
Abstract
In their search for novel molecules of therapeutic benefit during the 1990s, Minale and his team collected extracts from the shallow water lithistid marine sponge Callipelta sp. Subsequent testing of these specimens revealed promising activity in cytotoxic assays by inhibiting in vitro proliferation of KB and P388 cells. Whilst a closer inspection of the extract revealed callipeltins A–C as the major metabolites responsible for the observed activity, further analysis led to the discovery of three additional cytotoxic components: callipeltosides A, B and C (each differing in the attached sugar). At the time, these molecules represented a structurally unprecedented class of polyketides, rightly drawing the attention of the synthetic community. Although the connectivity of these molecules could be deduced by Minale, questions surrounding the absolute stereochemistry of the sugar moieties (C1’–C8’, d or l) and configuration of the trans-configured chlorocyclopropane with respect to the C1–C19 unit remained. Furthermore, the stereochemistry of the glycosidic linkage could not be conclusively determined. This chapter details the substantial effort of the synthetic community to elucidate the structure of these fascinating molecules from start to finish, describing nigh on 20 years of collective work by world leaders in the field.
Graphical Abstract

Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Isolation
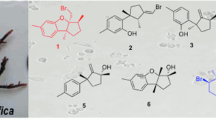
Extracts from marine sponges have provided a large number of complex molecules that exhibit a wide range of biological activities. In their search for novel compounds that display antitumour and antiviral properties, Minale and his team examined an extract from the shallow water lithistid marine sponge Callipelta sp., located off New Caledonia [1, 2]. This specimen was found to inhibit the in vitro proliferation of P388 and KB cells whilst also providing protection against HIV. Further study of the extract indicated that callipeltins A–C were the major metabolites responsible for the biological activity [1, 2]. However, additional analysis of the dichloromethane extract of this marine sponge (2.5 kg freeze dried) revealed callipeltoside A as a minor metabolite (3.5 mg isolated) [3] and later callipeltosides B and C (both ≤1.0 mg) (Fig. 1) [4]. At the time of isolation, these polyketides represented an unprecedented new class of natural products, which has now been expanded upon with the disclosure of the phorbasides, aurisides, dolastatins and others [5, 6].

The callipeltosides in their corrected forms
1.1 Structural Features and Assignment
1.1.1 Callipeltoside A
Minale and co-workers successfully deduced the connectivity of callipeltoside A through the use of COSY, HMQC and HMBC experiments. Further ROESY and nOe techniques provided an indication of the relative stereochemistry of the C1–C13 core whilst revealing the preferred chair conformation of the embedded C3–C7 pyran. Measurement of the 1H coupling constant between C20 and C21 (J20,21 = 3.1 Hz, having removed additional J21,22 splitting) gave strong evidence for a trans-configured chlorocyclopropane moiety. However, the configuration of the chlorocyclopropane with respect to the rest of the molecule could not be determined [3].
The attached 4-amino-4,6-dideoxy-2-O,3-C-dimethyl-α-talopyranosyl-3,4-urethane (later named callipeltose A) sugar was also without precedent. HMBC correlations between C5 and H1′ as well as H5 and C1′ provided the point of attachment for the glycosidic linkage, with nOe experiments giving evidence for the relative stereochemistry, suggesting an α-anomeric linkage. The nOe correlations also indicated that the pyran ring adopted a six-membered boat conformation, which was imparted by the five-membered cyclic carbamate [3].
Although the connectivity of callipeltoside A had been established, the absolute configuration could not be determined, and therefore the power of total synthesis was required to confirm the structure [3]. It was later shown by Trost [7, 8] (and also Evans [9, 10], Paterson [11], Panek [12], Hoye [13] and Ley [14, 15] vide infra) that the C1–C19 fragment and callipeltose A sugar were enantiomeric to that originally reported by Minale, whilst the chlorocyclopropane (C20–C22) was incorrectly configured with respect to the rest of the molecule (Fig. 2).

Originally disclosed structure of callipeltoside A and subsequent absolute assignment following total synthesis
1.1.2 Callipeltosides B and C
The structures of callipeltosides B and C were assigned in much the same way as callipeltoside A, with the authors suggesting them to contain ‘the same macrolide portion including stereochemistry’ as the parent molecule, the only difference being the attached sugar. Despite this comment, the isolation paper unusually depicts the structures of callipeltosides B and C as having an enantiomeric C1–C13 core to callipeltoside A (Fig. 3), also showing an oppositely configured chlorocyclopropane. Although Minale made no claim to know the absolute stereochemistry of callipeltosides A, B or C, the relative configuration described in the text and accompanying structures differed in their descriptions [4].

Originally disclosed structures of callipeltosides B and C and subsequent absolute stereochemical assignment following total synthesis
In keeping with the originally depicted structure of callipeltoside A, callipeltosides B and C were also shown to contain d-configured sugars. The configuration of these sugars was assigned on the basis that callipeltose C bore a likeness with the evalose sugar in everninomicin B [4].
The callipeltose B sugar was identified to contain an N-formyl group at the C4′H position, meaning that the molecule existed as ‘two inseparable conformers’ in a 4:1 ratio, whilst callipeltose C instead contained a secondary hydroxy group with opposite stereochemistry. Both the callipeltose B and C sugars were found to adopt chair conformations (following nOe experiments), with analysis of the 13C NMR chemical shifts (extrapolated from HMQC experiments) indicating that the anomeric linkage was β-equatorial in both cases (callipeltoside B, δ = 99.2; and callipeltoside C, 98.9 ppm) [4].
The first synthesis of callipeltoside C was completed by MacMillan in 2008 (Sect. 2.8), confirming that the absolute stereochemistry of the molecule in terms of the C1–C13 core, chlorocyclopropane unit and sugar configuration (l-configured rather than the original d-configuration suggested by Minale) were identical to callipeltoside A (Fig. 3). The configuration of the glycosidic linkage was tentatively suggested to be β-equatorial ‘based on the isolation studies for callipeltoside B’ [16].
The Ley group completed the synthesis of callipeltoside C in 2012 [14, 15], confirming the structural assignment by MacMillan [16]. Measurement of the 1JC–H coupling value of the glycosidic bond and thorough NOESY analysis of the sugar portion implied that the stereochemistry at C1′H was (R)-configured, in contrast to callipeltoside A. In the same work, this group also disclosed the first total synthesis of callipeltoside B, noting that the callipeltose B sugar was also l-configured, but with (S)-stereochemistry at the C1′H position. No further studies were performed, but it was reasonably assumed that the inversion of stereochemistry at the C1′H position for callipeltoside C is connected to the opposing configuration of the C4′H substituent (Fig. 3) [14, 15].
1.2 Biological Activity of the Callipeltosides
1.2.1 Studies by Minale
Preliminary studies by Minale indicated that callipeltoside A was moderately cytotoxic in assays against human bronchopulmonary non-small-cell lung carcinoma, affecting the NSCLC-N6 and P388 cell lines (IC50 = 11.26 and 15.26 μg/mL, respectively) [3]. Further research involving the NSCLC-N6 cell line showed a cell cycle-dependent effect, with cell division blocked at the G1 phase level (Table 1). However, the small amounts of isolated material prevented additional investigation into this process and hence determination of the exact mode of action [3].
Callipeltosides B and C also showed cytotoxic activity against the NSCLC-N6 cell line, albeit at a more modest level (IC50 values of 15.1 μg/mL and 30.0 μg/mL, respectively) [4].
1.2.2 Studies by Trost
Further investigations into the biological activity of callipeltoside A were carried out by Trost and co-workers [8]. During their synthesis of callipeltoside A, diastereomer 7, deschlorocallipeltoside A (8) and the callipeltoside aglycon (9) were also synthesised (Fig. 4).

Compounds synthesised and tested by Trost against the A2780 human ovarian carcinoma cell line
Treatment of A2780 human ovarian carcinoma cells (following 48 h incubation) with callipeltoside A as well as 7, 8 and 9 showed that the callipeltose A sugar was essential for biological activity, whilst the trans-chlorocyclopropane was less critical (Table 2, entries 1 and 3). Interestingly the introduction of the oppositely configured trans-chlorocyclopropane (7) resulted in improved biological activity relative to the natural product (Table 2, entry 2) [8].
2 Structural Investigations and Total Syntheses
The first total synthesis of callipeltoside A (1) was completed in 2002 by Trost [7, 8], following excellent initial work by Paterson who completed what proved to be the enantiomeric C1–C13 core (2001) [17].
The synthesis of callipeltoside A was also achieved thereafter by the groups of Evans (2002) [9, 10], Paterson (2003) [11], Panek (2004) [12], Hoye (2010) [13] and Ley (2012) [14, 15]. In addition, a formal synthesis of the callipeltoside aglycon has been disclosed by Marshall [18], partial syntheses by Olivo [19,20,21] and Yadav [22] as well as numerous contributions towards the callipeltose A sugar [23,24,25,26]. The absolute configuration of callipeltoside C (3) was initially determined by MacMillan (2008) [16], whilst the Ley group supported this assignment and completed the family with the first synthesis of callipeltoside B [14, 15]. Due to the page restriction imposed on this review, only the completed syntheses of these molecules and subsequent learnings will be reported.
2.1 Paterson Aglycon Synthesis (2001)
2.1.1 Retrosynthesis
Paterson’s approach to the callipeltoside aglycon involved disconnection along the C1–C13O ester linkage which, in the forward direction, would be formed by Yamaguchi macrolactonisation. Since the relative configuration of the trans-chlorocyclopropane with respect to the rest of the molecule was in doubt, attachment of each enantiomer was necessary. Therefore, the C17–C18 bond was chosen as a further key disconnection point, revealing vinyl iodide 11 and the cyclopropyl alkyne 12. Compound 11 was further disconnected at the pyran moiety, giving fragment 13, which in turn could be accessed through sequential aldol reactions. This strategy therefore allows the facile synthesis of both C13 epimers and allows for the relative configuration of the macrolide ring to be resolved (Scheme 1) [17].

Paterson’s retrosynthesis of the callipeltoside aglycon
2.1.2 Synthesis of the C1–C17 Vinyl Iodide
The synthesis began by union of known vinyl iodide 17 and the enolate of enal 16 by Lewis acid-mediated vinylogous aldol reaction [17, 27, 28]. This resulted in an intentional 1:1 mixture of epimeric products at C13, also installing the desired trisubstituted double bond present in the natural product scaffold [17]. TBS protection of the C13 alcohol followed by boron-mediated anti-aldol reaction with 15 set the C8 and C9 stereocentres [17, 29,30,31]. Diastereoselective reduction of the ketone functionality using the Evans–Tishchenko protocol formed the remaining C7 stereocentre in high selectivity. A further three synthetic manipulations provided advanced protected C5–C17 fragment 23 (Scheme 2) [17].

Synthesis of C5–C17 fragment 23. Reagents and conditions: (a) 16, PhMe, −78°C; LDA, then 17, 18, THF, −78°C, 80%; (b) TBSCl, imidazole, CH2Cl2, RT, 87%; (c) (i) Cy2BCl, Et3N, Et2O, −5°C, then 15, −78°C to −27°C; (ii) CH2Cl2–H2O, SiO2, RT, 99% (over 2 steps), dr > 98.5:1.5; (d) SmI2, EtCHO, THF, −20°C to −10°C, 92%, dr > 98.5:1.5
PMB protecting group removal and oxidation gave aldehyde 24, which underwent Mukaiyama aldol reaction with 25 to give linear precursor 26 in good diastereoselectivity (95:5) and yield (85%). Acid-catalysed deprotection of the TES group resulted in cyclisation and methyl ketal formation to give 27 in an excellent yield of 96% (Scheme 3) [17].

Manipulation of the C1–C17 fragment and formation of the C3–C7 pyran. Reagents and conditions: (a) (i) DDQ, CH2Cl2–pH 7 buffer (10:1), 40°C, 88%; (ii) Dess–Martin periodinane, CH2Cl2, RT, 86%; (b) 25, BF3•OEt2, CH2Cl2, −100°C, 85%, dr = 95:5; (c) PPTS, (MeO)3CH, MeOH, 20°C, 96%
The free hydroxy was protected as the TBS-ether and the C13OH revealed thereafter following selective removal of the TBS group using TBAF. This allowed for separation of the C13 epimeric alcohols, which could then be separately saponified and the requisite 12-membered rings formed by Yamaguchi macrolactonisation (Scheme 4) [17]. With the two macrocycles in hand, the Paterson group used molecular modelling and compared the nOe data collected by Minale [3]. The group concluded that macrocycle 31 was more likely the desired conformation, and so the unwanted isomer (28) was recycled by oxidation and diastereoselective reduction prior to saponification (Scheme 4) [17].

Formation of the proposed callipeltoside macrocycle and the C13 epimeric material. Reagents and conditions: (a) TBSCl, imidazole, CH2Cl2, RT; (b) TBAF, THF, RT, 87% (over 2 steps); (c) TPAP, NMO, 4 Å MS, CH2Cl2, RT; (d) NaBH4, CeCl3•7H2O, EtOH, −78°C to 0°C, 46% (over 2 steps); (e) Ba(OH)2•8H2O, MeOH, RT; (f) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, PhMe, 80°C, 53% (over 2 steps, 28 to 29) and 70% (over 2 steps, 30 to 31)
2.1.3 Synthesis of Enantiomeric trans-Chlorocyclopropyl Alkynes 12 and ent-12
The synthesis of enantiomeric trans-chlorocyclopropyl alkynes 12 and ent-12 was achieved using an asymmetric Simmons–Smith cyclopropanation developed by the Charette group [32,33,34]. Following this, oxidation, dibromoolefination and Corey–Fuchs reaction gave the trans-chlorocyclopropane 12 in short sequence (Scheme 5, one enantiomer shown) [17].

Access to enantiomeric cyclopropyl alkynes (one enantiomer shown; 12). Reagents and conditions: (a) n-BuLi, TMEDA, THF, −78°C to 0°C, 80%; (b) ZnEt2, CH2I2, 34, CH2Cl2, 0°C to RT, dr = 97.5:2.5; (c) (COCl)2, DMSO, CH2Cl2, −78°C, then Et3N, −78°C to 0°C; (d) Zn, pyridine, PPh3, CBr4, CH2Cl2, RT, 40% (over 3 steps); (e) n-BuLi, Et2O, −78°C, no reported yield, used directly in the next step
2.1.4 Synthesis of Diastereomeric Aglycons 39 and 41
Cyclopropyl alkynes 12 and ent-12 were cross-coupled with the C1–C17 vinyl iodide (31) by Sonogashira reaction to give 38 and 40. Each molecule underwent TBS deprotection, with treatment using PPTS in H2O installing the hemi-ketal functionality. The diastereomeric aglycons 39 and 41 were synthesised in 54% (over 3 steps) and 67% (over 3 steps), respectively (Scheme 6) [17].

Synthesis of diastereomeric aglycons 39 and 41. Reagents and conditions: (a) 12 or ent-12 [(PPh3)2PdCl2], CuI, i-Pr2NH, EtOAc, −20°C to RT; (b) TBAF, THF, RT; (c) PPTS, MeCN, H2O, RT, 54% (over 3 steps, 31 to 39), and 67% (over 3 steps, 31 to 41)
The 1H and 13C NMR data for diastereomeric aglycons 39 and 41 were found to be near identical, preventing any conclusive assignment. However, the trans-chlorocyclopropane influenced the optical rotation of these molecules, which was found to be significantly different in both sign and magnitude [39, [α]D20 = −97.8 (c = 0.19, CHCl3); 41, [α]D20 = +45.8 (c = 0.28, CHCl3)] [17]. This result outlined the importance of the optical rotation for the structural determination of callipeltoside A. However, at this point in time, attachment of the callipeltose A sugar was not reported by Paterson, and therefore the absolute configuration of callipeltoside A was not determined.
2.2 Trost Synthesis of Deschlorocallipeltoside A (8)
Prior to their synthesis of callipeltoside A, the Trost group completed deschlorocallipeltoside A (8) [35]. By targeting 8, Trost hoped to confirm the relative stereochemistry of the C1–C22 core with respect to the callipeltose A sugar and provide a convergent strategy to this family of molecules [8, 35]. Comparison of callipeltoside A with auriside B (42) revealed several structural similarities (Fig. 5), suggesting that the synthetic effort should focus on the synthesis of the enantiomeric C1–C13 portion (to that disclosed by Paterson) and an l-configured sugar [8, 35, 36].

Comparison of the callipeltoside scaffold with auriside B (42)
2.2.1 Retrosynthesis
Initial retrosynthetic disconnections involved the late-stage attachment of the callipeltose A sugar (43) by Schmidt glycosidation and Horner–Wadsworth–Emmons (HWE) coupling of the C1–C13 macrocycle with phosphonate 45 [8, 35]. Attachment of these fragments at a late stage meant that the stereochemical uncertainties associated with callipeltose A and the trans-chlorocyclopropane unit (for the synthesis of callipeltoside A) could be resolved at the end of the synthesis. Callipeltose A 43 was to be constructed from l-rhamnose (46), with phosphonate 45 by Sonogashira reaction between iodoalkyne 48 and cyclopropyl alkyne 49 followed by further synthetic manipulation [8, 35]. An acylketene cyclisation would provide the C1–C13 macrocycle, whilst a ruthenium-Alder-ene type coupling [37] and palladium-catalysed allylic alkylation [38] would be key to the synthesis of substrate 47 (Scheme 7) [8, 35].

Trost retrosynthesis of deschlorocallipeltoside A (8)
2.2.2 Synthesis of the C1–C13 Macrocycle 44
Synthesis of the C1–C13 macrocycle began from (S)-configured Roche ester 50, which was converted into alkyne 51 in 5 linear steps [8, 35]. A ruthenium-catalysed Alder-ene type reaction between 51 and 52 was implemented to construct 53, with the trisubstituted double bond formed with complete regioselectivity (Scheme 8) [8, 35, 37]. Incorporation of the Troc protecting group was key to obtaining high yield (85%), lower catalyst loading (5 mol%) and shorter reaction time (30 min) compared to the unprotected version (62%, 10 mol%, 2 h, respectively) [8, 35]. The regioselectivity of this reaction was thought to arise from ruthenium metallacycle 58 (Scheme 9), with a dative interaction between the non-bonding methoxy electrons and ruthenium essential to reduce the equilibration of the oxidative cyclisation and enhance the rate of the β-hydride elimination step [8, 37].

Synthesis of C7–C15 fragment 55. Reagents and conditions: (a) CpRu(MeCN)3PF6 (5 mol%), 52, acetone, RT, 85%; (b) p-methoxyphenol, (S,S)-54 (7.5 mol%), [Pd2dba3]•CHCl3 (2.5 mol%), TBAC, CH2Cl2, 79%, dr = 95:5, regioselectivity = 66:34 (2°:1°)

Proposed mechanism for the ruthenium-catalysed Alder-ene reaction [8]
The C13 stereocentre was installed via an asymmetric palladium-catalysed allylic alkylation reaction with (R,R)-diphenyl compound 61 identified as the chiral ligand of choice, providing 55 in excellent diastereoselectivity (95:5), good yield (79%) and reasonable branched-to-linear ratio (75:25) [8, 35, 38]. The proposed mechanism for this transformation is shown in Scheme 10 [8, 38]. It is postulated that following alkene coordination to the active palladium species, the corresponding palladium π-allyl species forms with loss of the OTroc group and formation of the putative cationic complex 62 [8]. It is suggested that the anti-π-allyl species is favoured over the syn-π-allyl form by virtue of the increased steric bulk of the substituted syn-π-allyl configuration [8]. The (R,R)-diphenyl ligand 61 is key for setting the C13 stereocentre, influencing the diastereofacial nucleophilic attack of p-methoxyphenol [8].

Proposed catalytic cycle for the palladium-catalysed allylic alkylation using (R,R)-61 [8]
However, further analysis of C13 stereocentre using the O-methylmandelate method [39] revealed that the undesired C13 configuration had been set. Fortunately, this unanticipated result could be rectified by use of the enantiomeric (S,S)-diphenyl ligand 54 (see Scheme 8) in excellent diastereoselectivity (95:5) and slightly lower branched to linear regioselectivity (66:34) [8].
With the C13 stereocentre in place, routine deprotection and oxidation of the primary alcohol gave 69. Addition of the E-lithium enolate of t-butylthiopropionate generated the C6 stereocentre in good diastereoselectivity (dr = 84:16) by means of a Cram chelation-controlled aldol reaction. Subsequent protection and reduction delivered aldehyde 70, which was further reacted with 71 under Felkin–Anh control and protected as TBS-ether 72. Treatment of 72 with CAN then liberated the C13 alcohol which, upon heating, cyclised to produce the C1–C13 macrocycle (44) in 82% yield via the acyl ketene (Scheme 11) [8, 35].

Formation of macrocycle 44. Reagents and conditions: (a) (i) t-butylthiopropionate, LDA, THF, −108°C, 82%, dr = 84:16; (ii) TBSOTf, 2,6-lutidine, CH2Cl2, 0°C, 86%; (iii) DIBAL-H, PhMe, −78°C, 79%; (b) (i) 71, BF3•OEt2, CH2Cl2, −78°C, 94%, dr > 95:5; (ii) TBSOTf, 2,6-di-t-butylpyridine, CH2Cl2, 0°C, 95%; (c) (i) CAN, acetone–H2O (4:1), 0°C, 82%; (ii) 0.5 mM in PhMe, 110°C, 82%
2.2.3 Completion of the Deschlorocallipeltoside Aglycon 75
Dihydroxylation and sodium periodate-mediated cleavage converted macrocycle 44 to aldehyde 73, with which the deschlorocallipeltoside sidechain 45Footnote 1 could be appended via a Horner–Wadsworth–Emmons reaction [8, 35]. This was achievable in low yield (40%), with the E-isomer predominating in a 4:1 ratio. TBS deprotection and transketalisation with PPTS in wet MeCN then afforded the deschlorocallipeltoside aglycon 75 (Scheme 12) [35].

Completion of the deschlorocallipeltoside aglycon 75. Reagents and conditions: (a) OsO4, NMO, THF–H2O (4:1), 0°C, then NaIO4, THF–H2O, RT, 80%; (b) 45, LiHMDS, THF, −78°C, 40%, E:Z = 4:1; (c) HF•pyridine, MeOH, RT, then PPTS, H2O, MeCN, RT, 60%
2.2.4 Synthesis of L-Callipeltose A (43) and Deschlorocallipeltoside A
The bicyclic ring system of callipeltose A (76) was synthesised according to previous methodology reported by Guiliano [25] starting from l-rhamnose. A three-step sequence then gave trichloroacetimidate 43, in preparation for coupling to the deschlorocallipeltoside aglycon (75) by Schmidt glycosidation (Scheme 13) [8, 35].

Synthesis of callipeltose A (43) and deschlorocallipeltoside A (8). Reagents and conditions: (a) ref [25]; (b) (i) TBSOTf, 2,6-lutidine, CH2Cl2, RT, 55%; (ii) MeI, Ag2O, DMF, RT, 69%; (c) (i) H2SO4, PPTS, Ac2O, RT, 81%; (ii) K2CO3, MeOH, RT, 89%; (iii) Cl3CCN, NaH, CH2Cl2, RT, 86%; (d) (i) 43, TMSOTf, 4 Å MS, dichloroethane, RT, 80%; (ii) TBAF, AcOH, THF, RT, 95%
Deschlorocallipeltoside A was completed in two steps by attachment of trichloroacetimidate 43 to glycosyl acceptor 75 and subsequent deprotection using TBAF. The resulting 1H and 13C NMR data showed a striking similarity with callipeltoside A (1), except in the regions where the chlorine substituent would be present. This provided evidence that the relative configuration of the C1–C13 fragment with respect to the callipeltose A sugar was correct and would likely translate to the natural product following attachment of the correct trans-chlorocyclopropane moiety. Whilst this was a major milestone, the absolute stereochemistry of the natural product could not be inferred as the sign and magnitude of the optical rotation differed significantly [[α]D = +45.0 (c = 0.50, MeOH) verses [α]D = −17.6 (c = 0.04, MeOH) for callipeltoside A (1)] following omission of the chlorine substituent (see Sect. 2.1.4 for comment on the influence of the trans-chlorocyclopropane on the measured optical rotation) [35].
2.3 Trost Synthesis of Callipeltoside A (2002)
Following the synthesis of deschlorocallipeltoside A [35], Trost completed the first total synthesis of callipeltoside A [7, 8]. Prior to this, two stereochemical issues needed to be resolved: (1) the correct enantiomer of the trans-chlorocyclopropane had to be determined by attachment to macrocycle 73 and (2) the absolute stereochemistry of the natural product deduced as a result [7, 8]. With this in mind, the initial synthetic strategy was executed in exactly the same way as for deschlorocallipeltoside A, with the only difference being the synthesis of the trans-chlorocyclopropane moiety.
2.3.1 Synthesis of Enantiomeric trans-Chlorocyclopropanes 84 and ent-84
Preparation of the trans-chlorocyclopropane commenced from bis-menthol compound 78, which was commercially available as either enantiomer [7, 8]. Sequential deprotonation using LiTMP and addition of bromochloromethane gave trans-configured cyclopropane 79 in high diastereoselectivity (>99:1) after recrystallisation [7, 8]. Hydrolysis of a single ester group and acid chloride formation gave substrate 80 which, upon submission to a modified Barton–Crich–Motherwell decarboxylation procedure [40], provided the requisite trans-chlorocyclopropane geometry in good yield (60%) and diastereoselectivity (dr = 97:3) [7, 8]. A further three synthetic steps provided compound 37, which underwent a one-pot Stille reaction/elimination sequence to incorporate the C16–C17 trans-double bond and internal alkyne [41]. Additional manipulation by means of an Appel reaction and displacement of the resulting bromide with P(OEt)3 gave phosphonate coupling partner 84 in 10 linear steps and 20% overall yield (Scheme 14, one enantiomer shown for clarity) [7, 8].

Synthesis of the trans-chlorocyclopropane (one enantiomer shown). Reagents and conditions: (a) LiTMP, BrCH2Cl, THF, −78°C, 87%, dr > 99:1 (following one crystallisation); (b) (i) NaOH, i-PrOH, 70°C; (ii) SOCl2, RT, 90% (over 2 steps); (c) 81, DMAP, TBAI, CCl4 (0.02 M), RT, then AIBN, 80°C, 60%, dr = 97:3; (d) (i) HCl•HNMe(OMe), i-PrMgCl, THF, −10°C; (ii) DIBAL-H, CH2Cl2, −78°C; (iii) CBr4, PPh3, CH2Cl2, RT, 80% (over 3 steps); (e) (i) 83, [Pd2dba3]•CHCl3, (4-OMeC6H4)3P, DIPEA, DMF, 80°C, 66%; (ii) CBr4, PPh3, CH2Cl2, −40°C, 90%; (iii) P(OEt)3, 100°C, 93%
2.3.2 Completion of Callipeltoside A (1)
Phosphonate 84 and its enantiomer were appended to 73 via a Horner–Wadsworth–Emmons reaction. Subsequent deprotection and concomitant cyclisation then afforded diastereomeric aglycons 9 and 86 [7, 8], proceeding in near identical yield and selectivity to that previously reported for deschlorocallipeltoside A [35]. Once again the callipeltose A sugar 43 was attached by Schmidt glycosidation (Sect. 2.2.4), with deprotection using TBAF revealing callipeltoside diastereomers 85 and 7 (Scheme 15). Comparison of the 1H and 13C NMR spectra generated for these compounds with the natural isolate disappointingly revealed no significant differences [7, 8]. However, as previously noted by Paterson (Sect. 2.1.4) [17], the presence of the trans-chlorocyclopropane unit meant significantly different optical rotations for each diastereomer. An optical rotation of [α]D22 = −19.2 (c = 1.0, MeOH) was recorded for diastereomer 85, whereas 7 was [α]D22 = +156.3 (c = 0.55, MeOH) [7, 8]. Comparison with the natural isolate [[α]D22 = −17.6 (c = 0.04, MeOH)] therefore suggested that diastereomer 85 was consistent with being callipeltoside A (1). Hence, the structural assignment of callipeltoside A was made possible purely by comparison of the sign and magnitude of the measured optical rotation [7, 8].

Completion of callipeltoside A. Reagents and conditions: (a) (i) LiHMDS, 84 or ent-84, THF, −78°C to −40°C to RT, E:Z = 4:1; (ii) HF•pyridine, MeOH, 0°C, 50% (over 2 steps in both cases); (b) (i) 43, TMSOTf, 4 Å MS, 1,2-dichloroethane, −30°C; (ii) TBAF, AcOH, THF, RT, 70% (over 2 steps in both cases)
Trost’s synthesis of callipeltoside A was completed in a total of 50 manipulations, with 22 steps making up the longest linear sequence (0.47% overall yield) [7, 8]. Although the stereochemical uncertainties for callipeltoside A had been resolved, the group wanted to improve the diene-yne synthesis, as only 4:1 E:Z ratio (at C14–C15) was obtained from the earlier Horner–Wadsworth–Emmons reaction.
2.3.3 Second-Generation Synthesis of the Callipeltoside Aglycon 86
The second generation began with the interception of alkene 44 (Sect. 2.2.3), which was converted to the corresponding hemi-ketal 87 following TBS deprotection. Cross metathesis with crotonaldehyde in the presence of Grubbs’ second-generation catalyst and subsequent Takai reaction provided vinyl iodide 88 in a significantly improved 8:1 E:Z ratio at C16–C17 (Scheme 16) [8]. At this point it was noted that volatile alkyne 12 (described by Paterson, Sect. 2.1.3) [17] was avoided for practical reasons, and so a one-pot reaction involving formation of the alkynyl stannane in situ and subsequent Stille coupling delivered 86. This sequence represented an improvement on the step count and diene (E) selectivity of the first-generation synthesis, but was only reported with the oppositely configured trans-chlorocyclopropane (ent-37) (Scheme 16) [8].

Second-generation coupling of the trans-chlorocyclopropane. Reagents and conditions: (a) (i) HF•pyridine, MeOH, 0°C; (ii) PPTS, MeCN–H2O (3:1), RT, 91% (over 2 steps); (b) crotonaldehyde, Grubbs’ II, CH2Cl2, 40°C, then CrCl2, CHI3, dioxane–THF (4:1), 0°C, E:Z = 8:1, 84%; (c) n-BuLi, ent-37, Me3SnCl, Et2O, −78°C to RT, then 88, (MeCN)2PdCl2 (3 × 10 mol%), DMF, RT, 70%
2.4 Evans Synthesis of Callipeltoside A (2002)
Shortly after Trost published the completion of callipeltoside A, Evans also disclosed the synthesis of the molecule [9, 10].
2.4.1 Retrosynthesis
The absolute configuration of callipeltoside A was not known at the start of the project, and so Evans applied a similar retrosynthetic approach whereby the trans-chlorocyclopropane was attached at a late stage. This was to be achieved by Horner–Wadsworth–Emmons reaction with sidechain 92, enabling the stereochemistry of the sidechain to be deduced at a late stage [9, 10]. Sequential aldol reactions would be key for the synthesis of the C1–C13 fragment 91, which would be completed by macrocyclisation to form the C13–O bond (Scheme 17).

Evans approach to callipeltoside A
2.4.2 Synthesis of C1–C13 Fragment 91
A copper-catalysed vinylogous aldol reaction between 95 and 96 set the (R)-configured C13 stereocentre and forged the C10–C11 trisubstituted double bond to give 98 as a single isomer in 93% yield and high enantioselectivity (er = 97.5:2.5) (Scheme 18) [9, 10, 42]. Aldehyde 99 was then synthesised in a three-step protection, reduction and oxidation sequence.

Synthesis of the C5–C14 fragment. Reagents and conditions: (a) 97, CH2Cl2, −78°C; 1 N HCl, EtOAc, RT, 93%, er = 97.5:2.5; (b) 100, Cy2BCl, EtNMe2, Et2O, 0°C to −78°C then RCHO, −78°C to −20°C
Interestingly, aldol reaction between (R)-aldehyde 99 and oxazolidone 100 gave very poor diastereoselection in favour of the desired anti-product 101 (dr = 55:45). This was not the case when (S)-configured enantiomer 99 was subjected to the same conditions, with excellent diastereoselectivity (dr = 92:8) observed. As a result, the undesired C13 configured product 102 was carried through the synthesis with the need to invert this stereocentre at a later stage (Scheme 18) [9, 10].
Further routine manipulations followed by the addition of Chan’s diene 104 under Felkin–Anh control resulted in 105 in excellent diastereoselectivity (dr > 95:5). Silylation, methanolysis and methylation then gave callipeltoside pyran 106. The C13 stereocentre was then inverted using a four-step procedure involving selective TBS deprotection, mesylation (to give 107), hydrolysis of the methyl ester and finally macrocycle formation. PMB-protected scaffold 91 was completed by treatment with TBAF (Scheme 19) [9, 10].

Synthesis of C1–C13 macrocycle 91. Reagents and conditions: (a) 104, BF3•OEt2, PhMe, −90°C, 88%, dr > 95:5; (b) (i) TBSOTf, 2,6-lutidine, CH2Cl2, −78°C; (ii) PPTS, MeOH, RT; (iii) MeOTf, 2,6-di-t-butylpyridine, CH2Cl2, RT, 50% (over 3 steps); (c) (i) TBAF, THF, RT; (ii) MsCl, Et3N, DMAP, CH2Cl2, 0°C; (iii) LiOH, H2O, MeOH–THF (1:1), RT, 67% (over 3 steps); (d) (i) Cs2CO3, 18-crown-6, PhMe, 110°C; (ii) TBAF, THF, RT, 66% (over 2 steps)
2.4.3 Synthesis of trans-Chlorocyclopropane Sidechain 116
Similar to the Trost synthesis [7, 8], Evans designed the route to the trans-chlorocyclopropane unit to be flexible enough such that both enantiomers could be accessed (Scheme 20, desired enantiomer shown) [9, 10, 43]. Oxidative cleavage of 108 using KIO4 provided aldehyde 109, from which compound 112 could be produced using either a one- or two-step procedure. Although the Takai protocol resulted in vinyl chloride 112 in good stereoselectivity (E:Z = 6.7:1), this process was not ideal since it was difficult to separate the two isomers and only resulted in poor isolated yield (< 40%). This was remedied by use of a two-step sequential homologation (to alkyne 111) and reduction sequence using the Ohira–Bestmann reagent (110), followed by Masuda’s one-pot hydroboration/chlorination methodology [44]. This not only resulted in better double bond selectivity (E:Z > 20:1) but also improved overall yield (70%, 111 to 112) [9, 10, 43].

Formation of trans-chlorocyclopropane fragment 116 (desired enantiomer). Reagents and conditions: (a) KIO4, KHCO3, H2O–THF (3:1), RT; (b) 110, K2CO3, MeOH, RT, 94% (over 2 steps); (c) CrCl2, CHCl3, THF, 70°C, < 40% (over 2 steps), E:Z = 6.7:1; (d) (i) Thexyl2BH, −15°C to 0°C, THF, (ii) CuCl2, H2O, HMPA, THF, 0°C to 70°C, 70% (over 2 steps), E:Z > 20:1; (e) ZnEt2, CF3COOH, CH2I2, CH2Cl2, 0°C to RT, 82%, dr > 98:2; (f) Dowex resin, MeOH, RT, 84%; (g) Pb(OAc)4, K2CO3, CH2Cl2, 0°C; (h) PPh3, CBr4, CH2Cl2, 0°C to RT, 94% (over 2 steps); (i) 94, Pd(PPh3)4, TlOEt, THF–H2O (3:1), RT, 85%; (j) DBU, PhMe, 110°C, 91%
The authors comment that cyclopropanation of vinyl chloride 112 was troublesome at first, with the traditional Simmons–Smith reaction as well as the modified variants developed by Furukawa (Et2Zn, CH2I2) [45] and Denmark (Et2Zn, CH2I2, ZnI2) [46] all resulting in little or no product. However, the conditions developed by Shi (Et2Zn, CH2I2, TFA) [47] gave 113 in good yield (82%) and excellent diastereoselectivity (dr > 98:2) [9, 10, 43]. Deprotection, oxidative cleavage of the resulting diol and dibromoolefination gave 37 (also used by Paterson [17] and Trost [7, 8]). This underwent Suzuki reaction with boronic acid 94 under Roush-modified conditions [48], with final elimination of HBr providing advanced fragment 116 in 33% overall yield (Scheme 20) [9, 10, 43].
2.4.4 l-Callipeltose A Sugar 90
Synthesis of the l-callipeltose A sugar began from d-threonine (93), which was converted into chiral substrate 118 in a three-step procedure [49]. The aldol reaction between 118 and the lithium enolate of 119 was performed in order to set the C2′ and C3′ stereocentres. However, whilst the desired C3′-stereocentre could be accessed using this method (consistent with Felkin–Anh selectivity), the undesired syn aldol adduct predominated (dr = 66:34), meaning poor selectivity for C2′ stereocentre formation (Scheme 21) [9, 10, 49]. This unexpected stereochemical outcome was hypothesised to be the result of chelation between the lithium and α-alkoxy substituent, thereby reinforcing the preference for the formation of an (E)-configured enolate. The relative configuration of the C2′ and C3′ stereocentres was determined by nOe studies following conversion of aldol adducts 120 and 122 to the corresponding lactones. In order to ensure formation of an enolate that could only exist in the (Z)-configuration (and therefore set the desired C2′ stereochemistry), the lithium enolate derived from 124 was studied. This resulted in formation of the correct Felkin–Anh anti-aldol adduct 125 in excellent yield (80%) and much-improved diastereoselectivity (dr = 94:6). With the desired C2′ stereochemistry set, conversion to lactone 123 followed by routine synthetic manipulations afforded thioglycoside 90 as a 50:50 mixture of α/β anomers (Scheme 21) [9, 10, 49].

Synthesis of l-callipeltose A (90). Reagents and conditions: (a) (i) 1 m NaOH, CbzCl, MeCN, 0°C to RT; (ii) MeI, K2CO3, DMF, 0°C to RT; (iii) TsOH•H2O, 2,2-dimethoxypropane, PhH, RT, 93% (over 3 steps); (b) (i) i-PrMgCl, HN(OMe)Me•HCl, THF, −78°C to −40°C; (ii) MeMgBr, THF, −40°C to RT, 61% (over 2 steps); (c) LDA, then 118, −78°C to −30°C, 56%, dr = 66:34; (d) HF, THF (no yield given); (e) LDA, then 118, −78°C to −30°C, 80%, dr = 94:6; (f) (i) AcOH–H2O (1.5:1), 70°C, 71%; (ii) Me3OBF4, DTBMP, CH2Cl2, RT, 82%; (g) DIBAL-H, CH2Cl2, −78°C, then Ac2O, pyridine, DMAP, −78°C, 93%; (h) (i) BF3•OEt2, PhSH, CH2Cl2, 0°C to RT, 81%, α:β = 50:50; (ii) NaH, THF, 0°C to RT, 97%; (i) TBSOTf, 2,6-lutidine, CH2Cl2, 0°C to RT, 92%
2.4.5 Completion of Callipeltoside A
In contrast to the callipeltoside A synthesis completed by Trost [7, 8], Evans appended the callipeltose A sugar prior to the trans-chlorocyclopropane [9, 10]. Glycosidation between thioglycoside 90 [as an anomeric mixture (α/β = 50:50)] and the C1–C13 core delivered the coupled material as a single product. Subsequent PMB deprotection and oxidation then gave aldehyde 129. Horner–Wadsworth–Emmons reaction between 129 and 130 resulted in moderate E:Z selectivity at the C14/C15 E-olefin (3:1). Treatment of this mixture with a catalytic amount of iodine was thereafter found to result in isomerisation of the undesired Z-isomer, improving the ratio to 11:1. Final TBS deprotection then provided callipeltoside A (1) in 25 steps and 4.0% overall yield. The optical rotation of this synthetic material [[α]D = −17 (c = 0.19, MeOH)] was in full agreement with both the callipeltoside A natural isolate (Minale [3]) and that prepared by Trost (Scheme 22) [7, 8]. For completion, oppositely configured trans-chlorocyclopropane diastereomer 7 was also produced in an analogous fashion (not shown, also synthesised by Trost, Sect. 2.3.2), with the measured optical rotation having both different sign and magnitude [[α]D = +140 (c = 0.05, MeOH)] [9, 10].

Completion of callipeltoside A. Reagents and conditions: (a) (i) 90 (α/β = 50:50), NIS, TfOH, DTBMP, 4 Å MS, CH2Cl2, −15°C to RT; (ii) DDQ, MeOH, CH2Cl2–H2O (1.3:1), RT, 83%; (b) SO3•pyridine, Et3N, DMSO, CH2Cl2, 0°C; (c) (i) LiHMDS, 130, THF, −78°C to RT; (ii) I2, CH2Cl2, RT, E:Z = 11:1; (iii) TBAF, AcOH, THF, RT, 56% (over 4 steps)
2.5 Paterson Synthesis of Callipeltoside A (2003)
Paterson’s synthesis of the enantiomeric callipeltoside aglycon 41 [17] (Sect. 2.1) was adapted for the synthesis of callipeltoside A. Key to this was the development of an asymmetric vinylogous Mukaiyama aldol reaction to set the C13 stereocentre. A chiral Lewis acid system requiring (R)-BINOL-Ti(Oi-Pr)2 [derived from (R)-BINOL and Ti(Oi-Pr)4] in the presence of CaH2 provided trisubstituted alkene 133 in 94% yield and high enantioselectivity (er = 97:3). Further manipulation resulted in aldehyde (R)-20, allowing for the interception of the analogous enantiomeric route (Sect. 2.1) (Scheme 23) [11].

Paterson’s asymmetric synthesis of (R)-20. Reagents and conditions: (a) CaH2, (R)-BINOL, 132, Ti(Oi-Pr)4, THF, RT, then −78°C, aldehyde 17, followed by silyl ketene acetal 131, 96%, er = 97:3
The callipeltoside aglycon was completed in the same manner as the enantiomeric version previously shown in Sect. 2.1.4. The callipeltose A sugar was synthesised following the procedure reported by Guiliano [25] and the corresponding trichloroacetimidate coupling partner 43 attached using Schmidt glycosidation conditions (analogous to Trost; see Sect. 2.2.4) [7, 8]. TBS deprotection with TBAF then gave callipeltoside A in 23 steps (longest linear) and 4.8% overall yield (Scheme 24) [11].

Completion of callipeltoside A. Reagents and conditions: (a) (i) 12, Pd(PPh3)2Cl2, CuI, i-Pr2NH, EtOAc, −20°C to 0°C, 83%; (ii) TFA, aq. THF, RT, 98%; (b) (i) 43, TMSOTf, CH2Cl2, 4 Å MS, −30°C; (ii) TBAF, AcOH, THF, RT, 76% (over 2 steps)
2.6 Panek Synthesis of Callipeltoside A (2004)
2.6.1 Retrosynthesis
The fourth synthesis of callipeltoside A was completed by Panek in 2004 [12]. Since the absolute and relative stereochemistry of callipeltoside A had been confirmed at this point, late-stage installation of the trans-chlorocyclopropane was not required. Instead, the Panek group sought to unite fully assembled C11–C22 fragment 136 with pyran core 135 by means of a Julia–Kocienski olefination, forming the trisubstituted C10/C11 olefin in the process. In contrast to previous syntheses, pyran 135 and callipeltose A sugar 137 were to be derived from the relevant dihydropyrans arising from [4 + 2] annulations using chiral organosilanes (Scheme 25) [12, 50].

Panek retrosynthesis of callipeltoside A
2.6.2 Formation of C1–C10 Pyran 135
The synthesis began with the installation of the C8 and C9 stereocentres via the anti-selective condensation of α-benzyloxyacetaldehyde 141 with chiral silane (S)-142 in the presence of SnCl4. This provided tetrahydrofuran 143 in high yield (87%) and diastereoselectivity (dr > 97:3). Treatment with SbCl5 resulted in elimination and the formation of the anti-homoallylic alcohol, which could be converted to aldehyde 144 by methylation and ozonolysis. At this point aldehyde 144 could be reacted with chiral organosilane 145 in the presence of TfOH to afford tetrahydropyran 138 in 85% yield and as a single diastereomer (dr > 97:3). This was then converted to advanced pyranone 146 in a further three steps (Scheme 26) [12].

Synthesis of the C1–C10 scaffold. Reagents and conditions: (a) 141, SnCl4, CH2Cl2, then 142, CH2Cl2, −78°C, 87%, dr > 97:3; (b) (i) SbCl5, CH2Cl2, −78°C to −50°C; (ii) Me3OBF4, proton sponge, 4 Å MS, CH2Cl2, RT; (iii) O3, MeOH–CH2Cl2 (3:1), Me2S, −78°C, 81% (over 3 steps); (c) 145, TfOH, CH2Cl2, −70°C, 85%, dr > 97:3
Luche reduction provided the C5 stereocentre, and the resulting hydroxy was protected as its TBS-ether (147). The C1–C10 scaffold was then further manipulated by ester saponification and Arndt–Eistert homologation to give 148 in four steps and 56% overall yield. Installation of the anomerically favoured ketal (MeOH/CSA), Bn deprotection and oxidation then afforded aldehyde 135 (Scheme 27) [12].

Completion of pyran 135. Reagents and conditions: (a) (i) CeCl3•7H2O, MeOH, −78°C, then NaBH4, MeOH, −78°C, 93%; (ii) TBSOTf, 2,6-lutidine, CH2Cl2, −78°C, 97%; (b) (i) LiOH, THF–MeOH–H2O (3:1:1), RT, then 5% HCl; (ii) (COCl)2, DMF, CH2Cl2; (iii) CH2N2, Et2O, 0°C, 70% (over 3 steps); (iv) PhCO2Ag, pyridine, MeOH, RT, 80%; (c) (i) CSA, MeOH, RT, 97%; (ii) H2, Pd/C, EtOAc; (iii) PDC, 4 Å MS, CH2Cl2, RT, 88% (2 steps)
2.6.3 Synthesis of the C11–C22 Fragment
Panek chose to assemble the entire C11–C22 fragment prior to coupling with aldehyde 135.
The synthesis again required dibromoolefin 37 as a key building block, which was synthesised as described by Paterson [11, 17] (Sect. 2.1.3). This underwent Stille reaction using the conditions developed by Shen [41] (and also used by Olivo [20] and Trost [8]) to give enyne 116, from which phosphonate 84 was prepared in a further two-step procedure (Scheme 28). Horner–Wadsworth–Emmons reaction between phosphonate 84 and aldehyde 139 under identical conditions to Trost [8] interestingly resulted in the formation of a single E,E-isomer, highlighting the sensitivity of this reaction to substrate choice (Trost observed a 4:1 E/Z mixture when the coupling was performed with the callipeltoside C1–C13 macrocycle). With the entire C11–C22 carbon scaffold in place, the TBDPS group was removed and stereocentre inverted by Mitsunobu reaction with 1-phenyl-1H-tetrazole-5-thiol. Oxidation to the corresponding sulfone followed by protecting group manipulation gave Julia–Kocienski coupling partner 136 (Scheme 28) [12].

Completion of the C11–C22 fragment. Reagents and conditions: (a) 83, Pd2dba3, (4-MeOC6H4)3P, DIPEA, DMF, 80°C, 68%; (b) LiHMDS, then 139, THF, −78°C, 89%; (c) (i) TBAF, THF, RT, 91%; (ii) 1-phenyl-1H-tetrazole-5-thiol, DIAD, PPh3, THF, 0°C to RT; (iii) (NH4)6Mo7O24, EtOH–acetone (2:1), RT, 66% (2 steps); (d) (i) TsOH, MeOH, RT; (ii) EVE, PPTS, RT, 90% (2 steps)
2.6.4 Synthesis of Callipeltose A
Organosilane methodology was also used to access the callipeltose A sugar [12, 51]. Stereoselective [4 + 2] annulation between acetaldehyde and chiral organosilane 151 (made in 5 steps, 55%) gave dihydropyran 140 in high yield and diastereoselectivity (dr = 10:1). This was converted to 152 in a further two steps, with selective reduction and methanolysis providing 153 as a single anomer. Advanced intermediate 155 was then constructed in short order by Sharpless dihydroxylation, selective methylation and carbamate formation. 1,5-C–H insertion using Rh2(OAc)4 then forged the requisite 5-membered ring present in callipeltose A in 93% yield (Scheme 29) [12, 51].

Synthesis of the callipeltose A scaffold (137). Reagents and conditions: (a) MeCHO, TMSOTf, CH2Cl2, −78°C, dr = 91:9, 80%; (b) DIBAL-H, CH2Cl2, −78°C, then CSA, MeOH; (c) AD-mix-α, NaHCO3, MeSO2NH2, OsO4, t-BuOH–H2O (1:1), RT, 65% (over 2 steps); (d) (i) t-BuOK, MeI, THF, 0°C; (ii) Cl3CCONCO, CH2Cl2, RT, then K2CO3, MeOH–H2O (10:1), 79% (over 2 steps); (e) Rh2(OAc)4, PhI(OAc)2, MgO, PhH, 80°C, 93%.
2.6.5 Completion of Callipeltoside A
Aldehyde 135 and sulfone 136 were coupled using a Julia–Kocienski olefination to afford 156 in low yield (20%) after acidic cleavage of the ethoxyethyl ether (EE) protecting group. The yield could not be improved despite further investigation involving different bases (KHMDS, NaHMDS) and more polar solvents (DME, DMF) [12].
Hydrolysis of the ester group in 156 was followed by Yamaguchi macrolactonisation, which gave 157 as well as eliminated product 158 in a 50:50 ratio These products were separated and 157 processed by deprotection and hemi-ketal installation to give the callipeltoside aglycon (9).Footnote 2 This was then coupled to trichloroacetimidate 43 using the literature known Schmidt glycosidation conditions to give callipeltoside A (1) in 1.4% overall yield and 25 steps (longest linear) (Scheme 30) [12].

Completion of callipeltoside A. Reagents and conditions: (a) (i) 136, LiHMDS, then 135, THF, −78°C to RT; (ii) PPTS, MeOH, 20% (over 2 steps); (b) (i) LiOH, MeOH–H2O–THF, 90%; (ii) 2,4,6-Cl3C6H2COCl, Et3N, DMAP, PhMe, 80°C, 90% (157 and 158, 1:1); [158 to 9 conversion: (i) PPh3•HBr, H2O–CH2Cl2 (11:1), RT, 97%; (ii) TBAF, THF, RT, 85%]; (c) (i) TBAF, THF, RT, 85%; (ii) PPTS, MeCN–H2O (3:1), RT, 89%; (d) (i) 43, TMSOTf, 4 Å MS, CH2Cl2, −30°C; (ii) TBAF, AcOH–THF (1:1), RT, 73% (over 2 steps)
2.7 Hoye Synthesis of Callipeltoside A (2010)
2.7.1 Retrosynthesis
The synthesis of callipeltoside A was disclosed by Hoye in 2010 [13]. Similar disconnections involving late-stage attachment of the callipeltose A sugar via a Schmidt glycosidation and Horner–Wadsworth–Emmons reaction with phosphonate 84 were key to the assembly of callipeltoside A. However, a dual macrocyclisation/hemi-ketal formation process from acylketene precursor 161 was anticipated to form the C1–C13 macrocycle in one step. This precursor was to be accessed by alkenyllithium addition to aldehyde 163 (Scheme 31) [13, 52, 53].

Hoye’s approach to callipeltoside A
2.7.2 Synthesis of the C1–C13 Fragment
Lithium-halogen exchange with vinyl iodide 162 followed by addition to aldehyde 163 gave a 50:50 mixture of separable C9 epimeric alcohols.Footnote 3 Methylation of the desired C9 alcohol afforded 164, which was converted to lactonisation precursor 161 in an additional five linear steps (Scheme 32) [13].

Synthesis of cyclisation precursor 161. Reagents and conditions: (a) (i) n-BuLi, 162, Et2O, −78°C, then 163, Et2O, dr = 50:50 (separation by SiO2 chromatography), 34% of each; (ii) MeI, NaH, DMF, RT, 80%; (b) (i) 9-BBN, THF, −78°C to RT, then NaOH (10%)–H2O2 (30%) (6:1), 70%, dr = 87.5:12.5 (at C6, separation by SiO2 chromatography); (ii) Dess–Martin periodinane, CH2Cl2, RT, 76%; (iii) 71, BF3•OEt2, CH2Cl2, −78°C, 90%; (iv) TMSCl, Et3N, CH2Cl2, RT; (v) DDQ, CH2Cl2–H2O (12:1), 78% (over 2 steps)
Substrate 161 underwent the desired dual macrolactonisation/hemi-ketal formation process to afford the C1–C13 macrocyclic core 160 in 76% yield. This reaction was proposed to proceed via intermediates 165, 166 and 167 (Scheme 33) [13, 53].

2.7.3 Completion of Callipeltoside A
Compound 168 was synthesised following a three-step protecting group manipulation. Subsequent oxidation to the aldehyde, coupling to phosphonate 84 and deprotection gave the familiar callipeltoside aglycon (9). In accordance with previous syntheses, the trichloroacetimidate of callipeltose A sugar 159 (although with an exposed NH this time) was attached to 9 using Schmidt conditions, delivering the natural product in 21 steps and 0.7% yield (Scheme 34) [13].

Completion of callipeltoside A. Reagents and conditions: (a) (i) SO3•pyridine, DMSO, Et3N, CH2Cl2, 0°C; (ii) 84, LiHMDS, THF, −78°C; (iii) TBAF, THF, 54% (over 3 steps); (b) 159, TMSOTf, 4 Å MS, CH2Cl2, 0°C, 70%
2.8 MacMillan Synthesis of Callipeltoside C (2008)
2.8.1 Retrosynthesis
The first synthesis of callipeltoside C was reported in 2008 by MacMillan [16]. At this stage the relative configuration of callipeltose C with respect to the callipeltoside aglycon had not been validated, and therefore the absolute configuration of the molecule was not known [4]. In contrast to previous approaches towards these molecules, organocatalytic processes featured heavily in MacMillan’s synthesis. Pyran 170 and callipeltose C sugar 169 were to be accessed using organocatalytic asymmetric aldol reactions, whilst the C13 stereocentre of 171 was to be set by an organocatalytic oxyamination reaction (Scheme 35) [16, 54,55,56,57,58].

Retrosynthesis of callipeltoside C
2.8.2 Synthesis of Pyran 170
The synthesis began with an l-proline-catalysed organocatalytic aldol reaction between propionaldehyde (176) and Roche ester-derived aldehyde 177. This gave aldehyde 174 in moderate yield (48%), but set the desired C6 and C7 stereocentres in both excellent diastereo- (dr = 92:8) and enantioselectivity (er > 99:1) [16]. The C5 stereocentre was then formed by propargylzinc addition to give the desired 1,3-anti-diol relationship (180, dr = 86:14). With the 4 contiguous C5–C8 stereocentres in place, a palladium-catalysed alkoxycarbonylation process developed by Marshall [59, 60] impressively gave advanced C1–C9 pyran 181 as a single diastereomer in one step (Scheme 36). The proposed mechanism for this transformation is described in Scheme 37 [59, 60]. Subsequent TBS protection of the C5 hydroxy, PMB removal and Parikh–Doering oxidation then gave 170 in six steps and 44% overall yield from aldehyde 177 (Scheme 36).

Synthesis of pyran 170. Reagents and conditions: (a) l-proline (10 mol%), DMSO, H2O, 4°C, 48% (75% brsm), dr = 92:8 (syn:anti), er > 99:1; (b) HCCCH2Br, Zn, THF, −100°C, 98%, dr = 86:14; (c) PdCl2(MeCN)2 (5 mol%), p-benzoquinone, CO, MeOH, 0°C, 75%, dr > 95:5; (d) (i) TBSCl, imidazole, DMF, RT; (ii) DDQ, pH 7 phosphate buffer, CH2Cl2, 0°C; (iii) SO3•pyridine, Et3N, CH2Cl2, DMSO, 0°C, 80% (over 3 steps)

2.8.3 Formation of Vinyl Iodide 171
Carboalumination of 4-pentyn-1-ol delivered the desired trisubstituted double bond as a single regioisomer which, following treatment with iodine, gave 188. Subsequent Swern oxidation and organocatalytic α-oxyamination then provided 189 in excellent enantioselectivity (er > 99:1) [57, 58]. Reduction of the aldehyde, N–O bond cleavage and bis-protection gave 171 in seven steps and 57% yield (Scheme 38) [16].

Organocatalytic oxyamination approach to vinyl iodide 171. Reagents and conditions: (a) (i) Me3Al, Cp2ZrCl2, I2, THF, −30°C; (ii) (COCl)2, Et3N, DMSO, CH2Cl2, −50°C, 92% (over 2 steps); (b) l-proline, (20 mol%), PhNO, DMSO, RT, er > 99:1; (c) (i) NaBH4, EtOH, RT; (ii) Zn, EtOH–AcOH (3:1), 77% (over 3 steps); (d) (i) Bu2Sn(OMe)2, PMBCl, TBAI, PhMe, Δ; (ii) TBSCl, imidazole, DMF, RT, 80% (over 2 steps), er > 99:1
2.8.4 Fragment Union and Completion of the Aglycon (9)
Vinylic iodide 171 (2.2 equiv.) was converted to the corresponding Grignard and added to aldehyde 170 to provide the C9 stereocentre in both high diastereoselectivity (dr = 94:6) and yield (98%). The addition of MgBr2 to substrate 170 prior to Grignard addition was found to be important for both yield and diastereoselectivity, with formation of a complex between MgBr2 and the pyran and aldehyde oxygens speculated. It is also noted that an intermolecular Nozaki–Hiyama–Kishi reaction was also attempted and in contrast gave the undesired C9 alcohol configuration [16].
With 191 in hand, methylation, PMB deprotection, oxidation and Horner–Wadsworth–Emmons olefination with trans-chlorocyclopropane-containing phosphonate 130 gave 193 [16]. Interestingly, attachment of 130 at this stage of the synthesis resulted in high E:Z selectivity (10:1–19:1) compared to Trost [7, 8], Evans [9, 10] and Paterson [11] and once again highlights the subtleties associated with substrate choice when performing reactions under similar conditions (see Sects. 2.2.3, 2.3.2 and 2.4.5; E:Z = 4:1).
Deprotection of C13-OTBS and saponification of the methyl ester gave seco-acid 193 which, when subjected to Yamaguchi macrocyclisation conditions, gave eliminated lactone 158 as well as the desired product (157, not shown; see Sect. 2.6.5). Treatment of the mixture with aqueous PPh3•HBr then installed the hemi-ketal functionality, whilst final TBS deprotection using TFA delivered the callipeltoside aglycon (9) in 18 steps and 19% overall yield (Scheme 39) [16].

Synthesis of callipeltoside aglycon (9). Reagents and conditions: (a) 171, t-BuLi, MgBr2•Et2O, Et2O, then 170, CH2Cl2, −78°C, 98%, dr = 94:6; (b) (i) MeOTf, 2,6-di-t-butylpyridine, CH2Cl2, RT; (ii) DDQ, pH 7 buffer, CH2Cl2, 0°C; (iii) SO3•pyridine, Et3N, DMSO, CH2Cl2, −10°C, 84% (over 3 steps); (c) (i) 130, LiHMDS, THF, −78°C, then 192, THF, −78°C, 84%, E:Z = 19:1; (ii) TBAF, THF, 0°C, 100%; (iii) Ba(OH)2•8H2O, MeOH, RT, 84% (over 3 steps); (d) 2,4,6-Cl3C6H2COCl, DIPEA, THF, RT, then addition to DMAP, PhMe, 60°C, 83%; (e) (i) PPh3•HBr, H2O, CH2Cl2, RT; (ii) TFA, THF–H2O (5:1), RT, 81% (over 2 steps)
2.8.5 Synthesis of d-Callipeltose C
The synthesis commenced with dimerisation of aldehyde 194 under organocatalytic conditions to provide 173 in good yield and excellent enantioselectivity (er > 99:1) (Scheme 40) [16, 54]. This was then subjected to the organocatalytic Mukaiyama aldol conditions developed in the MacMillan laboratory, with the Lewis acid (MgBr2) and solvent (CH2Cl2) choice critical for the formation of the desired sugar scaffold (195) [16, 55, 56]. The anomeric hydroxy was then protected as its benzyl ether with concomitant removal of the primary TIPS protecting group. Subsequent conversion of the primary alcohol to the corresponding phenyl thiocarbonate and deoxygenation under Barton–McCombie conditions provided the C6′ methyl group. Oxidation of the remaining C3′ secondary hydroxy to the ketone then gave 196. Further manipulation by means of diastereoselective methyl Grignard addition (dr = 95:5), removal of the benzyl group and formation of the trichloroacetimidate provided the d-callipeltose C sugar coupling partner 169. Overall, this fragment was synthesised in eight steps and 14.5% yield (Scheme 40) [16].

Organocatalytic approach to d-configured callipeltose C. Reagents and conditions: (a) d-proline, DMF, 75%, er > 99:1; (b) 172, MgBr2•Et2O, CH2Cl2, 47%, dr > 95:5; (c) (i) AcCl, BnOH, 110°C; (ii) PhOCSCl, pyridine, CH2Cl2; (iii) n-Bu3SnH, AIBN, PhH, 120°C; (iv) Dess–Martin periodinane, CH2Cl2, 0°C, 47% (over 4 steps); (d) (i) MgBr2•Et2O, MeMgBr, CH2Cl2, dr > 95:5; (ii) H2, Pd/C, EtOAc; (iii) Cl3CCN, Cs2CO3, CH2Cl2, 86% (over 3 steps), dr = 95:5
2.8.6 Completion of Callipeltoside C
The callipeltoside aglycon was coupled with d-callipeltose C (169) using the conditions developed by Tietze [16, 61] which, following TIPS deprotection, gave spectroscopic data inconsistent with the natural isolate [4]. Since previous syntheses of callipeltoside A had determined the absolute stereochemistry of the molecule, it was reasonably assumed that the problem lay with the callipeltose C sugar rather than the aglycon. As a result, the enantiomeric l-configured sugar (ent-169) was synthesised under the same conditions outlined in Scheme 40. Schmidt glycosidation and final TIPS deprotection gave compound 3, which this time had identical spectroscopic data to naturally occurring callipeltoside C [4, 16]. This result not only confirmed the absolute stereochemistry of callipeltoside C but also suggested that both callipeltosides A and C contain l-configured sugars (Scheme 41). This was in contrast to the original structures drawn in the isolation papers [3, 4].Footnote 4 However, it is noteworthy that the glycosidic linkage attaching callipeltose C to the aglycon was only tentatively assigned, with the MacMillan group assuming it to be the β-equatorial anomer by comparison with the originally reported structure of callipeltoside B (Figs. 3, 5) [16].

Completion of callipeltoside C. Reagents and conditions: (a) (i) 169, TMSOTf, 4 Å MS, CH2Cl2, −30°C; (ii) TASF, DMF, 40°C, 58% (over 2 steps); (b) (i) ent-169, TMSOTf, 4 Å MS, CH2Cl2, −30°C; (ii) TASF, DMF, 40°C, 63% (over 2 steps)
2.9 Ley Syntheses of Callipeltosides A, B and C (2012)
2.9.1 Retrosynthesis
The Ley group’s approach to the entire callipeltoside family involved a highly convergent strategy whereby the molecule(s) was split into three equally sized fragments: the C1–C9 pyran (170), C10–C22 vinylic iodide (197) and the relevant sugar moiety [14, 15]. The common callipeltoside aglycon would then be assembled by union of 170 and 197 via a diastereoselective Oppolzer–Radinov alkenylzinc addition [62], followed by Yamaguchi macrolactonisation. The C1–C9 pyran was to be formed using an AuCl3-catalysed cyclisation, which the group had reported previously [63]. Since other approaches to the callipeltosides had often struggled to produce the dien-yne as a single isomer, the group also chose to address this synthetic issue. With this in mind, the C15–C16 and C17–C18 bonds were disconnected, revealing bis-stannane linchpin 201, from which bidirectional Stille reactions could be performed. Pyranone 202 was considered to be a key building block from which each callipeltose sugar could be accessed (Scheme 42) [14, 15].

Ley group approach to the callipeltosides (callipeltoside C shown for clarity)
2.9.2 Synthesis of Pyran 170
Synthesis of pyran fragment 170 began with crotylation of known Roche ester-derived aldehyde 203 to give homoallylic alcohol 205 in good yield (70%) and diastereoselectivity (dr = 88:12). Conversion to the triol using catalytic osmium tetroxide followed by sodium periodate-mediated cleavage gave the requisite aldehyde, with which a chelation-controlled addition of propargyl zinc furnished diol 206 in 72% over three steps (dr = 85:15 at C5). Two further synthetic steps gave ynoate 208, enabling all minor diastereomers to be separated (208, dr > 95:5) and the 1,3-anti-relationship to be proven by X-ray crystallography (not shown). Subsequent deprotection afforded the diol, which smoothly underwent AuCl3-catalysed cyclisation in MeOH to give pyran 209 as a single, anomerically favoured diastereomer in 96% yield. Further routine synthetic manipulations gave pyran fragment 170 in 26% yield from 203 (11 linear steps) with minimal use of chromatography (Scheme 43) [14, 15].

Synthesis of pyran 170. Reagents and conditions: (a) crotylborane 204, 4 Å MS, PhMe, −78°C, 70%, dr = 88:12; (b) (i) OsO4, NMO, acetone–H2O (2:1), RT; (ii) NaIO4, THF–H2O (10:1), 0°C to RT; (iii) Zn, propargyl bromide, THF, 0°C to −100°C, 72% (over 3 steps), dr = 85:15 (at C5); (c) 2,2-dimethoxypropane, (±)-CSA, acetone, RT; (d) n-BuLi, THF, −40°C to −78°C, then ClCO2Me, 73% (over 2 steps), dr > 95:5; (e) (i) QP-SA, MeOH, RT, 95%; (ii) AuCl3 (2 mol%), MeOH, RT, 96%
2.9.3 Completion of the C10–C22 Fragment
In order to construct fragment 197 in a stereospecific fashion via sequential Stille cross-coupling reactions, the vinyl iodide functionality was masked in order to provide orthogonal reactivity. Work began from known vinyl iodide 210 [64, 65], which contained a vinyl silane moiety that could be converted to the corresponding vinyl iodide at a late stage. Lithiation of 210 followed by reaction with 211 gave the secondary alcohol with installation of the C13 stereocentre. A two-step protecting group manipulation provided 212 which, following oxidation, dibromoolefination and Corey–Fuchs reaction, gave alkyne 213. Subsequent Pd-catalysed hydrostannylation and tin–iodine exchange led to the desired (E)-vinyl iodide 200 as a single stereoisomer [14, 15].
Stille coupling partner 215 was completed by treatment of known dibromide 37 with TBAF to give the bromoalkyne, which was then coupled to bis-stannane 201 via a low temperature (−10°C) Stille reaction with stoichiometric Ag2CO3 as an additive. Fragments 200 and 215 were united by a second Stille reaction to yield advanced substrate 216 which, following treatment with NIS, provided 197 in a completely stereospecific manner in good yield (Scheme 44) [14, 15].

Completion of the C10–C22 Fragment 197. Reagents and conditions: (a) (i) t-BuLi, PhMe, −78°C, then 211, BF3•OEt2, PhMe; (ii) TBSOTf, 2,6-lutidine, CH2Cl2, −78°C, 53% (over 2 steps); (iii) DDQ, pH 7 phosphate buffer, CH2Cl2, 0°C, 89%; (b) (i) SO3•Py, Et3N, DMSO, CH2Cl2, 0°C to RT; (ii) PPh3, CBr4, CH2Cl2, then aldehyde, 2,6-lutidine, CH2Cl2, 0°C, 89% (over 2 steps); (iii) n-BuLi, THF, −78°C, then H2O, 85%; (c) (i) Pd(PPh3)2Cl2 (3 mol%), Bu3SnH, THF, 0°C; (ii) I2, CH2Cl2, −78°C, 49% (over 2 steps); (d) ZnEt2, CH2I2, CH2Cl2, 0°C, then 214, 33, CH2Cl2, 0°C to RT, 74%; (e) (i) PCC, Celite®, CH2Cl2, RT; (ii) PPh3, CBr4, CH2Cl2, 0°C, then aldehyde, CH2Cl2, 0°C to RT, 70% (over 2 steps); (f) (i) TBAF, DMF, 65°C; (ii) Pd2dba3 (10 mol%), AsPh3 (40 mol%), Ag2CO3 (1.0 equiv.), THF, dark, −10°C, then 201, 45% (over 2 steps); (g) Pd(PFur3)2Cl2 (15 mol%), DMF, dark, RT, 63%; (h) NIS, MeCN, RT, 84%
2.9.4 Union of Fragments 170 and 197 and Macrocycle Formation
In order to couple fragments 170 and 197, the Ley group turned to the work of Oppolzer and Radinov [62] as well as the studies of Marshall [66, 67], who had previously shown that the stereochemical information of an appropriate enantioenriched lithio N-methylephedrine alkoxide could be transferred to reactions of this type. The stereochemical model proposed in the literature revealed (1R,2S)-(–)-N-methylephedrine to be the reagent of choice; but in practice poor C9 diastereoselectivity was observed (34:66, not shown). The reaction was also performed with the enantiomeric ligand, (1S,2R)-(+)-N-methylephedrine, this time resulting in a much-improved C9 diastereomeric ratio of 91:9. Methylation of both diastereomeric mixtures allowed the group to intercept a known compound disclosed by MacMillan [16], meaning that the stereochemical outcome of these reactions could be straightforwardly determined. To their surprise, the diastereomerically enriched material using (1S,2R)-(+)-N-methylephedrine was found to be the desired compound, in contrast to the models disclosed by Oppolzer [62] and Noyori [68, 69]. It was proposed that the unexpected reversal in selectivity was due to bis-chelation between the active metal complex and the pyran oxygen, resulting in the exposure of the opposite diastereotopic face to reaction [70]. The authors note that a similar result was documented by Myers in his synthesis of the tetracycline antibiotics [70].
With the C9 stereocentre in place, the methylated derivative was deprotected and saponified to give seco-acid 193. Yamaguchi macrolactonisation, and a one-pot deprotection/hemi-ketal formation step afforded the callipeltoside aglycon 9 (Scheme 45).

Diastereoselective union of 170 and 197 and completion of the callipeltoside aglycon. Reagents and conditions: (a) 1. 197, (1.3 equiv.), t-BuLi, Et2O, −78°C; 2. ZnBr2, Et2O, −78°C to 0°C; 3. (1S,2R)-(+)-N-methylephedrine (1.1 equiv.), n-BuLi, PhMe, 0°C; 4. 170, PhMe, 0°C, 48%; (b) (i) MeOTf, DTBP, CH2Cl2, RT, 73%; (ii) TBAF, THF, RT, 74%; (iii) Ba(OH)2•8H2O, MeOH, RT, quant.; (c) (i) 2,4,6-trichlorobenzoyl chloride, Et3N, RT, then addition to DMAP, PhMe, 80°C; (ii) TFA, THF–H2O (5:1), RT, 58% (over 2 steps)
2.9.5 Preparation of Callipeltose A, B and C Thioglycosides
The synthesis of the callipeltose sugars commenced from pyranone 202, which could be straightforwardly accessed in two steps. Nosylation of the secondary alcohol, displacement and diastereoselective methyl addition provided 218, containing three of the stereocentres present in callipeltose A and B. Hydroxy-directed epoxidation, ring opening and selective 2° alcohol methylation gave 219, which could be diversified to each callipeltose sugar [24]. Callipeltose A thioglycoside was completed in an additional four steps as a single anomer. Despite being able to synthesise the callipeltose B sugar, it is noted that the N-formyl group prevented thioglycoside formation and hence coupling to the callipeltoside aglycon. As a result, compound 219 was TMS-protected and the thioglycoside formed to give callipeltose B precursor 221, which would have to be further manipulated once attached to the aglycon (9) [14, 15].
The callipeltose C sugar was also synthesised from 202, with the C3′ stereocentre once again formed by diastereoselective methyl addition, albeit this time by means of a complex-induced proximity effect from the neighbouring C4′ hydroxy [71]. TES protection, followed by the epoxidation, ring opening, methylation and thioglycosidation sequence mentioned previously, gave the callipeltose C thioglycoside (224) as a single anomer (Scheme 46) [14, 15].

Synthesis of callipeltoses A, B and C from common pyranone 202. Reagents and conditions: (a) (i) NsCl, pyridine, CH2Cl2, RT, 95%; (ii) n-Bu4NN3, CH2Cl2, 0°C, 72%; (iii) MeLi, THF, −100°C, 79%; (b) (i) m-CPBA, MeOH, 0°C to RT, 52%; (ii) KOt-Bu, MeI, THF, 0°C, 79%; (c) (i) H2, Pd(OH)2/C, EtOAc, RT, 93%; (ii) triphosgene, pyridine, THF, −78°C to RT, 72%; (iii) TIPSOTf, 2,6-lutidine, CH2Cl2, RT, 97%; (iv) PhSH, BF3•OEt2, CH2Cl2, 0°C to RT, 80%, single anomer; (d) ZnI2, TBAI, TMSSPh, DCE, 65°C, 60%, single anomer; (e) MeLi•LiBr, Et2O, −78°C, 78%; (f) (i) TESCl, pyridine, DMAP, CH2Cl2, RT; (ii) m-CPBA, MeOH, 0°C to RT, 45% (over 2 steps); (g) (i) KOt-Bu, MeI, THF, 0°C, 81%; (ii) ZnI2, TBAI, TMSSPh, DCE, 65°C, 86%, single anomer
2.9.6 Completion of Callipeltosides A, B and C
The callipeltoside aglycon was straightforwardly coupled with thioglycoside donors 220 and 224 using the conditions described by Evans [10]. Final deprotection of these two compounds then led to callipeltosides A and C, with spectroscopic data in full agreement with that previously reported [3, 4, 14, 15].
With the syntheses of both callipeltoside A and callipeltoside C completed, attention switched to callipeltoside B. Since there had been no previous synthesis of callipeltoside B, it was assumed that like callipeltosides A and C, it also contained an l-configured sugar. Attachment of azido sugar 221 (l-configured) proceeded without incident, and the azide moiety was selectively reduced using 1,3-propanedithiol [72]. Formylation of the amine using pentafluorophenyl formate (225) [73] and deprotection then delivered putative callipeltoside B (2) as a 4:1 rotameric mixture (by 1H NMR) (Scheme 47). The synthetic sample was in complete agreement with the spectroscopic data for the natural isolate disclosed by Minale [4, 14, 15].

Completion of callipeltosides A, B and C. Reagents and conditions: (a) (i) 220, 4 Å MS, DTBMP, CH2Cl2, RT, 50 min, then −15°C, NIS, TfOH, −15°C to RT; (ii) TBAF, THF, RT, 83% (over 2 steps); (b) (i) 224, 4 Å MS, DTBMP, CH2Cl2, RT, 50 min, then −15°C, NIS, TfOH, −15°C to RT; (ii) TASF, DMF, 40°C, 57% (over 2 steps); (c) (i) 221, 4 Å MS, DTBMP, CH2Cl2, RT, 50 min, then −15°C, NIS, TfOH, −15°C to RT, 56%; (ii) 1,3-propandithiol, Et3N, pyridine–H2O (10:1), RT; (iii) 225, CHCl3, RT; (iv) TASF, DMF, 40°C, 52% (over 3 steps), 4:1 rotameric mixture (by 1H NMR); (d) (i) ent-221, 4 Å MS, DTBMP, CH2Cl2, RT, 50 min, then −15°C, NIS, TfOH, −15°C to RT, 41%; (ii) 1,3-propandithiol, Et3N, pyridine–H2O (10:1), RT; (iii) 225, CHCl3, RT; (iv) TASF, DMF, 40°C, 57% (over 3 steps), 4:1 rotameric mixture (by 1H NMR)
In order to ensure that the correct sugar had indeed been attached, the enantiomeric thioglycoside (ent-221, d-configured) was synthesised in analogous fashion to that shown in Scheme 46 and attached to the callipeltoside aglycon (9). This was then also reduced, formylated and deprotected to afford diastereomer 226, which had a significantly different 1H NMR to the natural isolate [4]. This provided further evidence that all three callipeltose sugars were l-configured, with callipeltoside B completed for the first time [14, 15].
With the entire family of molecules completed, the configuration of the glycosidic linkages in callipeltosides B and C still needed to be determined.
2.9.7 Stereochemistry of the Glycosidic Linkages of Callipeltosides B and C
2.9.7.1 Callipeltoside C
Although the Ley group had completed the second total synthesis of callipeltoside C, the configuration of the glycosidic linkage was still under question, with the MacMillan group tentatively assigning the stereochemistry by comparison with the assumed glycosidic linkage found in callipeltoside B [16]. As a result, the Ley group attempted to determine the C1′ stereochemistry by analysis of the 1JC–H coupling constant [74,75,76,77,78] and NOESY data.
2.9.7.1.1 1JC–H Coupling Constant
Since measurement of the 1JC–H coupling constant [from a HSQC (heteronuclear single quantum coherence) experiment without 13C decoupling] had proven to be a useful technique for carbohydrate chemists to determine whether the proton at an anomeric centre is axial or equatorial, this was assessed initially. It is noted that a value of ~170 Hz suggests an equatorial proton at C1′H, whilst ~160 Hz is indicative of an axial proton [74,75,76,77,78]. Unfortunately, measurement of the 1JC–H coupling constant gave an inconclusive result, returning a value of 1JC-H = 166.5 Hz.
2.9.7.1.2 NOESY Data
As an alternative method to determine the stereochemistry of the glycosidic linkage, the Ley group conducted NOESY experiments and assessed all potential chair conformers (1)–(4) (Fig. 6; X-ray analysis of sugar intermediates suggested a chair configuration, not shown). Analysis of this averaged data indicated that structure (1) was the only chair conformer that accounted for all observed nOe interactions [14, 15]. With this in mind, the C1′ stereocentre was assigned to be (R)-configured, in agreement with the assignment previously suggested by MacMillan [16].

Key nOe’s for callipeltose C
2.9.7.2 Callipeltoside B
The C1′H stereochemistry of callipeltoside B was analysed in exactly the same way as for callipeltoside C. Once again, the 1JC–H coupling value gave no indication of stereochemistry, with a coupling constant of 1JC–H = 165.6 Hz measured. The stereochemistry was therefore inferred from NOSEY experiments (Fig. 7). This method revealed conformer (4) to be the only structure accounting for all observed nOe interactions. Therefore, callipeltoside B was noted to have a (S)-configured glycoside linkage (Fig. 7) [14, 15].

Key nOe’s for callipeltose B
The authors note that whilst the same glycosidic linkage is present in both callipeltosides A (1) and B (2) [(S)-configured], callipeltoside C (3) is oppositely configured [(R)-configured]. Since each callipeltose sugar was attached under identical glycosidation conditions, it was assumed that the stereochemical course of the reaction must be influenced by the C4′ substituent. However, at present no further work has been reported to further study this [14, 15].
3 Conclusion and Final Remarks
The completion of the callipeltosides represents nearly 20 years of synthetic work, spanning across multiple research groups. Early syntheses focused on determining the configuration of the trans-chlorocyclopropane ring with respect to the C1–C19 core, in the process determining the absolute stereochemistry of the callipeltoside aglycon. The synthesis of callipeltoside A was accomplished shortly thereafter. These routes evolved over time, with more elegant and convergent approaches implemented once the stereochemical ambiguities associated with the aglycon had been overcome. The final pieces of the puzzle concerned the absolute configuration of the callipeltose B and C sugars, whilst the stereochemistry of the anomeric linkage of these molecules remained uncertain. The MacMillan and Ley groups later provided evidence to suggest that all members of the family contained l-configured sugars (having both also synthesised the corresponding d-configured callipeltose B and C sugars), with the latter research group further analysing the stereochemistry at C1′H to complete the study.
The different approaches towards the callipeltosides highlighted in this review stand as a testament to the power of modern synthetic technologies in their ability to supplement classical natural product structural analysis. It provides access to novel analogues and, in the programme above on the callipeltosides, makes available significantly greater quantities of material than that which is obtained by extraction from natural sources.
Notes
- 1.
This was synthesised by Sonogashira reaction between alkynyl iodide 48 and cyclopropyl alkyne 49 followed by stereoselective reduction, Appel reaction and displacement of the primary bromide with P(OMe)3 [35].
- 2.
The undesired lactone (158) was recycled in two steps to provide additional quantities of the callipeltoside aglycon (9) (conditions shown in Scheme 30).
- 3.
The undesired C13 epimer was oxidised and diastereoselectively reduced to afford additional quantities of 164.
- 4.
References
Zampella A, D’Auria MV, Paloma LG, Casapullo A, Minale L, Debitus C, Henin Y (1996) J Am Chem Soc 118:6202–6209
D’Auria MV, Zampella A, Paloma LG, Minale L (1996) Tetrahedron 52:9589–9596
Zampella A, D’Auria MV, Minale L, Debitus C, Roussakis C (1996) J Am Chem Soc 118:11085–11088
Zampella A, D’Auria MV, Minale L (1997) Tetrahedron 53:3243–3248
Yeung K, Paterson I (2005) Chem Rev 105:4237–4313
Paterson I, Findlay AD (2008) Pure Appl Chem 80:1773–1782
Trost BM, Dirat O, Gunzner JL (2002) Angew Chem Int Ed 41:841–843
Trost BM, Gunzner JL, Dirat O, Rhee YH (2002) J Am Chem Soc 124:10396–10415
Evans DA, Hu E, Burch JD, Jaeschke G (2002) J Am Chem Soc 124:5654–5655
Evans DA, Burch JD, Hu E, Jaeschke G (2008) Tetrahedron 64:4671–4699
Paterson I, Davies RDM, Heimann AC, Marquez R, Meyer A (2003) Org Lett 5:4477–4480
Huang H, Panek JS (2004) Org Lett 6:4383–4385
Hoye TR, Danielson M, May AE, Zhao H (2010) J Org Chem 75:7052–7060
Frost JR, Peason CM, Snaddon TN, Booth RA, Ley SV (2012) Angew Chem Int Ed 51:9366–9371
Frost JR, Pearson CM, Snaddon TN, Booth RA, Turner RM, Gold J, Shaw DM, Gaunt MJ, Ley SV (2015) Chem Eur J 21:13261–13277
Carpenter J, Northrup AB, Chung D, Wiener JJM, Kim S, MacMillan DWC (2008) Angew Chem Int Ed 47:3568–3572
Paterson I, Davies RDM, Marquez R (2001) Angew Chem Int Ed 40:603–607
Marshall JA, Eidam PM (2008) Org Lett 10:93–96
Velazquez F, Olivo HF (2000) Org Lett 2:1931–1933
Olivo HF, Velazquez F, Trevisan HC (2000) Org Lett 2:4055–4058
Romero-Ortega M, Colby DA, Olivo HF (2002) Tetrahedron Lett 43:6439–6441
Yadav JS, Haldar A, Maity T (2012) Eur J Org Chem:2062–2071
Gurjar MK, Reddy R (1998) Carbohydr Lett 3:169–172
Pihko AJ, Nicolaou KC, Koskinen AMP (2001) Tetrahedron Asymmetry 12:937–942
Smith GR, Finley JJ, Giuliano RM (1998) Carbohydr Res 308:223–227
Toth A, Remenyik J, Bajza I, Liptak A (2003) ARKIVOC:28–45
Saito S, Shiozawa M, Ito M, Yamamoto H (1998) J Am Chem Soc 120:813–814
Saito S, Shiozawa M, Yamamoto H (1999) Angew Chem Int Ed 38:1769–1771
Paterson I, Goodman JM, Isaka M (1989) Tetrahedron Lett 30:7121–7124
Paterson I, Perkins MV (1992) Tetrahedron Lett 33:801–804
Paterson I, Norcross RD, Ward RA, Romea P, Lister MA (1994) J Am Chem Soc 116:11287–11314
Charette AB, Juteau H (1994) J Am Chem Soc 116:2651–2652
Charette AB, Prescott S, Brochu C (1995) J Org Chem 60:1081–1083
Wang T, Liang Y, Yu Z (2011) J Am Chem Soc 133:9343–9353
Trost BM, Gunzner JL (2001) J Am Chem Soc 123:9449–9450
Sone H, Kigoshi H, Yamada K (1996) J Org Chem 61:8956–8960
Trost BM, Toste FD, Pinkerton AB (2001) Chem Rev 101:2067–2096
Trost BM, Toste FD (1999) J Am Chem Soc 121:4545–4554
Trost BM, Belletire JL, Godleski S, McDougal PG, Balkovec JM (1986) J Org Chem 51:2370–2374
Barton DHR, Crich D, Motherwell WB (1983) Tetrahedron Lett 24:4979–4982
Shen W, Wang L (1999) J Org Chem 64:8873–8879
Evans DA, Kozlowski MC, Murray JA, Burgey CS, Connell B (1999) J Am Chem Soc 121:669–685
Evans DA, Burch JD (2001) Org Lett 3:503–505
Masuda Y, Hoshi M, Arase A (1992) J Chem Soc Perkin Trans 1:2725–2726
Furukawa J, Kawabata N, Nishimura J (1968) Tetrahedron 24:53–58
Denmark S, O’Conner SP (1997) J Org Chem 62:3390–3401
Yang Z, Lorenz JC, Shi Y (1998) Tetrahedron Lett 39:8621–8624
Frank SA, Chen H, Kunz RK, Schnaderbeck MJ, Roush WR (2000) Org Lett 2:2691–2694
Evans DA, Hu E, Tedrow JS (2001) Org Lett 3:3133–3136
Masse CE, Panek JS (1995) Chem Rev 95:1293–1316
Huang HB, Panek JS (2003) Org Lett 5:1991–1993
Hoye TR, Zhao H (1999) Org Lett 1:169–172
Hoye TR, Danielson ME, May AE, Zhao H (2008) Angew Chem Int Ed 47:9743–9746
Northrup AB, MacMillan DWC (2002) J Am Chem Soc 124:6798–6799
Northrup AB, Mangion IK, Hettche F, MacMillan DWC (2004) Angew Chem Int Ed 43:2152–2154
Northrup AB, MacMillan DWC (2004) Science 303:1752–1755
Brown SP, Brochu MP, Sinz CJ, MacMillan DWC (2003) J Am Chem Soc 125:10808–10809
Zhong G (2003) Angew Chem Int Ed 42:4247–4250
Marshall JA, Yanik MM (2000) Tetrahedron Lett 41:4717–4721
Semmelhack MF, Kim C, Zhang N, Bodurow C, Sanner M, Dobler W, Meier M (1990) Pure Appl Chem 62:2035–2040
Tietze LF, Fischer R, Guder HJ, Goerlach A, Meumann M, Krach T (1987) Carbohydr Res 164:177–194
Oppolzer W, Radinov RN (1991) Tetrahedron Lett 32:5777–5780
Diéguez-Vázquez A, Tzschucke CC, Crecente-Campo J, McGrath S, Ley SV (2009) Eur J Org Chem:1698–1706
Nicolaou KC, Chakraborty TK, Piscopio AD, Minowa N, Bertinato P (1993) J Am Chem Soc 115:4419–4420
Nicolaou KC, Piscopio AD, Bertinato P, Chakraborty TK, Minowa N, Koide K (1995) Chem Eur J 1:318–333
Marshall JA, Eidam P (2004) Org Lett 6:445–448
Layton ME, Morales CA, Shair MD (2002) J Am Chem Soc 124:773–775
Pu L, Yu H (2001) Chem Rev 101:757–824
Noyori R, Suga S, Kawai K, Okada S, Kitamura M, Oguni N, Hayashi M, Kaneko T, Matsuda Y (1990) J Organomet Chem 382:19–37
Brubaker JD, Myers AG (2007) Org Lett 9:3523–3525
Beak P, Meyers AI (1986) Acc Chem Res 19:356–363
Bayley H, Standring DN, Knowles JR (1978) Tetrahedron Lett 19:3633–3634
Kisfaludy L, Ötvös L (1987) Synthesis:510
Muller N, Pritchard DE (1959) J Chem Phys 31:768–771
Maciel GE, McIver Jr JW, Ostlund NS, Pople JA (1970) J Am Chem Soc 92:1–11
Bock K, Lundt I, Pedersen C (1973) Tetrahedron Lett 13:1037–1040
Bock K, Wiebe L (1973) Acta Chem Scand 27:2676–2678
Bock K, Pedersen C (1974) J Chem Soc Perkin Trans 1:293–299
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Frost, J.R., Ley, S.V. (2020). The Callipeltoside Story. In: Kiyota, H. (eds) Marine Natural Products. Topics in Heterocyclic Chemistry, vol 58. Springer, Singapore. https://doi.org/10.1007/7081_2020_40
Download citation
DOI: https://doi.org/10.1007/7081_2020_40
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-4636-2
Online ISBN: 978-981-16-4637-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)