
Abstract
After a short presentation of major variants of nucleophilic substitution of hydrogen, application of these reactions to introduction of substituents into aromatic and heteroaromatic rings and construction of heterocyclic systems are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Alkyl hydroperoxides
- Amination
- Ammonia
- Benzimidazole
- Benzisoxazole
- Carbanions
- Heterocycles
- Hydroxylation
- Indole
- Nitro compounds
- Nucleophiles
- Nucleophilic substitution
- Oxidation
- Phenazine
- Potassium permanganate
- Pyridine
- Quinoline
- Sulfones
- Vicarious
1 Introduction
Nucleophilic substitution of hydrogen in electron-deficient arenes is presently a well-established process proceeding in a few ways [1–5]. Although these reactions are of general character and great practical value for organic synthesis, particularly of heterocyclic systems, they have not been adequately recognized. The key step of these reactions is a fast and reversible addition of nucleophiles to the electron-deficient aromatic rings in positions occupied by hydrogen to form the so-called σH adducts. It should be stressed that when the electron-deficient rings contain halogens or other nucleofugal groups X in similarly activated positions, addition of nucleophilic agents in these positions, to form σX adducts, proceeds slower than formation of σH adducts. The relation of rates is shown in Scheme 1.
The initially formed σH adducts can be converted into products of nucleophilic substitution of hydrogen in a variety of ways: oxidation with external oxidants, conversion into nitrosoarenes according to intramolecular redox stoichiometry, vicarious substitution, cine- and tele-elimination, ANRORC, etc. These processes have been discussed in a concise way in our preceding reviews [4, 6–10]. The major message of those reviews is that nucleophilic substitution of hydrogen, in its many variants, is the main, primary process, whereas the conventional nucleophilic substitution of halogens X, the SNAr process, is just a secondary “ipso” reaction [9, 10].

Scheme 1
Here we intend to present a more detailed discussion of the three major ways of conversion of the σH adducts into the corresponding products of nucleophilic substitution of hydrogen in nitroarenes, particularly in electron-deficient heterocyclic systems, namely: vicarious nucleophilic substitution (VNS), oxidative nucleophilic substitution (ONSH), and conversion into nitrosoarenes according to intramolecular redox stoichiometry. Our main goal is to show that these reactions offer wide possibilities for the synthesis and modifications of heterocycles.
2 Vicarious Nucleophilic Substitution of Hydrogen
Amongst many variants of conversion of the σH adducts into the corresponding SNH products, the vicarious nucleophilic substitution is undoubtedly considered as one of the most versatile and practically important processes [4, 6]. It proceeds when nucleophiles contain nucleofugal groups L at the nucleophilic centers, as, for instance, in the case of α-halocarbanions. Addition of α-halocarbanions to nitroarenes in the ortho- or para-positions occupied by hydrogen results in the formation of σH adducts, which undergo base-induced β-elimination of HL to produce nitrobenzylic carbanions of the SNH products, isolated upon protonation. Since α-halocarbanions generated from substituted α-chloroalkanenitriles, carboxylic esters, etc. are rather unstable, the carbanion of chloromethyl phenyl sulfone has been chosen as the model nucleophile for investigation of the VNS reactions. Indeed, in the presence of a strong base, this carbanion reacts with nitrobenzene bearing a variety of substituents to replace hydrogen in the ortho- and (or) para-positions relative to the nitro group. The products, ortho- (or para-) nitrobenzyl phenyl sulfones, exist in the reaction media in the form of nitrobenzylic carbanions, which are not electrophilic anymore thus, the reaction proceeds exclusively as monosubstitution. It was shown that ortho- and para-halonitrobenzenes react with this carbanion according to the VNS pathway, resulting in displacement of hydrogen, without competing substitution of halogen (SNAr reaction). This can be exemplified by selective displacement of 2-hydrogen in 4-fluoronitrobenzene by action of chloromethyl phenyl sulfone under basic conditions (Scheme 2) [11, 12].

Scheme 2
However, under the conditions that favor dissociation of the σH adducts and disfavor β-elimination, namely, a higher temperature and absence of a strong base, the conventional SNAr of fluorine atom can be observed [13]. Interestingly, the reaction of meta-dinitrobenzene with an excess of this carbanion gave the disubstitution product, whereas with equimolar amounts of reactants only monosubstitution proceeds. It is evident that the anion of 2,4-dinitrobenzyl phenyl sulfone is still sufficiently active electrophile to react with the carbanion of chloromethyl phenyl sulfone (Scheme 3) [14].

Scheme 3
It was subsequently shown that carbanions generated from substituted α-chloroalkanenitriles [15] and alkyl α-chloroalkanoates [16, 17], chloroalkyl oxazolines [18, 19], chloroform [20], etc., although they are much less stable than the model sulfone carbanion, are able to react with nitroarenes to give the VNS products.
2.1 Mechanism
When analyzing plausible mechanisms of the VNS reactions of nitroarenes with α-chlorocarbanions, one should clarify a few key questions: how to proceed the addition and subsequent conversion of σH adducts and how other substituents may affect both of these steps – rate and orientation of the addition, rate of the elimination, etc. It is well known that nitroarenes are active electron acceptors, whereas carbanions are good electron donors; thus, these reactants can enter a single-electron transfer (SET) to form anion radicals of nitroarenes and radicals from carbanions [21, 22]. Further coupling of these electrophilic radicals with nucleophilic anion-radical species could give σH adducts. This SET pathway, alternative to the direct addition, is often favored by authors and the concept is sometimes abused, see [23] and rebuttal [24]. Nevertheless, numerous observations contradict participation of the SET mechanism in the VNS reactions:
-
Orientation of a nucleophile at the addition step can be efficiently controlled by the reaction conditions – namely, addition of the potassium salts of carbanions in DMF and DMSO proceeds in para- and ortho-positions, whereas in THF it occurs preferentially at the ortho-position to the nitro group, because a carbanion in the form of a tight ion pair with K+ cation is attracted to the ortho-position due to interaction of K+ with oxygen of the nitro group [25]. Such attraction could not operate in case of non-charged radicals, eventually produced from the carbanions via the SET mechanism.
-
The σH adducts of α-halocarbanions with nitroarenes can be detected by 1H NMR [26]. The 1H NMR spectra of a vast majority of σH adducts are well resolved and unambiguously interpreted, thus indicating that the formation of paramagnetic species via the SET mechanism is scarcely possible.
-
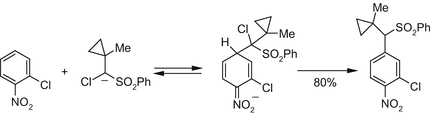
The most straightforward differentiation between the formation of σH adducts via two-step SET process and direct nucleophilic addition has been made by using the so-called “fast radical clock”. It has been shown that the carbanion of chloro(1-methylcyclopropyl)methyl phenyl sulfone reacts with nitroarenes along the VNS pathway without any complications (Scheme 4), whereas the ring opening rearrangement of the corresponding radical, generated separately, has been shown to proceed with a very high rate constant ~109 [27].
Scheme 4

The way of conversion of the σH adducts into the VNS products was clarified by taking into account the effects of strength and concentration of base on the rate of the reaction and also by measuring the kinetic isotope effect (KIE) of the reaction. Many observations have indicated that the rate of VNS is affected by the base concentration – mainly on the basis of intramolecular competition between VNS and SNAr of fluorine, as it has been observed, for instance, for para-fluoronitrobenzene (Scheme 2) [13]. For detailed studies of the competition between replacement of 2-F and 6-H, 2-F and 6-D, and 2-H and 6-D, respectively, 2-fluoro-4-bromo-nitrobenzene was chosen as the model compound (Scheme 5). These studies have revealed that kH/kF is a function of base concentration at low base concentrations whereas it appears to be constant at high base concentrations [28, 29]. Thus, at a low base concentration the dissociation of σH adducts does occur faster than the base-induced β-elimination, the system equilibrates, and the β-elimination is the rate-limiting step. On the other hand, at a high base concentration the σH adducts, once formed, undergo a fast β-elimination. Therefore, it is the addition that becomes the rate-limiting step and the k H/k F observed is equal to the ratio of the addition rates. These observations and conclusions are also in agreement with the value of k H/k D. KIE at low base concentrations proved to be ~4, whereas at high base concentrations the secondary KIE equal to ~0.9 was observed. The significant value of the primary KIE confirms that the second step, the base-induced β-elimination, is the rate-determining one, whereas the secondary KIE shows that the nucleophilic addition associated with sp 2 to sp 3 rehybridization of the carbon atom appears to be the rate-determining step at high base concentrations [29]. Thus, subtle features of the mechanism of VNS were clarified. Also it should be mentioned that σH adducts were observed by UV–VIS spectroscopy, which enabled to monitor the formation of the σH adducts in the reaction mixtures and their conversion into nitrobenzylic carbanions, the final VNS products [26].

Scheme 5
The effect of substituents on the rate of addition of carbanions to nitroarenes and the rate of β-elimination of HL from the σH adducts have also been studied [8, 30, 31]. The former effect is an important parameter, because it is, in fact, a measure of influence of substituents on electrophilic activity of nitroaromatic rings. The effect of substituents on rate of the SNAr reactions of o- and p-halonitrobenzenes has been thoroughly studied [2, 32]. However, since the SNAr of halogen is a secondary process, the obtained data cannot be used as a real measure of electrophilicity of halonitroarenes. We have determined the effects of substituents and the ring structure on the rate of the VNS reaction of nitroarenes with the carbanion of chloromethyl phenyl sulfone by using competitive experiments under the conditions, which assure a fast β-elimination of HL from the σH adducts [30, 31]. The values of VNS rates obtained under such conditions proved to correlate with those of the addition step. Selected values of the relative rate constants in relation to nitrobenzene as the standard are shown in Fig. 1.

Fig. 1
The VNS reactions always proceed in the ortho- or/and para-positions relative to the nitro group. When both of these positions are available for a nucleophilic attack, the ratio of ortho-/para-substitution is determined by the nature of carbanion and the reaction conditions. Less sterically demanding, secondary carbanions can react in both of these positions, whereas bulky tertiary carbanions react preferentially in the para-position to the nitro group. It has been however shown that even tertiary carbanions react preferentially in the ortho-positions under the kinetically controlled conditions, namely, at low temperatures and with a high concentration of a strong base [33]. On the other hand, it is the para-substitution pattern that dominates under thermodynamic control at higher temperature and low concentration of a weak base [30]. Several examples of the formation of kinetic and thermodynamic products depending on the reaction conditions are shown in Scheme 6.

Scheme 6
2.2 Scope of the Reaction
The VNS in nitroarenes with carbanions is presented in general in Scheme 7, thus, discussion of the scope and limitations of this reaction should clarify what kind of carbanions (nature of Y, L, and R) and nitroarenes (kind of Z) can enter the reaction.

Scheme 7
From the very mechanism it stems that L in the carbanions should be a nucleofugal group, which is able to be eliminated from the σH adducts as HL. Besides halogen atoms (Cl, Br), alkoxy, aryloxy, alkylthio, and arylthio groups and many other substituents can be eliminated in this way. There are practically no limitations concerning substituents R and groups Y, stabilizing the carbanions. Thus, the VNS is of wide scope, as far as carbanions are concerned. Similarly, the reaction has no restrictions in respect of substituents Z in the nitroaromatic ring, which might be halogens, alkoxyl, aryl, and any functional group. In fact, it is sufficient for the nitroarene ring to have at least one position, ortho- or para-, to the nitro group, which is occupied by hydrogen atom – and this hydrogen can be replaced with a functionalized carbon substituent via VNS. Only substituents that under highly basic reaction conditions are deprotonated into anions directly conjugated with an aromatic ring hinder the VNS reaction, because such rings are not electron-deficient anymore. Indeed, mono-nitrophenols and mono-nitrothiophenols fail to enter VNS reaction; however, dinitrophenols react satisfactorily [14]. This is the reason why the VNS proceeds in mononitroarenes selectively as monosubstitution, whereas dinitroarenes can form products of mono- and disubstitution.
A serious limitation of VNS is connected with its mechanism, namely, conversion of intermediate σH adducts into the VNS products via bimolecular base-induced β-elimination. To cause the reaction, it is therefore necessary that these σH adducts be produced in a reasonable concentration. Indeed, low nucleophilic carbanions, such as dimethyl chloromalonate, do not react with moderately electrophilic nitrobenzene because of unfavorable equilibrium of the addition step, but react nicely with more electrophilic nitrothiazoles (Scheme 8) [34].

Scheme 8
A plethora of electron-deficient arenes can enter the VNS reaction: carbocyclic and heterocyclic aromatic compounds activated by the nitro group and arenes that are active electrophiles due to their electronic configuration, such as azulene [35, 36], electron-deficient annulenes [37], tropylium cation [38], and particularly azines and azinium cations. Interestingly, η6-transition metal complexes of arenes, such as benzene tricarbonylchromium, do not enter the VNS reactions. Although the addition of carbanions to these electron-deficient rings proceeds efficiently, and these adducts can be oxidized to form the products of ONSH, the β-elimination of HCl from the σH adducts of α-halocarbanions does not occur [39, 40].
Besides carbanions, oxygen and nitrogen nucleophiles containing nucleofugal groups at the O- and N-nucleophilic centers can provide efficient hydroxylation and amination of nitroarenes according to the VNS pathway. Thus, anions of commercially available tert-butyl and cumyl hydroperoxides, although being moderately active nucleophiles, are able to add to nitroarenes of sufficient activity to form σH adducts. Subsequent base-induced β-elimination of the corresponding alcohols followed by protonation affords o- and p-nitrophenols [41]. Also in these cases the nucleophilic substitution of hydrogen proceeds faster than the conventional SNAr of halogen. For instance, anions of both of these hydroperoxides react with 2,4-dinitrochlorobenzene to produce 2,4-dinitro-5-chlorophenol in excellent yield (Scheme 9) [41, 42].

Scheme 9
Amination of nitroarenes with hydroxylamine, known for over 100 years [43], proceeds undoubtedly according to the VNS mechanism. Modern aminating agents, such as 4-amino-1,2,4-triazole [44–46], sulfenamides [47, 48], and O-methyl hydroxylamine [49, 50], are more versatile and efficient than hydroxylamine. 1,1,1-Trimethyl hydrazinium iodide proved to be particularly useful for this purpose [51–53]. Amination with this reagent proceeds via addition of the hydrazino moiety followed by a base-induced β-elimination of trimethylamine from the corresponding σH adducts.
Nitro derivatives of five-membered heterocycles are active partners in the VNS reactions [54]. It is interesting to compare the results of the VNS reactions with the model carbanion of chloromethyl phenyl sulfone, obtained for the following series of compounds: 2-nitrothiophene, N-methyl-2-nitropyrrole, and 2-nitrofuran (Scheme 10) [55]. Although in these 2-nitroheterocycles the position 3 appears to be the preferred addition site, a low yield of the VNS product derived from the reaction of 2-nitrofuran and the formation of 5-isomer from N-methyl-2-nitropyrrole requires rationalization [54]. These results indicate that the orientation is affected by conjugation of the electron pairs of these heteroatoms with the nitro group. Indeed, when 2-nitropyrrole was N-protected by phenylsulfonyl group (Z=NSO2Ph) to prevent such conjugation, the VNS reaction proceeded exclusively in position 3 [55]. The addition to 2-nitrofuran takes place in both 3- and 5-positions; however, decomposition of the σH adducts at C-5 via the ring opening appears to proceed faster than β-elimination, thus giving rise to the only product derived from the VNS in the position 3 [54].

Scheme 10
This reasoning is supported by the observation that the reaction of 2-nitrofuran with trichloromethyl carbanion proceeds in both 3- and 5-positions, because in this case the base-induced β-elimination of HCl from the intermediate σH adducts is a fast process [54]. Also the VNS reactions of nitro derivatives of other 5-membered heterocycles, imidazoles [20, 56] and thiazoles [34], with a variety of α-halogeno carbanions have been shown to proceed efficiently.
Pyridine is known to exhibit a significant electron-deficient character, thus being able to add strong nucleophiles, such as alkyl lithium or amide anion. However, its electrophilic activity is not sufficient to add moderately active nucleophiles, such as α-halocarbanions. On the other hand, all isomeric 2-, 3-, and 4-nitropyridines react smoothly with the carbanion of chloromethyl phenyl sulfone [57] and a variety of other α-halocarbanions [58–60], thus giving the expected VNS products. Electrophilic activity of the pyridine ring is strongly enhanced via the formation of pyridinium salts that can be exploited in intramolecular VNS reactions, leading to isothiazolo[4,3-b]pyridines [61]. Also N-pyridyl dicyanomethylides enter the VNS reaction with a variety of α-halocarbanions [62]. Some azines are active electrophiles per se and do not need activation by electron-withdrawing substituents. A convincing example is 1,2,4-triazine in which all three positions 3, 5, and 6 are vulnerable for a nucleophilic attack. The most active position in the 1,2,4-triazine ring is 5, then 3, and 6 [63, 64].
Some peculiar observations were made when the model carbanion of chloromethyl phenyl sulfone was reacted with quinoxaline (Scheme 11) [65]. Instead of the expected VNS product, the bis-aziridine derivative was formed. It was suggested that in the initially formed σH adducts the negative charge was mostly located on the vicinal nitrogen atom, thus facilitating the 1,3-intramolecular nucleophilic substitution, leading to mono-aziridine derivative [65]. Addition of the second molecule of the same carbanion to the C=N bond proceeds faster than the first addition to aromatic quinoxaline system, thus giving bis-aziridine compound as the final product. This reasoning was substantiated by the reaction of quinoxaline N-oxide with the same model carbanion, proceeding along the VNS pathway, because the negative charge of the σH adducts was located mostly on the oxygen atom (Scheme 11) [65].

Scheme 11
A variety of other azaaromatic compounds, pteridines [66], pyridazines [67] etc., enter the VNS reactions with the model carbanion of chloromethyl phenyl sulfone or other α-chlorocarbanions. In these reactions azine N-oxides are more active electrophiles than azines themselves. For instance, quinoline fails to enter the VNS reaction with the model carbanion, whereas quinoline N-oxide reacts rather smoothly [68]. Also 3-(chloromethylsulfonyl-amino)pyridine-N-oxide and its quinoline analogue are able to undergo intramolecular VNS reactions [61].
3 Oxidative Nucleophilic Substitution of Hydrogen
Since hydride anions are unable to depart spontaneously from the anionic σH adducts, they should be removed by external oxidants. However, possibilities for conversions of the σH adducts into products of oxidative nucleophilic substitution of hydrogen (ONSH) appear to be limited, since nucleophiles, and particularly carbanions, are usually sensitive to oxidation. Thus, ONSH can be feasible in two major cases:
-
a.
Nucleophiles are resistant toward oxidation.
-
b.
Addition of nucleophiles to electron-deficient rings, affording σH adducts, proceeds to completion.
Indeed, ONSH proceeds efficiently with nucleophiles resistant toward oxidation, such as hydroxide anion and ammonia. Many textbooks on organic chemistry describe the “hydrolysis” of para-chloronitrobenzene on heating with aqueous KOH, which in fact is the SNAr reaction, proceeding via intermediacy of σCl adducts. However, when this nitroarene is exposed to KOH and oxygen at low temperature in liquid ammonia, 2-nitro-5-chlorophenol is formed in high yield (Scheme 12) [69].

Scheme 12
Thus, it is evident that σH adducts with the hydroxide anion are formed much faster than isomeric σCl adducts, and at low temperature these species are long-lived enough to be oxidized by oxygen. A similar situation appears to occur in the reaction of halonitroarenes with ammonia. Since KMnO4 is well soluble in liquid ammonia, such solution can be used for oxidative amination. The system KMnO4/liquid ammonia proved to be a very useful tool for amination of electron-deficient heterocycles – the procedure, which is often termed as oxidative version of the Chichibabin reaction [70]. Also it has been unambiguously shown that ONSH is faster than the conventional SNAr of halogen, as illustrated in Scheme 13 [71].

Scheme 13
There are two major variants of ONSH with nucleophiles sensitive to oxidation: (a) addition is an irreversible process; and (b) equilibrium of the reversible addition is shifted in favor of the σH adducts. Nucleophilic organometallic compounds, alkyllithium and alkyl-magnesium reagents, are active enough to add irreversibly to nitroarenes in positions occupied by hydrogen to form the σH adducts [72]. Due to irreversibility of the addition, the SNAr reaction on treatment of ortho- and para-halonitrobenzenes with these C-nucleophiles is not observed. Further oxidation of the formed σH adducts with a variety of oxidants, preferably KMnO4, affords products of oxidative nucleophilic alkylation. This reaction appears to be an important method for direct incorporation of alkyl substituents into aromatic rings (Scheme 14) [72, 73].

Scheme 14
Equilibrium of the addition of nucleophiles to nitroarenes is a function of many factors, such as their nucleophilicity, electron deficiency of arenes, and their ability to stabilize σH adducts, as well as the reaction conditions. Thus, all these parameters are responsible for the feasibility of ONSH with nucleophiles sensitive to oxidation. Of substantial importance is temperature, since, due to the entropy factor, the equilibrium is shifted toward the adducts at a low temperature. For instance, addition of highly nucleophilic carbanion of 2-phenylpropionitrile to moderately active m-chloro nitrobenzene at −70°C in liquid ammonia or DMF/THF proceeds to completion, selectively in the para-position. Further oxidation of the formed σH adducts with KMnO4 in liquid ammonia, or with dimethyldioxirane in THF, gave 2-phenyl-2-(para-nitrophenyl)- and 2-phenyl-2-(para-hydroxyphenyl)-propionitriles, respectively (Scheme 15) [74, 75].

Scheme 15
The effect of temperature on the addition equilibrium can, for instance, be observed in the reaction of the carbanion of diethyl benzylphosphonate with 4-fluoronitrobenzene. At low temperature the addition proceeds exclusively at the position 2, and oxidation of the produced σH adduct affords the product of ONSH. On the other hand, at room or a higher temperature the SNAr of fluorine in the position 4 takes place [76]. Similarly, when the reaction of nitroarenes with the anion of diphenylphosphine is carried out at low temperature in liquid ammonia in the presence of KMnO4 diphenyl(nitroaryl)phosphine oxides are formed, as illustrated by the ONSH in 4-fluoronitrobenzene (Scheme 16) [77].

Scheme 16
The σH adducts of nitroarenes with various nucleophiles can be oxidized with a few oxidants, and oxygen is probably the most common oxidant, although it has a limited application. It oxidizes σH adducts resulted from the addition of OH− anion to nitroarenes to produce nitrophenols and also σH adducts of secondary and primary carbanions. Some observations and experiments lead to conclusion that for oxidation by oxygen the anionic σH adducts should first be deprotonated, so in fact, dianions are oxidized [78]. Oxidation of such σH adducts with oxygen appears to proceed via an electron transfer. On the other hand, oxidation of the σH adducts of nitroarenes with ammonia, the Grignard reagents, various carbanions, or diphenylphosphine by action of KMnO4 appears to proceed via direct abstraction of the hydride anion, as is suggested by high value of the kinetic isotope effect of the oxidation [79]. Oxidation of such σH adducts with dichlorodicyanoquinone (DDQ) also appears to proceed via abstraction of the hydride anions.
4 Conversion of σH Adducts into Nitrosoarenes
The third general way of converting the σH adducts of nucleophiles to nitroarenes involves elimination of water or other small molecules to form substituted nitrosoarenes, according to intramolecular redox stoichiometry. For example, phenylacetonitrile and other arylacetonitriles react with nitroarenes in the presence of KOH in protic media to form nitrosoarenes or products of their further transformations (Scheme 17) [80, 81].

Scheme 17
It appears that the reaction proceeds via protonation of the intermediate anionic σH adducts followed by elimination of water to form nitrosoarenes. Since nitrosoarenes are very active electrophiles, they can undergo further transformations by action of nucleophilic agents and usually are not isolated as such. However, when conversion of σH adducts is carried out as a separate step without base and nucleophiles, the substituted nitrosoarenes might be isolated, often in good yields. Due to high activity of the nitroso group in inter- and intramolecular reactions, this way of conversion of σH adducts becomes a versatile tool for organic synthesis, in particular for obtaining of heterocycles. For instance, treatment of a mixture of 3-phenylallyl phenyl sulfone and 6-methoxy-3-nitropyridine with DBU and t-butyldimethylsilyl chloride results in the formation of substituted naphthyridine (Scheme 18) [82]. The reaction proceeds via addition of the sulfone carbanion followed by conversion of the intermediate σH adducts into nitrosoarene and subsequent intramolecular condensation of the newly generated ambident carbanion with the formed nitroso group.

Scheme 18
Reactions of nitroarenes with anilines in the presence of a strong base, proceeding via intermediacy of the corresponding σH adducts (Scheme 19), are of particular interest since they provide a synthetic way to valuable 2-nitrosodiarylamines [83–86]. Thus, when p-chloro- or p-fluoronitrobenzene was reacted with anilines in the presence of t-BuOK in THF at low temperature (−60°C), 2-nitroso-5-chloro(or fluoro)phenyl arylamines were obtained in good yields (Scheme 19) [85]. Competing SNAr of halogen was not observed under these conditions. It should be mentioned that a simple mixing of these p-halonitrobenzenes with anilines at elevated temperatures results in SNAr of halogen [87].

Scheme 19
The reaction of 1-nitronaphthalene and other bicyclic nitroarenes, for instance, 5-nitroquinoline, with dimethyl phosphite in methanol in the presence of sodium methoxide proceeds via formation of the corresponding σH adducts with the phosphite anion, which are converted into substituted nitrosoarenes. Subsequent N-deoxygenation results in the formation of nitrenes that react further to give benzazepines and analogues (Scheme 20) [88].

Scheme 20
5 Introduction of Substituents into Electron-Deficient Heterocycles via Nucleophilic Substitution of Hydrogen
In the following sections we will present some recent examples of introduction of various substituents into heteroarenes via nucleophilic substitution of hydrogen. The full account of the previous results was given in our preceding reviews [4, 89, 90].
5.1 Carbon Substituents
The introduction of carbon substituents into electron-deficient aromatic and heteroaromatic rings is of great importance because products can be of interest per se and can also serve as valuable intermediates in further synthesis, particularly in heterocyclizations.
Alkyl substituents can be incorporated directly into nitroheteroaromatic rings via the VNS reactions with carbanions of alkyl trifluoromethyl sulfones (Scheme 21) [91].

Scheme 21
Diarylmethylation of nitroarenes can be performed efficiently via VNS, using carbanions of benzhydryl aryl sulfides [92]. Similarly, the VNS reaction of 4-ethoxy-3-nitropyridine with carbanion of 9-chlorofluorene results in incorporation of the fluorenyl fragment into the pyridine ring (Scheme 22) [93]. Also heteroarylmethyl substituents were introduced into nitroarenes via the VNS reactions with carbanions of chloromethyl derivatives of pyridine, thiazole, and benzothiazole [94].

Scheme 22
Alkyl substituents can be introduced into heterocyclic rings also via direct ONSH reaction with the Grignard reagents [72] or via the VNS reaction with carbanions of α-chloroalkyl carboxylic esters [17], followed by hydrolysis and decarboxylation [95–97]. For example, treatment of 5-nitroisoquinoline with the carbanion of ethyl chloroacetate under the standard VNS condition in DMF in the presence of t-BuOK gave the expected (5-nitroisoquinol-6-yl)acetate, which was transformed into 6-methyl-5-nitroisoquinoline via hydrolysis and decarboxylation (Scheme 23) [95].

Scheme 23
Synthesis of a variety of phenylethynylazines can be performed by using the methodology of direct nucleophilic replacement of hydrogen in the reactions of azine N-oxides with the carbanion of phenylacetylene (Scheme 24) [98].

Scheme 24
An exceptional example of introduction of alkenyl substituent via nucleophilic substitution of hydrogen is phenylethenylation of boron-dipyrromethene (BODIPY), which has been realized in the reaction of the latter with β-nitrostyrene catalyzed by the phenylthiolate anion (Scheme 25) [99]. The initial step of the reaction is the Michael-type addition of thiolate to nitrostyrene to form the nitronate anion which adds to the electron-deficient pyrrole ring, followed by base-induced elimination of nitrous acid. In the final step, elimination of the thiolate results in incorporation of phenylvinyl substituent and regeneration of the catalyst.

Scheme 25
Even weak C-nucleophiles, such as 2-nitropropenide anion, are able to add quantitatively to superelectrophilic nitrobenzofurazan and nitrobenzofuroxan. Further oxidation of the intermediate σH adducts with ammonium cerium(IV) nitrate (CAN) results in incorporation of α-nitroisopropyl substituent into these heterocyclic systems (Scheme 26) [100].

Scheme 26
We have described a two-step method for introduction of chloromethyl substituents into nitroarenes. This approach consists in the VNS of hydrogen with tert-butyl dichloroacetate anion [58] followed by one-pot hydrolysis and decarboxylation [101]. This approach has been used for the synthesis of (chloromethyl)nitroimidazole, a precursor of (nitroheteroaryl)methyl mustard, which was tested as hypoxia-selective cytotoxins (Scheme 27) [102].

Scheme 27
On the other hand, direct dihalomethylation of electron-deficient arenes via VNS with trihalomethyl carbanions generated by deprotonation of haloforms is a general process [20]. Due to facile hydrolysis of the dihalomethyl group, the reaction can be considered as nucleophilic formylation. This approach has already found wide application in the synthesis of heterocyclic aldehydes, which are difficult to obtain by other methods [103–105]. For example, the VNS reaction of 5-nitroquinoline with tribromomethyl carbanion affords 6-(dibromomethyl)-5-nitroquinoline, which is transformed by hydrolysis into the corresponding aldehyde, the starting material for the synthesis of biologically active coumarin derivative (Scheme 28) [103].

Scheme 28
The VNS of hydrogen in 1-benzyl-4-nitroimidazole by action of trichloromethyl carbanion results in the formation of 5-dichloromethyl derivative (Scheme 29). Hydrolysis and condensation of the resulting aldehyde with diethyl malonate afford the corresponding alkene that, upon reduction of the nitro group, undergoes cyclization into imidazopyridone [106].

Scheme 29
The synthesis of fluoroalkyl-substituted heterocycles is a subject of continuous interest; this challenging issue has been presented in details in reviews [107, 108]. It has been shown that trifluoromethyl carbanion, generated from (trifluoromethyl)trimethylsilane (the Ruppert reagent), adds easily to 2-chloro-3-nitropyridine. The produced σH adducts can be oxidized with dimethyldioxirane (DMD) to form two isomeric 2-chloro-4-(and 6-)trifluoromethyl-3-hydroxypyridines (Scheme 30) [109].

Scheme 30
Similarly produced trifluoromethyl carbanion, as well as perfluoroisopropyl carbanion generated by addition of fluoride anion to perfluoropropene, is able to add to a variety of N-(4-methoxy-benzyl) pyridinium and quinolinium salts. The obtained dihydroazines can easily be transformed into 2-perfluoroalkyl azines through oxidative dealkylation–aromatization with CAN or DDQ (Scheme 31) [110, 111].

Scheme 31
The VNS is the reaction of choice for incorporation of α-sulfonylalkyl substituents into nitroarenes and their heteroanalogues. Particularly accessible and useful are nitroarylmethyl phenyl sulfones and their heteroanalogues that are efficiently produced in the VNS reactions of carbanions of chloromethyl aryl sulfones with a great variety of nitroarenes and nitroheteroarenes. Nitro derivatives of heterocycles, such as pyrrole [54, 55], furan [54], thiophene [54], imidazole [106, 112, 113], pyrazole [114], pyridine [57], indole [115], indazole [116, 117], benzimidazole [118], benzotriazole [119], benzofuroxan [120], quinoline [121], and porphyrins [122, 123], have been shown to enter this reaction.
In search for new antiparasitic agents active against Trichomonas vaginalis, a number of 4-(arylsulfonylmethyl)-5-nitroimidazole derivatives have been prepared via the VNS reaction of 4-nitroimidazoles with substituted aryl chloromethyl sulfones (Scheme 32) [124].

Scheme 32
Introduction of arylsulfonylmethyl substituents into nitroheteroaromatic rings is of great practical value because these sulfones are versatile intermediates in organic synthesis. Nitrobenzyl aryl sulfones and their heterocyclic analogues can easily be transformed into the corresponding ethenyl derivatives by a simple alkylation with simultaneous elimination of arylsulfinate anion [125]. Diethyl methylenemalonate substituent can be introduced in the position 4- of 5-nitroimidazole via the VNS reaction of 5-nitroimidazole with the carbanion of chloromethyl phenyl sulfone [112, 124], followed by condensation of the obtained 4-(phenylsulfonyl)methyl derivative with diethyl bromomalonate or diethyl ketomalonate (Scheme 33) [126].

Scheme 33
Azolopyridazines bearing no nitro substituent are, nevertheless, sufficiently active electrophiles to enter the VNS reaction. However, similar to the series of quinoxalines [127] and pyridazinones [67], in the reactions of azolopyridazines with the carbanion of bromomethyl phenyl sulfone, two ways for conversion of the intermediate σH adducts are observed, depending on the structure of these heterocyclic compounds – β-elimination, leading to the VNS product, or intramolecular substitution, resulting in formation of the cyclopropane ring (Scheme 34) [128].

Scheme 34
The monochlorobenzosultam carbanion, generated through symproportionation of an equimolar mixture of benzosultam and its dichloro derivative, is capable of addition to 2-chloro-3-nitropyridine. Subsequent elimination of HCl from the intermediate σH adduct affords 3-(pyridin-2-yl)-substituted benzosultam according to the VNS mechanism (Scheme 35) [129].

Scheme 35
3-Nitroimidazo[1,2-a]pyridine reacts smoothly with the carbanion of ethyl chloroacetate to give the expected VNS product bearing ethoxycarbonylmethyl substituent at position 2 (Scheme 36) [130].

Scheme 36
Oxidative nucleophilic substitution of hydrogen in the reactions of nitroarenes with carbanions of protected amino acids offers an access to α-(nitroaryl)amino acids and their heteroanalogues. According to this protocol, nitropyridines react with carbanions of protected alanine, serine, and threonine esters to give the corresponding nitropyridyl α-amino acids [131–133]. For example, N-(1,3-dithiolane-2-ylidene)alanine isopropyl ester, which is readily available from the reaction of alanine ester with carbon disulfide and 1,2-dibromoethane, adds to 2-chloro-3-nitropyridine in THF in the presence of t-BuOK to form the σH adduct that upon oxidation with DDQ and hydrolysis gives the corresponding ester of α-nitroarylalanine (Scheme 37) [132].

Scheme 37
Also carbanions of serine and threonine esters, protected in the form of oxazolines, are capable of addition to nitropyridines to form the corresponding σH adducts that can be oxidized into α-(nitropyridyl) amino acid derivatives [132]. It should be mentioned that addition of the carbanion of the protected threonine to nitropyridine proceeds with a high diastereoselectivity, which is controlled by the second chiral center present in the oxazoline ring (Scheme 38) [133].

Scheme 38
Under much milder conditions, oxidative substitutions in BODIPY’s with malonic esters, acetophenone, and ethyl phenylacetate proved to take place (Scheme 39). For example, the reaction with tert-butyl malonate in the presence of potassium carbonate and oxygen proceeds at the position 3. When a twofold excess of nucleophile was used, 3,5-disubstituted product was obtained [134].

Scheme 39
1-Alkyl-5- and 1-alkyl-6-nitroindoles undergo the VNS substitution of hydrogen at the positions 4 and 7, respectively, by action of chloromethyl sulfones and (4-chlorophenoxy)acetonitrile to give the corresponding VNS products in high yields [115]. 1-Methoxy-6-nitroindole reacts in a similar manner, yielding the expected 7-indolylacetonitrile (Scheme 40) [135].

Scheme 40
3-Nitroimidazo[1,2-a]pyridine has been reported to react efficiently with carbanion of (4-chlorophenoxy)acetonitrile to give the expected VNS product containing the cyanomethyl group at position 2 (Scheme 41) [130].

Scheme 41
2-Arylphenylamines required for the synthesis of novel dopamine antagonists, containing the 1,3-benzodiazepine fragment [136], have been prepared from 5-chloro-4-methoxy-2-nitrophenylacetonitrile, derived from the reaction of 2-chloro-5-nitroanisole with cyanomethyl dimethyldithiocarbamate, proceeding via typical VNS procedure (Scheme 42) [15].

Scheme 42
Nitro derivatives of arylporphyrins, which contain the nitro group in the pyrrole rings, enter the VNS reaction with carbanions bearing a leaving group at α-position, thus giving the expected substitution products in good yields (Scheme 43) [123].

Scheme 43
5.2 Hydroxylation
Nitro derivatives of a variety of heteroaromatic compounds enter the VNS reactions with alkyl hydroperoxide anions to produce the expected hydroxylation products [41, 137–139]. For instance, the VNS hydroxylation of 2-chloro-5-nitropyridine with tert-butylhydroperoxide was shown to give 2-chloro-5-nitro-6-hydroxypyridine that exists in its tautomeric form of pyridone [41] (Scheme 44). It should be stressed that the SNAr of chlorine located in the highly activated position 2 was not competing with the VNS.

Scheme 44
The addition of nucleophiles to bicyclic heteroaromatic systems usually proceeds more easily; thus, activation of isoquinoline by the cyano group is sufficient for the VNS hydroxylation (Scheme 45) [41].

Scheme 45
tert-Butylhydroperoxide was used for the synthesis of 2-hydroxy-4-phenoxy-5-nitropyridine; the latter was subsequently converted into 18F-labeled PBR28 radiotracer (Scheme 46) [138].

Scheme 46
Also nitroquinolines undergo the direct methoxylation with potassium methoxide in THF, as exemplified in Scheme 47 [140].

Scheme 47
5.3 Amination
The VNS amination requires ammonia derivatives, bearing good leaving groups at nitrogen, as starting materials. Thus, derivatives of hydrazine (trimethylhydrazinium halides and 4-amino-1,2,4-triazole) and hydroxylamine (methoxyamine and arylsulfenamides) proved to be efficient aminating agents. The VNS amination of 3-nitropyridine and its substituted derivatives was observed to proceed efficiently with 4-amino-1,2,4-triazole [46, 60], hydroxylamine [46], and methoxyamine in the presence of zinc chloride [49]. Amination of 5-, 6-, 7-, and 8-nitroquinolines [141, 142], 4-nitroisoquinoline [46], 5- and 6-nitrobenzimidazoles [143], nitro-1,2,3- and nitro-1,2,4-triazoles [144], dinitropyrazole [145], dinitroquinazoline [146], and dinitroindazoles[147] with 1,1,1-trimethylhydrazinium iodide was also reported. Nitrophenyl fragments in porphyrins were aminated successfully with 2,4,6-trichlorophenyl sulfenamide [148, 149]. Triphenylporphyrin derivatives, in which the internal ring is activated by the carbonyl group, were aminated with 4-amino-1,2,4-triazole [150, 151]; similarly nitrocorroles were aminated, as shown in Scheme 48 [152].

Scheme 48
Oxidative amination of 3-nitropyridine with ammonia, alkyl- and dialkylamines, and KMnO4 as oxidant was reported by Bakke (Scheme 49) [60].

Scheme 49
The mechanism of oxidative alkylamination of 3-nitropyridine, quinazoline, and 1,3-dinitrobenzene with permanganate anion, including determination of the kinetic isotope effect for the oxidation step, was thoroughly studied [153]. Bis(pyridine)silver(I)permanganate AgPy2MnO4 [154–156] was found to be superior to KMnO4 for the reaction of higher alkyl and dialkylamines, inter alia, due to a low solubility of KMnO4 in these amines. However, in the presence of tetraalkylammonium chloride, which forms lipophilic tetraalkylammonium permanganate, the latter oxidant becomes equally active [153].
1,3,7-Triazapyrenes were aminated successfully into mono- and bis-dialkylamino compounds under mild conditions in aqueous solution with potassium hexacyanoferrate(III) as oxidant (Scheme 50) [157, 158].

Scheme 50
Aliphatic and cyclic dialkylamino groups can also be incorporated into 4-substituted-2-nitrothiophenes via the ONSH with dialkylamines and AgNO3, as oxidant (Scheme 51) [159].

Scheme 51
In oxidative amination of nitropyridines with 2-, 3-, and 4-aminopyridines, leading to N,N′-dipyridylamines, nitrobenzene proved to be effective as oxidant (Scheme 52) [160].

Scheme 52
3-Nitro-1,5-naphthyridines were oxidatively methylaminated in liquid methylamine as solvent in the presence of potassium permanganate [161]. Direct oxidative amination of 3-nitronicotinate with formanilide proceeds at the position 6 of the pyridine ring, resulting in the formation of anilinopyridine (Scheme 53) [162].

Scheme 53
An unusual reaction course was observed in the reaction of dialkylamines with 4-nitrofurazan [163]. Indeed, treatment of the latter with an excess of morpholine gave two products, one of them being derived from oxidative substitution of hydrogen in position 7, while the second, bis-morpholino compound, proved to be the result of the redox process (Scheme 54).

Scheme 54
Oxidative nucleophilic substitution of hydrogen in 2-chloro-3-nitropyridine by action of N-lithio-S,S-diphenylsulfilimines has been shown to be accompanied with the SNAr displacement of chloro atom (Scheme 55). Both of these products were oxidized with m-CPBA to form dinitropyridines [164, 165].

Scheme 55
6 Construction of Heterocyclic Compounds via Nucleophilic Substitution of Hydrogen
6.1 Indoles
The indole fragment is present in a great variety of biologically active compounds and other products of practical importance. It is no wonder that use of the reactions aimed at construction of indole skeleton is of significant value. Nucleophilic substitution of hydrogen opens a wide avenue for the synthesis of indoles, bearing a variety of substituents in both aromatic and heteroaromatic rings, as well as for obtaining of azaindoles and indoles condensed with other ring systems.
There are two key starting materials for the synthesis of indoles via nucleophilic substitution of hydrogen: meta-nitroaniline and its derivatives and nitroarenes or their heterocyclic analogues. In the first case nitrogen of the amino group is the precursor of the indole nitrogen, whereas in case of nitroaromatic compounds it is nitrogen of the nitro group.
The synthesis of 4- and 6-nitroindoles via the direct reaction of meta-nitroanilines with ketone enolates appears to be the simplest and the most efficient one in terms of atom economy. This method of the indole moiety construction, exemplified in Scheme 56, is of general character, considering ketones and meta-nitroanilines, which might bear a variety of substituents [166, 167]. This approach has enabled the synthesis of all kinds of substituted indole derivatives including cycloalkeno[b]indoles, tetrahydrocarbazoles, and tetrahydrocarbolines, when cyclic ketones were employed (Scheme 56) [167]. It should be mentioned that this versatile method can be applied to a large-scale synthesis [168].

Scheme 56
Despite simplicity and versatility of this new way of indole synthesis, there have been only few reports on application of this reaction to the synthesis of compounds of biological interest [168–170]. Thus, 2,3-dimethyl-4-nitroindole, obtained according to [167], was oxidized to nitroacetophenone derivative, which was used as a starting material for the synthesis of homocamptothecin derivatives, tested as potential inhibitors of DNA topoisomerase I (Scheme 57) [170].

Scheme 57
Similar heterocyclizations were shown to proceed between meta-nitroanilines and carbanions of alkanenitriles to produce 2-amino-4-(and -6-)nitroindoles. For example, the reaction of meta-nitroaniline with acetonitrile leads to 2-amino-4-nitroindole, while 6-nitroindole derivative is formed in the reaction with phenylacetonitrile (Scheme 58) [171].

Scheme 58
Both reactions proceed via the addition of carbanions to the nitroaromatic ring followed by oxidation of the intermediate σH adducts by atmospheric oxygen to form the corresponding nitrobenzyl nitriles, which undergo intramolecular cyclizations.
meta-Nitrobenzoisonitriles can readily be obtained from meta-nitroanilines. The VNS reaction of these isonitriles with carbanions of sulfones and nitriles, bearing good leaving groups, leads to ortho-isocyanobenzyl sulfones and cyanides, respectively. Under the reaction conditions intramolecular addition of the intermediate carbanions to the isocyano group takes place, resulting in the formation of substituted indoles (Scheme 59) [172].

Scheme 59
4-Nitro-2-oxo-2,3-dihydroindole derivatives (nitrooxindoles) can be obtained by intramolecular ONSH [173] and VNS [174] reactions of meta-nitroanilides of alkanoic and α-chloroalkanoic acids (Scheme 60).

Scheme 60
Introduction of functionalized carbon substituents in the ortho-position to the nitro group of nitroarenes provides even wider possibilities for the synthesis of indoles. One particularly useful pathway is direct cyanomethylation of nitroarenes with chloroacetonitrile or, more conveniently, aryloxyacetonitriles to produce ortho-nitroaryl acetonitriles that can further be converted into indoles in a few ways. It is worth to note that synthesis of indoles via catalytic hydrogenation of such nitriles has been well known for many years [175], however was of a limited value, because ortho-nitroaryl acetonitriles were not easily available. Facile synthesis of ortho-nitroaryl acetonitriles via the VNS methodology has opened a wide avenue to a variety of substituted indoles. Moreover, some halogen substituents (Cl, Br) in the nitroaromatic rings may not only improve effectiveness of the VNS reactions but also prevent introduction of cyanomethyl substituent into undesired positions [176, 177]. Such auxiliary substituents can be subsequently removed during hydrogenation. The general character and versatility of this approach to indoles is nicely illustrated by the synthesis of all isomeric 4-, 5-, 6-, and 7-methoxy-substituted indoles via the VNS cyanomethylation of isomeric nitroanisoles and their halogenated derivatives (Scheme 61) [176].

Scheme 61
Similarly, hydroxyindoles can be obtained via the VNS cyanomethylation of benzyl nitrophenyl ethers, eventually containing halogens in nitrophenyl rings followed by hydrogenation and simultaneous removal of the benzyl group and halogens [176, 177]. Recently the VNS cyanomethylation followed by hydrogenation has been used for synthesis of indoles containing pentafluorosulfanyl substituents (Scheme 62) [178].

Scheme 62
In the synthesis of eudistomins C and E, antiviral agents of marine origin containing 5-methoxyindole fragment, both indole units were prepared via vicarious nucleophilic substitution with aryloxyacetonitriles (Scheme 63) [179]. A proper choice of both nitroarene and cyanomethylating agent enabled synthesis of the prerequisite nitrophenyl acetonitriles to be performed. 4-Bromo-5-methoxy-2-nitrophenylacetonitrile, required for the synthesis of eudistomin C, was prepared via the VNS cyanomethylation of 2-bromo-4-nitroanisole with 2,4,6-trichlorophenoxyacetonitrile. The bulky leaving group in the carbanion appears to suppress substitution of hydrogen at sterically hindered position 3 in 2-bromo-4-nitroanisole. 6-Bromo-5-methoxyindole was synthesized by hydrogenation of this nitroaryl-substituted acetonitrile and then transformed in a few steps into eudistomin C [179]. A similar strategy has been used in the synthesis of eudistomin E from 2,6-dibromo-4-nitroanisole and 4-chlorophenoxyacetonitrile [179].

Scheme 63
Interestingly, the VNS reaction of 3-nitroanisole with 4-chlorophenoxyacetonitrile proceeds in the most hindered position 2 [176]. 2-Nitro-6-methoxyphenylacetonitrile obtained was then reduced and converted into 4-methoxyindole, which was then transformed in three steps into rapalexin A, an unusual isothiocyanate alkaloid derived from Brassica rapa (Scheme 64) [180].

Scheme 64
4-Hydroxyindole, a starting material for the synthesis of β-blockers, can be obtained analogously from 3-benzyloxynitrobenzene [176, 181]. Cyanomethylation of this nitroarene with 4-chlorophenoxyacetonitrile also proceeds at the position 2. Further catalytic hydrogenation of the obtained nitroarylacetonitrile provides 4-hydroxyindole [176]. Similarly, the VNS cyanomethylation of 3-benzyloxynitrobenzene with 4-chlorophenoxyacetonitrile containing 14C in the cyano group was used for the synthesis of 2-14C-labeled 4-hydroxyindole (Scheme 65), an intermediate for the synthesis of a pindolol analogue LY3688242 [182].

Scheme 65
Ortho-nitroaryl-substituted acetonitriles are relatively strong C–H acids, and their C-alkylation followed by hydrogenation leads to 3-substituted indoles [176]. It has been shown earlier that VNS in 2,4-dinitrophenol proceeds regiospecifically at the most hindered position 3 due to electronic configuration of the dinitrophenolate anion [14]. This orientation pattern has been employed for the synthesis of the precursor of damirone B from dinitroguaiacol, in which cyanomethylation proceeds exclusively at position 5 to form upon O-methylation 3,4-dimethoxy-2,6-dinitrophenylacetonitrile. Further alkylation of the nitrile carbanion with ethyl bromoacetate and hydrogenation provides the skeleton of damirone tricyclic system (Scheme 66) [183, 184].

Scheme 66
3,6-Dimethyl-5-methoxyindole prepared via the VNS cyanomethylation of 3-methyl-4-methoxynitrobenzene has been used as starting material for the synthesis of cyclopropano-annelated quinone methide, a reductive alkylating agent for in vitro studies of its interactions with deoxyguanosine-5′-monophosphate (Scheme 67) [185].

Scheme 67
The VNS cyanomethylation of 2-chloro-5-nitropyridine affords the corresponding nitropyridyl-substituted acetonitrile that undergoes hydrogenative cyclization into 5-chloro-6-azaindole (Scheme 68), a key starting material for the synthesis of potential Xa factor inhibitor [186].

Scheme 68
A similar VNS cyanomethylation of 3-nitropyridine and subsequent hydrogenation of the so-formed ortho-nitropyridyl-substituted acetonitriles provided 4- and 6-azaindoles. The VNS of hydrogen in 2-methoxy-5-nitropyridine with the carbanion of aryloxyacetonitrile leads to pyridylacetonitrile. Alkylation of the latter with bromoacetonitrile followed by a two-step reduction efficiently results in the formation of 5-azamelatonin (Scheme 69) [187]. Condensation of pyridyl-substituted acetonitriles with aromatic aldehydes followed by catalytic reduction gave 3-benzyl-4-azaindoles [187].

Scheme 69
There is one more way for conversion of ortho-nitroarylacetonitriles into indoles. Alkylation of such nitriles with allyl or benzyl halides followed by treatment of the compounds obtained with basic agents results in a multistep transformation, which is likely to proceed via intermediate nitrosoarenes, to produce 1-hydroxyindoles. For instance, alkylation of ortho-nitroarylacetonitriles with 3-phenylallyl bromide gives the compounds that in the presence of chlorotrimethylsilane and triethylamine undergo cyclization into 3-cyano-1-hydroxy-2-vinylindoles (Scheme 70) [188]. Presumably, this reaction proceeds via O-silylation of the nitronate anion and 1,5-elimination of trimethylsilanol from the intermediate trimethylsilyl nitronate, followed by cyclization and a hydrogen shift.

Scheme 70
Another example of the reaction proceeding in a similar manner is the conversion of 2-(5-chloro-2-nitrophenyl)-3-phenylpropionitrile into N-hydroxyindole derivative (Scheme 71) [189]. The intermediate vinyl nitroso compound undergoes electrocyclization, resulting in the formation of nitrone (2H-indole N-oxide), which is tautomerized into N-hydroxyindole.

Scheme 71
A peculiar way of formation of 1-hydroxyindole has been observed in the reaction of nitrobenzenes with dimethyl 2-cyanocyclopropane-1,1-dicarboxylate (Scheme 72) [190]. Treatment of 1-trifluoromethyl-4-nitrobenzene with this ester at low temperature results in the VNS of hydrogen, proceeding via opening of the cyclopropane ring. Quenching of the reaction mixture at low temperature leads to 4-cyano-4-arylbutyric acid derivative; however, when the reaction mixture was allowed to warm-up to 0°C, cyclization into 1-hydroxyindole takes place via an intramolecular addition of the carbanion to the nitro group.

Scheme 72
The Knoevenagel condensation of alkyl ortho-nitroarylacetates and ortho-nitroarylacetonitriles with aliphatic aldehydes proved to give the corresponding alkylidene nitriles and esters [191–194]. In the presence of a base these nitriles undergo cyclization into indole or quinoline derivatives, depending on the reaction conditions (Scheme 73) [195].

Scheme 73
Depending on the conditions, reduction of α-(2-nitroaryl)acrylonitriles with carbon monoxide can give two products (Scheme 74) [194]. Thus, reduction with palladium acetate–triphenylphosphine complex (neutral conditions) leads to indole-3-carbonitriles, while in the presence of DBU or t-BuOK (basic conditions) 4-cyanoquinoline was formed.

Scheme 74
The VNS in meta-dinitrobenzene by action of carbanions of α-haloketones leads to 2,4-dinitrobenzyl ketones, which can be reduced to 1-hydroxy-6-nitroindoles under mild conditions with tin(II) chloride (Scheme 75) [196].

Scheme 75
The VNS reaction of 4-nitroanisole with ethyl chloroacetate followed by the Knoevenagel condensation of the product with acetaldehyde affords α-(2-nitrophenyl)crotonate, which in the presence of t-BuOK in tert-butanol undergoes cyclization into N-hydroxyindole-3-carboxylate (Scheme 76). Further alkylation of the latter compound with methyl iodide results in N-methoxyindole. It is worth mentioning that in this reaction a partial loss of the alkene chain does happen to occur [194]. A similar phenomenon has been observed earlier in our laboratory [195].

Scheme 76
The reaction of 2-nitroarylacetonitriles and their heteroanalogues with trioxane has been reported to afford α-(2-nitroaryl)acrylates and their heteroanalogues, which can be reduced with carbon monoxide in the presence of palladium acetate–triphenylphosphine complex to give esters of the corresponding indole-3-carboxylic acids in high yields (Scheme 77) [193].

Scheme 77
The reaction of 3-nitropyridine with methyl chloroacetate under basic conditions provides ethyl nitropyridyl acetates, followed by their catalytic hydrogenation and cyclization into azaoxindoles (Scheme 78) [59, 60, 197].

Scheme 78
The VNS reaction of nitrobenzene with ethyl α-chloropropionate, proceeding in the para-position of the benzene ring, can be followed in situ by the SNAr of fluorine atom in subsequently added 1-fluoro-2,4-dinitrobenzene to give 2,4,4′-trinitrodiarylpropionate, which being hydrogenated is transformed into 3-aryloxindole derivative (Scheme 79) [198, 199].

Scheme 79
Phosphonium ylide, generated from allyl triphenylphosphonium chloride, is capable of addition to 1-nitronaphthalene or 5-nitro-8-methoxyquinoline in the presence of DBU and titanium tetraisopropoxide to form unstable N-hydroxyindole derivative, which is transformed by action of ethyl bromoacetate into benzo- or pyridoindoles (Scheme 80) [200].

Scheme 80
ortho-Nitrobenzyl aryl sulfones, readily available via the VNS reactions of nitroarenes with the carbanions of chloromethyl aryl sulfones, upon reduction and conversion of the amino group into imidate [201–203] or imine [204] functionality, are able to undergo cyclization into substituted indoles. This procedure is particularly useful due to the possibility to direct the VNS reaction selectively in ortho-position to the nitro group when the reaction is carried out in t-BuOK/THF (Scheme 81) [25]. This approach was used for the synthesis of 5- and 7-bromo-3-sulfonylindoles that were subsequently functionalized by the Stille coupling with tributyl(vinyl)tin. The obtained vinyl derivatives were then transformed into the corresponding amino compounds, tested as norepinephrine reuptake inhibitors and 5-HT2A receptor antagonists [202].

Scheme 81
Alternatively, N-substituted 3-phenylsulfonylindoles have been synthesized via reductive N-alkylation of ortho-aminobenzyl sulfones with ketones followed by condensation with dimethylformaldehyde dimethylacetal and cyclization (Scheme 82) [203].

Scheme 82
In this section we have presented numerous ways to construct indole derivatives via nucleophilic substitution of hydrogen. It has been shown that this approach is one of the simplest, versatile, and efficient ways to such ring systems, which can easily be adopted for large-scale operations. Another advantage of this methodology is that in contrast to the majority of modern methods no transition metals are used, so troublesome removal of their impurities from the final products is not necessary [205].
6.2 Quinolines
Since the quinoline ring system is often encountered in pharmaceuticals, plant protection agents, photoactive compounds, etc., a search for new synthetic pathways to quinoline derivatives continues to be of a substantial interest. There are numerous examples of synthesis of quinolines and their condensed analogues via nucleophilic substitution of hydrogen in nitroarenes. One can classify those into two broad categories:
-
a.
Direct syntheses, when the quinoline ring system is formed directly, or as result of the domino reaction from the intermediate σH adducts
-
b.
Products of nucleophilic substitution of hydrogen are subsequently converted into quinolines
Direct methods are based on the reactions of nitroarenes or nitroheteroarenes with carbanions affording the intermediate σH adducts that, under the reaction conditions, are converted into nitrosoarenes according to the intramolecular redox stoichiometry. The nitrosoarenes are known to be rather active electrophilic partners and are able to enter in situ further reactions to produce quinolines as the ultimate products.
An important example is the formal synthesis of eupolauramine, an alkaloid from the bark of the African plant Eupomatia laurina. This approach involves addition of the carbanion of allyl phenyl sulfone to 1-methoxy-4-nitronaphthalene followed by conversion of the σH adduct formed into the corresponding nitrosoarene. Further intramolecular condensation affords phenylsulfonyl-substituted azaphenanthrene (Scheme 83) [82]. The sulfone obtained was transformed into tricyclic azaaromatic acid, from which in turn the final alkaloid can be obtained following the known procedure [206].

Scheme 83
A novel pathway for the synthesis of substituted 3-aminoquinolines, proceeding via addition of the dianion of 3-aminocrotonates to nitroarenes, is exemplified by the reaction of ethyl N-pivaloyl-3-aminocrotonate with 2,4-dichloronitrobenzene. The intermediate σH adduct upon silylation or acylation is transformed into the corresponding ethyl 3-(N-pivaloyl amino)quinoline-2-carboxylate (Scheme 84) [207].

Scheme 84
There are numerous examples of construction of condensed pyridines (and also quinolines and acridines) via cascade reactions, involving conversion of the σH adducts of benzylic or allylic carbanions to nitroarenes followed by their intramolecular cyclization to form the pyridine ring. Thus, the reaction between 4-chloronitrobenzene and phenylacetonitrile, which is known to produce in protic media the corresponding 2,1-benzisoxazole via conversion of the intermediate σH adduct into nitrosoarene and its further condensation reaction [80], can proceed in aprotic media along another way. The same σH adduct formed in tetrahydrofuran, when treated with trialkylchlorosilanes or pivaloyl chloride, undergoes cyclization into acridine derivative (Scheme 85) [208].

Scheme 85
A similar reaction has been observed to proceed between 6-nitroquinoline and thienylmethyl tolyl sulfone in aprotic acetonitrile. The intermediate σH adduct, being treated with bis-trimethylsilyl acetamide, is converted into nitrosoarene, which undergoes intramolecular condensation to give thienophenanthroline in good yield (Scheme 86) [209].

Scheme 86
Analogous transformation was reported to proceed between arylacetonitriles and 3-nitroimidazo[1,2-a]pyridine, thus leading to pyrido-annelated imidazoquinolines (Scheme 87) that are of interest as highly fluorescent dyes [130].

Scheme 87
The reaction of the same nitroimidazopyridine with 3-indolylacetonitrile leads directly to pentacyclic azaaromatic system (Scheme 88) [210].

Scheme 88
Syntheses of quinolines from the products of the nucleophilic substitution of hydrogen proved to be also valuable. For instance, ortho-nitrobenzyl sulfones react with dialkyl maleates and fumarates to produce directly quinoline 2,3-dicarboxylate N-oxides [211]. The reaction proceeds via the Michael addition of the nitrobenzyl carbanion followed by elimination of benzenesulfinic acid and subsequent intramolecular addition of the allylic carbanion to the nitro group. This approach has recently been used for the synthesis of fluorine-substituted quinoline N-oxides (Scheme 89) [212].

Scheme 89
The reaction of ortho-nitroarylacetonitriles with the Vilsmeier–Haack reagent, prepared from N-methylpyrrolidone, followed by intramolecular cyclization, induced by diazabicycloundecene (DBU) in the presence of bis-trimethylsilylacetamide (BSA), leads directly to pyrrolo[3,2-b]quinoline derivatives (Scheme 90) [213].

Scheme 90
A versatile synthesis of pyrrolo-annelated quinolines has been reported to occur via alkylation of the VNS products, ortho-nitroaryl-substituted acetonitriles, with α-bromoketones. The obtained ketonitriles can be reduced under mild conditions with tin(II) chloride in ethyl acetate–ethanol mixture into quinoline-4-carbonitriles [214]. The same reaction sequence has been applied to 5-nitroindol-4-yl- and 4-nitroindol-5-yl-acetonitriles to obtain tricyclic 4-cyano-2-phenyl derivatives of pyrrolo[3,2-f]- and pyrrolo[2,3-h]-quinolines (Scheme 91) [214].

Scheme 91
Nitrobenzosultams obtained by intramolecular vicarious [215] or oxidative nucleophilic substitution of hydrogen [216] in N-chloromethylsulfonyl- or N-methylsulfonyl-substituted meta-nitroanilides have been reported to enter the Knoevenagel condensation with acetaldehyde [217]. The formed ethylidene sultam, when treated with DBU, undergoes cyclization into the tricyclic sultam system, bearing the quinoline N-oxide fragment (Scheme 92) [217]. The reaction appears to proceed via intramolecular addition of the allylic carbanion to the nitro group.

Scheme 92
6.3 2,1-Benzisoxazoles
The 2,1-benzisoxazole (anthranil) ring system is of interest as a key intermediate for the synthesis of other heterocycles. 2,1-Benzisoxazoles can be derived from the direct multistep domino reaction of some carbanions with nitroarenes or by conversion of the products of nucleophilic substitution of hydrogen in nitroarenes. As early as in 1960, Davis and Pizzini reported that the reaction of 4-chloronitrobenzene with phenylacetonitrile in the presence of potassium hydroxide in protic media affords 3-phenyl-5-chloro-2,1-benzisoxazole in high yield [80] (Scheme 17).
This is in fact a general way for the synthesis of 2,1-benzisoxazoles, which have found wide application as starting materials to obtain other heterocyclic systems: quinolines [218–221], polyquinolines [222–224], acridines [80, 225–227], and benzodiazepines [228]. The most common approach is reduction of anthranils to 2-aminobenzophenones, which are appropriate starting materials to build other heterocyclic systems.
ortho-Nitroarylacetic esters, nitriles, and ortho-nitrobenzylsulfones, available via the VNS methodology, are readily converted into 2,1-benzisoxazoles through condensation on treatment with chlorotrimethylsilane in the presence of triethylamine (Scheme 93) [229].

Scheme 93
Alternatively, ortho-nitroaryl-substituted acetonitriles and their heteroanalogues can be dehydrated into benzisoxazoles on treatment with concentrated sulfuric acid (Scheme 94) [230].

Scheme 94
The formation of 2,1-benzisoxazoles has also been observed when the VNS products derived from bicyclic nitroarenes and heteroarenes have been allowed to react with phenolate and thiolate anions (Scheme 95), as well as with some carbanions [231].

Scheme 95
Benzisoxazoles can readily be obtained by anaerobic, spontaneous transformation of carbanions of α-(ortho-nitroaryl)benzyl phosphonates, derived from the ONSH in nitroarenes with carbanion of diethyl benzylphosphonate, whereas oxygen oxidation of such carbanions gives nitrobenzophenones (Scheme 96) [232].

Scheme 96
6.4 Phenazines and Other Heterocyclic Compounds from 2-Nitrosodiphenylamines
It has recently been found in our laboratory that anilines react with nitroarenes in the presence of a strong base to form 2-nitrosodiphenylamines [86]. The reaction proceeds via addition of the N-anion of anilines to nitroarenes in the ortho-position to the nitro group, followed by conversion of the formed σH adduct according to an intramolecular redox stoichiometry (Scheme 97). The reaction is of general character; thus, a variety of 2-nitrosodiphenylamines become readily available. These compounds are versatile starting materials for the synthesis of heterocycles containing two nitrogen atoms: phenazines, benzimidazoles, quinoxalines, etc. Simple treatment of 2-nitrosodiphenylamines with acetic acid leads to phenazines in high yields.

Scheme 97
This two-step process is analogous to, but much more efficient than, the classic Wohl–Aue synthesis of phenazines [233]. The versatility of this approach has been demonstrated by the synthesis of 1-methoxyphenazine that can be obtained from two different pairs “nitroarene–aniline”, namely, nitrobenzene–meta-anisidine or meta-nitroanisole–aniline (Scheme 98) [84].

Scheme 98
Availability of nitroarenes and anilines opens almost unlimited simple and efficient access to phenazines, as well as to their derivatives condensed with an additional ring, as exemplified by the synthesis of pyrrolo[3,2-b]phenazine from 5-nitroindoles (Scheme 99) [234].

Scheme 99
Carbanions of benzyl aryl sulfones are able to react with 2-nitrosodiphenylamines to produce 1,2-diarylbenzimidazoles. The reaction appears to proceed via attack of these carbanions on the nitroso group followed by intramolecular addition– elimination process to give benzimidazoles (Scheme 100) [83].

Scheme 100
Condensation of 2-nitrosodiphenylamines with a variety of highly stabilized carbanions of dialkyl malonates, alkyl phenylacetates, and trialkyl phosphonoacetates is an efficient way for the synthesis of a great deal of substituted N-arylquinoxalin-2(1H)-ones (Scheme 101) [235, 236]. Double additions of anions of 2-cyanoalkyl carboxylates to 2-nitroso-4-alkylaminodiphenylamines result in the formation of pyrroloquinoxalinones [236].

Scheme 101
6.5 Miscellaneous Syntheses of Heterocycles
3-Arylsulfonylindazoles, novel 5-HT6 receptor antagonists, have been synthesized via the VNS reactions of para-substituted nitrobenzenes with carbanion of chloromethyl aryl sulfones followed by hydrogenation of the nitro group (Scheme 102). Further reaction of ortho-aminobenzyl sulfones with sodium nitrite-acetic acid gave the desired 5-substituted indazoles of potential biological activity [237, 238].

Scheme 102
Oxidative intramolecular amination of meta-nitro-substituted diaryl triazenes has been established to proceed under mild basic conditions in the presence of K2CO3 in DMF (Scheme 103) [239].

Scheme 103
Conversion of the initially formed σH adducts into intermediate nitrosoarenes appears to be involved in the reaction of nitroarenes with guanidines, leading to 3-amino-1,2,4-benzotriazines (Scheme 104) [240]. The mechanism of this transformation proposed by the authors includes oxidation of the intermediate σH adduct to form the corresponding 2-nitrophenyl-substituted guanidine, which undergoes cyclization into 3-amino-1,2,4-benzotriazine-1-oxide. An alternative mechanism can be suggested that involves reduction of the nitro group to the nitroso one, followed by cyclization resulting in the N=N bond formation. Partial reduction of the nitro group was observed earlier in the reactions of cyclic (3-nitrophenyl)guanidines [241].

Scheme 104
7 Conclusion
From the data presented it is evident that nucleophilic substitution of hydrogen is an efficient synthetic tool for introduction of a variety of substituents into heterocyclic rings and construction of heterocyclic systems. We do hope that numerous examples of nucleophilic substitution of hydrogen, as well as use of this versatile and general reaction for the synthesis of heterocyclic compounds will attract attention of researchers working in the field of organic synthesis.
References
Chupakhin ON, Charushin VN, van der Plas HC (1994) Nucleophilic aromatic substitution of hydrogen. Academic Press, San Diego
Terrier F (2013) Modern nucleophilic aromatic substitution. Wiley-VCH, Weinheim
Terrier F (1991) Nucleophilic aromatic displacement. Verlag Chemie, Weinheim
Mąkosza M, Wojciechowski K (2004) Chem Rev 104:2631
Orlov VY, Kotov AD, Rusanov AI (2010) Functionalization of carbo-, N- and O-containing heteroaromatic systems. Mir Publishing Company, Moscow
Mąkosza M, Winiarski J (1987) Acc Chem Res 20:282
Mąkosza M (1996) Russ Chem Bull 45:491
Mąkosza M, Kwast A (1998) J Phys Org Chem 11:341
Mąkosza M (2010) Chem Soc Rev 39:2855
Mąkosza M (2011) Synthesis 2341
Goliński J, Mąkosza M (1978) Tetrahedron Lett 19:3495
Mąkosza M, Goliński J, Baran J (1984) J Org Chem 49:1488
Glinka T, Mąkosza M (1983) J Org Chem 48:3860
Mąkosza M, Ludwiczak S (1984) J Org Chem 49:4562
Mąkosza M, Winiarski J (1984) J Org Chem 49:1494
Stahly GP, Stahly BC, Lilje KC (1984) J Org Chem 49:578
Mudryk B, Mąkosza M (1988) Synthesis 1007
Florio S, Lorusso P, Granito C, Luisi R, Troisi L (2004) J Org Chem 69:4961
Florio S, Lorusso P, Granito C, Ronzini L, Troisi L (2003) Eur J Org Chem 4053
Mąkosza M, Owczarczyk Z (1989) J Org Chem 54:5094
Russell GA, Janzen EG, Strom ET (1964) J Am Chem Soc 86:1807
Bilkis II, Shein SM (1975) Tetrahedron 31:969
Zhang X, Yang D, Liu Y (1993) J Org Chem 58:224
Mąkosza M, Podraza R, Kwast A (1994) J Org Chem 59:6796
Mąkosza M, Glinka T, Kinowski J (1984) Tetrahedron 40:1863
Lemek T, Mąkosza M, Stephenson DS, Mayr H (2003) Angew Chem Int Ed 42:2793
Mąkosza M, Kwast A (2004) Eur J Org Chem 2125
Mąkosza M, Lemek T, Kwast A (1999) Tetrahedron Lett 40:7541
Mąkosza M, Lemek T, Kwast A, Terrier F (2002) J Org Chem 67:394
Błażej S, Mąkosza M (2008) Chem Eur J 14:11113
Seeliger F, Błażej S, Bernhardt S, Mąkosza M, Mayr H (2008) Chem Eur J 14:6108
Miller J (1968) Aromatic nucleophilic substitution. Elsevier, Amsterdam
Mudryk B, Mąkosza M (1988) Tetrahedron 44:209
Mąkosza M, Rydz A, Wróbel Z (1995) Pol J Chem 69:918
Mąkosza M, Kuciak R, Wojciechowski K (1994) Liebigs Ann Chem 615
Mąkosza M, Podraza R (2000) Eur J Org Chem 193
Ostrowski S, Moritz RJ, Mudryk B (1995) Monatsh Chem 126:447
Ostrowski S, Mąkosza M (1989) Liebigs Ann Chem 1989:95
Ostrowski S, Mąkosza M (1989) J Organomet Chem 367:95
Müller P, Bernardinelli G, Motallebi S (1990) Helv Chim Acta 73:1242
Mąkosza M, Sienkiewicz K (1998) J Org Chem 63:4199
Pews RG, Lysenko Z, Vosejpka PC (1997) J Org Chem 62:8255
Angeli A, Angelico F (1901) Gazz Chim Ital 31:27
Katritzky AR, Laurenzo KS (1988) J Org Chem 53:3978
Szpakiewicz B, Grzegożek M (2008) Can J Chem 86:682
Bakke JM, Svensen H, Trevisan R (2001) J Chem Soc Perkin Trans 1:376
Mąkosza M, Białecki M (1998) J Org Chem 63:4878
Parent EE, Dence CS, Sharp TL, Welch MJ, Katzenellenbogen JA (2006) Nucl Med Biol 33:615
Seko S, Miyake K (1998) J Chem Soc Chem Commun 1519
Seko S, Miyake K, Kawamura N (1999) J Chem Soc Perkin Trans 1:1437
Pagoria PF, Mitchell AR, Schmidt RD (1996) J Org Chem 61:2934
Mitchell AR, Coburn MD, Schmidt RD, Pagoria PF, Lee GS (2002) Thermochim Acta 384:205
Rozhkov VV, Shevelev SA, Chervin II, Mitchell AR, Schmidt RD (2003) J Org Chem 68:2498
Mąkosza M, Kwast E (1995) Tetrahedron 51:8339
Kwast E, Mąkosza M (1990) Tetrahedron Lett 31:121
Bernard MK (2000) Tetrahedron 56:7273
Mąkosza M, Chylińska B, Mudryk B (1984) Liebigs Ann Chem 8
Mąkosza M, Sienkiewicz K, Wojciechowski K (1990) Synthesis 850
Andreassen EJ, Bakke JM (2006) J Heterocycl Chem 43:49
Bakke JM (2005) J Heterocycl Chem 42:463
Wojciechowski K, Kosiński S (2001) Tetrahedron 57:5009
Itoh T, Nagata K, Okada M, Ohsawa A (1993) Chem Pharm Bull 41:220
Mąkosza M, Goliński J, Rykowski A (1983) Tetrahedron Lett 24:3277
Charushin VN, Rusinov VL, Chupakhin ON (2008) Compr Heterocycl Chem 9:95
Goliński J, Mąkosza M, Rykowski A (1983) Tetrahedron Lett 24:3279
Mąkosza M, Ostrowski S (1988) J Prakt Chem 330:789
Ostrowicz A, Bałoniak S, Mąkosza M, Rykowski A (1992) Tetrahedron Lett 33:4787
Hamana M, Fujimura Y, Haradahira T (1987) Heterocycles 25:229
Kolesnichenko GA, Malykhin EV, Shteingarts VD (1985) Zh Org Khim 21:1150
van der Plas HC, Woźniak M (1986) Croat Chem Acta 59:33
Szpakiewicz B, Grzegozek M (2004) Russ J Org Chem 40:829
Bartoli G (1984) Acc Chem Res 17:109
Mąkosza M, Surowiec M (2001) J Organomet Chem 624:167
Mąkosza M, Staliński K (1997) Chem Eur J 3:2025
Adam W, Mąkosza M, Staliński K, Zhao C-G (1998) J Org Chem 63:4390
Mąkosza M, Sulikowski D (2009) J Org Chem 74:3827
Mąkosza M, Paszewski M, Sulikowski D (2008) Synlett 2938
Mąkosza M, Sypniewski M (1994) Tetrahedron 50:4913
Mąkosza M, Staliński K (1998) Tetrahedron Lett 39:3575
Davis RB, Pizzini LC (1960) J Org Chem 25:1884
Davis RB, Pizzini LC, Bara EJ (1961) J Org Chem 26:4270
Wróbel Z (2000) Eur J Org Chem 521
Wróbel Z, Stachowska K, Grudzień K, Kwast A (2011) Synlett 1439
Kwast A, Stachowska K, Trawczyński A, Wróbel Z (2011) Tetrahedron Lett 52:6484
Wróbel Z, Kwast A (2010) Synthesis 3865
Wróbel Z, Kwast A (2007) Synlett 1525
Kotsuki H, Kobayashi S, Matsumoto K, Suenaga H, Nishizawa H (1990) Synthesis 1990:1147
Danikiewicz W, Mąkosza M (1991) J Org Chem 56:1283
Mąkosza M, Wojciechowski K (1997) Liebigs Ann/Recueil 1805
Mąkosza M, Wojciechowski K (2001) Heterocycles 54:445
Wróbel Z, Mąkosza M (1990) Org Prep Proced Int 22:575
Mąkosza M, Surowiec M, Voskresensky S (2000) Synthesis 1237
Wojciechowski K (1988) Bull Pol Acad Sci Chem 36:235
Florio S, Lorusso P, Luisi R, Granito C, Ronzini L, Troisi L (2004) Eur J Org Chem 2118
Achmatowicz M, Thiel OR, Gorins G, Goldstein C, Affouard C, Jensen R, Larsen RD (2008) J Org Chem 73:6793
Hartz RA, Ahuja VT, Zhuo X, Mattson RJ, Denhart DJ, Deskus JA, Vrudhula VM, Pan S, Ditta JL, Shu Y-Z, Grace JE, Lentz KA, Lelas S, Li Y-W, Molski TF, Krishnananthan S, Wong H, Qian-Cutrone J, Schartman R, Denton R, Lodge NJ, Zaczek R, Macor JE, Bronson JJ (2009) J Med Chem 52:7653
Leahy DK, Li J, Sausker JB, Zhu J, Fitzgerald MA, Lai C, Buono FG, Braem A, de Mas N, Manaloto Z, Lo E, Merkl W, Su BN, Gao Q, Ng AT, Hartz RA (2010) Org Process Res Dev 14:1221
Prokhorov AM, Mąkosza M, Chupakhin ON (2009) Tetrahedron Lett 50:1444
Leen V, Van der Auweraer M, Boens N, Dehaen W (2011) Org Lett 13:1470
Goumont R, Jan E, Mąkosza M, Terrier F (2003) Org Biomol Chem 1:2192
Mąkosza M, Białecki M (1991) Synlett 181
Tercel M, Lee AE, Hogg A, Anderson RF, Lee HH, Siim BG, Denny WA, Wilson WR (2001) J Med Chem 44:3511
Couch GD, Burke PJ, Knox RJ, Moody CJ (2008) Tetrahedron 64:2816
Avila B, Solano DM, Haddadin MJ, Kurth MJ (2011) Org Lett 13:1060
Ellis D (2011) Synthetic Comm 41:963
Crozet MD, Suspene C, Kaafarani M, Crozet MP, Vanelle P (2004) Heterocycles 63:1629
Pace A, Buscemi S, Vivona N (2007) Org Prep Proced Int 39:1
Pace A, Buscemi S, Vivona N (2005) Org Prep Proced Int 37:447
Surowiec M, Mąkosza M (2004) Tetrahedron 60:5019
Loska R, Majcher M, Mąkosza M (2007) J Org Chem 72:5574
Loska R, Mąkosza M (2007) J Org Chem 72:1354
Crozet MD, Reacutemusat V, Curti C, Vanelle P (2006) Synthetic Comm 36:3639
Mąkosza M, Kwast E (1987) Bull Pol Acad Sci Chem 35:287
Bernard MK, Mąkosza M, Szafran B, Wrzeciono U (1989) Liebigs Ann Chem 1989:545
Wojciechowski K, Mąkosza M (1989) Synthesis 106
Dudzińska-Uzarewicz J, Wrzeciono U, Frankiewicz A, Linkowska E, Kohler T, Nuhn P (1988) Pharmazie 43:611
Bernard MK (1997) Pol J Chem 71:1413
Bernard MK (1995) Pol J Chem 69:1120
Bernard MK, Szoja C, Wrzeciono U (1990) Liebigs Ann Chem 755
Ostrowski S, Wojciechowski K (1990) Can J Chem 68:2239
Mąkosza M, Kinowski A, Danikiewicz W, Mudryk B (1986) Liebigs Ann Chem 69
Ostrowski S, Mikus A (2003) Mol Divers 6:315–321
Ostrowski S, Raczko AM (2005) Helv Chim Acta 88:974
Crozet MD, Botta C, Gasquet M, Curti C, Rémusat V, Hutter S, Chapelle O, Azas N, De Méo M, Vanelle P (2009) Eur J Med Chem 44:653
Mąkosza M, Tyrała A (1987) Synthesis 1142
Crozet MD, Perfetti P, Kaafarani M, Crozet MP, Vanelle P (2004) Lett Org Chem 1:326
Mąkosza M, Glinka T, Ostrowski S, Rykowski A (1987) Chem Lett 61
Katrusiak A (2006) Tetrahedron Lett 47:4259
Wojciechowski K, Modrzejewska H (2003) Synthesis 1503
Rahimizadeh M, Pordel M, Bakavoli M, Eshghi H, Shiri A (2009) Mendeleev Comm 19:161
Mąkosza M, Sulikowski D (2010) Synlett 1666
Mąkosza M, Surowiec M, Szczepańska A, Sulikowski D, Maltsev O (2007) Synlett 470
Sulikowski D, Mąkosza M (2010) Eur J Org Chem 4218
Leen V, Gonzalvo VZ, Deborggraeve WM, Boens N, Dehaen W (2010) Chem Commun 46:4908
Yamada K, Kawasaki T, Fujita T, Somei M (2001) Heterocycles 55:1151
Zhu Z, Sun Z-Y, Ye Y, McKittrick B, Greenlee W, Czarniecki M, Fawzi A, Zhang H, Lachowicz JE (2009) Bioorg Med Chem Lett 19:5218
Rouse MB, Seefeld MA, Leber JD, McNulty KC, Sun L, Miller WH, Zhang S, Minthorn EA, Concha NO, Choudhry AE, Schaber MD, Heerding DA (2009) Bioorg Med Chem Lett 19:1508
Damont A, Boisgard R, Kuhnast B, Lemee F, Raggiri G, Scarf AM, Da Pozzo E, Selleri S, Martini C, Tavitian B, Kassiou M, Dolle F (2011) Bioorg Med Chem Lett 21:4819
Stavenger RA, Cui HF, Dowdell SE, Franz RG, Gaitanopoulos DE, Goodman KB, Hilfiker MA, Ivy RL, Leber JD, Marino JP, Oh HJ, Viet AQ, Xu WW, Ye GS, Zhang DH, Zhao YD, Jolivette LJ, Head MS, Semus SF, Elkins PA, Kirkpatrick RB, Dul E, Khandekar SS, Yi T, Jung DK, Wright LL, Smith GK, Behm DJ, Doe CP, Bentley R, Chen ZX, Hu ED, Lee D (2007) J Med Chem 50:2
Kawakami T, Suzuki H (2000) J Chem Soc Perkin Trans 1:1259
Grzegożek M (2008) J Heterocyclic Chem 45:1879
Grzegożek M, Szpakiewicz B, Kowalski P (2009) Arkivoc 84
Titova IA, Vakul'skaya TI, Larina LI, Mizandrontsev MI, Volkov VA, Dolgushin GV, Lopyrev VA (2005) Russ J Org Chem 41:1306
Vakul’skaya T, Titova I, Larina L, Verkhozina O, Dolgushin G, Lopyrev V (2006) Chem Heterocycl Comp 42:1427
Schmidt RD, Lee GS, Pagoria PF, Mitchell AR, Gilardi R (2001) J Heterocycl Chem 38:1227
Millar RW, Philbin SP, Claridge RP, Hamid J (2004) Propellants Explosives Pyrotechnics 29:81
Bastrakov MA, Starosotnikov AM, Kachala VV, Glukhov IV, Shevelev SA (2009) Russ Chem Bull 58:414
Grzyb S, Ostrowski S (2012) Jordan J Chem 7:231
Ostrowski S, Grzyb S (2012) Tetrahedron Lett 53:6355
Richeter S, Jeandon C, Gisselbrecht JP, Graff R, Ruppert R, Callot HJ (2004) Inorg Chem 43:251
Richeter S, Jeandon C, Gisselbrecht J-P, Ruppert R, Callot HJ (2002) J Am Chem Soc 124:6168
Stefanelli M, Mandoj F, Mastroianni M, Nardis S, Mohite P, Fronczek FR, Smith KM, Kadish KM, Xiao X, Ou ZP, Chen P, Paolesse R (2011) Inorg Chem 50:8281
Verbeeck S, Herrebout WA, Gulevskaya AV, van der Veken BJ, Maes BUW (2010) J Org Chem 75:5126
Gulevskaya AV, Mae BUW, Meyers C, Herrebout WA, van der Veken BJ (2006) Eur J Org Chem 5305
Gulevskaya AV, Maes BUW, Meyers C (2007) Synlett 71
Gulevskaya AV, Verbeeck S, Burov ON, Meyers C, Korbukova IN, Herrebout W, Maes BUW (2009) Eur J Org Chem 2009:564
Borovlev I, Demidov O, Saigakova N, Pisarenko S, Nemykina O (2011) J Heterocycl Chem 48:1206
Demidov OP, Borovlev IV, Pisarenko SV, Nemykina OA, Saigakova NA (2010) Chem Heterocycl Comp 46:636
Bianchi L, Maccagno M, Petrillo G, Sancassan F, Tavani C, Morganti S, Rizzato E, Spinelli D (2007) J Org Chem 72:5771
Patriciu O-I, Pillard C, Finaru A-L, Sandulescu I, Guillaumet G (2007) Synthesis 3868
Grzegożek M, Szpakiewicz B (2006) J Heterocyclic Chem 43:425
Tjosaas F, Fiksdahl A (2007) J Organomet Chem 692:5429
Levinson FS, Varganov RV, Zakiev MM, Shakirova IM, Sheremetev AB, Dmitriev DE, Krivolapov DB, Litvinov IA, Sharafutdinova DR, Efremov YY (2008) Mendeleev Comm 18:329
Millar RW, Claridge RP, Sandall JPB, Thompson C (2002) Arkivoc 19
Claridge RP, Millar RW, Sandall JPB, Thompson C (1999) Tetrahedron 55:10243
Moskalev N, Mąkosza M (1999) Tetrahedron Lett 40:5395
Moskalev N, Barbasiewicz M, Mąkosza M (2004) Tetrahedron 60:347
Sulur M, Sharma P, Ramakrishnan R, Naidu R, Merifield E, Gill DM, Clarke AM, Thomson C, Butters M, Bachu S, Benison CH, Dokka N, Fong ER, Hose DRJ, Howell GP, Mobberley SE, Morton SC, Mullen AK, Rapai J, Tejas B (2012) Org Process Res Dev 16:1746
Timofeev EN, Kolganova NA, Smirnov IP, Kochetkova SV, Florentiev VL (2008) Russian J Bioorg Chem 34:201
Guo W, Miao Z, Sheng C, Yao J, Feng H, Zhang W, Zhu L, Liu W, Cheng P, Zhang J, Che X, Wang W, Luo C, Xu Y (2010) Eur J Med Chem 45:2223
Moskalev N, Mąkosza M (2000) Heterocycles 52:533
Wojciechowski K, Mąkosza M (1984) Tetrahedron Lett 25:4793
Mąkosza M, Paszewski M (2002) Synthesis 2203
Mąkosza M, Hoser H (1994) Heterocycles 37:1701
Walker GN (1955) J Am Chem Soc 77:3844
Mąkosza M, Danikiewicz W, Wojciechowski K (1988) Liebigs Ann Chem 203
Lerman L, Weinstock-Rosin M, Nudelman A (2004) Synthesis 3043
Iakobson G, Posta M, Beier P (2013) Synlett 24:855
Yamagishi H, Matsumoto K, Iwasaki K, Miyazaki T, Yokoshima S, Tokuyama H, Fukuyama T (2008) Org Lett 10:2369
Pedras MSC, Zheng Q-A, Gadagi RS (2007) Chem Commun 4:368
Groszek G, Bednarski M, Dybala M, Filipek B (2009) Eur J Med Chem 44:809
Czeskis BA, Clodfelter DK, Wheeler WJ (2002) J Labelled Compd Rad 45:1143
Mąkosza M, Stalewski J, Maslennikova O (1997) Synthesis 1131
Chackal S, Dudouit F, Houssin R, Hénichart J-P (2003) Heterocycles 60:615
Khdour O, Ouyang A, Skibo EB (2006) J Org Chem 71:5855
Yoshikawa K, Yokomizo A, Naito H, Haginoya N, Kobayashi S, Yoshino T, Nagata T, Mochizuki A, Osanai K, Watanabe K, Kanno H, Ohta T (2009) Bioorg Med Chem 17:8206
Jeanty M, Suzenet F, Guillaumet G (2008) J Org Chem 73:7390
Wróbel Z, Mąkosza M (1993) Synlett 597
Wróbel Z, Mąkosza M (1997) Tetrahedron 53:5501
Stalewski J (1998) Tetrahedron Lett 39:9523
Wróbel Z, Kwast A, Mąkosza M (1993) Synthesis 31
Mąkosza M, Tyrała A (1986) Synthetic Comm 16:419
Söderberg BCG, Banini SR, Turner MR, Minter AR, Arrington AK (2008) Synthesis 903
Banini SR, Turner MR, Cummings MM, Söderberg BCG (2011) Tetrahedron 67:3603
Wróbel Z, Mąkosza M (1993) Tetrahedron 49:5315
Bujok R, Wróbel Z, Wojciechowski K (2012) Synlett 23:1315
Andreassen EJ, Bakke JM, Sletvold I, Svensen H (2004) Org Biomol Chem 2:2671
Lawrence NJ, Davies CA, Gray M (2004) Org Lett 6:4957
Cao S, Wu JJ, Li L, Zhu LJ, Zhang J, Yu JL, Qian XH (2008) Org Biomol Chem 6:1293
Korda A, Wróbel Z (2003) Synlett 1465
Wojciechowski K, Mąkosza M (1986) Synthesis 651
Heffernan GD, Coghlan RD, Manas ES, McDevitt RE, Li Y, Mahaney PE, Robichaud AJ, Huselton C, Alfinito P, Bray JA, Cosmi SA, Johnston GH, Kenney T, Koury E, Winneker RC, Deecher DC, Trybulski EJ (2009) Bioorg Med Chem 17:7802
Bernotas RC, Antane S, Shenoy R, Le V-D, Chen P, Harrison BL, Robichaud AJ, Zhang GM, Smith D, Schechter LE (2010) Bioorg Med Chem Lett 20:1657
Wojciechowski K, Mąkosza M (1986) Bull Soc Chim Belg 95:671
Garrett CE, Prasad K (2004) Adv Synth Catal 346:889
Kawase M, Miyake Y, Sakamoto T, Shimada M, Kikugawa Y (1989) Tetrahedron 45:1653
Bujok R, Kwast A, Cmoch P, Wróbel Z (2010) Tetrahedron 66:698
Bobin M, Kwast A, Wróbel Z (2007) Tetrahedron 63:11048
Wróbel Z (2004) Synlett 1929
Rahimizadeh M, Pordel M, Ranaei M, Bakavoli M (2012) J Heterocycl Chem 49:208
Mąkosza M, Tyrała A (1992) Acta Chem Scand 46:689
Kotovskaya SK, Zhumabaeva GA, Charushin VN, Chupakhin ON (2009) Russ Chem Bull 58:170
Wróbel Z, Wojciechowski K, Kwast A, Gajda N (2010) Synlett 2435
Bujok R, Trawczyński A, Wróbel Z, Wojciechowski K (2012) Synlett 2682
Wojciechowski K (1992) Pol J Chem 66:1121
Wojciechowski K, Mąkosza M (1992) Synthesis 571
Wróbel Z (2001) Tetrahedron 57:7899
Angibaud PR, Venet MG, Filliers W, Broeckx R, Ligny YA, Muller P, Poncelet VS, End DW (2004) Eur J Org Chem 479
Fan XS, Zhang XY, Zhang YM (2005) Heteroatom Chem 16:637
Fan XS, Zhang YM (2002) Tetrahedron Lett 43:7001
Mamo A, Nicoletti S, Tat NC (2002) Molecules 7:618
Kim JL, Kim JK, Hong SI (1999) Polym Bull 42:511
Zhang X, Shetty AS, Jenekhe SA (1998) Acta Polymerica 49:52
Simas ER, Martins TD, Atvars TDZ, Akcelrud L (2009) J Lumin 129:119
Orlov VY, Kotov AD, Ganzha VV, Sokolov VG (2004) Mendeleev Comm 17:37
Orlov VY, Ganzha VV, Kotov AD, Sokolov VG (2007) Russ J Org Chem 43:1502
Rahimizadeh M, Pordel M, Bakavoli M, Bakhtiarpoor Z, Orafaie A (2009) Monatsh Chem 140:633
Sternbach LH (1979) J Med Chem 22:1
Wróbel Z (1997) Synthesis 753
Bakavoli M, Pordel M, Rahimizadeh M, Jahandari P, Seresht ER (2008) Heterocycles 75:165
Wróbel Z, Mąkosza M (1995) Heterocycles 40:187
Sulikowski D, Mąkosza M (2009) Acta Chim Slov 56:680
Wohl A, Aue W (1901) Chem Ber 34:2442
Bujok R, Więcław M, Wróbel Z, Wojciechowski K (2014) Monatsh Chem DOI: 10.1007/S00706-013-1087-3
Wróbel Z, Stachowska K, Kwast A, Gościk A, Królikiewicz M, Pawłowski R, Turska I (2013) Helv Chim Acta 96:956
Królikiewicz M, Cmoch P, Wróbel Z (2013) Synlett 24:973
Takahashi M, Suga D (1998) Synthesis 986
Haydar SN, Yun H, Andrae PM, Mattes J, Zhang J, Kramer A, Smith DL, Huselton C, Graf R, Aschmies S, Schechter LE, Comery TA, Robichaud AJ (2010) J Med Chem 53:2521
Zhou Z, Liu Q-L, Li W, Zhu Y-M (2011) Heterocycles 83:2057
Zhumabaeva GA, Kotovskaya SK, Perova NM, Charushin VN, Chupakhin ON (2006) Russ Chem Bull 55:1243
Esser F, Pook K-H (1992) Synthesis 596
Acknowledgements
This work was supported by grant N204193038 from Ministry of Higher Education and Science.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Mąkosza, M., Wojciechowski, K. (2013). Nucleophilic Substitution of Hydrogen in Arenes and Heteroarenes. In: Charushin, V., Chupakhin, O. (eds) Metal Free C-H Functionalization of Aromatics. Topics in Heterocyclic Chemistry, vol 37. Springer, Cham. https://doi.org/10.1007/7081_2013_115
Download citation
DOI: https://doi.org/10.1007/7081_2013_115
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-07018-6
Online ISBN: 978-3-319-07019-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)