
Abstract
Cholesterol synthesis is a fundamental process that contributes to cellular cholesterol homeostasis. Cells execute transcriptional and post-translational mechanisms to control the abundance of enzymes of the cholesterol synthesis pathway, consequently affecting cholesterol production. One such highly tuned enzyme is squalene monooxygenase (SM), which catalyzes a rate-limiting step in the pathway. A well-characterized mechanism is the cholesterol-mediated degradation of SM. Notably, lipids (cholesterol, plasmalogens, squalene, and unsaturated fatty acids) can act as cellular signals that either promote or reduce SM degradation. The N-terminal region of SM consists of the shortest known cholesterol-responsive degron, characterized by atypical membrane anchoring structures, namely a re-entrant loop and an amphipathic helix. SM also undergoes non-canonical ubiquitination on serine, a relatively uncommon attachment site for ubiquitination. The structure of the catalytic domain of SM has been solved, providing insights into the catalytic mechanisms and modes of inhibition by well-known SM inhibitors, some of which have been effective in lowering cholesterol levels in animal models. Certain human cancers have been linked to dysregulation of SM levels and activity, further emphasizing the relevance of SM in health and disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cholesterol synthesis
- Degron
- Endoplasmic reticulum-associated degradation (ERAD)
- Squalene
- Squalene monooxygenase
- Ubiquitin
1 Introduction
The mevalonate pathway leads to the formation of essential metabolites, including ubiquinone, dolichol and cholesterol (Brown and Sharpe 2016). Cellular regulation of enzymes within the pathway is achieved by control of gene expression and protein turnover; this allows the mevalonate pathway to be less or more active, depending on the metabolic needs of the cell (Brown and Sharpe 2016).
In addition, the pathway is of clinical importance in cardiovascular disease; statins (a class of cholesterol-lowering drugs) inhibit this pathway by inhibiting 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), the first rate-limiting enzyme of the pathway (Buhaescu and Izzedine 2007; Goldstein and Brown 2015). The pathway is also of significant interest for research related to cancers, immunity and Alzheimer’s disease (Buhaescu and Izzedine 2007; Mullen et al. 2016).
Genes encoding the enzymes of the mevalonate pathway are transcriptionally controlled by transcription factors known as the Sterol Regulatory Element Binding Proteins (Brown and Sharpe 2016; Shimano and Sato 2017). However, post-translational regulation mechanisms for these enzymes can differ substantially. Sterols which accelerate the degradation of one enzyme may not do so for another enzyme (Brown and Sharpe 2016; Chen et al. 2019). Also, how these enzymes sense sterols are determined by unique intrinsic protein domains (Sever et al. 2003; Howe et al. 2016; Luo et al. 2020). Finally, the protein effectors which mediate the degradation of these enzymes are quite distinct, adding another layer of complexity to the regulation of their half-lives (Zelcer et al. 2014; Menzies et al. 2018).
Here, we focus on squalene monooxygenase (SM, UniProt ID:Q14534, EC 1.14.14.17), a second rate-limiting enzyme located in the endoplasmic reticulum (ER) (Gill et al. 2011). This enzyme has gained increasing attention over the past decade given its link to human diseases [reviewed in (Chua et al. 2020)], including cancers (Stopsack et al. 2017; Cirmena et al. 2018; Garcia-Bermudez et al. 2019), cardiovascular disease (Belter et al. 2011), and Rett Syndrome (Buchovecky et al. 2013). Here, our specific focus is on the unique homeostatic mechanisms employed by cells to regulate SM levels and activity in response to diverse lipids. Thus, the main goal of our review is to describe post-translational mechanisms that regulate SM, encompassing endoplasmic reticulum-associated degradation (ERAD), protein quality control mechanisms, ubiquitination and lipid-regulated protein stability.
2 Advances in Squalene Monooxygenase Research
Inhibitor Development and Protein Structure
In the 1990s when mammalian SM was first cloned (Sakakibara et al. 1995), natural and synthetic mammalian SM inhibitors were examined in cell models, enzyme assays, and animals (Belter et al. 2011). These exploratory studies were aimed at lowering cholesterol levels by inhibiting SM, but none of these inhibitors have yet to be tested in humans (Belter et al. 2011; Padyana et al. 2019). Around the same time, terbinafine was developed to inhibit the yeast homolog of SM, known as Erg1p (Ryder 1992). Terbinafine is still used clinically to treat fungal infections. In 2019, the structure of the catalytic domain of SM was finally solved, providing insights into catalytic mechanisms of enzyme-substrate and enzyme-inhibitor interaction (Padyana et al. 2019).
Gene Regulation
Transcription of the gene encoding SM, SQLE, is controlled by the transcription factors, Sterol Regulatory Element Binding Protein-2, Sp1 and NF-Y (Howe et al. 2017). Independent studies since the early 2000s have mapped the sterol response elements within SQLE, identifying which nucleotide positions in the proximal promoter are responsible for the sterol-responsiveness of the SQLE gene (Nagai et al. 2002; Howe et al. 2017).
Regulation by Protein Degradation
Although there were some suggestions of SM being another rate-limiting enzyme in the 1970s (Gonzalez et al. 1979), it was only in 2011 that SM was formally shown to be regulated post-translationally (Gill et al. 2011). Excess cholesterol accelerates the proteasomal degradation of SM, accumulating the substrate, squalene (Gill et al. 2011), which notably is downstream from the classic rate-limiting enzyme HMGCR (DeBose-Boyd 2008; Goldstein and Brown 2015). Thus, SM positions itself as another rate-limiting step of the pathway, with cholesterol feeding back on its own de novo synthesis by signaling the destruction of a later enzyme in the pathway than HMGCR. This feedback process requires the first 100 amino acids of SM (termed SM N100) (Gill et al. 2011). Unlike mammalian SM, the yeast counterpart, Erg1p, undergoes accelerated degradation by lanosterol and lacks the mammalian SM N100 region (Gill et al. 2011; Foresti et al. 2013). Subsequently, several proteins of the ERAD pathway which facilitate the degradation of SM have been identified, including the E3 ubiquitin ligase MARCHF6 (Foresti et al. 2013; Zelcer et al. 2014), the ubiquitin-conjugating enzyme Ube2J2 (Stefanovic-Barrett et al. 2018; Chua et al. 2019a; Tan et al. 2019), and the ATPase valosin-containing protein (VCP)/p97 which assists the unfolding of ER proteins destined for degradation (Chua et al. 2019b).
3 Architecture of the Squalene Monooxygenase Protein
Broadly, SM is divided into two parts; the first part is the N-terminal region encompassing a regulatory domain (the SM N100 degron), with the second part being the C-terminal catalytic domain constituting the bulk of the protein (Brown et al. 2019; Padyana et al. 2019) (Fig. 1).

Primary sequence and illustration of the squalene monooxygenase domains. (a) The regulatory domain comprises an amphipathic helix (orange) and a re-entrant loop (red) capable of sensing cholesterol. Catalytically important residues are those involved in the interaction with the squalene monooxygenase inhibitor, NB-598 (purple), and also those that permit FAD binding (green). Two putative transmembrane regions are shown in the C-terminal end of the protein (brown). (b) Schematic of the primary sequence in (a) in the context of the endoplasmic reticulum membrane. The cholesterol-sensing membrane bound structures of the regulatory domain, the re-entrant loop and amphipathic helix, are facing the cytosol. The C-terminal region make up the bulk of the protein and have the soluble FAD co-factor binding site and a substrate binding domain. (a) was reprinted with slight modifications from Progress in Lipid Research, volume 79, Chua et al., Squalene monooxygenase: a journey to the heart of cholesterol synthesis (volume 79), 2020, with permission from Elsevier. Content of (b) was originally sourced from an article licensed under a Creative Commons Attribution 4.0 International License published by Brown and colleagues (Brown et al. 2019)
The SM N100 degron is found in many eukaryotic organisms such as humans and other mammals (Gill et al. 2011). However, it is absent in plants (such as Panax ginseng and Arabidopsis thaliana) and lower organisms such as Saccharomyces cerevisiae (Gill et al. 2011; Chua et al. 2020). Interestingly, although chicken, zebrafish and lamprey SM contain the SM N100 degron, the ability of the SM N100 degron to be degraded in response to excess cholesterol differs between these species (Chua et al. 2017), which suggests that sequence-specific properties enable this response in mammals, like humans and hamsters (Gill et al. 2011).
The C-terminal catalytic domain contains the binding pocket for FAD (a co-factor), squalene (the substrate) and SM inhibitors (NB-598 and Compound-4″) (Brown et al. 2019; Padyana et al. 2019) (Fig. 1). This entire domain is well-conserved across organisms functionally, but displays subtle amino acid differences that affect inhibitor binding (discussed in Sect. 3.1) (Padyana et al. 2019).
3.1 The Catalytic Domain of Squalene Monooxygenase
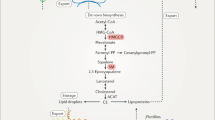
SM catalyzes the conversion of squalene to 2,3(S)-monooxidosqualene, which can then act again to introduce a second epoxide group, forming 2,3(S);22(S),23-dioxidosqualene, ultimately generating the oxysterol 24,25(S)-epoxycholesterol (Fig. 2). Synthesis of cholesterol and 24,25(S)-epoxycholesterol occur in parallel (Fig. 2), and the flux down these two branches is determined by the accumulation of 2,3(S)-monooxidosqualene (Gill et al. 2008). Typically, the majority of 2,3(S)-monooxidosqualene is converted to lanosterol by lanosterol synthase, directing the pathway towards cholesterol synthesis (Fig. 2). Levels of 24,25(S)-epoxycholesterol are usually very low, ranging from 0.1% to 1% with respect to cholesterol, depending on tissue and cell type (Brown 2009). Upon partial inhibition of lanosterol synthase, the reaction catalyzed by SM converting 2,3(S)-monooxidosqualene to 2,3(S);22(S),23-dioxidosqualene is favoured, resulting in greater flux down the shunt pathway to produce 24,25(S)-epoxycholesterol (Gill et al. 2008).

Simplified schematic of the cholesterol synthesis pathway. Squalene monooxygenase undergoes accelerated degradation upon cholesterol excess in cells. In addition to its role as a substrate of squalene monooxygenase, squalene (shaded green) can allosterically stabilize squalene monooxygenase. The pathway highlights the conversion of 2,3(S)-monooxidosqualene to either lanosterol or 2,3(S);22(S),23-dioxidosqualene which represents a divergence of the pathway to produce either cholesterol (shaded red) or 24(S),25-epoxycholesterol as the end product
To date, no SM recombinant proteins have been successfully co-crystalized with the squalene substrate, but there are structural and biochemical data showing the non-competitive binding mechanism of mammalian SM inhibitors, NB-598 and Compound-4″ (Padyana et al. 2019). Molecular docking analyses of the SM catalytic domain structure demonstrated that squalene binds to the same pocket as these inhibitors (Padyana et al. 2019).
Despite the high conservation of the catalytic domain between mammals and fungal species, terbinafine has more potent inhibitory effects on fungal Erg1p than mammalian SM (Padyana et al. 2019). As reported, this is a consequence of specific amino acid differences within the catalytic domain that are not conserved (Padyana et al. 2019). Three human SM amino acids (Phe-166, Ile-197 and Leu-324) are not conserved with fungal Erg1p, which affects the interaction of the binding pocket with the aromatic structure of terbinafine (Padyana et al. 2019). Smaller hydrophobic valine residues of Erg1p replace two human SM amino acids, Ile-197 and Leu-324, enabling optimal interactions between terbinafine and the Erg1p binding pocket (Padyana et al. 2019). Towards the C-terminal end of the catalytic domain are two putative transmembrane domains which have yet to be validated for anchoring SM to ER membranes (Brown et al. 2019; Padyana et al. 2019) (Fig. 1).
3.2 Cholesterol Induces Structural Changes in the Degron of Squalene Monooxygenase
The only validated ER-embedded structures of SM are the re-entrant loop (spanning amino acids Thr-11 to Ser-43) (Howe et al. 2015) and amphipathic helix (amino acids Gln-62 to Leu-73) (Chua et al. 2017) located within the SM N100 degron (Fig. 1). Notably, the hydrophobicity of the SM N100 degron has hampered the expression of recombinant full-length SM in multiple studies (Sakakibara et al. 1995; Gill et al. 2011), including the structural elucidation of the catalytic domain of SM (Padyana et al. 2019).
Both the re-entrant loop and amphipathic helix of SM N100 undergo structural changes upon cholesterol excess (Howe et al. 2015; Chua et al. 2017). The borders of the SM N100 re-entrant loop appear to be more buried in the membrane with cholesterol excess (Howe et al. 2015; Howe and Brown 2017). In comparison, the current model for the amphipathic helix is that it deforms with cholesterol excess, with the helix transitioning into a cytosolically exposed hydrophobic patch that increases protein disorder (Chua et al. 2017). These cholesterol-induced structural changes of the re-entrant loop and the amphipathic helix facilitate the degradation of SM by the ERAD machinery (Fig. 3, further discussed in Sect. 5), representing an example of allosteric misfolding (also termed “mallostery”) (Chua and Brown 2018; Wangeline and Hampton 2018).

Model highlighting mechanisms underlying the cholesterol-responsive degron in the endoplasmic reticulum-associated degradation of squalene monooxygenase. Squalene monooxygenase (SM) degradation relies on the cholesterol-responsive SM N100 degron. As cholesterol accumulates, this triggers conformational changes around the borders of the re-entrant loop and deforms the amphipathic helix of the SM N100 degron. This facilitates ubiquitination of serine amino acids near the deformed helix, catalyzed by the E3 ubiquitin ligase MARCHF6 and the E2 ubiquitin conjugating enzyme Ube2J2. Valosin-containing protein (VCP)/p97 acts as an extractor to enable the proteasomal degradation of squalene monooxygenase in the cytosolic compartment. A deubiquitinase (DUB) that could directly deubiquitinate the SM N100 degron and reduce cholesterol-mediated degradation of SM has not yet been identified. In addition, chaperones may also be involved to bind hydrophobic exposed regions of the degron
3.3 Squalene Monooxygenase Senses Cellular Lipid Levels
Evidence supporting direct binding of SM to cholesterol came from photoclickable cholesterol cross-linking experiments, although the precise binding location is still not known (Hulce et al. 2013). This binding likely occurs within the SM N100 degron as this region confers cholesterol-responsiveness to SM. Besides cholesterol, SM protein levels are also post-translationally regulated by other lipids (Honsho et al. 2015; Yoshioka et al. 2020).
Increasing plasmalogen levels in cells reduce cholesterol synthesis by promoting the degradation of SM (Honsho et al. 2015). This phenomenon is observed in vivo; mice deficient in plasmalogen synthesis in the cerebellum show higher SM levels (Honsho et al. 2019). These findings provide a link between plasmalogen and cholesterol homeostasis.
Unsaturated fatty acids such as oleate stabilize SM N100-GFP (SM N100 fused to green fluorescent protein as a model cholesterol-responsive degron) and reduces the cholesterol-accelerated degradation of the fusion protein (Stevenson et al. 2014). The substrate of SM, squalene, stabilizes SM and reduces the cholesterol-accelerated degradation of SM (Yoshioka et al. 2020).
How do these lipids exert their effects on SM? This may perhaps occur through membrane effects. For example, cholesterol can thicken the ER membrane (Hung et al. 2007) and induce conformational changes in the SM N100 degron as part of a stepwise process to enable accelerated degradation (Howe et al. 2015; Chua et al. 2017). Another possibility is that cholesterol induces conformational changes by binding directly to the SM N100 degron at a site yet to be determined, which might occur within the membrane embedded re-entrant loop (Howe et al. 2016). Whether the partially ER embedded amphipathic helix can bind cholesterol is still uncertain (Prinz 2017). An enantiomer of the natural form of cholesterol also elicits SM degradation, suggesting that membrane effects are in play (Kristiana et al. 2012).
Squalene binds to both the catalytic domain (Padyana et al. 2019) and the SM N100 degron (Yoshioka et al. 2020). While squalene binding at the catalytic domain permits epoxidation, binding at the membrane bound N-terminal SM N100 degron stabilizes the SM protein, introducing another scarce example of allosteric-mediated stabilization. Squalene excess stabilizes SM by blunting the interaction between the SM N100 degron and the E3 ubiquitin ligase MARCHF6, which reduces overall ubiquitination of SM, hindering SM degradation. It is possible the squalene-bound form may prevent exposure of degradation signals like the misfolded amphipathic helix (Nathan 2020). A plausible rationale for the evolution of squalene-mediated stabilization is that increased SM stability promotes cholesterol synthesis when the demand for cellular cholesterol is high. Thus, SM can be carefully tuned as the SM N100 degron constantly monitors fluctuating levels of lipids, notably the substrate (squalene) and the end-product of the pathway (cholesterol).
4 Non-canonical Ubiquitination of Squalene Monooxygenase
Most proteins that undergo degradation by the proteasome are post-translationally modified by ubiquitin (Hershko and Ciechanover 1998). Conjugated ubiquitin can act as a signal that facilitates proteolytic processing of the protein by the proteasome (Hershko and Ciechanover 1998). Ubiquitin is typically covalently attached to lysine amino acids on substrate proteins, a process catalyzed by E3 ubiquitin ligases (Hershko and Ciechanover 1998; McClellan et al. 2019). However, there are examples where ubiquitin can be conjugated to other amino acids such as serine, cysteine, and threonine (McClellan et al. 2019).
SM is ubiquitinated by MARCHF6, an E3 ubiquitin ligase which is stabilized by cholesterol (Sharpe et al. 2019). This is an efficient mechanism whereby the same lipid, cholesterol, reduces cholesterol synthesis through dual actions. Firstly, cholesterol favors a more unstable form of SM that is prone to degradation by ERAD (Chua et al. 2017), and secondly a cholesterol-stabilized form of MARCHF6 likely enables increased SM ubiquitination and subsequent degradation (Sharpe et al. 2019).
The SM N100 degron contains five lysine residues (Lys-15, Lys-16, Lys-82, Lys-90 and Lys-100) that could act as ubiquitination sites, but the degron still enables cholesterol-accelerated degradation despite having all lysines replaced with arginines (Gill et al. 2011). An alternative site for ubiquitination is the free N-terminus (McDowell and Philpott 2013), but blocking the N-terminus of the native SM N100 degron with a bulky mCherry reporter did not stabilize the SM N100 degron (Chua et al. 2019a).
From a series of systematic mutageneses, four serine residues (Ser-59, Ser-61, Ser-83 and Ser-87) were identified as essential for cholesterol-mediated degradation of the SM N100 degron, suggesting a possible role for non-canonical ubiquitination in the cholesterol-responsiveness of SM. Mass spectrometry confirmed serine 83 is ubiquitinated (Chua et al. 2019a). Interestingly, three of the four serines (Ser-59, Ser-61 and Ser-87) are not conserved in the chicken SM N100 degron. Introducing these human residues into the chicken SM N100 degron destabilized the protein, further supporting the involvement of non-canonical ubiquitination in SM degradation (Chua et al. 2019a). A number of other proteins also appear to be ubiquitinated on multiple amino acids (McDowell and Philpott 2013; McClellan et al. 2019), reflecting the versatility of E3 ubiquitin ligases to carry out their function (Mattiroli and Sixma 2014).
MARCHF6 and its yeast homolog, Doa10p, have been implicated in serine and threonine ubiquitination of substrate proteins (Weber et al. 2016; Chua et al. 2019a; McClellan et al. 2019). Furthermore, the E2 ubiquitin conjugating enzymes associated with MARCHF6 and Doa10p function, Ube2J2 and Ubc6p, are also linked to non-canonical ubiquitination of ERAD substrates, including SM (Wang et al. 2009; Weber et al. 2016; Chua et al. 2019a; McClellan et al. 2019) (Fig. 3).
Non-canonical ubiquitination examples are still limited and the list continues to expand slowly (McDowell and Philpott 2013; McClellan et al. 2019). Interestingly, the current examples are mostly represented by ERAD substrates (McClellan et al. 2019). Whether this indicates that non-canonical ubiquitination is common for ERAD substrates remains to be determined. Are other cholesterol synthesis enzymes ubiquitinated on serine and threonine residues? And if so, does this involve the MARCHF6/Ube2J2 pair, given its frequent involvement reported in other studies (McClellan et al. 2019)?
To date, it is unclear why the thermodynamically unfavorable process of serine and threonine ubiquitination occurs instead of lysine ubiquitination (McClellan et al. 2019). On the other hand, it is well known that serine and threonine residues can be phosphorylation sites. Further structural and biochemical studies are necessary to examine this aspect. The SM N100 degron represents a well-defined region targeted by MARCHF6 and Ube2J2, which could serve as a useful tool for studying non-canonical ubiquitination.
5 Protein Effectors in the Degradation of Squalene Monooxygenase
Like many other proteins in the ER, SM degradation by ERAD is carefully coordinated by a sequence of processes. A central process of ERAD is ubiquitination (Ruggiano et al. 2014; Christianson and Ye 2014). As discussed, SM ubiquitination requires MARCHF6 and Ube2J2 (Sect. 4). Additional proteins other than the ubiquitination machinery are needed to act on SM to drive ERAD as ubiquitination itself does not suffice to remove SM from the ER. VCP/p97 is a protein that forms a part of the ERAD process, specifically extraction, which dislocates SM from the ER (Chua et al. 2019b) (Fig. 3). VCP/p97 has the capacity to bind to ubiquitin on ERAD substrates via co-factors such as Ufd1 and Npl4 (Stach and Freemont 2017). Previous observations showed that these co-factors can bind Lys48-linked polyubiquitin chains (van den Boom and Meyer 2018). While not all ubiquitin-proteasome system substrates require VCP/p97 as part of their degradation, this requirement seems to stem from the fact that some substrates (like ERAD substrates) are embedded in organelles like the ER membrane (van den Boom and Meyer 2018).
An unexplored mechanism is whether there is an intermediate stage between SM ubiquitination by MARCHF6 and extraction by VCP/p97. The SM N100 degron is relatively hydrophobic, having a propensity to aggregate. The current idea is that cholesterol promotes ubiquitination by inducing conformational changes in SM N100 at the re-entrant loop and the amphipathic helix (Howe et al. 2015; Chua et al. 2017) (Fig. 3). This facilitates ubiquitination of accessible serine residues adjacent to the misfolded amphipathic helix (Chua et al. 2019a; Fig. 3). How are misfolded hydrophobic patches shielded from the aqueous cytosol in the interim? Yeast studies demonstrated that hydrophobic misfolded regions of ERAD substrates are shielded by a Hsp70 chaperone, prior to Doa10p-mediated ubiquitination and extraction by Cdc48p (yeast homolog of VCP) (Nakatsukasa et al. 2008; Needham et al. 2019). Perhaps such a chaperoning mechanism occurs during the ERAD of SM but has yet to be tested.
Finally, deubiquitination is one of the most unexplored processes of ubiquitin biology, and the general role for deubiquitinases in ERAD remain understudied (Sowa et al. 2009; Zhang et al. 2013; Christianson and Ye 2014). Protein levels for SM and the degron fusion protein, SM N100-GFP, increased upon treatment of cells by independent deubiquitinase inhibitors, PR-619 (broad-spectrum deubiquitinase inhibitor) or WP-1130 (targets five deubiquitinases) (Chua et al. 2019b). Inhibiting deubiquitinases should preserve ubiquitination and promote degradation, but SM showed the opposite effect whereby its levels decreased (Chua et al. 2019b). Such findings are difficult to explain given the inhibitors themselves likely perturb ubiquitination and degradation of the proteins mediating the ERAD of SM, such as MARCHF6, which also undergoes autoubiquitination and deubiquitination, thus affecting MARCHF6 stability (Hassink et al. 2005; Nakamura et al. 2014; Zattas et al. 2016).
6 Conclusions and Perspective for the Future
The past decade has seen major advances in our understanding of SM [as reviewed in (Chua et al. 2020)], but challenging questions remain.
How exactly does the SM N100 degron operate in three-dimensions? Does it associate with the putative transmembrane domains of the catalytic domain? Current improved structural biology methods may reveal new mechanisms underlying the regulatory actions of the SM N100 degron in the context of the full-length protein.
How can we map the interaction sites between the cholesterol-responsive SM N100 degron with cholesterol, or with other lipids that regulate it? Even if a structure of the SM N100 degron is obtained, there is no guarantee that lipid binding can be observed directly. Perhaps molecular dynamics would be necessary, as in the case of the crystal structure study where squalene binding to the catalytic domain of SM had to be modeled (Padyana et al. 2019).
Uncovering the in-depth mechanisms of how SM is degraded may offer strategies to degrade SM where it is implicated in diseases (Cirmena et al. 2018; Chua et al. 2020). The idea of degrading proteins for therapeutic purposes has been on the rise in recent years (Schapira et al. 2019). In the context of controlling cholesterol levels, this proposition has been tested with HMGCR, whereby a sterol-analog could potently induce HMGCR degradation, preventing atherosclerosis and lowering cholesterol levels in mice (Jiang et al. 2018).
Given that ubiquitination is reversible, what are the DUBs that deubiquitinate SM? And do they counteract or promote the cholesterol-mediated degradation of SM? While the role of ubiquitination in cholesterol homeostasis is already appreciated (Sharpe et al. 2014; Stevenson et al. 2016; van den Boomen et al. 2020), the fine balance between making or breaking ubiquitin chains in the context of cholesterol homeostasis remains largely unknown. This could form a complex mode of regulation to control cholesterol levels as exemplified by the regulation of low-density lipoprotein uptake which involves regulation of the low-density lipoprotein receptor by the DUB, USP2 and the E3 ubiquitin ligase, IDOL (Nelson et al. 2016).
With the advent of CRISPR screens, cryogenic electron microscopy and chemical biology tools as discovery frameworks, we anticipate answers to these challenging unanswered questions. We look forward to a wealth of further discoveries that will deepen our understanding of SM, an enzyme of cholesterol biosynthesis with far-reaching impacts in biotechnology and human health and disease.
References
Belter A, Skupinska M, Giel-Pietraszuk M et al (2011) Squalene monooxygenase – a target for hypercholesterolemic therapy. Biol Chem 392:1053–1075. https://doi.org/10.1515/BC.2011.195
Brown AJ (2009) 24(S),25-Epoxycholesterol: a messenger for cholesterol homeostasis. Int J Biochem Cell Biol 41:744–747. https://doi.org/10.1016/j.biocel.2008.05.029
Brown AJ, Sharpe LJ (2016) Chapter 11 – Cholesterol synthesis. In: Ridgway ND, McLeod RS (eds) Biochemistry of lipids, lipoproteins and membranes, 6th edn. Elsevier, Boston, pp 327–358
Brown AJ, Chua NK, Yan N (2019) The shape of human squalene epoxidase expands the arsenal against cancer. Nat Commun 10:888. https://doi.org/10.1038/s41467-019-08866-y
Buchovecky CM, Turley SD, Brown HM et al (2013) A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat Genet 45:1013–1020. https://doi.org/10.1038/ng.2714
Buhaescu I, Izzedine H (2007) Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 40:575–584. https://doi.org/10.1016/j.clinbiochem.2007.03.016
Chen L, Ma M-Y, Sun M et al (2019) Endogenous sterol intermediates of the mevalonate pathway regulate HMGCR degradation and SREBP-2 processing. J Lipid Res 60:1765–1775. https://doi.org/10.1194/jlr.RA119000201
Christianson JC, Ye Y (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol 21:325–335. https://doi.org/10.1038/nsmb.2793
Chua NK, Brown AJ (2018) Mallostery: filling a niche between quality and metabolic control. J Biol Chem 293:14951–14952. https://doi.org/10.1074/jbc.H118.005031
Chua NK, Howe V, Jatana N et al (2017) A conserved degron containing an amphipathic helix regulates the cholesterol-mediated turnover of human squalene monooxygenase, a rate-limiting enzyme in cholesterol synthesis. J Biol Chem 292:19959–19973. https://doi.org/10.1074/jbc.M117.794230
Chua NK, Hart-Smith G, Brown AJ (2019a) Non-canonical ubiquitination of the cholesterol-regulated degron of squalene monooxygenase. J Biol Chem 294:8134–8147. https://doi.org/10.1074/jbc.RA119.007798
Chua NK, Scott NA, Brown AJ (2019b) Valosin-containing protein mediates the ERAD of squalene monooxygenase and its cholesterol-responsive degron. Biochem J 476:2545–2560. https://doi.org/10.1042/BCJ20190418
Chua NK, Coates HW, Brown AJ (2020) Squalene monooxygenase: a journey to the heart of cholesterol synthesis. Prog Lipid Res 79:101033. https://doi.org/10.1016/j.plipres.2020.101033
Cirmena G, Franceschelli P, Isnaldi E et al (2018) Squalene epoxidase as a promising metabolic target in cancer treatment. Cancer Lett 425:13–20. https://doi.org/10.1016/j.canlet.2018.03.034
DeBose-Boyd RA (2008) Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res 18:609–621. https://doi.org/10.1038/cr.2008.61
Foresti O, Ruggiano A, Hannibal-Bach HK et al (2013) Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. eLife 2:e00953. https://doi.org/10.7554/eLife.00953
Garcia-Bermudez J, Baudrier L, Bayraktar EC et al (2019) Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567:118–122. https://doi.org/10.1038/s41586-019-0945-5
Gill S, Chow R, Brown A (2008) Sterol regulators of cholesterol homeostasis and beyond: the oxysterol hypothesis revisited and revised. Prog Lipid Res 47:391–404. https://doi.org/10.1016/j.plipres.2008.04.002
Gill S, Stevenson J, Kristiana I, Brown AJ (2011) Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab 13:260–273. https://doi.org/10.1016/j.cmet.2011.01.015
Goldstein JL, Brown MS (2015) A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161:161–172. https://doi.org/10.1016/j.cell.2015.01.036
Gonzalez R, Carlson JP, Dempsey ME (1979) Two major regulatory steps in cholesterol synthesis by human renal cancer cells. Arch Biochem Biophys 196:574–580. https://doi.org/10.1016/0003-9861(79)90310-2
Hassink G, Kikkert M, van Voorden S et al (2005) TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem J 388:647–655. https://doi.org/10.1042/BJ20041241
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479. https://doi.org/10.1146/annurev.biochem.67.1.425
Honsho M, Abe Y, Fujiki Y (2015) Dysregulation of plasmalogen homeostasis impairs cholesterol biosynthesis. J Biol Chem 290:28822–28833. https://doi.org/10.1074/jbc.M115.656983
Honsho M, Dorninger F, Abe Y et al (2019) Impaired plasmalogen synthesis dysregulates liver X receptor-dependent transcription in cerebellum. J Biochem (Tokyo) 166:353–361. https://doi.org/10.1093/jb/mvz043
Howe V, Brown AJ (2017) Determining the topology of membrane-bound proteins using PEGylation. In: Gelissen IC, Brown AJ (eds) Cholesterol homeostasis. Springer, New York, pp 201–210
Howe V, Chua NK, Stevenson J, Brown AJ (2015) The regulatory domain of squalene monooxygenase contains a re-entrant loop and senses cholesterol via a conformational change. J Biol Chem 290:27533–27544. https://doi.org/10.1074/jbc.M115.675181
Howe V, Sharpe LJ, Alexopoulos SJ et al (2016) Cholesterol homeostasis: how do cells sense sterol excess? Chem Phys Lipids 199:170–178. https://doi.org/10.1016/j.chemphyslip.2016.02.011
Howe V, Sharpe LJ, Prabhu AV, Brown AJ (2017) New insights into cellular cholesterol acquisition: promoter analysis of human HMGCR and SQLE, two key control enzymes in cholesterol synthesis. Biochim Biophys Acta Mol Cell Biol Lipids 1862:647–657. https://doi.org/10.1016/j.bbalip.2017.03.009
Hulce JJ, Cognetta AB, Niphakis MJ et al (2013) Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods 10:259–264. https://doi.org/10.1038/nmeth.2368
Hung W-C, Lee M-T, Chen F-Y, Huang HW (2007) The condensing effect of cholesterol in lipid bilayers. Biophys J 92:3960–3967. https://doi.org/10.1529/biophysj.106.099234
Jiang S-Y, Li H, Tang J-J et al (2018) Discovery of a potent HMG-CoA reductase degrader that eliminates statin-induced reductase accumulation and lowers cholesterol. Nat Commun 9:5138. https://doi.org/10.1038/s41467-018-07590-3
Kristiana I, Luu W, Stevenson J et al (2012) Cholesterol through the looking glass: ability of its enantiomer also to elicit homeostatic responses. J Biol Chem 287:33897–33904. https://doi.org/10.1074/jbc.M112.360537
Luo J, Yang H, Song B-L (2020) Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol 21:225–245. https://doi.org/10.1038/s41580-019-0190-7
Mattiroli F, Sixma TK (2014) Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat Struct Mol Biol 21:308–316. https://doi.org/10.1038/nsmb.2792
McClellan AJ, Laugesen SH, Ellgaard L (2019) Cellular functions and molecular mechanisms of non-lysine ubiquitination. Open Biol 9:190147. https://doi.org/10.1098/rsob.190147
McDowell GS, Philpott A (2013) Non-canonical ubiquitylation: mechanisms and consequences. Int J Biochem Cell Biol 45:1833–1842. https://doi.org/10.1016/j.biocel.2013.05.026
Menzies SA, Volkmar N, van den Boomen DJ et al (2018) The sterol-responsive RNF145 E3 ubiquitin ligase mediates the degradation of HMG-CoA reductase together with gp78 and Hrd1. eLife 7:e40009. https://doi.org/10.7554/eLife.40009
Mullen PJ, Yu R, Longo J et al (2016) The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer 16:718–731. https://doi.org/10.1038/nrc.2016.76
Nagai M, Sakakibara J, Nakamura Y et al (2002) SREBP-2 and NF-Y are involved in the transcriptional regulation of squalene epoxidase. Biochem Biophys Res Commun 295:74–80. https://doi.org/10.1016/S0006-291X(02)00623-X
Nakamura N, Harada K, Kato M, Hirose S (2014) Ubiquitin-specific protease 19 regulates the stability of the E3 ubiquitin ligase MARCH6. Exp Cell Res 328:207–216. https://doi.org/10.1016/j.yexcr.2014.07.025
Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL (2008) Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 132:101–112. https://doi.org/10.1016/j.cell.2007.11.023
Nathan JA (2020) Squalene and cholesterol in the balance at the ER membrane. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.2003388117
Needham PG, Guerriero CJ, Brodsky JL (2019) Chaperoning endoplasmic reticulum–associated degradation (ERAD) and protein conformational diseases. Cold Spring Harb Perspect Biol 11:a033928. https://doi.org/10.1101/cshperspect.a033928
Nelson JK, Sorrentino V, Avagliano Trezza R et al (2016) The deubiquitylase USP2 regulates the LDLR pathway by counteracting the E3-ubiquitin ligase IDOL. Circ Res 118:410–419. https://doi.org/10.1161/CIRCRESAHA.115.307298
Padyana AK, Gross S, Jin L et al (2019) Structure and inhibition mechanism of the catalytic domain of human squalene epoxidase. Nat Commun 10:97. https://doi.org/10.1038/s41467-018-07928-x
Prinz WA (2017) A cholesterol-sensing mechanism unfolds. J Biol Chem 292:19974–19975. https://doi.org/10.1074/jbc.H117.794230
Ruggiano A, Foresti O, Carvalho P (2014) ER-associated degradation: protein quality control and beyond. J Cell Biol 204:869–879. https://doi.org/10.1083/jcb.201312042
Ryder NS (1992) Terbinafine: mode of action and properties of the squalene epoxidase inhibition. Br J Dermatol 126:2–7. https://doi.org/10.1111/j.1365-2133.1992.tb00001.x
Sakakibara J, Watanabe R, Kanai Y, Ono T (1995) Molecular cloning and expression of rat squalene epoxidase. J Biol Chem 270:17–20. https://doi.org/10.1074/jbc.270.1.17
Schapira M, Calabrese MF, Bullock AN, Crews CM (2019) Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov 18:949–963. https://doi.org/10.1038/s41573-019-0047-y
Sever N, Yang T, Brown MS et al (2003) Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell 11:25–33. https://doi.org/10.1016/s1097-2765(02)00822-5
Sharpe LJ, Cook ECL, Zelcer N, Brown AJ (2014) The UPS and downs of cholesterol homeostasis. Trends Biochem Sci 39:527–535. https://doi.org/10.1016/j.tibs.2014.08.008
Sharpe LJ, Howe V, Scott NA et al (2019) Cholesterol increases protein levels of the E3 ligase MARCH6 and thereby stimulates protein degradation. J Biol Chem 294:2436–2448. https://doi.org/10.1074/jbc.RA118.005069
Shimano H, Sato R (2017) SREBP-regulated lipid metabolism: convergent physiology—divergent pathophysiology. Nat Rev Endocrinol 13:710–730. https://doi.org/10.1038/nrendo.2017.91
Sowa ME, Bennett EJ, Gygi SP, Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403. https://doi.org/10.1016/j.cell.2009.04.042
Stach L, Freemont PS (2017) The AAA+ ATPase p97, a cellular multitool. Biochem J 474:2953–2976. https://doi.org/10.1042/BCJ20160783
Stefanovic-Barrett S, Dickson AS, Burr SP et al (2018) MARCH6 and TRC8 facilitate the quality control of cytosolic and tail-anchored proteins. EMBO Rep 19. https://doi.org/10.15252/embr.201745603
Stevenson J, Luu W, Kristiana I, Brown AJ (2014) Squalene mono-oxygenase, a key enzyme in cholesterol synthesis, is stabilized by unsaturated fatty acids. Biochem J 461:435–442. https://doi.org/10.1042/BJ20131404
Stevenson J, Huang EY, Olzmann JA (2016) Endoplasmic reticulum-associated degradation and lipid homeostasis. Annu Rev Nutr 36:511–542. https://doi.org/10.1146/annurev-nutr-071715-051030
Stopsack KH, Gerke TA, Andrén O et al (2017) Cholesterol uptake and regulation in high-grade and lethal prostate cancers. Carcinogenesis 38:806–811. https://doi.org/10.1093/carcin/bgx058
Tan JME, Cook ECL, van den Berg M et al (2019) Differential use of E2 ubiquitin conjugating enzymes for regulated degradation of the rate-limiting enzymes HMGCR and SQLE in cholesterol biosynthesis. Atherosclerosis 281:137–142. https://doi.org/10.1016/j.atherosclerosis.2018.12.008
van den Boom J, Meyer H (2018) VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol Cell 69:182–194. https://doi.org/10.1016/j.molcel.2017.10.028
van den Boomen DJH, Volkmar N, Lehner PJ (2020) Ubiquitin-mediated regulation of sterol homeostasis. Curr Opin Cell Biol 65:103–111. https://doi.org/10.1016/j.ceb.2020.04.010
Wang X, Herr RA, Rabelink M et al (2009) Ube2j2 ubiquitinates hydroxylated amino acids on ER-associated degradation substrates. J Cell Biol 187:655–668. https://doi.org/10.1083/jcb.200908036
Wangeline MA, Hampton RY (2018) “Mallostery”—ligand-dependent protein misfolding enables physiological regulation by ERAD. J Biol Chem 293:14937–14950. https://doi.org/10.1074/jbc.RA118.001808
Weber A, Cohen I, Popp O et al (2016) Sequential poly-ubiquitylation by specialized conjugating enzymes expands the versatility of a quality control ubiquitin ligase. Mol Cell 63:827–839. https://doi.org/10.1016/j.molcel.2016.07.020
Yoshioka H, Coates HW, Chua NK et al (2020) A key mammalian cholesterol synthesis enzyme, squalene monooxygenase, is allosterically stabilized by its substrate. Proc Natl Acad Sci 117:7150–7158. https://doi.org/10.1073/pnas.1915923117
Zattas D, Berk JM, Kreft SG, Hochstrasser M (2016) A conserved C-terminal element in the yeast Doa10 and human MARCH6 ubiquitin ligases required for selective substrate degradation. J Biol Chem 291:12105–12118. https://doi.org/10.1074/jbc.M116.726877
Zelcer N, Sharpe LJ, Loregger A et al (2014) The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme a reductase and the cholesterol synthesis pathway. Mol Cell Biol 34:1262–1270. https://doi.org/10.1128/MCB.01140-13
Zhang Z-R, Bonifacino JS, Hegde RS (2013) Deubiquitinases sharpen substrate discrimination during membrane protein degradation from the ER. Cell 154:609–622. https://doi.org/10.1016/j.cell.2013.06.038
Acknowledgements
This work was supported by Australian Research Council Grant DP170101178 and NSW Health Investigator Development grant (to AJB).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Chua, N.K., Brown, A.J. (2020). The Degron Architecture of Squalene Monooxygenase and How Specific Lipids Calibrate Levels of This Key Cholesterol Synthesis Enzyme. In: Atassi, M.Z. (eds) Protein Reviews . Advances in Experimental Medicine and Biology(), vol 21. Springer, Cham. https://doi.org/10.1007/5584_2020_583
Download citation
DOI: https://doi.org/10.1007/5584_2020_583
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-67813-5
Online ISBN: 978-3-030-67814-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)