
Abstract
Porphyrins bearing nitro and amino substituents have been used as excellent synthons for further functionalization in order to obtain new compounds with adequate features for a wide range of applications. This chapter brings an update on the effort of several research groups to study the synthesis and reactivity features of meso-tetraarylporphyrins bearing those functionalities.
Graphical Abstract

Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
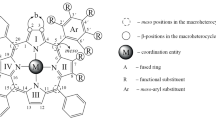


Keywords
1 Introduction
The extraordinary development observed in the porphyrin field after the structure elucidation of protoporphyrin IX by Fisher in 1929 [1] and the synthesis of chlorophyll a by Woodward in 1960 [2] shows that the scientific community has been having a great interest in the potentiality of these unique compounds. Today, it is accepted that porphyrin derivatives, besides their central role in respiration, photosynthesis, and other vital functions, have a promising future in several fields such as medicine [3], catalysis [4], and electronic materials [5]. Knowing that all those applications are strongly dependent on the structure of the macrocycle, there has been a considerable research directed towards the development of synthetic strategies to functionalize readily available porphyrins, especially meso-tetraarylporphyrins. Part of that work has been related to the functionalization of a primary group inserted in meso- or in β-pyrrolic positions of meso-tetraarylporphyrins. In this chapter, we highlight the most relevant and recent synthetic strategies concerning the functionalization of meso-tetraarylporphyrins through nitro or amino groups located at β-pyrrolic positions or in meso-phenyl groups. Occasionally, other porphyrin derivatives may also be discussed. The interest in nitro- and aminoporphyrins is mainly due to the very attractive reactivity and versatility of these two functional groups [6, 7].
The nitro group can improve the ability of porphyrin systems to act as radiosensitizers [8] and their usefulness in porphyrin functionalization has been demonstrated and is well documented. Nitroporphyrins themselves are widely used as starting materials: they can undergo direct nucleophilic addition and substitution reactions with a wide range of nucleophiles and displacement of the nitro group. The reduction of the nitro group to the amino group is a very useful reaction that extends the porphyrin potentialities for further functionalization via the amino group. In fact, a wide range of porphyrin derivatives with improved properties have been prepared via amide linkage, N-alkylation, nucleophilic substitution, diazotization, cycloaddition, palladium-catalyzed reactions, etc.
Besides the clear differences in reactivity of the β- and meso-aryl positions, the selective nitration of such positions can be controlled by the choice of the nitrating agent and the metal ion coordinated with the porphyrin macrocycle. The following sections cover useful nitration procedures and the exploitation of the nitro group in further functionalization of the porphyrin macrocycle. In the last topic it is highlighted the recent synthetic strategies concerning the functionalization of meso-tetraarylporphyrins through amino groups.
2 Synthesis and Reactivity of Nitroporphyrins
2.1 Synthesis of Meso-(Nitrophenyl)porphyrins
Meso-(Nitrophenyl)porphyrins can be obtained by the condensation of pyrrole with nitrobenzaldehydes (Scheme 1) or by nitration of meso-tetraarylporphyrins. The first strategy was used by Martell and coworkers [9, 10] to synthesize meso-tetrakis(4-nitrophenyl)porphyrin (1a). This synthesis was based on the Rothemund’s landmark conditions [11] to prepare meso-substituted porphyrins. The authors referred that the best yield (2.6%) was obtained when equimolar amounts of pyrrole and 4-nitrobenzaldehyde were heated at 120°C in a mixture of pyridine and methanol for 24 h. The synthetic improvements that appeared afterwards to obtain meso-tetraarylporphyrins were also considered in the synthesis of porphyrin 1a and its isomers 1b and 1c, respectively, with the nitro groups at para, meta, or ortho positions of the phenyl substituents [12, 13]. Porphyrin 1a, for instance, can be obtained in 19–22% by refluxing a solution of pyrrole and 4-nitrobenzaldehyde in propanoic acid containing acetic anhydride [14]. The same porphyrin can be obtained in 28% yield if the cyclocondensation is mediated by microwave irradiation in the presence of small amounts of propanoic acid [15]. Under the same microwave conditions, the cyclocondensation of equimolar amounts of pyrrole and 2-nitrobenzaldehyde afforded porphyrin 1c in 25% after ca. 5 min of reaction [15]. Under classical heating, porphyrin 1c, frequently used in the development of synthetic models for oxygen-binding hemoproteins, was obtained in 13% yield after refluxing pyrrole and 2-nitrobenzaldehyde in acetic acid for 20 min [16]. Much poor yields (4%) were reported for porphyrin 1b when 3-nitrobenzaldehyde and pyrrole were heated at reflux in propanoic acid containing acetic anhydride [14].

Synthesis of meso-(nitrophenyl)porphyrins
The cyclocondensation of pyrrole with a mixture of two aldehydes gives access to a wide variety of multifunctional porphyrins. Although not being considered an efficient and elegant strategy, due to the low yields (<5%) and the purification procedures required to separate the products mixture, this mixed-aldehyde approach is expeditious and is being largely exploited for the preparation of porphyrins bearing one or more meso-nitrophenyl groups. For instance, Tsuchida and coworkers [17–19] used that strategy for the preparation of the mono-(4-nitrophenyl)porphyrin 2 (Fig. 1). Using a 3:1 ratio of benzaldehyde and 4-nitrobenzaldehyde the desired porphyrin 2 was obtained in 2.7% yield; the related bis- (3 and 4) and tris(4-nitrophenyl) (5) substituted porphyrins were also isolated.

Structures of meso-(4-nitrophenyl)porphyrins
The same strategy was considered by Collman [20] to obtain the mono-(2-nitrophenyl) analogue and by Martell and coworkers [21] to obtain unsymmetrical 3-nitrophenyl substituted porphyrins. Little has also reported the synthesis of unsymmetrical porphyrins containing a 2,6-dinitrophenyl group, or a hydroxy-nitrophenyl group, as potential intermediates in the synthesis of difunctional “tailed-porphyrins” [22]. The mixed-aldehyde approach was also followed to prepare unsymmetrical porphyrins bearing o-nitrophenyl and p-carboxyphenyl substituents (6a–c, Fig. 2) [23]. The authors were able to optimize the benzaldehyde derivatives molar ratio in order to obtain the desired porphyrins in high yields. Other examples of unsymmetrical porphyrins prepared by this approach can be found in a review by Lindsey [12].

Unsymmetrical porphyrins bearing o-nitrophenyl and p-carboxyphenyl groups
As already mentioned, the direct nitration of meso-tetraarylporphyrins is another strategy to obtain porphyrins, mainly unsymmetrical ones, containing nitroaryl groups. This approach was considered for the first time by Kruper’s group in the nitration of meso-tetraarylporphyrins, namely TPP [24, 25]. The authors used an excess of red or yellow fuming nitric acid in different solvents (CHCl3, CH2Cl2, and AcOH) and referred higher yields for the mono-nitro derivative (ca 56%) in CHCl3; under these conditions the dinitro derivatives 3 and 4 (as a mixture of 2–3:1, respectively) were also isolated but the yields rarely exceed 5%. Better yields for the dinitro derivatives (28%) were obtained under conditions forcing the conversion of the mono-substituted derivative (use of 29 equivalents of red fuming nitric acid in CHCl3). The use of acetic acid as solvent allowed to obtain the tris(4-nitrophenyl)porphyrin 5 in 10% yield. The para regioselectivity was also observed in the nitration of porphyrins bearing 3-methyl- or 3-methoxyphenyl substituents.
Meng and coworkers [8] also studied the nitration of TPP but under slightly different conditions relatively to those reported by Kruper. The reactions were carried out by using a mixture of nitric acid and acetic acid and the degree of nitration was controlled by the reaction time. The 5-(4-nitrophenyl)-10,15,20-triphenylporphyrin 2 was obtained in 74% yield after 1 h of reaction. Significant improvements were also reported for the dinitro derivatives 3 and 4 (70% yield after 5 h of reaction) and for the trinitro derivative 5 (30% yield after 2 days of reaction). Attempts to obtain the tetrakis(4-nitrophenyl)porphyrin 1a via nitration of TPP failed and this was due to the degradation of the macrocycle during the time required for this lengthy reaction. Porphyrin 1a was obtained as a by-product (2%) during the tri-nitration conditions. The same authors studied the nitration of unsymmetrical porphyrins bearing phenyl and pyridyl groups. The nitration of these porphyrins requires higher reaction times than those used for TPP due to the protonation of the pyridyl groups; in certain cases the use of a mixture of acetic and sulfuric acids was required.
The selective nitration of TPP and other meso-tetraarylporphyrins at two neighboring aryl rings was described by Ostrowski and Lopuszynska [26]. Following a new protocol, which is based on the use of fuming yellow HNO3 in CHCl3, accompanied by a careful control of the reaction temperature, nitration of TPP afforded compound 3 in 42% yield. meso-Tetraarylporphyrins bearing 3-chlorophenyl, 3-methoxyphenyl or 3-methylphenyl groups afforded the corresponding 5,10-bis(4-nitroaryl)porphyrins in 30%, 37%, and 83% yields, respectively. The same group reported that under exhaustive nitration conditions tri-substituted derivatives can also be obtained in reasonable yields [27]. For instance, nitration of TPP afforded derivative 5 in 35% yield.
Smith and coworkers reported the selective nitration of the phenyl groups of TPP with sodium nitrite and trifluoroacetic acid [28]. The authors found that the degree of nitration can be efficiently controlled just by varying the amount of NaNO2/TFA used and the reaction time. Compound 2 was obtained in excellent yield (80–90%) after 3 min of reaction at room temperature in the presence of 1.8 equivalents of NaNO2 in TFA. The two isomeric bis(4-nitrophenyl)porphyrins 3 and 4 were obtained after 1.5 min of reaction in the presence of 8.1 equivalents of NaNO2 in a total yield of 63%, while the tris(4-nitrophenyl)porphyrin 5 required 1 h of reaction and 36.7 equivalents of NaNO2 to be isolated in 60% yield.
The nitration of the meso-phenyl groups of TPP with NO2BF4 has been also reported [29]. The authors found that the mode of addition is the key step for the success of this methodology. Mono-, bis-, and tris(4-nitrophenyl) derivatives were obtained in excellent yields (>90%) by dropwise addition of a sulfolane solution of 1.0, 2.9, or 4.6 equivalents, respectively, of that nitrating agent to a dichloromethane solution of TPP at room temperature. The authors have also reported the selectivity of the dinitration procedure for the 5,10-bis(4-nitrophenyl) isomer.
2.2 Synthesis of β-Nitro-Meso-Tetraarylporphyrins
Efficient nitrating procedures, based on electrophilic or radical conditions, are now well established for giving access to β-nitroporphyrins in excellent yields [30]. Most of the protocols are based on the use of metalloporphyrins. In fact, attempts to nitrate the free-base TPP using a mixture of nitric acid and sulfuric acid afforded only 2-nitro-meso-tetraphenylporphyrin 8a (M = 2H, Scheme 2) in low yield; this is due to the conversion of the starting porphyrin into the unreactive dication [31, 32]. The nitration of TPP (7a, M = 2H) under essentially neutral conditions was considered by Jackson and coworkers. Using nitronium tetrafluoroborate in a mixture of pyridine/chloroform at 140°C, the β-nitroporphyrin 8a (M = 2H) was isolated in 15% yield being accompanied by the pyridinium salt 9 (M = 2H) (18% yield). Attempted nitration of the zinc complex of TPP with nitronium tetrafluoroborate in pyridine afforded the pyridinium derivative 9 (M = Zn) in 80% yield [32].

Synthesis of β-nitro-meso-tetraarylporphyrins 8 and pyridinium salt 9
In contrast with the previous results, nitration of the copper, nickel, and palladium complexes of TPP with N2O4 occurs selectively at the β-pyrrolic positions, affording the corresponding complexes 8a (M = Cu, Ni or Pd) in quantitative yields [33]. The extension of this protocol to meso-tetraarylporphyrins 7b and 7c, (M = Cu), afforded the corresponding derivatives 8b and 8c in high yields (>90%), thus confirming the generality of the process. The presence of extra substituents at the β-pyrrolic position does not affect the site of nitration. For instance, the nitration of the copper complex of β-nitroporphyrin afforded an isomeric mixture of β,β′-disubstituted derivatives 10a–e (Fig. 3) in 85% total yield [34].

Structures of β-nitro-meso-tetraarylporphyrins
A procedure giving access to β-dinitro- and β-trinitro-meso-tetraphenylporphyrins using the controlled addition of fuming nitric acid to CuTPP was also reported [35]. The 2,12-dinitro- and 2,13-dinitro derivatives were obtained by the controlled addition of 0.7 mL of HNO3 to 100 mg of CuTPP in CHCl3 over a period of 1.2 min, while the 2,7-dinitro-, 2,8-dinitro-, and 2,18-dinitro derivatives were obtained by the addition of 1.0 mL of HNO3 during 1.0 min to the same amount of porphyrin. Yields of 20% for the pure products were reported. The trinitroporphyrins 11a–c (2,7,13-, 2,7,18-, and 2,8,12-trinitro) were obtained by increasing the amount of HNO3 up to 2.0 mL and maintaining the addition during the period of 1 min. The corresponding free-bases were obtained by demetallation with sulfuric acid. Electrochemical studies revealed that successive insertion of nitro groups at the β-positions shifts the one-electron ring oxidations anodically while the ring reduction occurs at a less cathodic potential relatively to the unsubstituted porphyrin free-bases.
Nitration of porphyrins coordinated with less electronegative metal ions, such as magnesium(II), zinc(II), chloroiron(III) and cobalt(II), and the β-nitro derivatives are accompanied by products resulting from reactions at the meso-position. In fact, the nitration of magnesium and zinc chelates of TPP by N2O4 afforded the corresponding β-nitro derivatives in low yields (ca. 25%) giving mainly the ring-opened bilinone 12 (Fig. 4) [36] and other non-porphyrin products resulting from reactions at the meso-positions. This metal ion dependent selectivity was justified by considering that the metalloporphyrin π-cation radicals obtained via oxidation by NO2· have different electron spin distributions (a1u or a2u) and, as a result, being the position of attack by further NO2· dependent on its spin density. The preferential attack at the meso-positions was also reported when the zinc complex of TPP was treated with thallium(III) nitrate or cerium(IV) ammonium nitrate followed by acid treatment [37]. Under these conditions the β-nitro derivative 8a (M = 2H) was isolated in low yields (15–28%) accompanied by porphodimethenes and the ring-opened bilinone 12.

Structure of the bilinone obtained by nitration of MgTPP or ZnTPP by N2O4
Callot and coworkers reported an excellent protocol to nitrate the copper complex of TPP based on the use of copper(I) nitrate in a mixture of chloroform, acetic acid, and acetic anhydride [38]. Using that nitrating mixture, Cavaleiro and coworkers prepared 8a (M = Cu) in 86% yield directly from TPP without the previous preparation of the copper complex [39]. The extension of this nitrating procedure to meso-tetrakis(pentafluorophenyl)porphyrin (7d, M = 2H) has provided access to the mono-nitroporphyrin 8d (M = 2H) and to a mixture of dinitro and trinitro isomers in quantitative yield [40]. Krishnan and coworkers also used copper(I) nitrate in chloroform to obtain the donor–acceptor porphyrins 8e (Fig. 5) for studies concerning quadratic nonlinear optics [41]. Callot and coworkers reported a mild procedure for the nitration of the nickel or copper complexes of meso-tetraarylporphyrins using lithium nitrate in CHCl3/Ac2O/AcOH, for 1.5 h at 40–45°C, affording the 2-nitro derivatives in 90–95% yield (aryl = phenyl, p-tolyl, and 3,5-di-tert-butylphenyl) [42].

Structure of porphyrin 8e
The selective β-mononitration of meso-tetraphenylporphyrin complexes can also be achieved using aqueous HNO3. Ostrowski et al. [43] found that several TPP complexes (7a, M = Zn, Cu, Ni, and Co) can be nitrated with adequate concentrations of HNO3 (ca. 25%) to afford the corresponding complexes 8a in very good yields (77% for M = Cu and 81% for M = Ni). A mixture of dinitro compounds (2–20%) is also detected in all cases. An extension of this work to other meso-tetraarylporphyrins (Ar = 3-NO2C6H4, 3-CH3C6H4, 3-ClC6H4, 2,6-Cl2C6H3, C6F5) afforded the corresponding mono-β-nitrated products in yields ranging from 74% to 93% [44]. These results show that the type of meso-aryl substituent does not change the site of nitration; it only affects the reaction yield. In fact, the systems less prone to electrophilic substitution require slightly drastic conditions (higher concentration of nitric acid and longer reaction times) to ensure high yields. Again, some dinitro compounds were also formed.
The nitration of the free-base meso-tetrakis(2,6-dichlorophenyl)porphyrin with red fuming nitric acid, at room temperature, afforded a 1:9 mixture of β-pentanitro- and β-hexanitroporphyrins in 70% yield [45]. Nitration of meso-tetrakis(pentafluorophenyl)porphyrin under similar conditions led to a mixture of regioisomers containing one nitro group on each pyrrole ring (55% yield). All attempts to obtain meso-tetrakis(2,6-dichlorophenyl)porphyrin substituted by more than six β-nitro groups or meso-tetrakis(pentafluorophenyl)porphyrin substituted by more than four β-nitro groups by using more HNO3, higher temperatures or longer reaction times in reactions between HNO3 and those porphyrins or their Zn(II) or Fe(III) complexes were unsuccessful [45]. Later, it was reported that the nitration of the zinc complex of meso-tetrakis(2,6-dichlorophenyl)porphyrin with red fuming nitric acid in the presence of nitromethane, acetic anhydride, and montmorillonite K-10, for 2 h at room temperature, affords the β-heptanitro derivative 13b (Fig. 6) in 50% yield [46]. An expeditious β-polynitration of meso-tetrakis(2,6-dichlorophenyl)porphyrin with montmorillonite K10-HNO3 using microwave irradiation has been described [47]. The microwave irradiation of that porphyrin with montmorillonite K10-HNO3 for 1.5 min selectively gave β-heptanitro derivative 13a in 72% yield. Similarly, the microwave irradiation of the Zn(II) and Cu(II) complexes with K10-HNO3 gave the β-heptanitro derivatives 13b and 13c in 81% and 75% yield, respectively.

β-Heptanitroporphyrin obtained by nitration of meso-tetrakis(2,6-dichlorophenyl)porphyrin with montmorillonite K10-HNO3 under microwave irradiation
3 Functionalization of Porphyrins via Nitro Groups
3.1 Functionalization of Meso-(Nitrophenyl)porphyrins
The nucleophilic aromatic substitution methodology has been used to functionalize meso-(nitrophenyl)porphyrins. Ostrowski and coworkers [48], for instance, reported that the copper and the zinc complexes of the meso-(4-nitrophenyl)porphyrin 2 react with carbanions 14 affording the corresponding products 15 in yields ranging from 50% to 67% (Scheme 3). These vicarious nucleophilic substitutions take place selectively at the ortho position to NO2 group; bulky carbanions of lower nucleophilicity do not react. Further studies showed that these reactions can also be performed using free-base porphyrins, with improved yields, if low temperatures are considered [49].

Vicarious nucleophilic substitution in meso-(4-nitrophenyl)porphyrins
The potentiality of meso-(nitrophenyl)porphyrins for further functionalization was shown in the synthesis of the porphyrin-fullerene dyad 16 (Fig. 7), a new artificial photosynthetic model [50].

Structure of a porphyrin-fullerene dyad
Ostrowski and coworkers also explored the activation of the nitro group towards the attack by nucleophiles to introduce the amino functionality in meso-tetraarylporphyrins bearing one or two nitrophenyl groups. For instance, the amino-functionalized porphyrins 17 (Fig. 8) were obtained from the reaction of the zinc, copper, and nickel complexes of porphyrin 2 with 1,1,1-trimethylhydrazinium iodide in the presence of KOH in DMSO [51, 52].

A porphyrin functionalized with a meso-(3-amino-4-nitrophenyl) group
The reaction of 5-(4-nitrophenyl)-10,15,20-triphenylporphyrin (2) and the corresponding zinc and copper complexes with other nucleophiles was also studied [53]. The authors reported that in the reaction of 2 with NaCN the substitution of the nitro group occurs and porphyrin 18 (Fig. 9) is obtained in reasonable yield. On the other hand, the reaction of 2 with phenoxides affords the diporphyrin derivative 19, while under the same conditions, but using the metal complexes, the nitro group is reduced to the amino group. A similar reduction occurs in the reaction with thiolates [53].

Structures of porphyrins 18 and 19
3.2 Functionalization of β-Nitroporphyrins
As already mentioned, nitroporphyrins are excellent starting materials to prepare new derivatives with improved features for specific applications. In fact, a nitro group is particularly useful to activate the pyrrole unit where it is inserted towards the attack by nucleophiles, dienes or 1,3-dipoles and also to direct electrophilic substitutions to the antipodal pyrrole ring. The alkene-type reactivity of β-nitro-meso-tetraarylporphyrins can be justified by the preferential localization of the double bond adjacent to the electron-withdrawing group; that double bond is not involved in the major aromatic delocalization pathway.
The possibility of using β-nitro-meso-tetraarylporphyrins for further functionalization at the β-pyrrolic positions was firstly considered by Crossley and coworkers [54, 55] who found that β-nitro-meso-tetraphenylporphyrin (8a, M = 2H) reacts with benzenethiolate and ethanethiolate to afford the corresponding β-thioethers. This pioneering work was followed by other publications exploring the reaction of β-nitro-meso-tetraarylporphyrins with nucleophiles to insert a variety of substituents at the β-pyrrolic positions. This topic has been comprehensively reviewed by Jaquinod [30].
The attack by nucleophiles to a β-nitro-meso-tetraarylporphyrin can occur either at the carbon atom containing the nitro substituent (ipso-attack) or at the adjacent β-position (α-attack) (Scheme 4). In fact, based on deuterium labeling experiments, Crossley and coworkers found that “soft” nucleophiles such as thiolates and the anion of benzaldoxime lead to products of type C, resulting from an ipso-attack [54, 56]. They also found that, in general, “hard” nucleophiles such as oxyanions [57], hydride [58], acylamide ions [59], Grignard and organolithium reagents [60] attack the β-pyrrolic position next to the nitro group. The outcome of these reactions is dependent on the nucleophile, coordinated metal ion, and temperature.

Representative reactivity of β-nitro-meso-tetraarylporphyrins with nucleophiles
The direct displacement of the nitro group was also observed when phenoxide ion and other phenols were used in reactions with the free-base 2-NO2TPP and with the corresponding Cu(II) and Ni(II) complexes [61, 62]. It was found that the type of product (2-aryloxy- or 2-hydroxyaryl-) can be controlled by the choice of solvent. This methodology was recently explored to build the ditopic chemosensor 22 (Scheme 5) [63]. This compound interacts selectively with histamine when compared with l-histidine and nicotine. Following the same protocol, Chen et al. [64] found that 8a and its Ni(II), Cu(II), and Zn(II) complexes react with 2-naphthoxide in protic solvents (2-naphthol, diglycol, and diglycol monomethyl ether), at 150°C, to afford only the C-coupling products 23 with yields varying from 50% to 81% (Scheme 6). In aprotic solvents (DMF or DMSO), at 150°C, the O-coupling products 24 are also obtained but at room temperature only compounds 23 are formed.

Reaction of 2-NO2TPP with phenoxides

Reaction of 2-NO2TPP with 2-naphthoxide
Following a synthetic methodology similar to the one developed by Crossley, Pan and coworkers [65] reported the synthesis of the β-(2,5-dihydroxyphenyl)porphyrins 25 (Scheme 7). The preliminary biological activity studies showed that the zinc(II) derivative has photo-toxicity on human chronic myelogenous leukemia cell and is able to cleave supercoiled DNA (pBR 322 DNA) while the copper(II) complex has lower biological activity.

Reaction of β-nitroporphyrins with hydroquinone
The use of anilines as nucleophiles in the reaction with β-nitro-meso-tetraarylporphyrins was considered by Cavaleiro and coworkers [66]. For instance, the reaction of 8a with aniline, at reflux temperature, gives the 2-(phenylamino) porphyrin 26a as the major product (53% yield), but the novel N-phenylquinolino[2,3,4-at]porphyrin 27a (6% yield) and the chlorin 28 are also formed (Scheme 8). When this reaction is performed in refluxing o-dichlorobenzene, porphyrin 27a is the main product (26% yield). This solvent was also adequate to obtain derivative 27b when p-toluidine was selected as the nucleophile. The oxidative cyclization of 2-arylaminoporphyrins 26 to the corresponding N-arylquinolino[2,3,4-at]porphyrins 27 can be done in excellent yields in nitrobenzene. This synthetic strategy is not efficient with anilines with electron-withdrawing substituents.

Reaction of 2-NO2TPP with anilines
Smith and coworkers explored the nitroalkene character of β-nitro-meso-tetraarylporphyrins to prepare β-fused pyrroloporphyrins 29 (Fig. 10) via the Barton–Zard condensation of the nickel complex of 2-NO2TPP with α-isocyanoacetic esters in the presence of DBU [67]. Interestingly, when the zinc complex of 2-NO2TPP was used, the cyclopropyl-annulated chlorin 30 was obtained.

Structures of porphyrins 29–31
Based on the conjugate addition of active methylene compounds, such as malonates or malononitrile, to β-nitro-meso-tetraarylporphyrins in the presence of a base, the same group was able to prepare a wide range of reduced porphyrins such as trans-nitrochlorins, cyclopropachlorins, or disubstituted trans-chlorins such as 31 [68, 69]. The product distribution can be controlled by the size of the carbanion, reaction time, and/or temperature as well as the use of free-bases or chelates.
Cavaleiro and coworkers reported that 1,3-diketones and 3-ketoesters such as acetylacetone and ethyl acetoacetate can act as efficient nucleophiles in Michael additions with β-nitro-meso-tetraarylporphyrins affording the corresponding derivatives 32 as the only products (Scheme 9) [70]. The central metal ion or the meso-aryl-substituents do not affect significantly the reactivity of the system, nor in terms of yields (72–88%) nor in terms of reaction times (40–50 min). The 1,3-dicarbonyl derivatives 32a,b showed to be excellent C3 synthons for the synthesis of porphyrins bearing an heteroaromatic group at the β-position [70].

Reaction of β-nitro-meso-tetraarylporphyrins with 1,3-diketones and 3-ketoesters
Cavaleiro and coworkers also considered the use of β-nitro-meso-tetraarylporphyrins as precursors to the novel [1,2,3]triazolo[4,5-b]porphyrins 33 (Scheme 10) [71]. Knowing that N-unsubstituted 1H-1,2,3-triazoles can be obtained from the reaction of sodium azide with alkenes bearing strongly electron-withdrawing, it was anticipated that the reaction β-nitroporphyrins with sodium azide could afford such type of compounds. As expected, the best yield (80%) was obtained with the porphyrin bearing electron-withdrawing groups at the meso positions.

Synthesis of [1,2,3]triazolo[4,5-b]porphyrins
A β-nitro-meso-tetraarylporphyrin was also used by Richeter et al. [72] to synthesize a porphyrin with an additional imidazole ring fused to a β,β′-pyrrolic bond (Scheme 11). The powerful amination reagent 4-amino-4H-1,2,4-triazole described by Callot and coworkers [73] was used to prepare intermediate 34 which, after reduction of the nitro group followed by reaction with formic acid and cyclization with trifluoroacetic acid, afforded the imidazo[4,5-b]porphyrin 35. The same authors found that compound 35 can be obtained in better yield (70%) if trimethyl orthoformate is used as an alternative to formic acid [74].

Synthesis of imidazo[4,5-b]porphyrins
Treatment of 2-nitro-meso-tetraarylporphyrins with excess triethyl phosphite at 155°C in 1,2-dichlorobenzene affords the corresponding cyclic enamines 36 in 70–75% yield (Scheme 12) [42].

Reaction of 2-nitro-meso-tetraarylporphyrins with triethyl phosphite
Cavaleiro and coworkers demonstrated that meso-tetraarylporphyrins can participate as dienophiles in Diels–Alder reactions affording adducts with important biological significance [75]. The same group found that they also participate as dipolarophiles in 1,3-dipolar cycloaddition reactions [76, 77]. As observed in other types of reactions, the presence of a β-nitro group also activates the porphyrin macrocycle towards cycloaddition reactions. This effect is evident in the reaction of β-NO2TPP 8a with diazomethane (Scheme 13) [78]. In fact, the cycloaddition occurs selectively at the substituted pyrrolic unit affording the pyrazoline-fused chlorin 37 (in 41% yield) accompanied by two minor compounds (38 and 39). It was shown that chlorin 37 is the precursor of the two minor products.

Reaction of 2-NO2TPP with diazomethane
The benefit of NO2 as a substituent to activate the β,β′-double bond where it is inserted was also considered in Diels–Alder reactions. Ostrowski et al. [79] revisited the reaction of porphyrins with the highly reactive diene ortho-benzoquinodimethane but now using β-NO2TPP as the dienophile (Scheme 14). The expected chlorin 40 was isolated as the main product in 54% yield accompanied by the naphthoporphyrin 41 and the dinaphthoporphyrin 42. When the non-functionalized TPP was used the expected chlorin was isolated in only 26% yield [75].

Diels-Alder reaction of 2-NO2TPP with ortho-benzoquinodimethane
4 Synthesis of Aminoporphyrins
In the last thirty years, porphyrins functionalized with amino groups have become popular starting materials for further functionalization. The possibility of using these versatile intermediates has been facilitated by their easy access in multigram scale from meso-tetraarylporphyrins, namely through well-known reduction procedures starting from adequate nitroporphyrins. Most of the protocols are based on the use of Sn/HCl or Sn/HCl /ultrasound [39], SnCl2/HCl [34, 80], NaBH4–Pd/C [54], HCOONH4–Pd/C [39], HCOONH4–Zn [81], or H2–Pd/C [82]. Other strategies consider the acid hydrolysis of adequate acetamidoporphyrins, prepared by condensation of pyrrole with acetamidobenzaldehydes under acidic conditions [83].
Nitrogen nucleophiles bearing a potential leaving group such as hydroxylamine, hydrazine, tosylhydrazine, and hydroxylamine O-sulfonic acid, 4-amino-4H-1,2,4-triazole are also being used to introduce the amino functionality directly in electrophilic centers of the porphyrinic core according to Callot procedures [73]. This approach was already mentioned for the amination of porphyrins bearing nitro groups and it is an important alternative to a previous approach involving also the reaction of 2-nitroporphyrins but with acylamide ions at the 3-position followed by hydrolysis of the amide bond [59].
The nucleophilic aromatic substitution of the para-F atoms of 5,10,15, 20-tetrakis(pentafluorophenyl)porphyrin (TF5PP) by amines, discovered by Kadish in 1990 [84], is also considered an efficient strategy to introduce different amino functionalities on that versatile platform. A comprehensive mini-review dedicated to this topic was recently published [85].
5 Functionalization of Aminoporphyrins
A wide range of porphyrin derivatives with improved properties have been prepared by functionalization of amino porphyrins, namely via amide linkage, alkylation, nucleophilic substitution, diazotization, cycloaddition reactions, and palladium-catalyzed transformations.
5.1 Via Amide Bonds
In the 1970 decade, meso-tetraarylporphyrins bearing aminophenyl groups, especially ortho-aminophenyl groups, were largely explored as excellent synthons in the construction of biomimetic models of heme proteins. The original work described by Collman in 1973 [86] was followed by the synthesis of a series of porphyrins named with fancy names such as “picket-fence”, “strapped”, or “pocket” porphyrins. Most of these compounds were constructed via amide bonds, and this topic has been the subject of exhaustive reviews [30, 87, 88].
The α,α,α,α-isomer of 5,10,15,20-tetrakis(o-aminophenyl)porphyrin 43 (Scheme 15) considered by Collman, also became very popular for the development of chiral catalysts and receptors for specific binding [89]. Most of the synthetic strategies reported on the development of receptors are based on the structural modification of porphyrin 43 using standard strategies or via its previous conversion into synthons 44 and 45 [90, 91].

Synthesis of “picket-fence” precursors
Porphyrins 46–54 described below are examples of picket-fence porphyrin-type receptors prepared from the α,α,α,α-isomer 43 [92]. The α,α,α,α-tetrakis(o-isocyanatophenyl)porphyrin 44 was considered in the synthesis of the anion sensors 46 and 47 using the adequate 2-(aminomethyl)phenol derivatives (Fig. 11) [93]. Compounds 46 were described as exhibiting good selectivity for AcO− and H2PO4 − while the p-nitrophenylazo derivatives 47 showed a selective coloration for F−, H2PO4 − and AcO−. Porphyrin derivatives 48 (Fig. 12), bearing different amino acid residues, were prepared by a similar approach [94]. These compounds showed promising features in sugar recognition.

“Picket-fence” porphyrins

“Picket-fence” porphyrins bearing amino acid residues
The reaction of α,α,α,α-tetrakis(o-chloroacetamidophenyl)porphyrin 45 with sodium imidazolate afforded porphyrin 49 (Fig. 13) containing imidazolium subunits [95]. UV/visible spectroscopic studies revealed that this receptor is selective for sulfate anions. Cyclic and square wave voltammetry studies demonstrate the receptor’s ability of compounds 45 and 49 to sense a variety of anions electrochemically via significant cathodic perturbations of the respective porphyrin’s first oxidation wave.

“Picket-fence” porphyrins containing imidazolium subunits
The reaction of 43 with 3,4-dimethoxybenzoyl chloride afforded the picket-fence porphyrin 50 and the corresponding complexes 51 were prepared using standard literature methods (Scheme 16). The anion binding ability of these compounds was evaluated and the best results were obtained with the cadmium and mercury complexes that showed to bind anions strongly in highly competitive solvent mixtures [96].

Synthesis of “picket-fence” porphyrin 50 and complexes 51
The disulfide and dithiocarbamate functionalized porphyrins 52 (Scheme 17) and 54 (Scheme 18) were considered in the synthesis of gold nanoparticles. The nanoparticles show to be more efficient to recognize anions than the free receptors in solution. The tetraamide 52 was prepared by reaction of 43 with thioctic acid, in the presence of EDC and HOBt (1-hydroxybenzotriazole), followed by the addition of Zn(AcO)2 (Scheme 17). The synthesis of the dithiocarbamateporphyrin 54 involved the reaction of synthon 45 with hexylamine, followed by reaction with carbon disulfide and Zn(AcO)2 (Scheme 18) [97, 98].

Synthesis of a “picket-fence“ porphyrin bearing disulfide groups

Synthesis of a “picket-fence” porphyrin bearing dithiocarbamate groups
meso-Tetraarylporphyrins bearing amino groups in para positions of the phenyl substituents were also considered in the design of receptors via amide bond. The cationic porphyrin 55 (Fig. 14), obtained by reacting 5,10,15,20-tetrakis(p-aminophenyl)porphyrin with 3-(pyridin-3-yl)propanoic acid in the presence of HOBt and HBTU (O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium hexafluorophosphate) followed by quaternization of the pyridyl nitrogens with methyl iodide and metallation with manganese(III), was described as showing a much higher preference for G-quadruplexes as opposed to duplex DNA [99].

Structure of the tetracationic porphyrin 55
The asymmetric 5-(4-aminophenyl)-10,15,20-triphenylporphyrin was used to prepare the porphyrin-functionalized [2]rotaxane host molecule 59 (Scheme 19). The synthesis involved the condensation of two equivalents of the aminoporphyrin with pyridine-3,5-dicarboxylic acid using adequate coupling agents, followed by cationization with methyl iodide. Then, a ring-closing metathesis mediated cyclization of the adequate bis-vinyl-functionalized benzene-1,3-dicarboxamide 58 in the presence of Grubbs 2nd generation catalyst afforded 59. This rotaxane exhibits a high binding affinity and general selectivity for chloride anions [100].

Synthesis of a porphyrin-functionalized [2]rotaxane
meso-Tetraarylporphyrins bearing aminophenyl groups have been used in the preparation of porphyrin derivatives conjugated to other bioactive compounds via amide bonds. A recent review covering this type of covalent attachment of porphyrins to peptides and proteins was recently published [101].
5.2 Via Alkylation of the Amino Group
The cationization of the amino groups was explored by several research groups to improve porphyrin DNA binding, photodynamic effect, solubility in physiological fluids, and selectivity to cancer cells. For instance, the cationic β-tetra- and β-octasubstituted porphyrins 64–67 were prepared by cationization of the corresponding aminoporphyrins 60–63 (Scheme 20) [102]. The synthetic approach to β-tetrasubstituted porphyrins 60 and 61 involved the mononitration of the 2,3,12,13-tetrabromoTPP with fuming HNO3, followed by Suzuki coupling with the adequate boronic acids and then reduction of the nitro group with SnCl2. A similar strategy was used to obtain the octasubstituted porphyrins 62 and 63 although the nitration of the phenyl groups preceded the bromination step.

Cationization of meso-(4-aminophenyl)porphyrins
Alkylation of 5-(4-aminophenyl)-10,15,20-triphenylporphyrin with 6-iodo-1,2: 3,4-di-O-isopropylidene-α-d-galactopyranose, followed by methylation with methyl iodide, afforded the cationic glycoporphyrin derivative 68 (Fig. 15) [103]. Removal of the isopropylidene groups from 68 by acid treatment afforded glycoporphyrin 69.

Cationic glycoporphyrins
5.3 Via Transition Metal Catalysis
Modification of amino groups mediated by transition metal complexes, such as palladium(0), is an interesting alternative to bromoporphyrins for carbon–nitrogen bond formation [13]. Van Lier and coworkers reported for the first time, but without experimental details, that 2-aminoporphyrins react with aryl halides affording 2-(arylamino)porphyrins [104]. Based on that methodology, usually known as Buchwald–Hartwig amination, Cavaleiro and coworkers were able to synthesize 2-arylaminoporphyrins 71 in excellent yields by reacting 2-NH2-NiTPP (70) with bromobenzene derivatives, even with electron-withdrawing substituents (Scheme 21) [66].

Synthesis of (2-arylamino)porphyrins via Buchwald–Hartwig amination
Cavaleiro and coworkers also used the Buchwald–Hartwig amination conditions to synthesize the porphyrin–phthalocyanine dyads 72 and 73 [105] and the porphyrin–porphyrin dyads 74 and 75 [106] (Fig. 16) where the two chromophores are linked by a nitrogen atom.

Structures of porphyrin-phthalocyanine dyads and porphyrin dyads obtained using Buchwald-Hartwing amination reactions
Dyad 73 was prepared by two complementary routes (Scheme 22). One of them involved the direct coupling of 2-NH2-NiTPP and the iodophthalocyanine 77 in the presence of Pd(OAc)2, rac-BINAP and KOtBu. The other approach involved the statistical cross-condensation of porphyrin-2-ylaminophthalonitrile 78 with 4-t-butylphthalonitrile. Phthalonitrile 78 was obtained from 2-aminoTPP and 4-iodophthalonitrile, using the same coupling conditions [Pd(OAc)2, rac-BINAP and KOtBu].

Synthetic routes to a porphyrin-phthalocyanine dyad
Similar catalytical conditions were used to couple 2-NH2-NiTPP with 2-Br-NiTPP (79) and 5-I-NiTPP (80) (Scheme 23). The electronic spectra of the dimers 75 and 81 are typical of highly delocalized systems and electrochemistry studies have shown that the first oxidation step occurs on the connecting amine function.

Synthesis of porphyrin dyads
The cyclic enamine 82 [42] (structurally related to 2-NH2-NiTPP) reacts with iodobenzene under Ullmann amination conditions (copper iodide, l-proline, potassium carbonate) to give access to the N-phenylquinolino[2,3,4-at]porphyrin 83 in good yield (Scheme 24) [107]. Electrochemical studies with the free-base and the corresponding Ni, Cu, and Pd complexes have shown that the presence of the N-phenyl group is responsible for the formation of stable radical cations.

N-Arylation of a quinolino[2,3,4-at]porphyrin
5.4 Via Diazonium Salts
The synthetic value of diazonium salts was considered in several synthetic approaches for further functionalization of the porphyrin core. These important synthons can be accessible from adequate aminoporphyrins using different diazotization conditions such as sodium nitrite and tetrafluoroboric acid at −5°C [108], sodium nitrite, and sulfuric acid [39] or with isoamyl nitrite [109].
The in situ decomposition of the diazonium salt obtained from 5-(4-aminophenyl)-10,15,20-triphenylporphyrin with isoamyl nitrite in the presence of single-walled carbon nanotubes (SWNTs) was considered in the synthesis of nanohybrids where the porphyrin was covalently attached to the nanotube (Scheme 25) [109]. The new materials showed better solubility and dispersion stability in organic solvents and superior optical limiting effects than SWNTs and C60.

Synthesis of pophyrin-SWNT nanohybrids
The diazonium salt of the nickel complex of 5-(4-aminophenyl)-10,15, 20-triphenylporphyrin was used to graft the corresponding complex to glassy carbon and gold and indium tin oxide surfaces via reduction of the diazonium moiety. Nitrosium tetrafluoroborate (NOBF4) was selected as the diazotizing agent. The characterization of the resulting materials confirms that the metallated porphyrin is intact, stably attached to the surface but with highly solvent-dependent electrochemistry [110].
The diazonium salts of the nickel or copper complexes of 2-amino-meso-tetraphenylporphyrin 72 are efficient intermediates to new 2-substituted porphyrins. Several β-alkyloxy substituted porphyrins 84 were obtained from the reaction of the in situ generated diazonium salt 83 (M = Cu) with alcohols or alkoxides (Scheme 26) [39, 111].

Synthesis of β-alkyloxy substituted porphyrins via diazonium salts
The diazotization of the 2-aminoporphyrins 72a–c with NaNO2 and sulfuric acid in tetrahydrofuran containing hydroperoxide gave rise to 2-diazo-3-oxo-tetraphenylchlorins 85 (Scheme 27) [112]. The photochemical induced dediazoniation of metallo 2-diazo-3-oxo-tetraphenylchlorins 85 in the presence of alcohols afforded the corresponding 2-alkyloxy derivatives 86 and other compounds that were justified by the existence of different reaction pathways after the formation of ketocarbenes by dediazoniation [112].

Synthesis of 2-diazo-3-oxo-tetraphenylchlorins
The reaction of porphyrin diazonium salts with sodium azide afforded porphyrins bearing azido substituents in excellent yields (ex: 87 and 88, Fig. 17). These compounds are important synthons for click chemistry [113–115].

Structures of β- and meso-azidoporphyrins
The porphyrin diazonium salt 83 (M = Ni) was also used as a pseudo-halide in Heck reactions [116]. The reactions were performed in the presence of methyl acrylate, propenal, and methyl vinyl ketone and afforded the expected unsaturated 2-substituted porphyrins 89a–c (Fig. 18). Depending on the α,β-unsaturated carbonyl compound used, the minor products 89d–f were also obtained. The formation of pyridoporphyrins 90 was justified by the reaction of the unchanged 2-aminoporphyrin with the α,β-unsaturated carbonyl compounds [117].

Structures of porphyrins 89–90
The extension of the previous studies to 3-sulfolene gave access, after isomerization and thermal extrusion of sulfur dioxide, to porphyrin 92 bearing a buta-1,3-dien-2-yl group in the β-pyrrolic position (Scheme 28) [118].

Synthesis of β-butadienyl porphyrin 92
The β-butadienyl porphyrin 92 showed to be an efficient diene in Diels–Alder reactions with a wide range of dienophiles such as [60]fullerene, N-phenylmaleimide, 1,4-benzoquinone and 1,4-naphthoquinone and fumaronitrile affording the expected adducts and/or the dehydrogenated ones in good yields. The adduct obtained from the reaction of 92 with fumaronitrile was used as precursor to a porphyrin–phthalonitrile that gave access to a series of novel porphyrin–phthalocyanine dyads bearing a rigid arrangement of the two units in close proximity [119].
Using the principles of the diazotization reaction, Igarashi’s group [120] developed a highly sensitive porphyrin-based spectrophotometric method for the determination of nitrite ion. This methodology uses the ability of the amino group of 5,10,15,20-tetrakis(4-aminophenyl)porphyrin to form a diazo group in the presence of nitrite ion in acidic conditions. The formation of a quinoid structure is responsible for a significant decrease in the absorbance relatively to the initial porphyrin. Latter, the same group used a porphyrin with only one amino group, the [5-(4-aminophenyl)-10,15,20-tris(4-pyridyl)porphyrin, in this sensing methodology [121].
5.5 The Dual Behavior of Aminoporphyrins
The dual behavior of 2-aminoporphyrins to act as an aromatic amine or as enamine gave access to an interesting number of new derivatives namely heterocyclic-fused porphyrins.
In 1997 Cavaleiro and coworkers reported that the reaction of the nickel(II) complex of 2-aminoTPP 81 with propenal and methyl vinyl ketone afforded, in the presence of H2SO4 and Pd(AcO)2, the fused pyridoporphyrins 90 [117]. The authors referred that a Michael addition, imine formation, and a dehydrogenation took place in products formation. The extension of those studies by the same group to other α,β-unsaturated carbonyl compounds such as 1,4-benzoquinone, 1,4-naphthoquinone and 2-hydroxy-1,4-naphthoquinone in the presence of catalytic amount of sulfuric acid, gave access to a plethora of new porphyrin–quinone dyads and π-extended heterocycle-fused porphyrin derivatives 93–98 (Scheme 29). The type of products obtained was dependent on the quinone used and was justified based on of dual behavior of the aminoporphyrin 81. The aromatic character can explain the formation of products 94, 99–101 while the others (93, 95–98) can be justified by its enamine character. The adaptation of the Nenitzescu reaction allowed the authors to elucidate the formation of the π-extended heterocyclic fused porphyrin derivatives 93 and 98.

Reaction of 2-aminoTPP with quinones
The products obtained from the reaction of 2-amino-meso-tetraphenylporphyrin with acryloyl chloride, also reflects the dual behavior of 2-aminoporphyrins [122]. While the amide 102 was the result of N-acylation, the formation of the dihydro-2-pyridone fused porphyrin 103 was justified through the aza-annulation reaction initiated by the Michael addition of the enamine followed by an intramolecular N-acylation (Scheme 30). Compound 103 is easily oxidized with DDQ to the corresponding 2-pyridone. The failure to obtain the dihydro-2-pyridone from the reaction with cinnamoyl chloride was explained by considering steric or electronic effects due to the phenyl group present on the β-carbon of the acyl chloride.

Reaction of 2-aminoTPP with acryloyl chloride
5.6 β-Iminoporphyrins as Heterodienes
The possibility of using the β-iminoporphyrins 104 as heterodienes in hetero-Diels–Alder reactions was reported by Cavaleiro and coworkers [123, 124]. A three component reaction involving β-aminoTPP, an aromatic aldehyde and an electron-rich dienophile (3,4-dihydro-2H-pyran or 2,3-dihydrofuran) catalyzed by lanthanum triflate leads to the expected tetrahydropyridine-fused derivatives 105 accompanied by the corresponding pyrido[2,3-b]porphyrins 106 (Scheme 31). A probable pathway to compounds 106 involves the aromatization of the pyridine ring, with the opening of the pyranyl ring, followed by the addition of another molecule of the enol ether. In the presence of La(OTf)3 and the enol ether, compounds 105 are converted into the corresponding pyrido[2,3-b]porphyrins 106.

Reaction of β-iminoporphyrin 104 with cyclic enol ethers
Pyrido[2,3-b]porphyrins bearing two vicinal hydroxyalkyl groups (107 and 108) were prepared through a two component domino reaction where an enol ether was used to generate the iminic heterodiene and also to act as the dienophile (Scheme 32) [125]. Treatment of the crude reaction mixture with a methanolic solution of p-toluenesulfonic acid is a key step in order to improve the yield of the desired products and to facilitate the purification process. The esterification of hydroxyalkyl groups in template 107a with succinic anhydride and dodecanoyl chloride afforded the corresponding esters in almost quantitative yields. The crystal structure of the most hydrophobic one showed that these porphyrin derivatives form one-dimensional supramolecular structure in the solid state.

Synthesis of pyrido[2,3-b]porphyrins 107–108
5.7 Aminoporphyrins as Carbene Acceptors
The catalytic insertion of ethyl diazoacetate into the amino group of 5-(4-aminophenyl)-10,15,20-triphenylporphyrin in the presence of an Rh-based catalyst was recently investigated [126]. Besides the formation of one compound resulting from the insertion of two carbene units, two other unexpected amides were isolated (Scheme 33). The formation of these amides was also observed when the 2-(4-aminophenyl)porphyrin was reacted with ethyl glycolate in the presence of the same catalyst. Derivative 110 crystallizes in an unusual chiral supramolecular metalloporphyrin chain, forming a right-handed helix arrangement.

Reaction of 5-(4-aminophenyl)-10,15,20-triphenylporphyrin with carbenes

Reaction of β-iminoporphyrin 112 with carbenes
The β-iminoporphyrin 112 reacts with carbenes generated from ethyl diazoacetate in the presence of catalytic amounts of lanthanum triflate (Scheme 34) [127]. The cis- and trans-aziridine 113 were obtained as the main products and the β-amino-α,β-unsaturated esters 114 and 115 as minor products (Scheme 35).

Synthesis of porphyrin-2,3-diones
Porphyrin-2,3-diones 116 are another example of porphyrin derivatives that can be obtained from 2-aminoporphyrins. Traditionally, the route to porphyrin-2,3-diones is the photo-oxidation of a 2-aminoporphyrin followed by hydrolysis of the resulting keto-imino chlorin [128], or the oxidation of 2-hydroxyporphyrins with the Dess–Martin periodinane (DMP) [129]. Burn reported that DMP is also efficient for the oxidation of 2-aminoporphyrins to porphyrin-2,3-diones 116 (Scheme 35) [130]. This method allows the reaction to be carried out on a large scale and it is easier than the classic photo-oxidation procedure. The porphyrin-2,3-diones are commonly used as building blocks for conjugated porphyrin arrays in the development of organic materials and molecular wires [131].
5.8 Aminoporphyrins in the Construction of New Assemblies
The design of molecular assemblies based on porphyrins self-association or aggregation is considered a simple method to afford supramolecular systems with a wide range of applications from models of enzyme active sites with relevance in catalysis to light-energy conversion and nanostructured components of electronic and optoelectronic devices.
In 1995 Gautam et al. [132] were able to conclude, from the coordination behavior of 5,10,15,20-tetrakis(3-aminophenyl)porphyrin and of the corresponding nitro precursor towards several metal(II) ions, such as Mg(II), Co(II), Zn(II) and Ag(II), that the amino derivatives are more prone to form aggregates than the nitro derivatives. These conclusions were based on the significant red shifts observed for the absorption and emission bands of the metallated amino derivatives when compared with the ones of the corresponding nitro derivatives. Similar red shifts were obtained for the nitro derivatives in the presence of dimethylaminopyridine supporting the existence of aggregated species in which metal ions are axially coordinated with the peripheral amino groups. Based on the model studies, possible structures were proposed and the authors refer that the amino group in the meta position is a key feature for the adequate binding of this group to the metal ion in the adjacent porphyrin.
The coordination ability of porphyrin derivatives bearing an amino group in conjugation with a keto group had also a great success in the construction of new assemblies connected by metal ions. The first studies [133, 134] involved the use of the enaminoketone porphyrins 118 obtained from the reaction of the corresponding ketone derivatives 117 with nitrogen nucleophiles bearing a potential leaving group (hydroxylamine, hydrazine, tosylhydrazine, and hydroxylamine O-sulfonic acid or 4-amino-4H-1,2,4-triazole) (Scheme 36). An alternative to the previous route used for obtaining ketone 117a that involves the formylation of a metalloporphyrin under Vilsmeier–Haack conditions followed by acid-catalyzed cyclization of the aldehyde and demetallation of the porphyrin [135–137] was suggested for derivative 117b. The new synthetic strategy is based on the hydrolysis of the ester group of the nickel complex of a porphyrin bearing a o-methoxycarbonylphenyl group and three 3,5-di-tert-butylphenyl groups, followed by acid chloride formation and an intramolecular Friedel–Crafts reaction. The authors refer the superiority of 4-amino-4H-1,2,4-triazole in the amination process when compared with the other nitrogen nucleophiles, since it can be used in the amination of free-bases and also of nickel, palladium, and copper complexes.

Synthesis of enaminoketone porphyrins
Porphyrin dimers 119 were assembled by several routes namely by the selective coordination of a metal ion to the external sites of two enaminoketone free-bases, followed by metallation of the internal sites or by coordination of two molecules of the metalloenaminoketone on a selected metal ion (Scheme 37). In general, this last approach showed to be more adequate due to the instability of the initial dimer obtained from the free-bases. The trans arrangement around the metal was confirmed by NMR experiments and the electronic spectra (intensified red-shifted Q bands) and electrochemical behavior (oxidation potentials split into two redox steps substantially lowered when compared with the monomeric units) of the dimers were indicative of a strong interaction between the two units introduced through the connecting metal; the coplanar coordination to the porphyrin ring is indicated by the authors as the main reason for the large interactions displayed in the ground state.

Synthesis of porphyrin dimers by coordination of a metal ion to the external sites of two enaminoketones
The extension of the previous studies to bis-enaminoketones allowed a stepwise preparation of polymetallic oligomers of type 120 and 121 (Fig. 19) connected by metal centers [73, 138].

Polymetallic porphyrin oligomers
These studies were extended to the synthesis of a series of nickel bis-enaminoketone isomers and by thionation of these derivatives to bis-enaminothioketone analogues [139]. The authors refer that by controlling the amount of the Lawesson’s reagent used in the thionation process it was possible to isolate the monothionated analogues affording derivatives with mixed external chelating groups (NO/NS). The electrochemistry behavior of the new derivatives was also studied and it is in good agreement with the recorded electronic spectra.
Porphyrins bearing other peripheral chelating groups fully conjugated with the porphyrin core, such as the enaminoaldehydes 122 (Scheme 38) and the enaminoketones 125 (Scheme 39), were also developed by the same group and were used in the preparation of porphyrin dimers linked by metal ions [42].

Synthesis of enaminoaldehyde functionalized porphyrins

Synthesis of enaminoketone porphyrins
The enaminoketone ligands 125 were obtained from 2-formyl-meso-tetraarylporphyrins 79 according to Ishkov conditions [140] followed by amination with 4-amino-4H-1,2,4-triazole. All ligands, namely the corresponding thioanalogues obtained by thionation with Lawesson’s reagent, were metallated with palladium affording the corresponding dimers. From the structural characterization the authors were able to conclude that in the enamino aldehyde and thioaldehyde series the cis isomer is thermodynamically favored and strong porphyrin–porphyrin interactions were also detected in this new series of ligands.
6 Conclusions
This chapter brings an update to the synthesis and reactivity features of nitro- and aminoderivatives of meso-tetraarylporphyrins. Such derivatives can be obtained by well-established synthetic and derivatization methodologies. The nitro and the amino substituents play a key role in the functionalization of the corresponding porphyrin macrocycles into a variety of other ones which have already demonstrated significant applications.
References
Fisher H, Zeile K (1929) Synthese des hämatoporphyrins, protoporphyrins und hämins. Liebigs Ann Chem 468:98
Woodward RB, Ayer WA, Beaton JM, Bickelhaupt F, Bonnett R, Buchschacher P, Closs GL, Dutler H, Hannah J, Hauck FP, Itô S, Langemann A, Le Goff E, Leimgruber W, Lwowski W, Sauer J, Valenta Z, Voltz H (1960) The total synthesis of chlorophyll. J Am Chem Soc 82:3800
Pandey RK, Zheng G (2000) Porphyrins as photosensitizers in photodynamic therapy. In: Kadish KM, Smith KM, Guilard R (eds) The porphyrin handbook. Academic, San Diego
Aida T, Inoue S (2000) Metalloporphyrins as catalysts for precision macromolecular synthesis. In: Kadish KM, Smith KM, Guilard R (eds) The porphyrin handbook. Academic, San Diego
Chou J-H, Kosal ME, Nalwa HS, Rakow NA, Suslick KS (2000) Applications of porphyrins and metalloporphyrins to materials chemistry. In: Kadish KM, Smith KM, Guilard R (eds) The porphyrin handbook. Academic, San Diego
Ono N (2002) The nitro group in organic synthesis. Wiley-VCH, New York
Ricci A (2008) Amino group chemistry: from synthesis to the life sciences. Wiley-VCH, Weinheim
Meng GG, James BR, Skov KA (1994) Porphyrin chemistry pertaining to the design of anti-cancer drugs; part 1, the synthesis of porphyrins containing meso-pyridyl and meso-substituted phenyl functional groups. Can J Chem 72:1894
Thomas DW, Martell AE (1956) Tetraphenylporphine and some para-substituted derivatives. J Am Chem Soc 78:1335
Thomas DW, Martell AE (1956) Absorption spectra of para-substituted tetraphenylporphines. J Am Chem Soc 78:1338
Rothemund P (1935) Formation of porphyrins from pyrroles and aldehydes. J Am Chem Soc 57:2010
Lindsey JS (2000) Synthesis of meso-substituted porphyrins. In: Kadish KM, Smith KM, Guilard R (eds) The porphyrin handbook. Academic, Boston
Cavaleiro JAS, Tomé AC, Neves MGPMS (2010) meso-Tetraarylporphyrin derivatives: new synthetic methodologies. In: Kadish KM, Smith KM, Guilard R (eds) Handbook of porphyrin science. World Scientific, Singapore
Bettelheim A, White BA, Raybuck SA, Murray RW (1987) Electrochemical polymerization of amino-, pyrrole-, and hydroxy-substituted tetraphenylporphyrins. Inorg Chem 26:1009
Chauhan SMS, Sahoo BB, Srinivas KA (2001) Microwave-assisted synthesis of 5,10,15, 20-tetraarylporphyrins. Synth Commun 31:33
Collman JP, Gagne RR, Christopher AR, Halbert TR, Lang G, Robinson WT (1975) “Picket fence porphyrins”. Synthetic models for oxygen binding hemoproteins. J Am Chem Soc 97:1427
Haesegawa E, Nemoto J, Kangyama T, Tsuchida E (1978) Oxygenation of polymer-covalently bonded metalloporphyrins. Macromolecules 11:947
Haesegawa E, Nemoto J-I, Kanayama T, Tsuchida E (1978) Syntheses and properties of vinyl monomers containing a meso-tetraphenylporphin ring and their copolymers. Eur Polym J 14:123
Tsuchida E (1979) Approaches to artificial macromolecular oxygen carriers. J Macromol Sci-Chem A13:545
Collman JP, Brauman JI, Doxsee KM, Halbert TR, Bunnenberg E, Linder RE, LaMar GN, Del Gaudio J, Lang G, Spartalian K (1980) Synthesis and characterization of “tailed picket fence” porphyrins. J Am Chem Soc 102:4182
Sun Y, Martell AE, Tsutsui M (1986) The synthesis and proton nuclear magnetic resonance study of some nitro- and amino-unsymmetrically meta-substituted tetraphenylporphyrins. J Heterocycl Chem 23:561
Little RG (1981) Notes on the synthesis of meso-substituted porphyrins from pyrryl carbinols and the mechanism of the Rothemund reaction. Heterocycl Chem 18:129
Schiavon MA, Iwamoto LS, Ferreira AG, Iamamoto Y, Zanoni MVB, Assis MD (2000) Synthesis and characterization of a novel series of meso(nitrophenyl) and meso(carboxyphenyl) substituted porphyrins. J Braz Chem Soc 11:458
Kruper WJ, Chamberlin TA (1988) US Patent 4,746,735
Kruper WJ, Chamberlin TA, Kochanny M (1989) Regiospecific aryl nitration of meso-substituted tetraarylporphyrins: a simple route to bifunctional porphyrins. J Org Chem 54:2753
Ostrowski S, Lopuszynska B (2003) Preparation of meso-tetraarylporphyrins nitrated in two neighboring aromatic rings. Synth Commun 33:4101
Ostrowski S, Mikus A, Lopuszynska B (2004) Synthesis of highly substituted meso-tetraarylporphyrins. Tetrahedron 60:11951
Luguya R, Jaquinod L, Fronczek FR, Vicente MGH, Smith KM (2004) Synthesis and reactions of meso-(p-nitrophenyl)porphyrins. Tetrahedron 60:2757
Smith NW, Dzyubam SV (2010) Efficient nitration of meso-tetraphenylporphyrin with nitronium tetrafluoroborate. Arkivoc vii:10
Jaquinod L (2000) Functionalization of 5,10,15,20-tetra-substituted porphyrins. In: Kadish KM, Smith KM, Guilard R (eds) The porphyrin handbook. Academic, San Diego
Jonhson AW, Winter M (1975) Meso-addition reaction of meso-tetraphenylporphyrin. Chem Ind (London) 351
Cavaleiro JAS, Neves MGPMS, Hewlins MJE, Jackson AH (1986) Reactions of porphyrins with nitronium tetrafluoroborate in pyridine. J Chem Soc Perkin Trans 1:575
Catalano MM, Crossley MJ, Harding MM, King, LC (1984) Control of reactivity at the porphyrin periphery by metal ion co-ordination: a general method for specific nitration at the β-pyrrolic position of 5,10,15,20-tetraarylporphyrins. J Chem Soc Chem Commun 1535
Crossley MJ, Govenlock LJ, Prashar JK (1995) Synthesis of porphyrin-2,3,12,13- and −2,3,7,8-tetraones: building blocks for the synthesis of extended porphyrin arrays. J Chem Soc Chem Commun 2379
Dahal S, Krishnan V, Nethaji M (1998) Coordination aggregation of meso-aryl porphyrins. Proc Indian Acad Sci (Chem Sci) 110:37
Cavaleiro JAS, Hewlins MJE, Jackson AH, Neves MGPMS (1986) Structures of the ring-opened oxidation products from meso-tetraphenylporphyrin. J Chem Soc Chem Commun 142
Evans B, Smith KM, Cavaleiro JAS (1978) Bile pigment studies. Part 4. Some novel reactions of metalloporphyrins with thallium(III) and cerium(IV) salts. Ring cleavage of meso-tetraphenylporphyrin. J Chem Soc Perkin Trans 1 768
Giraudeau A, Callot HJ, Jordan J, Ezhar I, Gross M (1979) Substituent effects in the electroreduction of porphyrins and metalloporphyrins. J Am Chem Soc 101:3857
Hombrecher HK, Gherdan VM, Ohm S, Cavaleiro JAS, Neves MGPMS, Condesso MF (1993) Synthesis and electrochemical investigation of β-alkyloxy substituted meso-tetraphenylporphyrins. Tetrahedron 49:8569
Vicente MGH, Neves MGPMS, Cavaleiro JAS, Hombrecher HK, Koll D (1996) Electrochemical and spectroscopic properties of Cu(II) β-nitro-meso-tetra(pentafluorophenyl)porphyrins. Tetrahedron Lett 37:261
Sen A, Ray PC, Das PK, Krishnan V (1996) Metalloporphyrins for quadratic nonlinear optics. J Phys Chem 100:19611
Richeter S, Jeandon C, Gisselbrecht J-P, Graff R, Ruppert R, Callot H (2004) Synthesis of new porphyrins with peripheral conjugated chelates and their use for the preparation of porphyrin dimers linked by metal ions. Inorg Chem 43:251
Ostrowski S, Szerszen D, Ryszczuk M (2005) Electrophilic nitration of meso-tetraarylporphyrin complexes at the beta-pyrrolic position. Synthesis 819
Wyrebek P, Ostrowski S (2007) Synthesis of some β-nitro-meso-tetraphenylporphyrin derivatives. J Porphyr Phthalocyanines 11:822
Bartoli JF, Battioni P, De Poor WR, Mansuy D (1994) Synthesis and remarkable properties of iron β-polynitroporphyrins as catalysts for monooxygenation reactions. J Chem Soc Chem Commun 23
Ozette K, Battioni P, Leduc P, Bartoli J-F, Mansuy D (1998) A new manganese-β-heptanitro-porphyrin with extreme redox potentials: spectral electrochemical and catalytic properties. Inorg Chim Acta 272:4
Chauhan SMS, Kumar A, Srinivas KA (2004) β-Polynitration of 5,10,15,20-tetrakis (2,6-dichlorophenyl)porphyrins with HNO3 and Cu(NO3)2 on clay using microwave irradiation. Synth Commun 34:2673
Ostrowski S, Shim YK (2001) Selective derivatization of TPP on one phenyl ring by nitration and subsequent nucleophilic substitution of hydrogen. Bull Korean Chem Soc 11:1
Ostrowski S, Urbańska N, Mikus A (2003) Nucleophilic substitution of hydrogen in meso-nitroaryl-substituted porphyrins unprotected at the NH-centers in the core ring. Tetrahedron Lett 44:4373
Ostrowski S, Mikus A (2003) A new approach to the synthesis of porphyrin-fullerene dyads. Mol Divers 6:315
Ostrowski S, Grzyb S (2007) Transformation of nitro-5,10,15,20-tetraarylporphyrins into their amino/nitro-functionalized derivatives. Jordan J Chem 2:297
Ostrowski S, Grzyb S, Mikus A (2007) Direct amination of meso-tetraarylporphyrin derivatives – easy route to A3B-, A2BC-, and A2B2-type porphyrins bearing two nitrogen-containing substituents at the meso-positioned phenyl groups. Helvetica Chim Acta 90:2000
Zhi-jun S, Yang-yan O, Qin H, Dan-li Z, Zhang-ping C (2005) Novel substitution reactions of 5-(4-nitrophenyl)-10,15,20-triphenylporphyrin with nucleophilic reagents. Wuhan Univ J Nat Sci 10:919
Baldwin JE, Crossley MJ, DeBernardis J (1982) Efficient peripheral functionalization of porphyrins. Tetrahedron 38:685
Catalano MM, Crossley MJ, Harding MM, King LC (1984) Control of reactivity at the porphyrin periphery by metal ion co-ordination: a general method for specific nitration at the β-pyrrolic position of 5,10,15,20-tetraarylporphyrins. J Chem Soc Chem Commun 1537
Crossley MJ, King LG, Pyke SM (1987) A new and highly efficient synthesis of hydroxyporphyrins. Tetrahedron 43:4569
Crossley MJ, King LG (1996) Reaction of metallo-2-nitro-5,10,15,20-tetraphenylporphyrins with oxyanions. Temperature-dependent competition between nucleophilic addition and single-electron transfer processes. J Chem Soc Perkin Trans 1 1251
Crossley MJ, King LG (1993) A new method for regiospecific deuteration and reduction of 5,10,15,20-tetraphenylporphyrins: nucleophilic reaction of borohydride ion with 2-nitro-5,10,15,20-tetraphenylporphyrins. J Org Chem 58:4370
Crossley MJ, King LG, Newsom IA, Sheehan CS (1996) Investigation of a 'reverse' approach to extended porphyrin systems. Synthesis of a 2,3-diaminoporphyrin and its reactions with α-diones. J Chem Soc Perkin Trans 1 2675
Crossley MJ, Harding MM, Tansey CW (1994) A convenient synthesis of 2-alkyl-5,10,15, 20-tetraphenylporphyrins: reaction of metallo-2-nitro-5,10,15,20-tetraphenylporphyrins with Grignard and organolithium reagents. J Org Chem 59:4433
Crossley MJ, Gosper JJ, King LG (1988) C-hydroxyarylation of tetraphenylporphyrin – convenient introduction of a functionality which is oriented towards the porphyrin centre. Tetrahedron Lett 29:1597
Crossley MJ, King LG, Simpson JL (1997) Solvent-dependent ambident nucleophilicity of phenoxide ion towards nitroporphyrins: synthesis of 2-hydroxyaryl- and 2-aryloxy-5,10,15,20-tetraphenylporphyrins by displacement of a nitro group. J Chem Soc Perkin Trans 1 3087
Guo Q-N, Li Z-Y, Chan W-H, Lau K-C, Crossley M (2010) Appending zinc tetraphenylporphyrin with an amine receptor at β-pyrrolic carbon for designing a selective histamine chemosensor. Supramol Chem 22:122
Chen Z, Hu Q, Zhai B, Jiang Z, Qin W (2007) Nucleophilic substitution on porphyrin ring: synthesis of 2-(2-hydroxynaphthyl)-5,10,15,20-tetraphenylporphyrin. Chin J Chem 55:251
Huang Q, Pan Z, Wang P, Chen Z, Zhang X, Xu H (2006) Zinc(II) and copper(II) complexes of β-substituted hydroxylporphyrins as tumor photosensitizers. Bioorg Med Chem Lett 16:3030
Pereira AMVM, Alonso CMA, Neves MGPMS, Tomé AC, Silva AMS, Paz FAA, Cavaleiro JAS (2008) A new synthetic approach to N-arylquinolino[2,3,4-at]porphyrins from β-arylaminoporphyrins. J Org Chem 73:7353
Jaquinod L, Gros C, Olmstead MM, Antolovich M, Smith KM (1996) First syntheses of fused pyrroloporphyrins. Chem Commun 1475
Jaquinod L, Gros C, Khoury RG, Smith KM (1996) A convenient synthesis of functionalized tetraphenylchlorins. Chem Commun 2581
Shea KM, Jaquinod L, Smith KM (1998) Dihydroporphyrin synthesis: new methodology. J Org Chem 63:7013
Giuntini F, Faustino MAF, Neves MGPMS, Tomé AC, Silva AMS, Cavaleiro JAS (2005) Synthesis and reactivity of 2-(porphyrin-2-yl)-1,3-dicarbonyl compounds. Tetrahedron 61:10454
Lacerda PSS, Silva AMG, Tomé AC, Neves MGPMS, Silva AMS, Cavaleiro JAS, Llamas-Saiz AL (2006) [1,2,3]Triazolo[4,5-b]porphyrins: new building blocks for porphyrinic materials. Angew Chem Int Ed 45:5487
Richeter S, Hadj-Aïssa A, Taffin C, van der Lee KA, Leclercq D (2007) Synthesis and structural characterisation of the first N-heterocyclic carbene ligand fused to a porphyrin. Chem Commun 2148
Richeter S, Jeandon C, Ruppert R, Callot HJ (2002) A modular approach to porphyrin oligomers using metal ions as connectors. Chem Commun 266
Lefebvre J-F, Leclercq D, Gisselbrecht J-P, Richeter S (2010) Synthesis, characterization, and electronic properties of metalloporphyrins annulated to exocyclic imidazole and imidazolium rings. Eur J Org Chem 1912
Tomé AC, Lacerda PSS, Neves MGPMS, Cavaleiro JAS (1997) meso-Arylporphyrins as dienophiles in Diels–Alder reactions: a novel approach to the synthesis of chlorins, bacteriochlorins and naphthoporphyrins. Chem Commun 1199
Silva AMG, Tomé AC, Neves MGPMS, Silva AMS, Cavaleiro JAS (1999) meso-Tetraarylporphyrins as dipolarophiles in 1,3-dipolar cycloaddition reactions. Chem Commun 1767
Tomé AC, Neves MGPMS, Cavaleiro JAS (2009) Porphyrins and other pyrrolic macrocycles in cycloaddition reactions. J Porphyr Phthalocyanines 13:408
Silva AMG, Tomé AC, Neves MGPMS, Cavaleiro JAS (2002) Porphyrins in 1,3-dipolar cycloaddition reactions: synthesis of a novel pyrazoline-fused chlorin and a pyrazole-fused porphyrin. Synlett 1155
Ostrowski S, Wyrębek P (2006) The first example of Diels–Alder cycloaddition of ortho-xylylenes to meso-tetraarylporphyrins containing electron-deficient β, β-double bonds. Tetrahedron Lett 47:8437
Rose E, Kossanyi A, Quelquejeu M, Soleilhavoup M, Duwavran F, Bernard N, Lecas A (1996) Synthesis of biomimetic heme precursors: the “double picket fence” 5,10,15, 20-tetrakis(2′,6′-dinitro-4′-tert-butylphenyl)porphyrin. J Am Chem Soc 118:1567
Weimin S, Qi S, Yucheng W, Lihong L, Jingchao T (2010) An alternative approach to amino porphyrins. J Heterocycl Chem 47:1221
Lipinska ME, Teixeira DMD, Laia CAT, Silva AMG, Rebelo SLH, Freire C (2013) β-Functionalized zinc(II)aminoporphyrins by direct catalytic hydrogenation. Tetrahedron Lett 54:110
George CD, Richardson T, Hofton ME, Vale CM, Neves MGPMS, Cavaleiro JAS (1999) Chlorine gas sensing using thin films of meso-tetra(p-stearamidophenyl)porphyrin. Mater Sci Eng C 8–9:559
Kadish KM, Araullo-McAdams C, Han BC, Franzen MM (1990) Syntheses and spectroscopic characterization of (T(p-Me2N)F4PP)H2 and (T(p-Me2N)F4PP)M where T(p-Me2N)F4PP = the dianion of meso-tetrakis(o, o, m, m-tetrafluoro-p-(dimethylamino)phenyl) porphyrin and M = cobalt(II), copper(II), or nickel(II). Structures of (T(p-Me2N)F4PP)Co and meso-tetrakis(pentafluorophenyl)porphinatocobalt(II), (TF5PP)Co. J Am Chem Soc 112:8364
Costa JIT, Tomé AC, Neves MGPMS, Cavaleiro JAS (2011) 5,10,15,20-Tetrakis(pentafluorophenyl)porphyrin: a versatile platform to novel porphyrinic materials. J Porphyr Phthalocyanines 15:1117
Collman JP, Gagne RR, Halbert TR, Marchon JC, Reed CA (1973) Reversible oxygen adduct formation in ferrous complexes derived from a picket fence porphyrin. Model for oxymyoglobin. J Am Chem Soc 95:7868
Baldwin JE, Perlmutter P (1984) Bridged, capped and fenced porphyrins. In: Vögtle F, Weber E (eds) Topics in current chemistry. Springer, Berlin
Tomé AC, Silva AMS, Alkorta I, Elguero J (2011) Atropoisomerism and conformational aspects of meso-tetraarylporphyrins and related compounds. J Porphyr Phthalocyanines 15:1
Sessler JL, Karnas E, Sedenberg E (2012) Porphyrins and expanded porphyrins as receptors. In: Philip A, Jonathan W (eds) Supramolecular chemistry: from molecules to nanomaterials. Wiley, Chichester
Collman JP, Boitrel B, Fu L, Galanter J, Straumanis A, Rapta M (1997) The chloroacetamido group as a new linker for the synthesis of hemoprotein analogues. J Org Chem 62:2308
Collman JP, Wang Z, Straumanis A (1998) Isocyanate as a versatile synthon for modular synthesis of functionalized porphyrins. J Org Chem 63:2424
Lindsey JS (1980) Increased yield of a desired isomer by equilibriums displacement on binding to silica gel, applied to meso-tetrakis(o-aminophenyl)porphyrin. J Org Chem 45:5215
Lee C, Lee DH, Hong J-I (2001) Colorimetric anion sensing by porphyrin-based anion receptors. Tetrahedron Lett 42:8665
Ladomenou K, Bonar-Law RP (2002) Urea porphyrins as simple receptors for sugars. Chem Commun 2108
Cormode DP, Murray SS, Cowley AR, Beer PD (2006) Sulfate selective anion recognition by a novel tetra-imidazolium zinc metalloporphyrin receptor. Dalton Trans 5135
Cormode DP, Drew MGB, Jagessar R, Beer PD (2008) Metalloporphyrin anion sensors: the effect of the metal centre on the anion binding properties of amide-functionalised and tetraphenyl metalloporphyrins. Dalton Trans 6732
Beer PD, Cormode DP, Davis JJ (2004) Zinc metalloporphyrin-functionalised nanoparticle anion sensors. Chem Commun 414
David P, Cormode DP, Drew MGB, Jagessar R, Beer PD (2008) Anion sensing porphyrin functionalized nanoparticles. J Inorg Organomet Polym 18:32
Dixon IM, Lopez F, Tejera AM, Esteve J, Blasco MA, Pratviel G, Meunier B (2007) A G-quadruplex ligand with 10000-fold selectivity over duplex DNA. J Am Chem Soc 129:1502
Brown A, Beer PD (2012) Porphyrin-functionalised rotaxanes for anion recognition. Dalton Trans 41:118
Giuntini F, Alonso CMA, Boyle RW (2011) Synthetic approaches for the conjugation of porphyrins and related macrocycles to peptides and proteins. Photochem Photobiol Sci 10:759
Chen B, Li A, Liang F, Zhou X, Cao X, He Z (2006) Synthesis of some multi-β-substituted cationic porphyrins and studies on their interaction with DNA. Tetrahedron 62:5487
Silva EMP, Serra VV, Ribeiro AO, Tomé JPC, Domingues P, Faustino MAF, Neves MGPMS, Tomé AC, Cavaleiro JAS, Ferrer-Correia AJ, Iamamoto Y, Domingues MRM (2006) Characterization of cationic glycoporphyrins by electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom 20:3605
Khan MM, Ali H, van Lier JE (2001) Synthesis of new aminoporphyrins via palladium-catalysed cross-coupling reactions. Tetrahedron Lett 42:1615
Soares ARM, Martínez-Díaz MV, Bruckner A, Pereira AMVM, Tomé JPC, Alonso CMA, Faustino MAF, Neves MGPMS, Tomé AC, Silva AMS, Cavaleiro JAS, Torres T, Guldi DM (2007) Synthesis of novel N-linked porphyrin-phthalocyanine dyads. Org Lett 9:1557
Pereira AMVM, Neves MGPMS, Cavaleiro JAS, Jeandon C, Gisselbrecht J-P, Choua S, Ruppert R (2011) Diporphyrinylamines: synthesis and electrochemistry. Org Lett 13:4742
Pereira AMVM, Neves MGPMS, Cavaleiro JAS, Gisselbrecht J-P, Jeandon C, Ruppert R (2011) A new approach to N-phenylquinolino[2,3,4-at]porphyrins. Electrochemical and photochemical studies. J Porphyr Phthalocyanines 15:575
Fuhrhop J-H (1978) Irreversible reactions on the porphyrin periphery (excluding oxidations, reductions, and photochemical reactions). In: Dolphin D (ed) The porphyrins, vol 2. Academic, New York
Guo Z, Du F, Ren D, Chen Y, Zheng J, Liuc Z, Tian J (2006) Covalently porphyrin-functionalized single-walled carbon nanotubes: a novel photoactive and optical limiting donor–acceptor nanohybrid. J Mater Chem 16:3021
Gross AJ, Bucher C, Coche-Guerente L, Labbé P, Downard AJ, Moutet J-C (2011) Nickel (II) tetraphenylporphyrin modified surfaces via electrografting of an aryldiazonium salt. Electrochem Commun 13:1236
Hombrecher HK, Gerdan VM, Cavaleiro JAS, Neves MGPMS (1994) An efficient synthesis of β-alkyloxy substituted porphyrins. J Prak Chem-Chem Ztg 336:542
Cavaleiro JAS, Gerdan VM, Hombrecher HK, Neves MGPMS, Silva A (1997) Synthesis and characterization of new 2-diazo-3-oxo-5,10,15,20-tetraphenylchlorins. Heterocycl Commun 3:253
Shen D-M, Liu C, Chen Q-Y (2007) Synthesis and versatile reactions of β-azidotetraarylporphyrins. Eur J Org Chem 1419
Shen D-M, Liu C, Shen Q-Y (2007) Synthesis and versatile reactions of β-azidotetraarylporphyrins. Eur J Org Chem 2888
Séverac M, Pleux LL, Scarpaci A, Blart E, Odobel F (2007) Synthesis of new azido porphyrins and their reactivity in copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction with alkynes. Tetrahedron Lett 48:6518
Ludke K, Alonso CMA, Neves MGPMS, Silva AMS, Cavaleiro JAS, Hombrechner HK (1997) A new approach to the synthesis of unsaturated porphyrins. Heterocycl Commun 3:503
Alonso CMA, Neves MGPMS, Silva AMS, Cavaleiro JAS, Hombrecher HK (1997) Reaction of β-amino-meso-tetraphenylporphyrin with α, β-unsaturated carbonyl compounds: an approach to fused pyridinoporphyrins. Tetrahedron Lett 38:2757
Alonso CMA, Neves MGPMS, Tomé AC, Silva AMS, Cavaleiro JAS (2000) Synthesis and Diels-Alder reactions of 2-(buta-1,3-dien-2-yl)-5,10,15,20-tetraphenylporphyrin. Tetrahedron Lett 41:5679
Tomé JPC, Pereira AMVM, Alonso CMA, Neves MGPMS, Tomé AC, Silva AMS, Cavaleiro JAS, Martinez-Diaz MV, Torres T, Rahman GMA, Rarney J, Guldi DM (2006) Synthesis and photophysical studies of new porphyrin–phthalocyanine dyads with hindered rotation. Eur J Org Chem 257
Kawakami T, Igarashi S (1996) Highly sensitive spectrophotometric determination of nitrite ion using 5,10,15,20-tetrakis(4-aminophenyl)porphine for application to natural waters. Anal Chim Acta 333:175
Kawakami T, Igarashi S (1997) Asymmetric porphyrin derivative having one reactive site as a highly sensitive spectrophotometric reagent for nitrite. Anal Chim Acta 354:159
Alonso CMA, Neves MGPMS, Tomé AC, Silva AMS, Cavaleiro JAS (2006) Reaction of 2-amino-5,10,15,20-tetraphenylporphyrinatonickel(ii) with α, β-unsaturated acyl chlorides: synthesis of 2-pyridone-fused porphyrin derivatives. J Mex Chem Soc 50:100
Alonso CMA, Neves MGPMS, Tomé AC, Silva AMS, Cavaleiro JAS (2001) Hetero-Diels–Alder reactions of β-imino-meso-tetraphenylporphyrin derivatives: a new approach to pyrido[2,3-b]porphyrins. Tetrahedron Lett 42:8307
Alonso CMA, Neves MGPMS, Tomé AC, Silva AMS, Cavaleiro JAS (2004) β-Imino-meso-tetraphenylporphyrin derivatives in hetero-Diels-Alder reactions. Eur J Org Chem 3233
Alonso CMA, Serra VV, Neves MGPMS, Tomé AC, Silva AMS, Paz FAA, Cavaleiro JAS (2007) An easy synthetic approach to pyridoporphyrins by domino reactions. Org Lett 9:2305
Gomes ATPC, Paz FAA, Neves MGPMS, Tomé AC, Silva AMS, Souza MCBV, Ferreira VF, Cavaleiro JAS (2011) Synthesis and characterization of new porphyrin/4-quinolone conjugates. Tetrahedron Lett 37:4741
Alonso CMA, Neves MGPMS, Tomé AC, Silva AMS, Cavaleiro JAS (2006) Beta-imino-meso-tetraphenylporphyrin derivatives: reactivity towards ethyl diazoacetate. Jordan J Chem 1:95
Crossley MJ, King LJ (1984) Novel heterocyclic systems from selective oxidation at the β-pyrrolic position of porphyrins. J Chem Soc Chem Commun 920
Beavington R, Rees PA, Burn PL (1998) A study on the oxidation of 2-hydroxyporphyrins to porphyrin-α-diones. J Chem Soc Perkin Trans 1 2847
Promarak V, Burn PL (2001) A new synthetic approach to porphyrin-α-diones and a-2,3,12,13-tetraone: building blocks for laterally conjugated porphyrin arrays. J Chem Soc Perkin Trans 1 14
Khoury T, Crossley MJ (2009) Expansion of the porphyrin π-system: stepwise annelation of porphyrin β, β′-pyrrolic faces leading to trisquinoxalinoporphyrin. New J Chem 33:1076
Gautam P, Krishnan V (1995) Coordination aggregation of meso-aryl substituted porphyrins. Proc Indian Acad Sci 107:477
Richeter S, Jeandon C, Ruppert R, Callot HJ (2001) Porphyrins acting as external and internal ligands: preparation of conjugated trimetallic dimeric porphyrins. Chem Commun 91
Richeter S, Jeandon C, Gisselbrecht JP, Ruppert R, Callot HJ (2002) Syntheses and optical and electrochemical properties of porphyrin dimers linked by metal ions. J Am Chem Soc 124:6168
Henrick K, Owston PG, Peters R, Tasker PA, Dell A (1980) Cyclisation involving a meso-phenyl substituent of a metalloporphyrin: X-ray structure of 5,10,15-triphenyl{22-oxo-benzo[2324]cyclohexa[a, b]porphinato(2-)}copper(II). Inorg Chim Acta 45:161
Callot HJ, Schaeffer E, Cromer R, Metz F (1990) Unexpected routes to naphtoporphyrin derivatives. Tetrahedron 46:5253
Ishkov YV, Zhilina ZI (1995) Porphyrins and their derivatives. 17. Intramolecular cyclization of 2-formyl-5,10,15,20-tetraphenylporphyrin complexes. Zh Org Khim 31:136
Richeter S, Jeandon C, Sauber C, Gisselbrecht JP, Ruppert R, Callot HJ (2002) Preparation, mass spectrometry and electrochemical studies of metal connected porphyrin oligomers. J Porphyr Phthalocyanines 6:423
Richeter S, Jeandon C, Gisselbrecht JP, Ruppert R, Callot HJ (2007) Synthesis, structural characterization, and electrochemical studies of nickel porphyrins bearing two peripheral conjugated chelating groups. Inorg Chem 46:10241
Ishkov YV (2001) Porphyrins and their derivatives: XXII. A new product of intramolecular cyclization of 5,10,15,20-tetraphenyl-2-formylporphyrin copper complex. Russ J Org Chem 37:288
Acknowledgments
Authors are grateful to “Fundação para a Ciência e a Tecnologia – FCT”, Lisbon, for funding QOPNA (the Research Unit in Organic Chemistry and projects). Thanks are also due to FCT for grants awarded to PhD and Post-Doc students. Thanks are also due to the University of Aveiro and to students involved in the work carried out in Aveiro.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Serra, V.I.V., Pires, S.M.G., Alonso, C.M.A., Neves, M.G.P.M.S., Tomé, A.C., Cavaleiro, J.A.S. (2013). Meso-Tetraarylporphyrins Bearing Nitro or Amino Groups: Synthetic Strategies and Reactivity Profiles. In: Paolesse, R. (eds) Synthesis and Modifications of Porphyrinoids. Topics in Heterocyclic Chemistry, vol 33. Springer, Berlin, Heidelberg. https://doi.org/10.1007/7081_2013_101
Download citation
DOI: https://doi.org/10.1007/7081_2013_101
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-38532-2
Online ISBN: 978-3-642-38533-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)