protocols developed almost half a decade ago to the recent ones rather involve stepwise differentiations involving various cytokines and could generate as high as 25 % functional insulin-positive cells in vitro. Most advanced protocols for β islet differentiations from human pluripotent stem cells focused on 3D culture conditions, which reportedly produced 60–65 % functional β islet cells. Here, we describe the protocol for differentiation of human pluripotent stem cells into functional β cells under both 2D and 3D culture conditions.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Keywords:
- Human embryonic stem cells
- Human pluripotent stem cells
- β islet differentiation
- Insulin
- 2D differentiation
- 3D differentiation
1 Introduction
As pluripotent stem cells (PSC) have the enormous potential to generate all three lineages, they are prospective candidates for biomedical research. Applications of PSC in biomedical research involve cell therapy/regenerative medicine and drug screening models. Diseases of global importance wanting regenerative medicine based cure are diabetes, stroke, Parkinson’s disease, spinal cord injury, hematological malignancies, blindness deafness, osteoarthritis, kidney failure. Hence, generation of scalable quantities of various cell types from PSC make use of various in vitro differentiation protocols will have enormous applications in regenerative medicine. Furthermore, differentiations of patient-specific PSCs have also found applications in basic research for various in vitro disease models (1–3).
1.1 Correlation Between In Vivo Development and Protocols for Differentiating Pluripotent Stem Cells into All Three Lineages, Ectoderm, Mesoderm, and Endoderm
The main goal of any in vitro differentiation protocol is to recapitulate the in vivo developmental ontogeny. Importantly, the design of in vitro cellular transitions leading to the development of ectoderm, mesoderm, or endoderm is based on the identification of intrinsic factors governing early embryonic development (4–7). These intrinsic factors are cytokines that are secreted from the different parts of the developing embryo that, in turn, influence the sequential expression of a cohort of genes (8). For example, the single layered blastula, during the early phase of embryonic development, first gives rise to the primitive streak, the body axis in mammals. The formation of primitive marks the beginning gastrulation (9). In the gastrulation stage, embryo gets organized for the first time into tri-germ lineage, ectoderm, mesoderm, and endoderm during the third week of gestation in humans. The physical events taking place during gastrulation involve the ingression of cells from the epithelial epiblast (embryonic ectoderm) through the primitive streak to give rise to the mesoderm and endoderm cell layers. The cellular events include epithelial–mesenchymal transitions (EMT) of the epiblast epithelia in which the epithelial phenotype of the epiblast cells get downregulated to give rise to a migratory mesenchymal phenotype (10). The mesoderm cells thus maintain a migratory phenotype and the endoderm cells reestablish cell–cell junctions to form a contiguous layer at the base of the primitive streak. The fibroblast growth factors (FGFs) are the important cytokines involved in mesoderm inductions and hence play a significant role in gastrulation (4). FGF receptors are involved in upregulating early mesodermal genes like brachyury and Tbx6 (4). Also, under the influence of FGF-signaling there takes place a concomitant decrease in epithelial genes like E-Cadherin and Snail (4).
The methods employed to differentiate pluripotent stem cells (PSC) into various lineages involve the first step either as the formation of three-dimensional aggregates known as embryoid bodies (EBs), or culture of PSC monolayers on various extracellular matrix proteins or culture of PSCs directly on supportive stromal layers (11). However, the first step of the majority of in vitro differentiation protocols from pluripotent stem cells (PSCs) involves the formation of 3D PSC aggregates, in the absence of anti-differentiation agents. Such 3D PSC aggregates are called embryoid bodies (EBs). EBs exhibits an interesting phenomenon of spontaneous differentiation into all three germ layers, and hence can be considered at par with the gastrula of an embryonic development. Indeed, spontaneous differentiation of EB involves FGF signaling like gastrulation (12). The initial events in EB differentiation is characterized by the primitive endoderm fate specification of cells of EBs located at the exterior (13). Under in vivo situations during mouse development, presence of Fgfr−/− progenitor cells are associated with the improper development of mesodermal and endodermal cells during gastrulation (14). Importantly, homozygous null Fgfr−/− embryos, die in during gastrulation (15, 16). On similar lines, targeted disruption of Fgfr in EBs reportedly blocked the maturation of visceral endoderm and cavitations in mouse embryoid bodies (13).
As the primitive streak divides the embryo into the rostro-caudal axis, the mesendoderm signals of Nodal/activin and FGF families are restricted only to the caudal end (17). So the non-caudal end gives rise to ectoderm cells: the central nervous system (CNS), the cranial placodes and neural crest cells, which together form the peripheral nervous system (PNS) and the skin. Also, during the initial patterning of the ectoderm, Wnt signaling comes at the top of the regulatory cascade and inhibits BMP activity in the epidermal ectoderm and FGF activity in the neural ectoderm (18). Similarly, the approaches used for in vitro neuronal differentiation from ESC/PSC aim at either generating regionally specified neural progenitor cells and/or differentiated neuronal/glial subtypes. Some of these protocols go via EB formation, similar to endodermal differentiation protocols while some of them are direct differentiation protocols. EB formation is carried out in the presence of retinoic acid (RA) (19). Also, RA is a developmentally regulated morphogen and has been reported to induce primary neurons in Xenopus (20, 21) hindbrain specification (22) and also motor neuron specification (23). Furthermore, during PSC differentiation into neuronal lineage, interplay of other cytokines and RA, which in turn, maintains a sustained activity of RA are most crucial events for the generation of neurons (24). As Wnt is the first cytokine responsible for neuronal development, activation of Wnt via overexpression of β catenin or treatment of the ES cells with Wnt 3a conditioned media resulted in the generation of neurons (25). Most of the neuronal differentiation protocols from human PSC require the supplements like b-FGF and Noggin (26). Noggin is responsible for the inhibition of BMP signaling, a prerequisite for neurogenesis (18, 27). Also, similar to the post-gastrulation events, the neuroepithelial cells in differentiation protocols are directed to form various neural cell types (27).
In PSC differentiation protocols, development of the primitive streak (PS) like population of gastrula and mesoderm induction is monitored by the expression of Brachyury (T). Cytokines like BMP4, when added to the EBs or directly to the PSCs, indirect differentiation protocols, generates Brachyury positive PS like cells followed by FLk1+ mesodermal progenitors (28–31). On the other hand, if Wnt signaling responsible for mesodermal patterning, when blocked in the differentiation protocols, resulted in the loss of T+ PS-like cells and mesodermal progenitors. Thus, loss of T+ cells in the in vitro PSC differentiation protocols upon blocking of Wnt signaling post PS development correlates with the active role of Wnt signaling in PS and mesoderm development (32, 33). Alternatively, addition of Wnt in the PS-like induced PSC induced cardiac mesoderm formation (34).
It is activin/Nodal signaling that plays a significant role in the development of definitive endoderm. Accordingly, all the endodermal in vitro differentiation protocols from PSC use Activin A for the induction of definitive endoderm (35–40). However, previous to DE, activin treatment in PS-like cells obtained from PSC results in the formation of mesoderm and endoderm (mesendoderm) as evident by the co-expression of Brachyury, FoxA2, and Gossecoid (41, 42). Interestingly, the GSC positive cells are capable of giving rise to mesoderm and endoderm, and hence are called mesendoderm progenitors. Furthermore, the DE cells can be induced into hepatic and pancreatic lineage in a stepwise fashion using a cocktail of cytokines.
1.2 Evolution of Protocols for Insulin-Producing β Islets from PSC and Their Respective Strategies and Success
Mimicking in vivo pancreas development in PSC differentiation protocols and obtaining scalable quantities of β islets can be quite complex. This complexity of differentiation protocols is attributed to the complexity of an adult pancreas, which comprises of exocrine (produce digestive enzymes) and endocrine (α, β, and pancreatic polypeptide producing cells) (43). Earliest attempts to obtain functional pancreatic β islets did not mimic the chronological in vivo events in pancreas organogenesis. Rather, the protocol involved a straightforward “Nestin selection pathway,” which made use of neuronal cues (44). The underlying reasons for using the “Nestin selection pathway” was due to the fact that pancreatic endoderm and neural ectoderm co-express a large number of markers suggesting a probable common pathway or cross talk between neuronal and pancreatic differentiation. For example, inhibitory sonic hedgehog from notochord promotes pancreatic organogenesis (45), innervations of β islets by neurons (46), Schwann cells surrounding β islets (47), and influence of such neural crest-derived neurons and Schwann cells on proliferation and maturation of β islets (48). Later on, this protocol exhibited serious flaws and was rejected because the insulin-positive cells were not because of de novo insulin synthesis, rather because of the uptake of insulin from the culture medium (49). Recently, Arntfield et al. (50) reported the presence of developmentally distinct progenitors of neural crest and pancreatic origin, in an adult mammalian pancreas.
Second generation of β islet differentiation protocols involved the overexpression of β transcription factors. After the discovery of transcriptional hierarchy of factors responsible for β cell organogenesis, scientists tried to perform forced expression of such factors. β cell transcription factors like Pdx1, Pax4 and Nkx2.2, upon overexpression in ESC resulted in the formation of β cells (51–53). Such protocols involved transfection methods and produced low percentage ~1 % insulin-positive cells (53).
Third generation of β islet differentiation protocols from PSC adopted an approach thereby mimicking the stepwise pancreatic organogenesis. The various steps involved in such protocols were definitive endoderm followed by sequentially priming the cells into primitive gut tube, posterior foregut, pancreatic endoderm, and endocrine precursors. Various protocols using this stepwise pancreas organogenesis approach were published by several research groups worldwide. (38–40, 54, 55, 57–59). However, all these protocols either involved a 2D/monolayer or a 3D approach to obtain β islets. In 3D protocols, 3D approach was used to obtain the functional β islets in the last step of differentiation, and proved to be more efficient as compared to the 2D protocols. The differences amongst all these protocols were the use of cytokines in various steps in the differentiation. The first step in definitive endoderm (DE) differentiation from PSC for most of these protocols involved the use of Activin A/a TGFβ family member, alone or in combination of other molecules. The choice of Activin A to induce DE was based on the studies of early embryonic development in different model systems that emphasized the role of nodal, a soluble molecule of TGFβ/Activin signaling family during gastrulation. Nodal promoted DE and mesoderm and a higher level of nodal was reported to facilitate endoderm specification in these model systems (60–63). D’Amour et al. (37, 54) used a combination of Activin A and Wnt3a; Jiang et al. (57) used Activin A and sodium butyrate in the complete absence of serum; Philips et al. (64), however, used BMP4 along with Activin A; Shi et al. (55) used Activin A and Retinoic acid; and Zhang et al. (65) had several other components like 0.2 % BSA, N2, B27, along with Activin A to induce DE. Bose et al. (40) used a combination Activin A and Retinoic acid in the first step for to obtaining DE cells. However, one protocol did not use Activin A for DE differentiation from hESC. Rather, this group used the old protocol of Nestin selection, in combination with Exendin-4, a GLP-1 analog for maturation of pancreatic progenitors (66). Interestingly, the protocol of Mao et al. (66) did not produce DE cells and the cells were directly enriched for pancreatic progenitors. Also, more recently, Rezania et al. (59) have not used Activin A for the induction of definitive endoderm. Instead, GDF8 (a TGFβ family member) was used in combination of GSK3β inhibition, in accordance with the concept of Naujok et al. (67). As per Naujok et al. (67), early steps of DE formation from hPSC require high levels of canonical Wnt signaling, GSK3β inhibition and low levels of Activin A.
In the next step to obtain primitive gut tube or posterior foregut, the growth factors used were a combination of cyclopamine (sonic hedgehog/shh inhibitor), noggin, b-FGF, and retinoic acid in the most of the aforementioned differentiation protocols that mimicked pancreatic organogenesis. The decision to use cyclopamine, noggin, b-FGF and retinoic acid to induce posterior foregut was based on reports from developmental biology and successes of various differentiation protocols from PSC that emphasized the role of such molecules in pancreas organogenesis (68–71).
Finally, for obtaining functional pancreatic endocrine progenitors and β cells, most of the protocols have used Exendin-4 in the culture media to stimulate insulin secretion, β cell proliferation (37, 54, 55, 64, 65) alone or in combination with betacellulin and Hepatocyte growth factor (HGF). The decision to use exendin-4, a GLP-1 analog was made based on previous reports about the role GLP-1 to promote insulin secretion from β cells (72) betacellulin to promote β cell proliferation (73–75) and HGF also to promote β cell growth and increase insulin production by β cells (76, 77).
Amongst all the third-generation 2D β islet differentiation protocols, different percentages of functional insulin-producing cells were obtained. The earlier protocols by Shi et al. (55) and D’Amour et al. (37) showed limited glucose sensitivity but showed sensitivities to a variety of secretagogues. β islets obtained in pancreatic differentiation protocol by Kroon et al. (38) exhibited high levels of in vivo glucose responsiveness. More recent pancreatic differentiation protocol of Nostro et al. (39) reported a generation of 25 % insulin-positive cells. 3D differentiation protocols evolved after the 2D differentiation protocols and were essentially able to mimic the in vivo conditions and strengthen the cellular interactions probably by improved secretory dynamics and electrical coupling of insulin-producing cells (78). Also, β cell proliferation and functions reportedly increase due to cell–cell interactions and signaling with extracellular matrices as available under 3D differentiation conditions (79–81). Indeed, the 3D pancreatic differentiations from mouse ESC/iPSC and human PSC gave rise to a high percentage of functional insulin-producing β cells. 60 % insulin producing cells were reported in case of pancreatic differentiations from mouse ES, iPSC and mouse fetal pancreas (82, 83). 65 and 50 % insulin producing cells were, however, reported in case of pancreatic differentiations from hESC (40) and (Rezania et al., 2014) (59) respectively. Interestingly, the differentiation protocol by Rezania et al. (2014) (59) had adopted a 3D approach right from the early stage of pancreatic endoderm.
1.3 Details of Our Protocol for In Vitro PSC Differentiation into β Islets
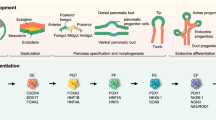
Our method of β islet differentiation from hESC also followed the sequential in vivo pancreatic organogenesis like all other third generation β islet differentiation protocols. This method is a 42-day protocol, a combination of monolayer 2D followed by a 3D differentiation. All the initial steps from DE induction till the generation of β islets were carried out under 2D monolayer conditions until 32 days (Fig. 1). Last 10 days of the protocol involved the culturing of 2D β islets under 3D conditions for β cell maturation. The first 32 days of 2D differentiation resulted in the formation of ~24.5 % functional β islets (Bose et al. (40)). However, upon 10 days of further maturation of these 2D β islets under 3D conditions, ~65 % of functional β islets were obtained (Bose et al. (40)). Amongst the first 32 days of differentiation, a combination of 2D and 3D approach was taken where spontaneously differentiated EBs, definitive endoderm, pancreatic endoderm and β islets were obtained. Spontaneous tri-lineage differentiation was first carried out in 3D by making of embryoid bodies (EBs) from hESC for first 48 h (step 1, Figs. 1 and 2). EBs were then plated onto Matrigel and subjected to sequential treatment of cytokines to induce the DE, pancreatic endoderm and finally the pancreatic endocrine/β cells (steps 2–4, Figs. 1 and 2). EBs plated onto Matrigel and treated with Activin A and retinoic acid for 6 days generated DE cells, which were characterized by the presence of Sox 17, CXCR4 and Fox A2 as DE marker and loss of Oct3/4 as a proof of concomitant loss of pluripotency (step 2, Figs. 1, 2, and 3). The DE cells were then induced with Noggin and b-FGF to form pancreatic endoderm (PE) for 12 days (step 3, Fig. 1). The successful conversion of cells into PE was evident from the expression of PDX1 (the first transcription factor in pancreatic organogenesis) along with NGN3, NKX6.1, and flattened morphology of cells (step 3, Figs. 2 and 3). The presence of E-Cadherin is the hallmark of a cell with an epithelial behavior (Fig. 2).The PE cells were then induced with b-FGF, nicotinamide and GLP-1 for pancreatic endocrine/β cell development, formation of islet-like clusters (ILC) in 2D (step 4, Figs. 1 and 2). The co-localization of insulin and c-peptide confirmed the presence of ILC in step 4 (Fig. 3). Further maturation of pancreatic endocrine cells was done under 3D conditions using CMRL media, which is protein free, vitamins and nucleoside rich media, used otherwise, for maintaining human β islets in culture. Liraglutide, a long-lived GLP-1 agonist also complemented for the maturation of β islets in 3D as evident from presence of mature β cell markers like MafA and exendin-1, a vasoactive peptide stimulating the release of insulin from the mature β cells (step 5, Figs. 1, 2, and 3).

Scheme of pancreatic differentiation protocol (Bose et al. 2012 (40), Reproduced with permission from Wiley)

Morphologies acquired by the cells during sequential differentiation process (Bose et al. 2012 (40). Supplementary information. Reproduced with permission from Wiley)

Expression of markers by immunofluorescence during steps 1–5 of the differentiation (Bose et al. 2012 (40), Reproduced with permission from Wiley)
2 Materials
2.1 Cell Lines, Media, and Supplements
-
1.
ReliCell hES1 (Human embryonic stem cell lines ReliCell hES1 (Mandal et al. 2006), BGO1 (ATCC)) (84), BGO1 (ATCC), Mouse Embryonic Fibroblast (Mandal et al.) (84).
-
2.
Culture Dish (35 mm, Nunc 153066).
-
3.
Ultra-Low Attachment culture dishes (60 mm, Corning, CLS3261).
-
4.
Gelatin (Sigma G1393).
-
5.
PBS.
-
6.
0.05 % Trypsin–EDTA (GIBCO, Life Technologies, 25300-054).
-
7.
Matrigel (BD Biosciences, San Jose, CA, 354230).
-
8.
Mouse embryonic fibroblast medium (Store at 4 °C).
DMEM-high glucose (GIBCO, Life Technologies, 11995-065)
500 mL
FBS (Hyclone, SH30071.03, Logan, UT)
50 mL
l-Glutamine (GIBCO, Life Technologies, 25030-081)
5 mL
MEM-Non Essential Amino acids (GIBCO, Life Technologies, 11140-050)
5 mL
0.1 mM 2-mercaptoethanol (GIBCO, Life Technologies, 21985-023)
500 μL
-
9.
Human embryonic stem cell medium (Store at 4 °C).
DMEM/F-12 (GIBCO, Life Technologies, 11320-033)
500 mL
FBS (Hyclone, SH30071.03, Logan, UT)
75 mL
Knockout serum replacement (GIBCO, Life Technologies, 10828-028)
25 mL
l-Glutamine (GIBCO, Life Technologies, 25030-081)
5 mL
MEM-non essential amino acids (GIBCO, Life Technologies, 11140-050)
5 mL
0.1 mM 2-mercaptoethanol (GIBCO, Life Technologies, 21985-023)
500 μL
-
10.
Supplements for Human embryonic stem cells.
bFGF (R&D Systems, 233-FB-025): Stock solution at 25 μg/mL in 0.1 % (w/v) BSA/PBS. Aliquot into 100 μL and store at −80 °C. Once thawed, keep at 4 °C. Add to Human embryonic stem cell medium at a final concentration of 4 ng/mL.
-
11.
Differentiation Medium for Human embryonic stem cells.
– Embryoid body formation media and Basal media for definitive endoderm differentiation (store at 4 °C)
-
DMEM/F-12 (GIBCO, Life Technologies, 11320-033)
500 mL
Knockout serum replacement (GIBCO, Life Technologies, 10828-028)
50 mL
l-Glutamine (GIBCO, Life Technologies, 25030-081)
5 mL
MEM-non essential amino acids (GIBCO, Life Technologies, 11140-050)
5 mL
50 mM 2-mercaptoethanol (GIBCO, Life Technologies, 21985-023)
500 μL
-
-
12.
Supplements for definitive endoderm differentiation medium.
Activin A (Sigma, A4941): Prepare a stock solution at 50 μg/mL in 0.1 % (w/v) BSA/PBS. Aliquot into 100 μL and store at −80 °C. Once thawed, keep at 4 °C. Add to human definitive endoderm differentiation basal medium at a final concentration of 100 ng/mL.
All trans-Retinoic Acid (Sigma, R2625): Prepare a stock solution at 10 mM in DMSO (Sigma, D2650). Aliquot into 100 μL and store at −80 °C. Once thawed, keep at 4 °C with protection from light. Add to human definitive endoderm differentiation basal medium to make a final concentration of 1 mM.
-
13.
Human pancreatic endoderm specification basal media (store at 4 °C).
DMEM/F-12 (GIBCO, Life Technologies, 11320-033)
500 mL
Knockout serum replacement (GIBCO, Life Technologies, 10828-028)
50 mL
l-Glutamine (GIBCO, Life Technologies, 25030-081)
5 mL
MEM-non essential amino acids (GIBCO, Life Technologies, 11140-050)
5 mL
50 mM 2-mercaptoethanol (GIBCO, Life Technologies, 21985-023)
500 μL
-
14.
Supplements for pancreatic endoderm specification medium.
bFGF (R&D Systems, 233-FB-025): Stock solution at 25 μg/mL in 0.1 % (w/v) BSA/PBS. Aliquot into 100 μL and store at −80 °C. Once thawed, keep at 4 °C. Add to human pancreatic endoderm specification medium at a final concentration of 20 ng/mL.
Noggin (Sigma, H6416): Stock solution at 25 μg/mL in 0.1 % (w/v) BSA/PBS. Aliquot into 100 μL and store at −80 °C. Once thawed, keep at 4 °C. Add to human pancreatic endoderm specification medium at a final concentration of 100 ng/mL.
-
15.
Media for the generation of Islet like clusters (store at 4 °C).
DMEM/F-12 (GIBCO, Life Technologies, 11320-033) 500 mL.
-
16.
Supplements for the media for the generation of Islet like cluster.
1 % N2 supplement (GIBCO, Life Technologies, 17502-048), 2 % B27 supplement (GIBCO, Life Technologies, 10889-038) added to the media accordingly.
bFGF (R&D Systems, 233-FB-025): Stock solution at 25 μg/mL in 0.1 % (w/v) BSA/PBS. Aliquot into 100 μL and store at −80 °C. Once thawed, keep at 4 °C. Add to human pancreatic endoderm specification basal medium to a final concentration of 20 ng/mL.
Nicotinamide (Sigma, N0636): Stock solution at 1 M in PBS. Aliquot into 5 mL and store at −20 °C. Once thawed, keep at 4 °C. Add to human pancreatic endoderm specification basal medium to a final concentration of 10 mM.
Liraglutide (Novo Nordisk): Prepare a stock solution at 10 mM in PBS. Aliquot into 100 μL and store at −80 °C. Once thawed, keep at 4 °C with protection from light. Add to human pancreatic endoderm specification basal medium to a final concentration of 10 nM.
-
17.
Media for the generation of 3D Islet like clusters (store at 4 °C).
CMRL Medium-1066 (GIBCO, Life Technologies, 11530-037)
500 mL
Knockout serum replacement (GIBCO, Life Technologies, 10828-028)
50 mL
Glutamax (GIBCO, Life Technologies, 35050-061)
5 mL
HEPES (GIBCO, Life Technologies, 15630-080)
5 mL
Nicotinamide (Sigma, N0636): Stock solution at 1 M in PBS. Aliquot into 5 mL and store at −20 °C. Once thawed, keep at 4 °C. Add to 3D Islet like clusters generation basal medium to a final concentration of 10 mM.
Zinc Sulfate Monohydrate (Sigma, 96495): Stock solution of 0.1 M in PBS. Filter sterilize and aliquot into 1 mL and store at −20 °C. Add to the 3D islet cluster generation basal media to a final concentration of 0.1 mM.
Exendin-1 Peptide (Abbiotec, 350215): Stock solution of 1 mg/mL (~0.2 mM) in sterile PBS. Aliquot 100 μL and store at −20 °C. Add to the 3D islet cluster generation basal media to a final concentration of 20 nM.
Liraglutide (Novo Nordisk): Prepare a stock solution at 10 mM in PBS. Aliquot into 100 μL and store at −80 °C. Once thawed, keep at 4 °C with protection from light. Add to 3D Islet like clusters generation basal medium to a final concentration of 10 nM.
HGF (R&D Systems 294-HGN): Stock solution at 10 μg/mL in 0.1 % (w/v) BSA/PBS. Aliquot into 100 μL and store at −80 °C. Once thawed, keep at 4 °C. Add to 3D Islet like clusters generation basal medium to a final concentration of 20 ng/mL.
2.2 Gene Expression Analysis by qRT-PCR
-
1.
RNeasy Mini Kit (Qiagen, Cat. No 74104 Germany).
-
2.
Random hexamers and Superscript II (Invitrogen, Life Technologies Cat. No: 11904-018).
-
3.
SYBR Green Platinum SYBR Green qPCR Supermix-UDG (Invitrogen, Life Technologies Cat. No: 11733-046).
-
4.
CFX-96/Real Time system C1000-Thermal cycler (Bio-Rad).
-
5.
Primers used at a stock concentration of, 100 μm (Sigma), Primers (Table 1) reconstituted in DNase-RNase free water (Ambion, Life Technologies, AM-9937) at 5 μm and stored at −20 °C freezer.
Table 1 List of primers used and their respective sequences
2.3 Immunofluorescence Staining of Cultured Cells
-
1.
Eight-well chamber Permanox slides (Thermo Scientific 177445).
-
2.
Fixation Buffer: 4 % (w/v) paraformaldehyde (PFA) (Sigma P6148).
-
3.
Washing buffer: PBS with 100 mM glycine (Sigma G8898), 0.3 % (v/v).
-
4.
Triton X-100 (Sigma T9284).
-
5.
Blocking buffer: PBS with 5 % Normal donkey serum (Sigma D9663).
-
6.
Primary antibodies with recommended dilution have given in Table 2.
Table 2 List of primary antibodies and their respective dilutions used for immunocytochemistry and flow cytometric analysis -
7.
Secondary antibodies with recommended dilution have given in Table 3.
Table 3 List of secondary antibodies and their dilutions used -
8.
Mounting media with DAPI (Sigma F6057).
2.4 Flow Cytometry
-
1.
FC Fixation Buffer: 4 % (v/v) PFA in PBS.
-
2.
FACS Buffer: PBS with 2 % (w/v) fetal bovine serum (Hyclone, SH30071.03, Logan, UT).
-
3.
Triton X-100 (Sigma T9284) 0.1 % (v/v).
-
4.
Primary antibodies with recommended dilution have given in Table 2. Secondary antibodies (Invitrogen) with recommended dilution have given in Table 3.
2.5 In Vitro Functional Assay (Insulin and C-Peptide Assay)
-
1.
Low attachment 6-well dishes (Corning Cat. No 3471).
-
2.
DMEM (Glucose free) (Invitrogen, Life Technologies: 11966-025).
-
3.
Kreb’s ringer bicarbonate buffer (130 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.5 mM CaCl2, 20 mM Hepes, and 0.1 % BSA (w/v)).
-
4.
Glucose (Sigma G8270).
-
5.
Tolbutamide (Sigma T0981).
-
6.
Nifedipine (Sigma N7634).
-
7.
Diazoxide (Sigma 9035).
-
8.
Human insulin ELISA kit and C-peptide ELISA kit (Mercodia, Sweden Catalog # 10-1172-01).
2.6 Immunohistochemistry of Tissues
-
1.
Fixation Buffer: 4 % (v/v) PFA in PBS for fixing cells.
-
2.
10 % Neutral Buffered formalin for fixation of the tissues.
-
3.
Paraffin block for embedding tissues.
-
4.
Microtome (Leica, RM2245).
-
5.
Ethanol Grades (100, 95, 80, and 50 %, respectively).
-
6.
Citrate buffer (Sigma W302600) for antigen retrieval.
-
7.
Blocking buffer: 1 % BSA (Sigma) in PBS.
-
8.
Permeabilization Buffer: 0.2 % Triton X-100.
-
9.
Primary antibody and secondary antibodies given in Tables 2 and 3.
-
10.
Mounting media with DAPI (Sigma F6057).
-
11.
Fluorescence microscope (Zeiss).
2.7 In Vivo Preclinical Validation of In Vitro Derived β Islets
-
1.
NOD-SCID mice (Jackson Labs).
-
2.
Streptozotocin (Sigma S0130-100MG).
-
3.
Surgical instruments.
-
4.
Permanent glue.
-
5.
Antibiotic ointment.
-
6.
Anesthesia: Avertin (1:10 solution of Tribromoethanol in tertiary amyl alcohol).
-
7.
Electrical shaver.
-
8.
1 mL syringe with 25G needle (BD 305903).
-
9.
Portable Glucometer (Lifescan Inc.).
3 Methods
3.1 Derivation of Mouse Embryonic Fibroblasts (MEFs)
-
1.
MEFs were derived from 13.5 days post-coitum fetuses at stages 21 and 22 (Theiler et al.) (85) of inbred C57BL6J mice.
-
2.
MEFs were cultured in MEF media till confluency in 5 % CO2 and 3 % oxygen (please refer to Section 4.2, point 6). Then, they were cryopreserved in 10 % DMSO at a cell density of 5 × 106 cells/mL until further use. Before using as feeders MEFs were mitomycin-C (10 μg/mL) treated to arrest the cell division. Mitomycin-C treated fibroblasts were prepared in large batches and stored in bulk at a concentration of 2 × 106 cells/mL.
-
3.
The inactivated cells were then plated on 0.2 % gelatin-coated 35 mm culture dishes in MEF media at a concentration of 0.2 × 106/dish. These MEF dishes used for derivation and maintenance of hESCs.
3.2 Cell Growth and Differentiation of Human Embryonic Stem Cells
Differentiation of hESC into functional β islets was carried out as per the differentiation scheme given in Fig. 1
-
1.
hESC undifferentiated colonies mechanically passaged on the MEF feeder cells, and the cultures were grown at 37 °C and 5 % CO2 (please refer to Section 4.2, points 1–3 and Section 4.3, point 1 for ensuring health of the undifferentiated cells and Section 4.2 point 4, and Section 4.3, point 2 to ensure a high propensity of endodermal differentiation by hPSC).
-
2.
Step 1: Embryoid bodies (EB) were formed by manually passaging the hESC colonies (100–150 cells per clump). These clumps were seeded on to low adherent 60 mm dishes in serum-free EB media for 48 h under normoxic conditions at 5 % CO2 (please refer to Section 4.2, point 7 and Section 4.3, point 3 for ensuring optimal conditions for differentiation).
-
3.
Step 2: At the end of 48 h, EBs were collected and plated in 35 mm matrigel coated culture dishes in EB medium and were allowed to adhere overnight. Simultaneously, EBS was also plated in matrigel coated 8-well Permanox slides for immunofluorescent staining at different steps of differentiation (refer to Section 4.1 for troubleshooting issues at this step). EBs were then given fresh media change with human definitive endoderm differentiation media containing KSR: DMEM with 100 ng/mL activin A and 1 mM all trans-RA and were allowed to grow in this media for another 6 days with media change given every alternate day. At the end of this step, definitive endoderm cells were obtained. A portion of the cells was taken for characterization by qRT-PCR Markers like CXCR4, SOX17, FOXA2 are expressed by these cells. Also, one of the Permanox chamber slides were fixed and stained for the aforementioned DE markers (refer to Section 4.1 for troubleshooting issues to obtain a high percentage of DE cells at this step).
-
4.
Step 3: Definitive endoderm cells obtained in the previous step were fed with pancreatic endoderm media containing KSR: DMEM/F12 with 20 ng/mL of bFGF and 100 ng/mL Noggin for another 12 days with media change given every alternate day to obtain pancreatic endoderm. A portion of the cells were taken for qRT-PCR characterization, markers like PDX1, NGN3, NKX6.1 seen expressed by these cells. One Permanox slide was fixed and assessed for the markers mentioned above by immunocytochemistry (please refer to Section 4.2, point 8 to ensure a high percentage of pancreatic endoderm).
-
5.
Step 4: To obtain insulin-producing precursors/islet-like clusters in culture, ILC (2D), the cells obtained in previous step, were further cultured in DMEM: F12, with N2 (1 %) and B27 (2 %) supplements, 1 mg/mL laminin, 20 ng/mL bFGF, and 10 mM nicotinamide, 10 nM liraglutide for 12 days with media change given on alternate days (please refer to Section 4.1 to troubleshoot issues related to the optimal morphologies of 2D ILC). One 35 mm plate from the 2D culture was harvested by trypsinization, a portion of the cells were taken for qRT-PCR characterization for markers like Pdx1 insulin, c-peptide, Glut 2 and flow-cytometric analysis for the estimation of insulin, C-peptide, and Pdx1. Also, one of the chamber slides from step 4 of differentiation was fixed with 4 % PFA and subjected to immunofluorescence staining for insulin and c-peptide.
-
6.
Step 5: The islet-like clusters (ILC) obtained in 2D culture, were carefully picked up under a stereomicroscope and transferred to low attachment tissue culture dishes for culturing under them under 3D conditions. The 3D cultures of ILC were supplemented with amino acid rich media, CMRL media 1066, supplemented with KSR, 10 mM nicotinamide, ITS (insulin transferring selenium), 0.1 mM zinc sulfate, glutmax, Hepes, 10 nM Liraglutide, 20 nM Exendin-1 and 20 ng/mL HGF (hepatocyte growth factor). The islets were cultured in 3D for another 10 days with media change given on alternate days before they were harvested for molecular characterization, in vitro functional assays and animal transplantation. For immunofluorescence analysis, the 3D ILC were plated onto Matrigel coated Permanox chamber slides in 3D islet media, allowed to adhere overnight, fixed and stained for the mature β cell markers.
3.3 Quantitative RT-PCR
-
1.
Undifferentiated cells and cells from all stages of differentiation were trypsinized; washed twice in PBS and the cell pellets were initially stored in RLT buffer provided in the RNeasy mini kit at −80 °C so that RNA can be isolated at the same time from the samples of different steps of differentiation.
-
2.
Total RNA extraction was done using RNeasy mini kit as per manufacturer’s instructions. RNA (1 μg) was reverse transcribed into cDNA-specific transcripts using random hexamers and superscript II (Invitrogen) as per manufacturer’s instructions. Real-time PCR was performed with SYBR Green: Platinum SYBR green qPCR Supermix-UDG in Bio-Rad CFX 96 C 1000 Real time PCR equipment. Relative gene expression was calculated by the δCt method compared with undifferentiated cells (day 0), using GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as housekeeping gene. Primer sequences are summarized in Table S1. Gene expression analysis was done for undifferentiated hESC and hESCs differentiated into pancreatic islet-like clusters in all the five steps of differentiations. RNA sample from human fetal pancreas was used as positive control (Bose et al. (40), Supplementary data).
-
3.
qRT-PCR reactions were run in triplicates for quantitative analysis for each experimental gene and sample along with the reference gene GAPDH using the Platinum SYBR green qPCR Supermix-UDG. 5 μL of SYBR Green qPCR Master Mix, 0.5 μL each of forward and reverse primers for each gene, 2 μL of cDNA and 2 μL water was mixed to obtain 10 μL of reaction volume for each reaction on thermal cycler.
-
4.
Gene specific primers and their amplicon sizes are listed in Table 1. The annealing temperature for all reactions is 60 °C, and 40 cycles are sufficient to detect signal. The thermal cycler is used to monitor fluorescence throughout the reaction and conduct melt curve analysis of all samples to verify specificity.
-
5.
Thermal cycler program is listed below.
-
(a) Hot start incubation—15 min at 95 °C. (b) Denaturation—15 s at 94 °C. (c) Annealing—30 s at 60 °C. (d) Extension and data acquisition—30 s at 72 °C. (e) Repeat steps (b–d) 39 times to complete reaction.
-
(b) The Thermal Cycler software generates cycle threshold (Ct) values for all samples; Relative gene expression (to GAPDH) is expressed as δCt. δCt ≥ 16 was considered to be the non-expression of a gene. Lower δCt corresponded to a high expression of a gene.
-
3.4 Immunofluorescence Staining
-
1.
Culture medium from the chamber slides was aspirated and the cells were washed with 1× PBS carefully, so as to avoid cell detachment.
-
2.
Cells were fixed for 10 min at room temperature with 4 % paraformaldehyde (PFA). 4 % PFA was aspirated and the cells were washed twice with PBS and then incubated with permeabilization buffer for 10 min at room temperature.
-
3.
Cells were then blocked using blocking buffer for 1 h at room temperature.
-
4.
Primary antibodies were prepared in fresh blocking buffer at appropriate dilution (see Section 2) and were incubated overnight at 4 °C.
-
5.
Cells were washed thrice with PBS for 5 min each. Appropriate secondary antibodies of dilutions of 1:1,000 in PBS were used and the cells were incubated for 60 min at room temperature.
-
6.
Post secondary antibody washes were carried out thrice with PBS for 5 min each. Cells were then mounted using immunofluorescence mounting medium with DAPI and viewed under a fluorescent microscope and photographed.
3.5 Flow Cytometry
-
1.
Undifferentiated and differentiated cells were trypsinized and collected as single cell suspension. Two washes in PBS were given; cells were centrifuged at 1,000 × g for 5 mineach to collect the cell pellet.
-
2.
Cell pellet was fixed in 4 % PFA for 20 min at 4 °C and processed immediately. 1 mL of PFA was used to fix 3 × 106 cells. Alternatively, the PFA fixed cells were stored at 4 °C and analyzed within 15 days of harvesting.
-
3.
For analysis, the PFA fixed cells were centrifuged, PFA decanted out and the cell pellet washed twice with 1× PBS. The cell pellet was then resuspended in 2 mL FACS buffer. For staining, 0.1 × 106 cells were aliquoted in 1.5 mL microfuge tubes. Cells were then centrifuged, supernatant aspirated out and the cell pellet containing 0.1 million cells was resuspended in 100 μL FACS buffer. 0.1 Million cells each of unstained control, isotype control and the respective primary antibody were processed in identical manner in three separate microfuge tubes.
-
4.
Primary antibodies were added to each cell suspension of 0.1 million cells and mixed well by pipetting. Primary antibody incubation was carried out at room temperature for 1 h. Concentration of the isotype control was kept same as that of the respective primary antibody.
-
5.
Each of the samples was washed twice after the primary antibody incubation using FACS buffer. Again 100 μL of cell suspension was left at the bottom of the tube.
-
6.
Respective secondary antibodies were added to the cell suspension of 100 μL from step 5, and incubation was carried out for 30–45 min at 4 °C in dark. Post secondary antibody incubation, each sample was washed with FACS buffer, centrifuged; supernatant aspirated leaving the stained cell suspension in 100 μL volume.
-
7.
Each stained sample was stained in approximately 300 μL of FACS buffer, transferred to FACS tube kept in ice until analysis.
-
8.
Samples were analyzed on a BD FACS Canto or any flow cytometer capable of excitation at 488 and 630 nm wavelengths. After excluding the dead cells and debris by applying forward scatter vs. side scatter gates, appropriate positive populations were obtained. Isotype controls were used for compensation controls and appropriate gating. Gated populations enabled quantitative comparisons of marker expression in samples collected from various steps of differentiation.
-
9.
The data was analyzed using the FACS Diva software (BD Biosciences). 10,000 events were acquired per sample and results were represented in the form of histogram and/or dot plots.
3.6 In Vitro Functional Assay (Insulin and C-Peptide Assay)
-
1.
Islet like clusters (ILC) in 3D form was cultured in low attachment 6-well dishes (Nunc) at a density of 15, 3D ILC per cm2 for 48 h.
-
2.
On the third day, the culture medium was changed to media with no glucose and then incubated overnight.
-
3.
The next day, the cells were washed with Krebs–Ringer bicarbonate buffer for 2 h and treated with different concentrations of Glucose (3.3 and 16.7 mM) alone and also with combination with 10 mM Tolbutamide, 50 mM Nifedipine and 250 mM Diazoxide and incubated for 2 h respectively.
-
4.
Culture supernatants were collected from different treatments. Insulin levels were estimated using human insulin ELISA kit and C-peptide levels were estimated using the C-peptide ELISA kit, both purchased from Mercodia, Sweden as per manufacturer’s instructions.
-
5.
Human plasma was run as a control along with the samples. Protein was estimated using the Bradford assay (Bio-Rad). Insulin concentration was normalized to the total amount of protein and expressed as mU/mg of total protein. C-peptide concentrations were expressed as pmol/mg of total protein.
3.7 In Vivo Preclinical Validation of In Vitro Derived Functional β Islets
-
1.
NOD-SCID mice purchased from Jackson labs were bred, and male mice of 10 week old were used for the study. Total 12 mice were used, 6 control group and 6 transplanted group.
-
2.
Streptozotocin stock was prepared in Citrate buffer, pH 4.5 immediately before administration.
-
3.
All the animals were weighed and checked for blood glucose levels prior to STZ injections.
-
4.
Animals were rendered diabetic with two doses of STZ, first dose 100 mg/kg body weight and second dose 40 mg/kg of body weight interspersed over a period of 72 h.
-
5.
Diabetic state of the animals was confirmed by a steady state fasting glucose level of ≥300 mg/dL over a period of 1 week.
-
6.
Diabetic animals were injected insulin till the transplantation of in vitro derived islets to prevent mortality.
-
7.
On the day of transplantation, ~1,200 3D ILC were collected, centrifuged at a speed of 500 g and resuspended at 200 ILC/20 μL of step 5 differentiation media. Surgeries were carried out under a laminar flow hood under aseptic conditions.
-
8.
The animals, one at a time, were anesthetized using avertin injected intraperitonially at a dose of 10 μL/mg of body weight.
-
9.
The animal was laid gently on a sterile dissection board with the ventral side facing up. The fur of the trunk portion was carefully shaved using an electric shaver.
-
10.
One horizontal incision was made on the left side of the animal through which the kidney was gently pushed out on the surface of the skin. The kidney was kept hydrated by irrigation with PBS, applied using a wet earbud, during the surgery.
-
11.
20 μL of ILC suspension containing 200 3D ILC from step 7 was gently placed underneath the kidney capsule using a 1 mL syringe with the bent needle. Leakage of the cells was prevented (please refer to Section 4.1 to troubleshoot the issues related to leakage of the cells). The control animals received 20 μL of step 5 differentiation media without any cells.
-
12.
The kidney was then gently pushed inside the abdominal cavity, the incision sealed with the help of glue. Antibiotic ointment was applied on the sealed incision.
-
13.
The animals were then allowed to recover from anesthesia by placing the cage under an infrared lamp.
-
14.
The animals were followed for blood glucose levels, body weight and mortality till 96 days post-transplantation.
3.8 Immuno-histochemistry of Tissues
-
1.
The animals were sacrificed and the kidneys were harvested and fixed in formalin-based fixative for 48 h. Only the area of interest was excised and the tissue sections were embedded into paraffin blocks (please refer to Section 4.1 to address the troubleshooting issues related to proper assessment of area of interest during sectioning).
-
2.
Sections of 10 μm thickness were taken from taken from paraffin embedded tissue with the help of a microtome and prepared on to glass slides.
-
3.
Tissues were then deparaffinized by incubation in xylene with 2–3 changes, 5 min each. Rehydration of the sections was done by passing them through different grades of alcohol (100, 95, 80, and 50 %, respectively).
-
4.
The sections were rinsed in PBS. HIER (heat-induced epitope retrieval) through microwave irradiation was done for antigen retrieval in citrate buffer for 10 min.
-
5.
Sections were blocked in 1 % BSA/PBS at room temperature for 1 h. Overnight incubation of 1:20 dilution of anti-insulin primary antibody was done at 4 °C.
-
6.
After two brief washes, incubation with the FITC conjugated secondary antibody (as detailed in Tables S3 and S4) was done at a dilution of 1:500 for 1 h in dark.
-
7.
Similarly staining was done for C-peptide.
-
8.
After two washes for 5 min each at room temperature, the sections were mounted using mounting media and coverslips. The slides were then visualized and photographed in dark under a Zeiss fluorescence microscope.
4 Notes
Experiments | Problems | Probable reasons | Solutions |
|---|---|---|---|
In vitro differentiation | Oct3/4 continues to persist after step 2 of differentiation. | Incomplete initiation of differentiation. | Plate the EBs sparsely onto the Matrigel coated plates to ensure proper contact of the cells with the DE inducers. |
In vitro differentiation | Cells show no inter-islet connections in step 4 of differentiation. | Incomplete β islet differentiation. Most of the cells might be at DE or PE stage. | Increase the duration of treatment by 3–4 days by 2D-ILC inducers.If the problem continues to persist, start a new batch of differentiation. |
In vivo islet transplantation | Absence of transplanted cells in the tissue sections. | Excision of incorrect area.Or, the cells were not transplanted properly and have leaked out during transplantation. | Ensure excising the correct area containing transplanted cells for immunocytochemical assessment.Ensure correct transplantation of cells underneath the kidney capsule using extra precautions and a good source of light during surgery. |
-
1.
Always ensure the health of pluripotent stem cells before using them for in vitro differentiation experiments. Healthy PSC can be identified by checking their morphology and sharp borders of PSC colonies with high Oct3/4 and telomerase activity.
-
2.
Ensure a mycoplasma and endotoxin free MEFs and PSC cultures before starting the in vitro differentiation experiments.
-
3.
Handle the cells gently.
-
4.
Also, ensure a quick propensity of endodermal differentiation (≥60 % Sox 17 positive cells) of the PSC cell line used for the study. Sox 17 positive cells can be obtained by 72 h treatment of the PSC lines directly plated onto Matrigel coated plates and treated with Activin A to a final concentration of 100 ng/mL.
-
5.
Always maintain the hESC/PSC in low oxygen (3 %) incubator for maintaining healthy PSC with least oxidative damage to the cells.
-
6.
Maintain primary cultures of MEFs in low oxygen (3 %) incubator.
-
7.
Carry out the EBs differentiation as well as the entire differentiation under normoxic conditions in a regular CO2 cell culture incubator.
-
8.
Ensure ≥60 % of PDX-1 positive cells in the end of step 3 of differentiation, or else, start a new batch of differentiation.
-
1.
Do not proceed with the in vitro differentiation if the PSC lines are unhealthy.
-
2.
Do not proceed further with the in vitro pancreatic differentiation if the cell lines used have low propensity for endodermal differentiation (≤60 %).
-
3.
Do not carry out the in vitro differentiation under hypoxic conditions.
References
Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ (2008) Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451:141–146
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920
Ciruna B, Rossant J (2001) FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell 1:37–49
Hardy KM, Garriock RJ, Yatskievych TA, D’Agostino SL, Antin PB, Krieg PA (2008) Non-canonical Wnt signaling through Wnt5a/b and a novel Wnt11 gene, Wnt11b, regulates cell migration during avian gastrulation. Dev Biol 320:391–401
Yang X, Chrisman H, Weijer CJ (2008) PDGF signalling controls the migration of mesoderm cells during chick gastrulation by regulating N-cadherin expression. Development 135:3521–3530
Hardy KM, Yatskievych TA, Konieczka J, Bobbs AS, Antin PB (2011) FGF signalling through RAS/MAPK and PI3K pathways regulates cell movement and gene expression in the chicken primitive streak without affecting E-cadherin expression. BMC Dev Biol 11:20
Alev C, Wu Y, Kasukawa T, Jakt LM, Ueda HR, Sheng G (2010) Transcriptomic landscape of the primitive streak. Development 137:2863–2874
Stern CD (ed) (2004) Gastrulation: from cells to embryo. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp 219–232
Nakaya Y, Sheng G (2009) An amicable separation: chick’s way of doing EMT. Cell Adh Migr 3:160–163
Murry CE, Keller G (2008) Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell 132:661–680
Chen Y, Li X, Eswarakumar VP, Seger R, Lonai P (2000) Fibroblast growth factor (FGF) signaling through PI 3-kinase and Akt/PKB is required for embryoid body differentiation. Oncogene 19:3750–3756
Esner M, Pachernik J, Hampl A, Dvorak P (2002) Targeted disruption of fibroblast growth factor receptor-1 blocks maturation of visceral endoderm and cavitation in mouse embryoid bodies. Int J Dev Biol 46:817–825
Ciruna BG, Schwartz L, Harpal K, Yamaguchi TP, Rossant J (1997) Chimeric analysis of fibroblast growth factor receptor-1 (Fgfr1) function: a role for FGFR1 in morphogenetic movement through the primitive streak. Development 124:2829–2841
Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J (1994) fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev 8:3032–3044
Deng C, Bedford M, Li C, Xu X, Yang X, Dunmore J, Leder P (1997) Fibroblast growth factor receptor-1 (FGFR-1) is essential for normal neural tube and limb development. Dev Biol 185:42–54
Coolen M, Sakura-Spengler T, Nicolle D, Le-Mentec C, Lallemand Y, DaSilva C, Plouhinec L, Robert B, Wincker P, Shi DL, Mazan S (2007) Evolution of axis specification mechanism in jawed vertebrates: insights from chondrichthyan. PLoS One 2:e374
Wilson SI, Rydstrom A, Trimborn K, Willert K, Nusse R, Jessell TM, Edlund T (2001) The status of Wnt signaling regulates neural and epidermal fates in the chicken embryo. Nature 411:325–330
Bain G, Kitchens D, Yao M, Huetner JE, Gottlieb DI (1995) Embryonic stem cells express neuronal properties in vitro. Dev Biol 168:342–357
Sharpe C, Goldstone K (2000) The control of Xenopus embryonic primary neurogenesis is mediated by retinoid signaling in the neuroectoderm. Mech Dev 9:69–80
Franco PG, Paganelli AR, López SL, Carrasco AE (1999) Functional association of retinoic acid and hedgehog signaling in Xenopus primary neurogenesis. Development 126:4257–4265
Begemann G, Meyer A (2001) Hindbrain patterning revisited: timing effects of retinoic acid signalling. Bioessays 23:981–986
Novitch BG, Wichterle H, Jessell TM, Sockanathan S (2003) A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 40:81–95
Tonge PD, Andrews PW (2010) Retinoic acid directs neuronal differentiation of human pluripotent stem cell lines in a non-cell-autonomous manner. Differentiation 80:20–30
Otero JJ, Fu W, Kan L, Cuadra AE, Kessler JA (2004) Beta-catenin signaling is required for neural differentiation of embryonic stem cells. Development 131:3545–3557
Trilck M, Hübner R, Frech MJ (2014) Generation and neuronal differentiation of patient-specific induced pluripotent stem cells derived from niemann-pick type C1 fibroblasts. Methods Mol Biol Dec 18 [Epub ahead of print]
Brafman DA. (2014). Generation, expansion, and differentiation of human pluripotent stem cell (hPSC) derived neural progenitor cells (NPCs). Methods Mol Biol Jul 26. [Epub ahead of print]
Wiles MV, Johansson BM (1999) Embryonic stem cell development in a chemically defined medium. Exp Cell Res 247:241–248
Park C, Afrikanova I, Chung YS, Zhang WJ, Arentson E, Fong Gh G, Rosendahl A, Choi K (2004) A hierarchical order of factors in the generation of FLK1- and SCL-expressing hematopoietic and endothelial progenitors from embryonic stem cells. Development 131:2749–2762
Ng ES, Azzola L, Sourris K, Robb L, Stanley EG, Elefanty AG (2005) The primitive streak gene Mixl1 is required for efficient hematopoiesis and BMP4-induced ventral mesoderm patterning in differentiating ES cells. Development 132:873–884
Nostro M, Cheng X, Keller GM, Gadue P (2008) Wnt, activin and BMP signaling regulate distinct stages in the developmental pathway from embryonic stem cells to blood. Cell Stem Cell 2:60–71
Lindsley RC, Gill JG, Kyba M, Murphy TL, Murphy KM (2006) Canonical Wnt signaling is required for development of embryonic stem cell-derived mesoderm. Development 133:3787–3796
Naito AT, Shiojima I, Akazawa H, Hidaka K, Morisaki T, Kikuchi A, Komuro I (2006) Developmental stage-specific biphasic roles of Wnt/beta catenin signaling in cardiomyogenesis and hematopoiesis. Proc Natl Acad Sci U S A 103:19812–19817
Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE (2007) Biphasic role for Wnt/ beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A 104:9685–9690
Kubo A, Shinozaki K, Shannon JM, Kouskoff V, Kennedy M, Woo S, Fehling HJ, Keller G (2004) Development of definitive endoderm from embryonic stem cells in culture. Development 131:1651–1662
Yasunaga M, Tada S, Torikai-Nishikawa S, Nakano Y, Okada M, Jakt LM, Nishikawa S, Chiba T, Era T, Nishikawa S (2005) Induction and monitoring of definitive and visceral endoderm differentiation of mouse ES cells. Nat Biotechnol 23:1542–1550
D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE (2006) Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 24:1392–1401
Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J (2008) Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 26:443–452
Nostro MC, Sarangi F, Ogawa S, Holtzinger A, Corneo B, Li X, Micallef SJ, Park IH, Basford C, Wheeler MB, Daley GQ, Elefanty AG, Stanley EG, Keller G (2011) Stage-specific signaling through TGFb family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development 138:861–871
Bose B, Shenoy PS, Konda S, Wangikar P (2012) Human embryonic stem cell differentiation into insulin secreting β-cells for diabetes. Cell Biol Int 36:1013–1020
Gouon-Evans V, Boussemart L, Gadue P, Nierhoff D, Koehler CI, Kubo A, Shafritz DA, Keller G (2006) BMP-4 is required for hepatic specification of mouse embryonic stem cell-derived definitive endoderm. Nat Biotechnol 24:1402–1411
Tada S, Era T, Furusawa C, Sakurai H, Nishikawa S, Kinoshita M, Nakao K, Chiba T, Nishikawa S (2005) Characterization of mesendoderm: a diverging point of the definitive endoderm and mesoderm in embryonic stem cell differentiation culture. Development 132:4363–4374
Slack JM (1995) Developmental biology of pancreas. Development 121:1569–1580
Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R (2001) Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 292:1389–1394
Hebrok M, Kim SK, Melton DA (1998) Notochord repression of endodermal Sonic hedgehog permits pancreas development. Genes Dev 12:1705–1713
Kirchgessner AL, Gershon MD (1990) Innervations of the pancreas by neurons in the gut. J Neurosci 10:1626–1642
Sunami E, Kanazawa H, Hashizume H, Takeda M, Hatakeyama K, Ushiki T (2001) Morphological characteristics of Schwann cells in the islets of Langerhans of the murine pancreas. Arch Histol Cytol 64:191–201
Plank JL, Mundell NA, Frist AY, LeGrone AW, Kim T, Musser MA, Walter TJ, Labosky PA (2011) Influence and timing of arrival of murine neural crest on pancreatic beta cell development and maturation. Dev Biol 349:321–330
Rajagopal J, Anderson WJ, Kume S, Martinez OI, Melton DA (2003) Insulin staining of ES cell progeny from insulin uptake. Science 299:363
Arntfield ME, Van der Kooy D (2013) The adult mammalian pancreas contains separate precursors of pancreatic and neural crest developmental origins. Stem Cell Dev 22:2145–2157
Blyszczuk P, Czyz J, Kania G, Wagner M, Roll U, St-Onge L, Wobus AM (2003) Expression of Pax4 in embryonic stem cells promotes differentiation of nestin-positive progenitor and insulin-producing cells. Proc Natl Acad Sci U S A 100:998–1003
Miyazaki S, Yamato E, Miyazaki J (2004) Regulated expression of Pdx-1 promotes in vitro differentiation of insulin producing cells from embryonic stem cells. Diabetes 53:1030–1037
Shiroi A, Ueda S, Ouji Y, Saito K, Moriya K, Sugie Y, Fukui H, Ishizaka S, Yoshikawa M (2005) Differentiation of embryonic stem cells into insulin-producing cells promoted by Nkx2.2 gene transfer. World J Gastroenterol 11:4161–4166
D’Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE (2005) Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol 23:1534–1541
Shi Y, Hou L, Tang F, Jiang W, Wang P, Ding M, Deng H (2005) Inducing Embryonic stem cells to differentiate into pancreatic beta cells by a novel three step approach using activin A and all-trans retinoic acid. Stem Cells 23:93–110
Madsen OD, Serup P (2006) Towards cell therapy for diabetes. Nat Biotechnol 24:1481–1483
Jiang J, Au M, Lu K, Eshpeter A, Korbutt G, Fisk G, Majumdar AS (2007) Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells 25:1940–1953
Jiang W, Shi Y, Zhao D, Chen S, Yong J, Zhang J, Qing T, Sun X, Zhang P, Ding M, Li D, Deng H (2007) In vitro derivation of functional insulin-producing cells from human embryonic stem cells. Cell Res 17:333–344
Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, Yang YH, Johnson JD, Kieffer TJ (2014) Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32:1121–1133
Conlon FL, Lyons KM, Takaesu N, Barth KS, Kispert A, Herrmann B, Robertson EJ (1994) A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development 120:1919–1928
Osada SI, Wright CV (1999) Xenopus nodal-related signaling is essential for mesendodermal patterning during early embryogenesis. Development 126:3229–3240
Lowe LA, Yamada S, Kuehn MR (2001) Genetic dissection of nodal function in patterning the mouse embryo. Development 128:1831–1843
Vincent SD, Dunn NR, Hayashi S, Norris DP, Robertson EJ (2003) Cell fate decisions within the mouse organizer are governed by graded Nodal signals. Genes Dev 17:1646–1662
Phillips BW, Hentze H, Rust WL, Chen QP, Chipperfield H, Tan EK, Abraham S, Sadasivam A, Soong PL, Wang ST, Lim R, Sun W, Colman A, Dunn NR (2007) Directed differentiation of human embryonic stem cells into the pancreatic endocrine lineage. Stem Cells Dev 16:561–578
Zhang D, Jiang W, Liu M, Sui X, Yin X, Chen S, Shi Y, Deng H (2009) Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res 19:429–438
Mao GH, Chen GA, Bai HY, Song TR, Wang YX (2009) The reversal of hyperglycemia in diabetic mice using PLGA scaffolds seeded with islet like cells derived from human embryonic stem cells. Biomaterials 30:1706–1714
Naujok O, Diekmann U, Lenzen S (2014) The generation of definitive endoderm from human embryonic stem cells is initially independent from activin A but requires canonical Wnt-signaling. Stem Cell Rev 10:480–493
Kim SK, Melton DA (1998) Pancreas development is promoted by cyclopamine, a hedgehog signaling inhibitor. PNAS U S A 95:13036–13041
Huang W, Wang G, Delaspre F, Vitery Mdel C, Beer RL, Parsons MJ (2014) Retinoic acid plays an evolutionarily conserved and biphasic role in pancreas development. Dev Biol 394:83–93
Kumar SS, Alarfaj AA, Munusamy MA, Singh AJ, Peng I, Priya SP, Hamat RA, Higuchi A (2014) Recent developments in β-cell differentiation of pluripotent stem cells induced by small and large molecules. Int J Mol Sci 15:23418–23447
Mfopou JK, Chen B, Mateizel I, Sermon K, Bouwens L (2010) Noggin, retinoids, and fibroblast growth factor regulate hepatic or pancreatic fate of human embryonic stem cells. Gastroenterology 138:2233–2245
Drucker DJ (1998) Glucagon-like peptides. Diabetes 47:159–169
Huotari MA, Palgi J, Otonkoski T (1998) Growth factor-mediated proliferation and differentiation of insulin-producing INS-1 and RINm5F cells: Identification of betacellulin as a novel β-cell mitogen. Endocrinology 139:1494–1499
Li L, Seno M, Yamada H, Kojima I (2001) Promotion of β-cell regeneration by betacellulin in ninety percent-pancreatectomized rats. Endocrinology 142:5379–5385
Li L, Seno M, Yamada H, Kojima I (2003) Betacellulin improves glucose metabolism by promoting conversion of intra islet precursor cells to β-cells in streptozotocin-treated mice. Am J Physiol Endocrinol Metab 285:E577–E583
Otonkoski T, Cirulli V, Beattie GM, Mally MI, Soto G, Rubin JS, Hayek A (1996) A role for hepatocyte growth factor/scatter factor in fetal mesenchyme-induced pancreatic β-cell growth. Endocrinology 137:3131–3139
Mashima H, Shibata H, Mine T, Kojima I (1996) Formation of insulin-producing cells from pancreatic acinar AR42J cells by hepatocyte growth factor. Endocrinology 137:3969–7396
Speier S, Gjinovci A, Charollais A, Meda P, Rupnik M (2007) Cx36-mediated coupling reduces beta-cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes 56:1078–1086
Hammar EB, Irminger JC, Rickenbach K, Parnaud G, Ribaux P, Bosco D, Rouiller DG, Halban PA (2005) Activation of NF-kB by extracellular matrix is involved in spreading and glucose-stimulated insulin secretion of pancreatic b cells. J Biol Chem 280:30630–30637
Weber LM, Hayda KN, Anseth KS (2008) Cell-matrix interactions improve b-cell survival and insulin secretion in three-dimensional culture. Tissue Eng Part A 14:1959–1968
Parnaud G, Hammar E, Ribaux P, Donath MY, Berney T, Halban PA (2009) Signaling pathways implicated in the stimulation of b-cell proliferation by extracellular matrix. Mol Endocrinol 23:1264–1271
Wang X, Ye K (2009) Three dimensional differentiations of embryonic stem cells into islet-like insulin producing clusters. Tissue Eng Part A 15:1941–1952
Saito H, Takeuchi M, Chida K, Miyajima A (2011) Generation of glucose responsive functional islets with a three-dimensional structure from mouse fetal pancreatic cells and iPS cells in vitro. PLoS One 6:e28209
Mandal A, Tipnis S, Pal R, Ravindran G, Bose B, Patki A, Rao MS, Khanna A (2006) Characterization and in vitro differentiation potential of a new human embryonic stem cell line, ReliCellhES1. Differentiation 74:81–90
Theiler K (1989) The house mouse development and normal stages from fertilization to 4 weeks of age, 2nd edn. Springer, Berlin
Acknowledgements
The authors earlier published the original research article corresponding to this Protocol chapter in Bose et al. (40), where the funding source was duly acknowledged. The authors also greatly acknowledge the contributions of Dr(s) Sudhakar Konda and Pralhad Wangikar, who were also the coinvestigators of the original study. Making of this protocol chapter does not involve any funding from any sources.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this protocol
Cite this protocol
Bose, B., Sudheer, P.S. (2015). In Vitro Differentiation of Pluripotent Stem Cells into Functional β Islets Under 2D and 3D Culture Conditions and In Vivo Preclinical Validation of 3D Islets. In: Turksen, K. (eds) Embryonic Stem Cell Protocols. Methods in Molecular Biology, vol 1341. Humana Press, New York, NY. https://doi.org/10.1007/7651_2015_230
Download citation
DOI: https://doi.org/10.1007/7651_2015_230
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2953-5
Online ISBN: 978-1-4939-2954-2
eBook Packages: Springer Protocols