
Abstract
The main achievements on palladium-catalyzed carbonylative synthesis of six-membered heterocycles based on aryl halides as the substrates have been summarized and discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Six-membered ring system is widely present in many natural products, pharmaceuticals, agrochemicals, and functional materials [1, 2]. Specifically, the oxygen-, nitrogen-, and sulfur-containing six-membered fused ring scaffolds are of great interest for the drug industry. For example, quinolones, pyrimidines, and quinazolines are present in many of the best-selling drugs (Fig. 1a) [3]. In 2009, in the highly cited paper titled Heteroaromatic Rings of the Future, Pitt el al. described a virtual exploratory heterocyclic library based on a learning computer program, which would be a powerful tool in drug discovery [4]. Many fused six-membered heterocyclic scaffolds are found in the library (Fig. 1b). Owing to their great importance for the human beings, the chemists have tried a lot to devise various methods to obtain these heterocyclic compounds. Look at the structures in Fig. 1, one could find that all the compounds contain a carbonyl group (-CO-). These types of carbonyl-incorporated six-membered rings are widely present in numerous other pharmaceutically interesting molecules. In the past decades, catalytic carbonylation reactions have found many applications in the synthesis of such type of six-membered heterocycles [5]. In this chapter, we mainly review the palladium-catalyzed formation of oxygen- and nitrogen-containing rings from organo halides. Some closely related ring formation from non-halide starting materials will also be mentioned.

Six-membered heterocycles in important drugs/their candidate structures
2 Chromone and Flavonoid Derivatives
Chromones and flavones (flavonoids) are an important class of biologically and pharmacologically interesting heterocycles (Fig. 2) [6–9]. For example, flavones have many activities such as antioxidant, antitumor, antimicrobial, and anti-inflammatory, and they could affect the behavior of some mammalian enzymes [10–16]. In clinical setting, flavonoid drugs have been used in the treatment of cancer, diabetes, cardiovascular disease, neurodegenerative disorders, etc. [17–20]. Although more than 4,000 naturally occurring flavonoids have been reported, chemists are still trying to synthesize more structurally versatile ones to explore their therapeutic applications. Besides the traditional procedures for their synthesis [8, 21], palladium-catalyzed carbonylations have provided a more straightforward route to flavonoid derivatives.

Structure of chromone and flavonoids
The first synthesis of chromones and flavones using palladium-catalyzed carbonylation reaction was reported in 1990 by Kalinin and coworkers [22]. In their report, 2-iodophenols and two equivalents of alkynes were converted into substituted chromone derivatives catalyzed by 1 mol% of PdCl2(dppf). However, only five examples were provided and the yields were moderate to good (Scheme 1). One drawback of this earliest procedure is the employment of diethylamine both as the solvent and base. The yields were much lower in other solvents like THF, DMF, benzene, and anisole (46–55%). In 1993, the substrate scope of this reaction was extended under the same reactions by the same group [23].

First synthesis of chromones and flavones by Pd-catalyzed carbonylation
In 2005, Yang and coworkers developed a PdCl2/PPh3-catalyzed carbonylative Sonogashira cross-coupling with the same starting materials at room temperature [24]. The key feature of this reaction was that water was used as the green solvent and the atmospheric pressure of CO. They used this new procedure to synthesis several flavones in good to excellent yields (Scheme 2). By using 4 mol% of Pd(PPh3)4 as the catalyst, several 2-ferrocenyl flavones were synthesized in 72–80% yields (Scheme 3) [25]. In their procedure, 4 mol% of CuI was needed as the cocatalyst and with K2CO3 as the base.

Pd-catalyzed carbonylative synthesis of flavones in water

Carbonylative synthesis of 2-ferrocenyl flavones
In 2009, Awuah and Capretta developed a microwave-assisted synthesis of flavones through one-pot Sonogashira–carbonylation–annulation reaction [26]. Starting from commercially available aryl bromides or iodides and trimethylsilylacetylene, flavones were obtained without isolation of the Sonogashira coupling intermediates. In their procedure, 1.5 mol% of Pd2(dba)3 was used as the catalyst, and PA–Ph (1,3,5,7-tetramethyl-2,4,8-trioxa-6-phenyl-6-phosphaadamantane) was used as the ligand (Scheme 4).

Microwave-assisted synthesis of flavones
In 2010, Yang and Alper reported a ligand-free carbonylative synthesis of flavones from 2-iodophenols and alkynes in phosphonium salt ionic liquids [27]. They used 5 mol% of PdCl2 as the sole catalyst and triethylamine as the base. Under atmospheric CO pressure, 64–96% yields of various substituted flavones were obtained. One drawback of this procedure was that the reaction must be performed at 110°C, and the catalyst loadings were high (5 mol%).
In 2011, Li and coworkers reported a Pd–carbene-catalyzed carbonylations of 2-iodophenols. Using 0.5 mol% [PdBr2(iPr2-bimy)L] (L=N-phenyl imidazole) as the catalyst and diethylamine as the base, a series of flavones were synthesized in good yields (Scheme 5).

Pd–NHC-catalyzed carbonylation of 2-iodophenols
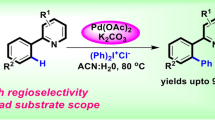
Using a new type of palladium–thiourea–dppp complex as the catalyst, Miao and Yang reported a regiospecific carbonylative annulation of 2-iodophenol to form flavones in moderate yields (Scheme 6) [28]. The authors found that significant amount of aurone 3 was observed in all cases (10–20%). Besides, the conversion of 2-iodophenols remained 80–90% even though the reaction time was extended to more than 2 days. To overcome these problems, acetylated 2-iodophenols were used as the substrates to afford the flavonoid products in 70–92% yields. The formation of aurones resulted from the oxidative addition of phenol O–H bond to Pd(0), which could not happen when the hydroxyl was protected by acetyl group.

Carbonylative annulation of 2-iodophenol acetates with high regioselectivity
The construction of flavones from 2-iodophenols also found many applications in total synthesis. Martin and coworkers reported the total synthesis of luteolin in 2011 (Scheme 7) [29]. Using PdCl2(PPh3)2 as the catalyst, Rixson et al. [30] realized a facile synthesis of some novel anthrapyran-2-ones by the carbonylative cross-coupling between 1-hydroxy-2-iodo-dihydroanthrancene and terminal alkynes. These structures may have been reported with antibacterial or antitumor activities (Scheme 8).

Carbonylation of 2-iodophenol as key step in total synthesis of luteolin

Pd-catalyzed carbonylative synthesis of anthrapyran-2-ones
Alper and coworkers reported the first carbonylation reaction with 2-iodophenols and allenes [31]. Using 5 mol% of Pd(OAc)2/dppb as the catalyst and iPr2NEt as the base, the reaction favored isomer A in high regioselectivity, especially when terminal allenes (R1=H) were employed. The regioselectivity resulted from the more favorable OH attacking on the less hindered terminus of the π-allyl palladium intermediate (Scheme 9).

Pd-catalyzed carbonylation of 2-iodophenols with allenes
Also started from 2-iodophenols and allenes, Grigg and coworkers performed the reaction under atmospheric CO pressure [32]. Using Pd(PPh3)4 as the catalyst and K2CO3 as the base, various functionalized chroman-4-ones were obtained in good to excellent yields (Scheme 10). Besides, 2-iodoanilines were also converted to quinol-4-ones under the same conditions.

Chroman-4-ones via Pd-catalyzed carbonylation–allene insertion
In 2000, Kadnikov and Larock reported the carbonylation of 2-iodophenols and internal alkynes (Scheme 11) [33]. A variety of substituted coumarins were synthesized in good yields in the presence of 5 mol% of Pd(OAc)2 under 1 atm of CO. Owing to the large over amount of alkynes (5 equiv.) and low CO pressure, the insertion of the internal alkyne occurs in preference to the insertion of CO. When asymmetric internal alkynes were used, two regioisomers were obtained in moderate selectivity. In 2014, Zhao, Cai et al. used a highly efficient and reusable PdCl2(PPh3)2/PEG-2000/H2O system for the carbonylative Sonogashira coupling (Scheme 12) [34]. This system could also been used for the synthesis of flavone under very mild conditions (1 atm CO and 25°C). In 2005, Cao and Xiao performed this reaction using Mo(CO)6 as the CO source under microwave irradiation, and a series of chromen-2-one derivatives was obtained (Scheme 13) [35].

Pd-catalyzed synthesis of coumarins from 2-iodophenols and internal alkynes

Pd-catalyzed carbonylative synthesis of coumarins in aqueous system

Microwave-promoted carbonylative synthesis of chromen-2-ones using Mo(CO)6 as CO source
In 2012, Gøgsig, Skrydstrup et al. reported a palladium-catalyzed synthesis of 1,3-diketones through carbonylative α-arylation in a two-chamber reactor using 9-methyl-9H-fluorene-9-carbonyl chloride as the solid CO source [36]. Starting from MOM-protected 2-iodophenol, the 13C-labeled 1,3-diketone was obtained in a 90% yield. Subsequent hydrolysis of the ethoxymethyl ether protecting group and dehydrative cyclization formed the 3-methyl-flavone in 99% yield (Scheme 14).

CO-free carbonylative synthesis of 3-methyl-flavone using two-chamber reactor
All the above carbonylative procedures toward flavones started from expensive iodophenols and terminal alkynes or allenes. In 2012, Wu et al. reported a carbonylative synthesis of flavones from 2-hydroxyacetophenones and aryl bromides (Scheme 15) [37]. This alternative procedure employed only 2 mol% Pd(OAc)2/dppb as the catalyst and two equivalent UBU as the base, which allowed for the convenient synthesis of various 2-(hetero)aryl-substituted flavones in moderate to good yields. Besides, when 2-propenyl phenol was used as the substrate, 3-methyl-flavone could be obtained in 61% yield.

Pd-catalyzed synthesis of flavones from 2-hydroxyacetophenones and aryl bromides
In the next year, Wu and coworkers developed a palladium-catalyzed carbonylative synthesis of chromenones from salicylic aldehydes and benzyl chlorides in good to excellent yields (Scheme 16) [38]. Regarding the reaction pathway, the benzyl chloride firstly underwent phenoxycarbonylation with salicylic aldehydes to form 2-formylphenyl 2-phenyl-acetate, which led to the products through intramolecular condensation. 2-Hydroxyacetone could also afford methyl-substituted chromenone but the yield was much lower.

Carbonylative synthesis of chromenones from salicylic aldehydes and benzyl chlorides
In 2014, Wu and coworkers provided three examples of palladium-catalyzed synthesis of chromenones starting from 2-bromofluorobenzenes and 1,2-diphenylethan-1-one under 50 bar of CO [39]. The combination of carbonylation and nucleophilic substitution afforded the desired product in 30–40% yields (Scheme 17).

Pd-catalyzed synthesis of chromenones from 2-bromofluorobenzenes
3 Valeralactone, Pyran-2-One, and Their Derivatives
As early as 1980, Cowell and Stille reported the synthesis of γ-lactones by palladium-catalyzed carbonylation of halo alcohols [40] (Fig. 3). In this paper, one example of δ-lactone, 1H-2-benzopyran-3(4H)-one, was also synthesized from (2-(bromomethyl)phenyl)methanol in the presence of 1.6 mol% PdCl2(PPh3)2 (Scheme 18). Owing to the industrial interest as a precursor for various pharmaceuticals and plant protection agents, Lindsell et al. reinvestigated this reaction in 2005 [41]. Although they failed to reproduce Stille’s procedure, they obtained near-quantitative yield of the same product under different reaction conditions. In 2006, Preston et al. patented a palladacycle catalyst to realize this reaction [42]. CO was bubbled through the reaction mixture under mild conditions, and quantitative yield of isochroman-3-one was obtained.

Structure of six-membered lactones

Synthesis of γ-lactones by Pd-catalyzed carbonylation
In 1983, Brunet et al. described Co2(CO)8-catalyzed SRN1 carbonylations of the same substrates under sunlamp-irradiated phase-transfer conditions [43]. Two benzolactones were synthesized in excellent yields (Scheme 19). In this procedure, Co2(CO)8 was used as both the catalyst and the CO source. When the amines were used instead of alcohols, the corresponding benzolactam was obtained albeit in somewhat lower yields.

Co2(CO)8-catalyzed radical carbonylative synthesis of benzolactones
In 2004, Alterman and coworkers reported a palladium-catalyzed carbonylation of 2-(o-bromophenyl)ethanol using molybdenum hexacarbonyl as the CO source [44]. At high temperature (180°C), isochroman-1-one was obtained in 74% yield (Scheme 20).

CO-free carbonylative synthesis of isochroman-1-one
In 2007, Alper and coworkers reported an interesting synthesis of highly substituted endocyclic enol lactones (Scheme 21) [45]. In this method, 3 mol% of PdCl2(PPh3)2 and 6 mol% dppp were used as the catalyst, and ionic liquid [bmim][Tf2N] was used as the reaction medium. Under 200 psi of CO, a series of β-diketones and alkynes were converted to the desired products in reasonably good regioselectivity and yields. More importantly, the system could be recycled five times with only modest loss of its catalytic activity. The authors also proposed a mechanism for this transformation. In the first step, PdCl2(PPh3)2 was reduced to Pd(0) species in the presence of CO. The in situ formed Pd(0)Ln oxidatively added into the enol –OH of the β-diketone to give Pd-hydride intermediate A. Then the alkyne would coordinate to A and CO insertion to form the intermediate B. Next, the alkyne was regioselectively inserted to the Pd–acyl bond to form C, which easily underwent reductive elimination to form D and regenerated Pd(0). D is rearranged through intramolecular cyclization of the vinyl acetate on the activated double bond to give the final product E (Scheme 22).

Pd-catalyzed carbonylative synthesis of substituted endocyclic enol lactones

Proposed mechanism for the transformation in Scheme 21
Based on the similar strategy, Wu and Hua described an efficient synthesis of 3,4,7,8-tetrahydro-2H-chromene-2,5(6H)-dione derivatives via a [3+2+1] cyclocarbonylative coupling of 1,3-cyclohexanediones, terminal alkynes, and CO (Scheme 23) [46]. Using 5 mol% Pd(PPh3)4 as the catalyst and THF as the solvent, the products were obtained in excellent selectivity. The route for the formation of chromene-2,5-dione derivatives was proposed to involve the similar steps in Scheme 22.

Pd-catalyzed cyclocarbonylative synthesis of chromene-2,5-dione derivatives
4 Saturated Oxygen-Containing Six-Membered Rings
In 2015, Yang and coworkers [47] described a Pd(II)-catalyzed intramolecular carbonylative cyclization reaction of aryl alkenes and aryl alkenols (Scheme 24). Using 10 mol% of PdCl2(PPh3)2 as the catalyst and 3.5 equiv. of CuCl2 as the oxidant, chromane derivatives were obtained under atmospheric CO pressure. When external alcohols were added as nucleophiles, α-(chroman-4-yl) acetate esters were obtained in high yields and stereoselectivity. When aryl alkenols were used as the substrates, five-membered lactone rings were constructed by intramolecular alkoxycarbonylation. It was noticeable that the only electron-rich aromatic rings led to the products in good yields. The authors also propose a mechanism to explain the formation of the fused ring products (Scheme 25). At the first step, the allylic alcohol reacted with palladium carbonyl complex to form complex B. Next, a cis-intramolecular nucleopalladation led to an intermediate palladacycle C. The migratory CO insertion led to complex D, which released the product by reductive elimination. The resultant Pd(0) was then reoxidized by CuCl2.

Pd-catalyzed intramolecular carbonylative cyclization toward chromanes

Proposed mechanism for the Pd-catalyzed carbonylative cyclization of aryl alkenols
5 Quinolinone Derivatives
Quinolinone and its derivatives have diverse chemical and pharmacological properties and thus attracted much interest of both synthetic and pharmaceutical chemists [48–53] (Fig. 4). Palladium-catalyzed carbonylative construction of these structures has become a very unique method because it employed some readily available starting materials and CO as the carbonyl source.

Structure of quinolinones
In 1990, Torii et al. reported an efficient synthesis of 3-substituted 3-(2-haloarylamino)prop-2-enoates catalyzed by 5 mol% Pd(OAc)2 and 20 mol% PPh3 under CO pressure (Scheme 26) [54]. When lactone substrates were employed, the desired products were obtained in very good yields.

First example of carbonylative synthesis of quinolin-4(1H)-ones
Two years later, Kalinin and coworkers synthesized 2-substituted quinolin-4-ones from 2-iodoanilines and terminal alkynes (Scheme 27) [55]. They used 5 mol% PdCl2(PPh3)2 as the catalyst and diethylamine as the solvent. Under 20 bar CO pressure, 61–95% yields of the products were isolated. As a synthetic application, Haddad et al. accomplished convergent synthesis of the key quinolone intermediate for protease inhibitor BILN 2061 by using 1.5 mol% PdCl2(dppf) as the catalyst [56]. In 2015, Larhed’s group reported a similar carbonylative synthesis of 4-quinolones using Co(CO)6 as the carbonyl source (Scheme 28) [57]. Under microwave conditions, this method yielded the products after only 20 min of microwave heating at 120°C. Using Pd(OAc)2/PtBu3·HBF4 as the catalytic system, the reaction could run in a one-pot two-step sequence at room temperature, and some sensitive substituents in the substrates could be well tolerated.

Carbonylative synthesis of 2-substituted quinolin-4-ones from 2-iodoanilines and terminal alkynes

Synthesis of 4-quinolones using Mo(CO)6 as the carbonyl source
In 2005, Alper and coworkers developed an intramolecular carbonylation procedure with recyclable palladium-complexed dendrimers on silica for the synthesis of some fused ring system [58]. In this report, they gave one example of six-membered ring synthesis. Starting from 2′-iodo-[1,1′-biphenyl]-2-amine, 6(5H)-phenanthridinone was obtained in 97% yield catalyzed by G1-palladium dendrimer (Scheme 29).

Recyclable carbonylation palladium catalyst for the phenanthridinone synthesis
In 2007, Alper’s group synthesized a series of methylene-2,3-dihydro-1H-quinolin-4-ones through the carbonylative cross-coupling between o-iodoanilines and allenes in ionic liquid. They used only 2 mol% Pd2(dba)3·CHCl3 and dppb as the catalyst, and both terminal and internal allenes were applicable as the substrates (Scheme 30). Under relatively mild conditions (5 bar of CO and 90°C), up to 82% yield of the products was obtained. When the asymmetric enteral allenes were used, two region isomers were isolated in a ca. 2:1 to 7:1 ratio. Notably, cyclononene could be incorporated into the dihydro-1H-quinolin-4-one ring system.

Pd-catalyzed carbonylative cross-coupling between o-iodoanilines and allenes
Quinazolinones are another important class of six-membered heterocyclic compounds containing two isolated nitrogen atoms. Until now, many methods had been reported for their synthesis [59–61]. As early as in 1987, Tilley and coworkers reported a carbonylative synthesis of pyrido[2,1-b]quinazoline derivatives starting from N-(2-bromophenyl)pyridine-2-amines (Scheme 31) [62]. The authors proposed that this reaction initialized with the oxidation of N-(2-bromophenyl)pyridine-2-amine onto Pd(0) to form the aryl-palladium bond. Then CO insertion and nucleophilic attack by the pyridine nitrogen afforded the desired product.

Pd-catalyzed aminocarbonylation of N-(2-bromophenyl)pyridin-2-amines
Inherently, 2-aminopyridine (pK a = 6.86) exists s as a mixture of tautomeric amino and imino isomers with the predominance of the amino form over the imino form (1000:1). By utilization of this unique tautomerization nature of 2-aminopyridine, several syntheses of nitrogen heterocycles were reported through carbonylation. In 2014, Wu, Beller and their coworkers reported a synthesis of quinazolinones by carbonylation/nucleophilic aromatic substitution sequence [63]. Started from 2-bromofluorobenzene and 2-aminopyridines, they obtained both linear and angular fused quinazolinones using Pd(OAc)2/n-BuPAd2 as the catalytic system (Scheme 32). In this regio-switchable procedure toward two isomers, the basicity difference between Et3N (pK a = 10.8) and DBU (pK a = 12) played a key role. In 2015, Xu and Alper updated Torii’s procedure (Scheme 33) reaction by using Pd(OAc)2 and DIBPP as the catalytic system [64]. Under their optimized conditions, nine examples of pyrido[2,1-b]quinazolin-11-ones and six examples of dipyrido[1,2-a:2,3:b]pyrimidin-5-ones were synthesized through Pd/DIBPP-catalyzed dearomatizing carbonylation. It was notable that the starting materials could be prepared from Pd-catalyzed amination of the 1,2-dibromides without isolation.

Regio-switchable procedure toward linear and angular fused quinazolinones

Pd-catalyzed dearomatizing carbonylation to form quinazolin-11-ones and pyrimidin-5-ones
In 1999, Cacchi et al. described a one-pot process synthesis of 6-aryl-11H-indolo[3,2-c]quinolines through a straightforward palladium-catalyzed carbonylative cyclization of o-(o′-aminophenylethynyl)trifluoroacetanilide with aryl iodides [65]. In this procedure, 2-(2-aminophenyl)-3-aroyl-1H-indol was formed as the primary product, which underwent intramolecular condensation to form the six-membered ring system (Scheme 34).

One-pot process synthesis of 6-aryl-11H-indolo[3,2-c]quinolones
The above strategy was also employed in the carbonylative synthesis of 3-substituted 4-aroylisoquinolines by Dai and Larock [66]. In the presence of 5 mol% of Pd(PPh3)4 as the catalyst and 5 equiv. of K2CO3 as the base, 2-(1-alkynyl)benzaldimines were cyclized to the desired products in moderate to good yields (Scheme 35). In the first step of the reaction, aroylpalladium complexes (A) were formed from aryl bromides or iodides by sequential oxidative addition and CO insertion. Then the aroylpalladium(II) species coordinate to the alkynes, and subsequent nucleophilic attack from the imine nitrogen produced the intermediate C. Primary product D was then released from C by reductive elimination and further deprotection of the tertiary butyl group in the presence of base to give the final products.

Pd-catalyzed carbonylative cyclization of 2-(1-alkynyl)benzaldimines to form 3-substituted 4-aroylisoquinolines
In 2000, Larksarp and Alper disclosed a catalyst system comprising Pd(OAc)2-dppf for the cyclocarbonylation of o-iodoanilines with heterocumulenes at 70–100°C for 12–24 h to give the corresponding 4(3H)-quinazolinone derivatives in good to excellent yields (Scheme 36) [67, 68]. Heterocumulenes such as isocyanates, carbodiimides, and ketenimines could be used in their procedures to prepare different substituted products.

Cyclocarbonylation of o-iodoanilines with various heterocumulenes
Based on these earlier works of their own group, Zeng and Alper later reported a highly practical and efficient method for the synthesis of 2-heteroquinazolin-4(3H)-ones through palladium-catalyzed tandem reaction [69] in good to excellent yields (Scheme 37). Using the same catalytic system, the authors developed a domino process for the synthesis of quinazolino[3,2-a]quinazolinones (Scheme 38) [70]. The starting carbodiimides were efficiently prepared by metathesis reactions of the corresponding isocyanates with N-(o-iodoaryl)triphenyliminophosphoranes. It was noteworthy that five new bonds and two fused heterocycles were formed in a single step. Less bulky alkyl amines led very good yields and steric hindrance in the amines decreased the yields. When N,N′-dimethylethylenediamine was used as the amine, the desired product with a nine-membered ring structure was not observed.

Pd-catalyzed tandem reaction to synthesize 2-heteroquinazolin-4(3H)-ones

Pd-catalyzed tandem reaction to synthesize quinazolino[3,2-a]quinazolinones
In 2008, Zheng and Alper developed a palladium-catalyzed cyclocarbonylation of 2-iodoanilines with imidoyl chlorides under 500 psi of CO [71]. The reaction was believed to proceed via in situ formation of an amidine, followed by oxidative addition, CO insertion, and intramolecular cyclization to give the substituted quinazolin-4(3H)-ones in 63–91% yields (Scheme 39).

Pd-catalyzed cyclocarbonylation of 2-iodoanilines with imidoyl chlorides
In 2013, Wu, Beller, and their coworkers developed a novel palladium-catalyzed four-component carbonylative coupling system for the selective construction of 4(3H)-quinazolinones in a one-pot fashion [72]. Starting from 2-bromoanilines, trimethyl orthoformate, and amines under 10 bar of CO, the desired products were isolated in good yields catalyzed by 2 mol% of Pd(OAc)2 and n-BuPAd2. Notably, the process tolerated the presence of various reactive functional groups and was very selective for quinazolinones (Scheme 40).

Pd-catalyzed four-component carbonylative coupling to form 4(3H)-quinazolinones
Later on, Wu’s group reported the synthesis of quinazolinones from 1-bromo-2-fluorobenzenes and amidines with 2 mol% of Pd(OAc)2 and 6 mol% n-BuPAd2 as the catalyst (Scheme 41) [39]. The reaction was believed to initialize from the aminocarbonylation to form the N-(amino(phenyl)methylene)-2-fluorobenzamide intermediate. The nucleophilic substitution and rearrangement led to the final products. As an evidence, 1,2-diphenylquinazolin-4(1H)-one was obtained in 70% yield when N-phenylbenzimidazole hydrochloride was used as the amidine.

Pd-catalyzed quinazolinones synthesis from 1-bromo-2-fluorobenzenes and amidines
In the same year, Wu, Beller, and their coworkers reported a domino synthesis of quinazolinediones through a double carbonylation process. Starting from commercially available 2-bromobenzonitriles and 2-bromoanilines series, 20 examples of isoindolo[1,2-b]quinazoline-10,12-dione structures were synthesized in the presence of Pd(OAc)2 and PtBu3·HBF4 as the catalyst (Scheme 42). After determination of the final product by X-ray crystalline structure and the validation of 2-phenyl-3-iminoisoindolin-1-one as the isomerization intermediate, the authors proposed a two-sequential aminocarbonylation reaction pathway in Scheme 42.

Synthesis of isoindolo[1,2-b]quinazoline-10,12-diones through double carbonylation
In Zeng and Alper 2008 [73] (Scheme 43), a wide variety of substituted isoquinolin-1(2H)-ones was synthesized in reasonable to good yields by the palladium-catalyzed cyclization of diethyl(2-iodoaryl)malonates with imidoyl chlorides and carbon monoxide in tetrahydrofuran. A palladium-catalyzed carbonylation–decarboxylation process may be involved in the one-step synthesis of the isoquinolin-1(2H)-ones. Later on, Okuro and Alper reported a palladium-catalyzed intermolecular cyclocarbonylation of 2-iodoanilines with diethyl ethoxycarbonylbutendienoate [74]. By the similar procedure involving Michael addition and subsequent carbonylation, 2,3,3-triethoxycarbonyl-2,3-dihydro-4(1H)-quinolinone derivatives were obtained in moderate to good yields (Scheme 44).

Pd-catalyzed carbonylative cyclization of diethyl(2-iodoaryl)malonates with imidoyl chlorides

Pd-catalyzed carbonylative synthesis of 2,3,3-triethoxycarbonyl-2,3-dihydro-4(1H)-quinolinone
In 2008, Broggini’s group [75] reported a carbonylative synthesis of 4-[(methoxycarbonyl) methyl]-3,4-dihydroisoquinolin-1-ones from N-allylamides of 2-iodobenzoic acids under high pressure of CO (Scheme 45). The reaction stared with the oxidative addition of aryl iodides with in situ generated Pd(0). The following intramolecular carbopalladation gave (σ-alkyl)Pd complexes, which underwent CO insertion and nucleophilic attack by MeOH to form the final products.

Pd-catalyzed coupling between N-allylamides of 2-iodobenzoic acids
In 2001, Kang and Kim reported a reaction between γ-allenic sulfonamides and aryl iodides in the presence of Pd(PPh3)4 (5 mol%) to form 3-aroyl-2- or 3-pyrrolines [76]. When started from δ-allenic sulfonamides, this procedure afforded two examples of piperidine-substituted enones in moderate yields (Scheme 46).

Pd(PPh3)4-catalyzed carbonylative coupling between γ-allenic sulfonamides and aryl iodides
In 2008, Alper’s group developed an efficient and simple synthesis of biologically fused ring isoquinolines by utilizing a palladium-catalyzed carboxamidation reaction and aldol condensation reaction cascade protocol (Scheme 47) [77]. The first step of the reaction was the oxidative addition of the aryl iodides with Pd(0) to give aryl-palladium species, and then CO insertion formed aroylpalladium complex which was then attacked by the amide nitrogen to give the imide intermediate. Finally, the intramolecular condensation led to the final product.

Pd-catalyzed carbonylative synthesis of fused ring isoquinolines
Larock’s group developed a palladium-catalyzed synthesis of 3,4-disubstituted 2-quinolones through the annulation of alkynes and N-substituted o-iodoanilines under 1 atm of CO [78, 79]. The best results were obtained using alkoxycarbonyl, p-tolylsulfonyl, and trifluoroacetyl substituents. The nitrogen protection groups were lost during the course of the reaction resulting in the formation of N-unsubstituted 2-quinolones in moderate to good yields (Scheme 48).

Pd-catalyzed synthesis of 3,4-disubstituted 2-quinolones from alkynes and N-substituted o-iodoanilines
In Tadd and Willis (2009) [80], palladium-catalyzed intermolecular aminocarbonylation/intramolecular amidation cascade sequences can be used to convert a range of 2-(2-haloalkenyl)aryl halide substrates efficiently and selectively to the corresponding 2-quinolones. Delaying the introduction of the CO atmosphere allows amination/carbonylation sequence and the preparation of an isoquinoline (Scheme 49).

Pd-catalyzed intermolecular cascade sequences to the 2-quinolones
In 1996, Cacchi, Marinelli et al. reported a palladium-catalyzed reaction of o-iodoaniline with unsaturated halides or triflates [81]. In the presence of K2CO3 and catalytic amounts of Pd(PPh3)4 under an atmosphere of carbon monoxide, 2-aryl- and 2-vinyl-4H-3,1-benzoxazin-4-ones were obtained in good to high yields (Scheme 50). It was noteworthy that when R was androsta-3,5-dienes, the corresponding products were also obtained in 68–78% yields.

Pd-catalyzed carbonylative synthesis of 4H-3,1-benzoxazin-4-ones from 2-iodoanilines and aryl halides and triflates
In 1999, Larksarp and Alper [82] reported a one-pot carbonylative regioselective synthesis of 2-substituted-4H-3,1-benzoxazin-4-ones from o-iodoanilines and acid chlorides (Scheme 51). In the presence of Pd(OAc)2 and diisopropylethylamine, the desired products were obtained in good to excellent yields. The reaction was believed to proceed via in situ amide formation from an o-iodoaniline and an acid chloride, followed by oxidative addition to Pd(0), CO insertion, and intramolecular cyclization to form the 2-substituted-4H-3,1-benzoxazin-4-one derivatives. In 2010, Salvadori et al. [83] realized the same reaction using Pd/C as the heterogeneous catalyst under microwave dielectric heating conditions (Scheme 52).

Pd-catalyzed carbonylative coupling with o-iodoanilines and acid chlorides

Pd/C-catalyzed carbonylative synthesis of 4H-3,1-benzoxazin-4-ones
Ács et al. transformed 2-iodoanilines to the corresponding 2-aryl-benzo[d][1,3]oxazin-4-one derivatives via double carbon monoxide insertion (Scheme 53) [84]. This reaction required a high CO pressure (100 bar).

Pd-catalyzed double carbon monoxide insertion of 2-iodoanilines
In 2011, Wu et al. described an alternative synthesis of 2-arylbenzoxazinones from commercially available 2-bromoanilines and aryl bromides (Scheme 54) [85]. The first step was the chemoselective aminocarbonylation of the aryl bromides to form the N-aroyl-2-bromoaniline intermediate. The second CO insertion into the aryl-palladium bond furnished the final product. In 2014, Li and Wu reported a palladium-catalyzed carbonylative synthesis of benzoxazinones from N-(ortho-bromoaryl)amides using paraformaldehyde as the carbonyl source [86]. Under this CO-free conditions, various substituted benzoxazinones were obtained in good yields under optimized conditions (Scheme 55). Using 13C-labeled paraformaldehyde, 4-13C-labeled benzoxazinone derivative was obtained, which has many important applications in pharmaceutical and biological topics.

Pd-catalyzed synthesis of 2-arylbenzoxazinones from 2-bromoanilines and ArBr

Pd-catalyzed carbonylative synthesis of benzoxazinones using paraformaldehyde as CO source
In 2015, Konishi et al. reported a synthesis of 4H-3,1-benzoxazin-4-one derivatives from N-(ortho-iodoaryl)amides by using phenyl formate as the CO source (Scheme 56) [87]. Under basic conditions, phenyl formate decomposed to phenol and carbon monoxide. The authors proposed that the reaction proceeded through the phenoxycarbonylation of the aryl iodide intermediates and basic condensation of the intermediates led to the desired products in moderate to good yield.

Carbonylative synthesis of 4H-3,1-benzoxazin-4-ones using phenyl formate as CO source
6 Other Six-Membered Heterocycles
Palladium-catalyzed carbonylation of C–X bonds has also found some application in some other six-membered heterocycle synthesis. In 1999, Xiao and Alper reported a regioselective heteroannulation of 2-iodothiophenols with allene CO pressure in the presence of palladium catalyst to form various thiochroman-4-one derivatives (Scheme 57) [88]. In 2008, the same group reported a novel synthesis of another type of sulfur-containing heterocycles, 3-substituted-3,4-dihydro-2H-1,3-benzothiazin-2-ones, by palladium-catalyzed carbonylation reaction of 2-substituted-2,3-dihydro-1,2-benzisothiazoles in pyridine (Scheme 58) [89]. The authors suggested that the reaction is initialized with the oxidation of S–N bonds with Pd(0) and then CO insertion into the S–Pd bonds or N–Pd bonds to form the product-releasing intermediates. Finally, the reductive elimination gave the products.

Pd-catalyzed carbonylative synthesis of thiochroman-4-one derivatives

Pd-catalyzed carbonylation reaction of 2-substituted-2,3-dihydro-1,2-benzisothiazoles
In 2001, Brown et al. gave an example of carbonylative synthesis of chromane derivative from 1-iodo-2-((3-methylbut-3-en-1-yl)oxy)benzene in the presence of two equivalents of silane (Scheme 59) [90].

Reductive carbonylation to form 2-(4-methylchromen-4-yl)acetaldehyde
In 2012, Ryu’s group gave several examples of the synthesis of functionalized δ-lactam and δ-lactones through Pd/light-accelerated atom-transfer carbonylation reactions (Scheme 60) [91]. These three-component coupling reactions involved a hybrid organometallic-radical pathway and showed the potential of the utilization of alkyl halides in carbonylative heterocycle synthesis.

Pd/light-accelerated atom-transfer carbonylation reactions to form lactams and lactones
7 Summary
Palladium-catalyzed synthesis of heterocycles from carbon–halide bonds in the presence of CO has been applied in many types of six-membered heterocyclic compounds. The combination of amino- or alkoxycarbonylation with various intramolecular condensations, nucleophilic substitutions, or further carbonylation reactions was effective for the construction of various carbonyl-derived six-membered rings and fused ring system. We believe that the combination of carbonylation with other types of intramolecular reaction would be a useful tool for the synthesis of more advanced heterocyclic compounds.
References
Katritzky AR, Rees CW, Scriven EFW (eds) (1996) Comprehensive heterocyclic chemistry II, vol 7. Pergamon, Oxford
Garrido Montalban A (2011) Heterocycles in natural product synthesis. Wiley, Weinheim, pp 299–339
Baumann M, Baxendale IR (2013) Beilstein J Org Chem 9:2265–2319
Pitt WR, Parry DM, Perry BG, Groom CR (2009) J Med Chem 52:2952–2963
Wu X-F, Neumann H, Beller M (2013) Chem Rev 113:1–35
Singh M, Kaur M, Silakari O (2014) Eur J Med Chem 84:206–239
Verma AK, Pratap R (2010) Nat Prod Rep 27:1571–1593
Gaspar A, Matos MJ, Garrido J, Uriarte E, Borges F (2014) Chem Rev 114:4960–4992
Havsteen B (1983) Biochem Pharmacol 32:1141–1148
Valdameri G, Kenski JCN, Moure VR, Trombetta-Lima M, Martinez GR, Sogayar MC, Winnischofer SMB, Rocha MEM (2014) Nat Prod Commun 9:1457–1460
Li J, Chen R, Wang R, Liu X, Xie D, Zou J, Dai J (2014) ChemBioChem 15:1672–1680
Inomata Y, Terahara N, Kitajima J, Kokubugata G, Iwashina T (2013) Biochem Syst Ecol 51:123–129
Caruso IP, Vilegas W, de Souza FP, Fossey MA, Cornelio ML (2014) J Pharm Biomed Anal 98:100–106
Ji W, Guo L, Lian J, Gong B (2008) J Pharm Pharmacol 60:1207–1212
Tamura H, Yoshioka M, Hasegawa M, Hosoda A, Matsugi M, Akamatsu M (2013) Bioorg Med Chem 21:2968–2974
Narwal M, Haikarainen T, Fallarero A, Vuorela PM, Lehtiö L (2013) J Med Chem 56:3507–3517
Li-Weber M (2009) Cancer Treat Rev 35:57–68
Walle T (2007) Semin Cancer Biol 17:354–362
Walle T (2007) Mol Pharm 4:826–832
Bibby MC, Double JA (1993) Anticancer Drugs 4:3–17
Kshatriya RB, Shaikh YI, Nazeruddin GM (2013) Orient J Chem 29:1475–1487
Kalinin VN, Shostakovsky MV, Ponomaryov AB (1990) Tetrahedron Lett 31:4073–4076
Torii S, Okumoto H, Xu LH, Sadakane M, Shostakovsky MV, Ponomaryov AB, Kalinin VN (1993) Tetrahedron 49:6773–6784
Liang B, Huang MW, You ZJ, Xiong ZC, Lu K, Fathi R, Chen JH, Yang Z (2005) J Org Chem 70:6097–6100
Ma W, Li X, Yang J, Liu Z, Chen B, Pan X (2006) Synthesis 2006:2489–2492
Awuah E, Capretta A (2009) Org Lett 11:3210–3213
Yang Q, Alper H (2010) J Org Chem 75:948–950
Miao H, Yang Z (2000) Org Lett 2:1765–1768
O’Keefe BM, Simmons N, Martin SF (2011) Tetrahedron 67:4344–4351
Rixson JE, Skelton BW, Koutsantonis GA, Gericke KM, Stewart SG (2013) Org Lett 15:4834–4837
Okuro K, Alper H (1997) J Org Chem 62:1566–1567
Grigg R, Liu A, Shaw D, Suganthan S, Woodall DE, Yoganathan G (2000) Tetrahedron Lett 41:7125–7128
Kadnikov DV, Larock RC (2000) Org Lett 2:3643–3646
Zhao H, Cheng M, Zhang J, Cai M (2014) Green Chem 16:2515–2522
Cao H, Xiao WJ (2005) Can J Chem 83:826–831
Gøgsig TM, Taaning RH, Lindhardt AT, Skrydstrup T (2012) Angew Chem Int Ed 51:798–801
Wu X-F, Neumann H, Beller M (2012) Chem Eur J 18:12595–12598
Wu X-F, Wu L, Jackstell R, Neumann H, Beller M (2013) Chem Eur J 19:12245–12248
Chen J, Natte K, Neumann H, Wu X-F (2014) Chem Eur J 20:16107–16110
Cowell A, Stille JK (1980) J Am Chem Soc 102:4193–4198
Lindsell WE, Palmer DD, Preston PN, Rosair GM, Jones RVH, Whitton AJ (2005) Organometallics 24:1119–1133
Preston PN, Lindsell WE, Whitton A, Palmer D, Jones R, Fieldhouse R (2006) WO 2006010885 A1, Heriot-Watt University, UK (Chem Abstr 2006, 144: 192381)
Brunet JJ, Sidot C, Caubere P (1983) J Org Chem 48:1166–1171
Wu X, Mahalingam AK, Wan Y, Alterman M (2004) Tetrahedron Lett 45:4635–4638
Li Y, Yu Z, Alper H (2007) Org Lett 9:1647–1649
Wu B, Hua R (2010) Tetrahedron Lett 51:6433–6435
Li S, Li F, Gong J, Yang Z (2015) Org Lett 17:1240–1243
Brown DJ (2004) Quinoxalines (supplement II). Wiley, Hoboken
Banno K, Fujioka T, Kikuchi T, Oshiro Y, Hiyama T, Nakagawa K (1988) Chem Pharm Bull 36:4377–4388
Yamamoto M, Maehara Y, Sakaguchi Y, Kusumoto T, Baba H, Sugimachi K (1997) Int J Oncol 10:53–57
Fujita M, Tsuchida T, Fujita T, Higashino K (1999) Oncol Rep 6:353–357
Koga Y, Kihara Y, Okada M, Inoue Y, Tochizawa S, Toga K, Tachibana K, Kimura Y, Nishi T, Hidaka H (1998) Bioorg Med Chem Lett 8:1471–1476
Kumar S, Bawa S, Gupta H (2009) Mini Rev Med Chem 9:1648–1654
Torii S, Okumoto H, Xu LH (1990) Tetrahedron Lett 31:7175–7178
Kalinin VN, Shostakovsky MV, Ponomaryov AB (1992) Tetrahedron Lett 33:373–376
Haddad N, Tan J, Farina V (2006) J Org Chem 71:5031–5034
Åkerbladh L, Nordeman P, Wejdemar M, Odell LR, Larhed M (2015) J Org Chem 80:1464–1471
Lu S-M, Alper H (2005) J Am Chem Soc 127:14776–14784
Connolly DJ, Cusack D, O’Sullivan TP, Guiry PJ (2005) Tetrahedron 61:10153–10202
Horton DA, Bourne GT, Smythe ML (2003) Chem Rev 103:893–930
Bergman J, Witt A (2003) Curr Org Chem 7:659–677
Tilley JW, Coffen DL, Schaer BH, Lind J (1987) J Org Chem 52:2469–2474
Chen J, Natte K, Spannenberg A, Neumann H, Langer P, Beller M, Wu X-F (2014) Angew Chem Int Ed 53:7579–7583
Xu T, Alper H (2015) Org Lett 17:1569–1572
Cacchi S, Fabrizi G, Pace P, Marinelli F (1999) Synlett 1999:620–622
Dai GX, Larock RC (2002) J Org Chem 67:7042–7047
Larksarp C, Alper H (1999) J Org Chem 64:9194–9200
Larksarp C, Alper H (2000) J Org Chem 65:2773–2777
Zeng F, Alper H (2010) Org Lett 12:1188–1191
Zeng F, Alper H (2010) Org Lett 12:3642–3644
Zheng Z, Alper H (2008) Org Lett 10:829–832
He L, Li H, Neumann H, Beller M, Wu X-F (2014) Angew Chem Int Ed 53:1420–1424
Zheng Z, Alper H (2008) Org Lett 10:4903–4906
Okuro K, Alper H (2012) J Org Chem 77:4420–4424
Ardizzoia GA, Beccalli EM, Borsini E, Brenna S, Broggini G, Rigamonti M (2008) Eur J Org Chem 2008:5590–5596
Kang S-K, Kim K-J (2001) Org Lett 3:511–514
Chouhan G, Alper H (2008) Org Lett 10:4987–4990
Kadnikov DV, Larock RC (2003) J Organomet Chem 687:425–435
Kadnikov DV, Larock RC (2004) J Org Chem 69:6772–6780
Tadd AC, Matsuno A, Fielding MR, Willis MC (2009) Org Lett 11:583–586
Cacchi S, Fabrizi G, Marinelli F (1996) Synlett 1996:997–998
Larksarp C, Alper H (1999) Org Lett 1:1619–1622
Salvadori J, Balducci E, Zaza S, Petricci E, Taddei M (2010) J Org Chem 75:1841–1847
Acs P, Muller E, Rangits G, Lorand T, Kollar L (2006) Tetrahedron 62:12051–12056
Wu X-F, Schranck J, Neumann H, Beller M (2011) Chem Eur J 17:12246–12249
Li W, Wu X-F (2014) J Org Chem 79:10410–10416
Konishi H, Nagase H, Manabe K (2015) Chem Commun 51:1854–1857
Xiao WJ, Alper H (1999) J Org Chem 64:9646–9652
Rescourio G, Alper H (2008) J Org Chem 73:1612–1615
Brown S, Clarkson S, Grigg R, Thomas WA, Sridharan V, Wilson DM (2001) Tetrahedron 57:1347–1359
Fusano A, Sumino S, Nishitani S, Inouye T, Morimoto K, Fukuyama T, Ryu I (2012) Chem Eur J 18:9415–9422
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Li, W., Wu, XF. (2015). Palladium-Catalyzed Carbonylative Synthesis of Six-Membered Heterocycles from Aryl Halides. In: Wu, XF., Beller, M. (eds) Transition Metal Catalyzed Carbonylative Synthesis of Heterocycles. Topics in Heterocyclic Chemistry, vol 42. Springer, Cham. https://doi.org/10.1007/7081_2015_150
Download citation
DOI: https://doi.org/10.1007/7081_2015_150
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-24961-2
Online ISBN: 978-3-319-24963-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)