
Abstract
Epithelial splicing regulatory protein 1 (ESRP1) and 2 (ESRP2) are members of the hnRNP family of RNA binding proteins that regulate alternative splicing events associated with epithelial phenotypes. These proteins play crucial roles during organogenesis, including craniofacial and epidermal development as well as branching morphogenesis in the lungs and salivary glands. Recent reports have also addressed their roles during cancer progression. Expression of ESRP proteins is low in normal epithelium but upregulated in carcinoma in situ and advanced carcinomas. Intriguingly, they are downregulated in invasive fronts. The plastic nature of ESRP expression suggests dual roles for them in cancer progression. Consistently, it has been shown that ESRPs suppress motility and anchorage-independent growth of cancer cells while supporting cell survival by enhancing resistance to reactive oxygen species. Regulatory circuits that fine-tune ESRP gene expression have recently emerged. Here, we summarize recent findings on the molecular mechanisms by which ESRPs exert positive as well as negative effects on cancer progression.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Epithelial splicing regulatory protein
- Alternative splicing
- Cell motility
- δ-crystallin enhancer binding protein
- CD44
1 ESRP1 and ESRP2: Splicing Regulatory Proteins Specifically Expressed in Epithelial Cells
Alternative splicing generates mRNA variants that encode protein isoforms with diverse or even opposite functions from pre-mRNAs transcribed from a single gene (David and Manley 2010; Braunschweig et al. 2013; Warzecha and Carstens 2012; Kaida et al. 2012). The basic machinery for pre-mRNA splicing represents the spliceosome, a large complex composed of snRNPs (U1, U2, U4, U5, and U6) and additional 150–200 proteins. Recruitment of the spliceosome to alternative splice sites is tightly regulated by RNA-binding proteins associated with cis-regulatory elements in pre-mRNAs, thus ensuring cell-type specific formation of mRNA variants. RNA-binding proteins that regulate alternative splicing events are classified into two families, serine/arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs). In general, SR proteins interact with exonic or intronic splicing enhancers to promote exon inclusion, whereas hnRNPs interact with exonic or intronic splicing suppressors to promote exon skipping (Warzecha and Carstens 2012; Kaida et al. 2012). The balance between these positive and negative regulators determines the extent of exon inclusion in target pre-mRNAs.
Fibroblast growth factor receptors (FGFRs) are proteins with two isoforms, namely epithelial FGFR-IIIb and mesenchymal FGFR-IIIc. These isoforms have distinct ligand binding properties but share a common intracellular signaling domain. They are produced by cell-type specific alternative splicing (Eswarakumar et al. 2005). The transition between FGFR isoforms affects the behavior of cells by altering their sensitivity to various fibroblast growth factor (FGF) ligands present in surrounding tissue microenvironments. One such example involves isoform switching of FGFRs during the epithelial-mesenchymal transition (EMT), a process by which epithelial cells lose their polarity and acquire motile and invasive phenotypes (Kalluri and Weinberg 2009). Epithelial cells are usually insensitive to FGF2 that is abundant in tumor tissues because they only express the FGFR-IIIb isoform. In cells that undergo EMT, FGFR-IIIb is downregulated while FGFR-IIIc is upregulated to confer sensitivity to FGF2, thus inducing even more aggressive phenotypes in the cells in the presence of FGF2 (enhanced EMT) (Shirakihara et al. 2011).
By genome-wide cDNA expression screening, Warzecha et al identified two RNA binding proteins belonging to the hnRNP family, RBM35A and RBM35B, as regulators of alternative splicing of the FGFR2 pre-mRNA (Warzecha et al. 2009a). Expression of RBM35A and 35B is well correlated to that of the epithelial FGFR2-IIIb isoform in various cell lines. In addition, in situ hybridization analysis of whole postnatal and adult mice revealed that their expression is epithelium-specific in various tissues and organs. They thus renamed RBM35A and B as epithelial splicing regulatory protein 1 (ESRP1) and 2 (ESRP2). Subsequently ESRPs were shown to bind preferentially to UGG-rich repeats and regulate epithelial specific splicing of a diverse array of target pre-mRNAs (Warzecha et al. 2009b; Warzecha et al. 2010; Dittmar et al. 2012), suggenting that ESRPs function to maintain the epithelial phenotypes of cells. Notably, some of the RNA splice variants regulated by ESRPs have been implicated in regulating cytoskeleton reorganization and cell adhesion (Warzecha et al. 2009b).
2 ESRP Expression Is Upregulated in Cancer Cells
The mechanisms that restrict expression of ESRPs in epithelial cells remain to be fully understood. δ-crystallin enhancer binding protein (δEF1, also called zinc finger E–box binding homeobox 1, ZEB1), Smad interacting protein 1 (SIP1, also called ZEB2), and Snail, transcriptional repressors that are expressed in cells with mesenchymal phenotypes, were shown to inhibit ESRP expression (Horiguchi et al. 2012; Reinke et al. 2012). Notably, δEF1 and SIP1 directly interact with the promoter regions of the ESRP genes as revealed by chromatin-immunoprecipitation assays (Horiguchi et al. 2012). By contrast, Grhl2 (grainyhead-like-2), a transcription factor expressed in epithelial cells, was reported to upregulate ESRP1 expression in breast cancer cells (Xiang et al. 2012). This upregulation may not be due to a direct effect of Grhl2 because Grhl2 and δEF1 mutually repress each other (Cieply et al. 2013; Werner et al. 2013), suggesting that Grhl2 induces ESRP1 expression by downregulating δEF1 expression.
Determining ESRP expression in human patient specimens would give valuable information on the role of ESRPs in pathological processes. In normal human pancreas, ESRP1 is only weakly expressed in pancreatic ductal cells but it is abundantly expressed in well-to-moderately differentiated adenocarcinoma although downregulated in poorly differentiated adenocarcinoma (Ueda et al. 2014). In normal human oral squamous epithelium, ESRP1 and 2 are weakly expressed in the basal layer (Ishii et al. 2014). In carcinoma in situ and advanced carcinomas, they are highly expressed. Importantly, ESRPs disappear from invasive fronts while they are re-expressed in cells that have metastasized to a lymph node. These findings indicate that the expression of ESRPs is plastic during cancer progression.
3 ESRPs Negatively Regulate Cell Motility Through Multiple Mechanisms
Downregulation of ESRPs in invasive fronts suggests negative regulatory roles for ESRPs in cancer invasion and metastasis. Consistently, ESRPs have been shown to suppress cell motility in vitro: Knockdown of ESRP1 or ESRP2 promotes cell motility in human mammary epithelial cells (Warzecha et al. 2010), pancreatic cancer cells (Ueda et al. 2014), head and neck squamous cell carcinoma (HNSCC) cells (Ishii et al. 2014), and clear-cell renal cell carcinoma cells (Mizutani et al. 2015). Conversely, the motility and invasiveness of pancreatic cancer cells are attenuated following overexpression of ESRP1 (Ueda et al. 2014).
Lu et al showed that ESRP1 is involved in the alternative splicing of Exo70, which produces two alternatively spliced protein isoforms: epithelially expressed Exo70-E and mesenchymally expressed Exo70-M (Lu et al. 2013). During EMT, the repression of Exo70-E coincides with the repression of ESRP1. Knockdown of ESRP1 in MCF7 breast cancer cells increased the expression of Exo70-M, which can interact with a complex of actin-related proteins, Arp2/3, to stimulate actin polymerization, resulting in promotion of cell migration and invasion. However, it remains to be demonstrated if Exo70-M is indispensable for increased cell motility upon knockdown of ESRP1.
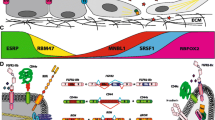
Ishii et al found that knockdown of either ESRP1 or ESRP2 enhanced the motility of HNSCC cell lines (SAS, HSC4) to the similar extent (Ishii et al. 2014). They found that ESRP2 knockdown caused cell dissociation accompanied with downregulation of E-cadherin while ESRP1 knockdown triggered formation of long filopodia. Increased cell motility with long filopodia formation by ESRP1 knockdown was attributed to induction of Rac1b, an alternatively spliced isoform of the Rac1 protein. Rac1 is a small G-protein that regulates reorganization of the actin cytoskeleton. Rac1b, often expressed in cancer cells, is a constitutively active form of Rac1 that contains a 19 amino acid residue insertion (Jordan et al. 1999; Schnelzer et al. 2000; Fiegen et al. 2004). Downregulation of E-cadherin as well as increased cell motility caused by ESRP2 knockdown is due to upregulation of EMT-associated transcription factors, including δEF1 and SIP1, that are known to repress E-cadherin (Comijn et al. 2001; Eger et al. 2005) and a cell motility-inhibitory protein RGS16 (regulator of G-protein signaling 16) (Hoshi et al. 2016). Thus, ESRP1 and 2 suppress cell motility of HNSCC cells via distinct mechanisms (Fig. 1). Intriguingly, δEF1 and SIP1 suppress the expression of ESRP1 and 2 as described above (Horiguchi et al. 2012), indicating that a double negative feedback circuit is formed between δEF1/SIP1 and ESRP2. This regulatory circuit may facilitate the immediate transition of cellular states as well as plastic expression of ESRPs in response to certain stimuli.

Molecular network that regulates motility of HNSCC cells. ESRP1 and ESRP2 suppress cell motility by downregulating Rac1b and δEF1/SIP1 expression, respectively (Ishii et al. 2014). δEF1/SIP1 in turn repress ESRP1 and ESRP2 expression (Horiguchi et al. 2012), thus forming a double negative feedback circuit between ESRP2 and δEF1/SIP1. The negative effect of ESRP1 on Rac1b expression appears to be due to regulation of the alternative splicing event generating Rac1 and Rac1b mRNAs. *The mechanism by which ESRP2 downregulates δEF1/SIP1 remains to be elucidated. **δEF1 and SIP1 enhance cancer cell motility by downregulating RGS16 in breast cancer cells (Hoshi et al. 2016), but this has not yet been demonstrated in HNSCC cells
The mechanisms by which ESRP2 downregulates δEF1 and SIP1 remain unclear. ESRP2 may downregulate δEF1/SIP1 through alternative splicing events of certain target pre-mRNAs or through a splicing-independent mechanism in HNSCC cells. Intriguingly, ESRP1 was shown to regulate the translation of pluripotency-related factors in embryonic stem cells by interacting with the 5’-untranslated region of target mRNAs in the cytoplasm (Fagoonee et al. 2013). In breast cancer cells, ectopic expression of ESRP1 and 2 resulted in higher expression of E-cadherin without affecting the expression levels of δEF1, SIP1, and Snail that downregulate E-cadherin mRNA (CDH1) (Horiguchi et al. 2012). The effects of ESRP2 on δEF1/SIP1 expression may therefore be context-dependent. Recently, Preca et al proposed a model for linking ESRP1 and δEF1 expression that is mediated by isoform switching of CD44 in breast cancer cells (Fig. 2) (Preca et al. 2015). CD44 is a transmembrane glycoprotein that can interact with extracellular matrices, including hyaluronan. Switching between the standard CD44 isoform (CD44s) and variant isoforms (CD44v) is regulated by alternative splicing (Ponta et al. 2003). ESRP1 expression increases CD44v levels in epithelial cells. During EMT, upregulated δEF1 represses ESRP1 expression, triggering the isoform switching of CD44 from CD44v to CD44s. The CD44s isoform, in turn, enhances δEF1 expression by an unknown mechanism, thus forming a positive feedback circuit. In the case of HNSCC cells, however, ESRP2 represses δEF1/SIP1 expression without affecting CD44 isoform switching (Ishii et al. 2014). Further studies are required to resolve these conflicting observations.

A regulatory circuit comprising ESRP1, δEF1 and CD44s in breast cancer cells. ESRP1 and δEF1 form a double negative feedback loop mediated by CD44s signaling in breast cancer cells (Preca et al. 2015). ESRP1 affects alternative splicing of the CD44 pre-mRNA to upregulate CD44v while downregulate CD44s that enhances expression of δEF1. δEF1 transcriptionally downregualtes ESRP1, resulting in induction of CD44s that in turn enhances δEF1 expression. *It remains to be elucidated how CD44s, but not CD44v, induces δEF1 expression
4 Dual Roles of ESRPs on Cancer Progression
Horiguchi et al reported that expression of ESRP1 and ESRP2 is inversely related to cancer malignancy: they are poorly expressed in ‘basal-like’ subtype of breast cancer cells exhibiting high malignancy, while highly expressed in luminal-type breast cancer cells exhibiting low malignancy (Horiguchi et al. 2012). Basal-like MDA-MB-231 cells that express ESRPs ectopically changed their morphology from spindle to cobble stone–like shape, accompanied by E-cadherin expression, and failed to proliferate efficiently in soft agar (Horiguchi et al. 2012). Leontieva & Ionov also reported that ectopic expression of ESRP1 in LS180 colon cancer cells attenuated anchorage-independent growth in vitro and tumorigenic potential in vivo (heterotopic xenograft model) (Leontieva and Ionov 2009). Ueda et al showed that pancreatic cancer cells that overexpress ESRP1 exhibit decreased metastasis to the liver and the lung when they were orthotopically implanted in mice (Ueda et al. 2014). Ueda et al further determined the survival rate in pancreatic ductal adenocarcinoma cases based on their immunohistochemical data. Both overall and the disease-free survival rates of the “ESRP1-high” group were higher than those of the “ESRP1-low” group (Ueda et al. 2014). Consistently, Preca et al reported that pancreatic ductal adenocarcinomas with poor outcome and recurrence expressed low levels of ESRP1 (Preca et al. 2015). These findings based on experiments using ESRP overexpression all suggest that ESRP1 has negative impacts on cancer progression.
Conversely, Yae et al reported that ESRP1-silenced 4T1 breast cancer cells that are orthotopically transplanted exhibit decreased incidence of lung metastasis, thus suggesting a positive role for ESRP1 in metastasis (Yae et al. 2012). The underlying mechanism appears to be as follows: ESRP1 expression results in increase in expression of the CD44v isoform. CD44v, but not CD44s, interacts with and stabilizes the xCT subunit of a glutamate-cystine transporter, leading to the enhanced uptake of cysteine to facilitate glutathione synthesis (Ishimoto et al. 2011). Thus, ESRP1 supports cancer cell proliferation by increasing levels of cellular glutathione that can serve as an antioxidant and confer resistance to reactive oxygen species to cells. They also utilized a public database to determine that breast cancer patients expressing high levels of ESRP1 mRNA exhibited a lower rate of overall survival (Yae et al. 2012). A positive role for ESRP1 may be related to the immunohistochemical findings that it is upregulated in carcinoma in situ and advanced carcinomas (Ueda et al. 2014; Ishii et al. 2014). However, it has also been reported that CD44s, but not CD44v, confers cells with resistance to cisplatin through activating the phosphoinositide-3-kinase/Akt pathway and CD44s mRNA is enriched in high-grade breast cancers (Brown et al. 2011). The conflicting data may be due to different experimental systems used in these studies.
ESRPs thus appear to have dual roles in cancer progression depending on the context of microenvironments surrounding cancer cells. In some situations, ESRP expression is favored as it supports cell survival; in other situations, downregulation of ESRPs is favored as this facilitates cell invasion. Therefore, cancer cells that are successful at fine-tuning ESRP expression to adapt surrounding circumstances would be able to undergo further progression.
5 Possible Functional Differences Between ESRP1 and ESRP2
The functional differences between ESRP1 and ESRP2 remain to be clearly elucidated. Both ESRP1 and ESRP2 harbor three RNA-recognition motifs (RRMs) and the amino acid sequences of each motif are well conserved between the two proteins (80–90 % identity) (Fig. 3). Interestingly, a point mutation in ESRP2 at the second RRM (Arg353Gln) is reported in breast cancers (Horvath et al. 2013). This mutation was shown to impair the ability of ESRP2 to bind to a cis-regulatory motif in the FGFR2 pre-mRNA, suggesting a role for RRM2 in recognizing target pre-mRNAs.

Schematic structures of ESRP1 and ESRP2. ESRP1 and ESRP2 share the N-terminal DnaQ-like exonuclease domain (DnaQ) and three tandem repeat RNA recognition motifs (RRMs) with a high degree of sequence conservation. The RRM2 and RRM3 of ESRP2 are implicated in target pre-mRNA recognition (Mizutani et al. 2015; Horvath et al. 2013). In the C-terminal region, ESRP1 has a proline-rich region that shares homology with DAZAP2 whereas ESRP2 has a region that shares homology with FAM70. The roles that these domains play in ESRP functions remain unclear
Knockdown of ESRP1, but not ESRP2, altered the isoform switching of CD44 from CD44v to CD44s in HNSCC cells, which suggests distinct roles for both ESRPs (Ishii et al. 2014). However, in normal mouse mammary gland epithelial (NMuMG) cells that lack ESRP1 expression, ESRP2 knockdown resulted in the isoform switching of CD44 (Horiguchi et al. 2012). Thus the target preference of both ESRPs is likely to be context-dependent. It can be affected by endogenous expression levels of ESRPs and target pre-mRNAs. Alternatively, it may be regulated by posttranslational modification of ESRP proteins. Recently, Mizutani et al reported that the splicing activity of ESRP2 is enhanced by K27-linked polyubiquitination of its RRM2 and RRM3 by a ubiquitin ligase Arkadia (Mizutani et al. 2015).
There is also evidence for distinct functions for both proteins in vivo. Germline knockout of Esrp1 resulted in abnormal craniofacial development and neonatal lethality (Bebee et al. 2015). Intriguingly, high expression of Esrp2 but not Esrp1 is observed in mouse liver, and similarly in adult human livers for the corresponding human orthologs. Esrp2-null mice display an increased number of diploid as well as tetraploid hepatocytes with smaller sizes, suggesting that mouse ESRP2 plays a role in postnatal liver development (Bhate et al. 2015). However, double knockout mouse embryos exhibit more severe phenotypes, including defects in branching morphogenesis in the lungs and salivary glands as well as epidermal hypoplasia and reduced hair follicles (Bebee et al. 2015). These findings suggest some functional redundancy between ESRP1 and ESRP2.
6 Concluding Remarks
Recent findings have revealed that ESRPs can either positively or negatively impact cancer progression. Thus, the plastic nature of their expression as well as the fine-tuning of their activities appears to be prerequisites for successful cancer progression. Several possible molecular mechanisms for regulating the expression and activity of ESRPs, have emerged.
First, ESRPs expression is regulated in complex circuits comprising transcriptional repressors and miRNAs. Notably, a double negative feedback circuit between ESRPs and δEF1/SIP1 to turn on/off the switch of ESRP expression in some types of cells can play a crucial role in the plastic expression of ESRPs (Fig. 1). Identification of internal or external cues that affect the balance in the circuit would help further our understanding of how this intricate regulatory system operates in vivo. The underlying mechanisms may not be uniform, as the circuit does not appear to operate in other cells.
Second, the activity of ESRPs can also be modulated through multiple mechanisms. Recently hnRNPM was reported to be a functional antagonist of ESRPs (Xu et al. 2014). In contrast to ESRPs, hnRNPM is a splicing regulatory protein that is highly expressed in mesenchymal cells but downregulated in epithelial cells. It drives splicing programs that oppose those promoted by ESRPs. There may be other functionally antagonistic splicing regulators that remain to be identified. Post-translational modifications of ESRPs appear to be important but only poorly understood. Thus far, only the K27-linked polyubiquitination of ESRP2 by Arkadia has been shown to enhance the splicing function of ESRP2 (Mizutani et al. 2015). Other covalent modifications of ESRP proteins could modulate their functions as well, which remain to be elucidated.
Given the dual functions of ESRPs in cancer progression, simple enhancement or inhibition of their activities would have unfavorable outcomes in patients. ESRPs themselves may therefore not be suitable for therapeutic molecular targets. The same argument can be applied to the use of ESRPs as prognostic markers, which has given controversial results thus far (Ueda et al. 2014; Mizutani et al. 2015; Preca et al. 2015; Yae et al. 2012). In addition to the plastic nature of ESRP1/2 expression during cancer progression (Ueda et al. 2014; Ishii et al. 2014), post-translational modification of ESRP2 is required at least under some conditions (Mizutani et al. 2015), indicating that expression of ESRP2 does not always correspond with its activity. The same may be true for ESRP1. Rather, proteins downstream of ESRPs could serve as prognostic markers (Mizutani et al. 2015) or molecular targets for therapeutic inhibition of cancer progression. Key molecular effectors downstream of ESRPs during cancer progression need to be further explored in the near future.
Abbreviations
- δEF1:
-
δ-crystallin enhancer binding protein
- EMT:
-
epithelial-mesenchymal transition
- ESRP:
-
epithelial splicing regulatory protein
- FGF:
-
fibroblast growth factor
- FGFR:
-
fibroblast growth factor receptor
- hnRNP:
-
heterogenous nuclear ribonucleoprotein
- HNSCC:
-
head and neck squamous carcinoma
- RRM:
-
RNA-recognition motif
- SIP1:
-
Smad interacting protein 1.
References
Bebee TW, Park JW, Sheridan KI, Warzecha CC, Cieply BW, Rohacek AM, Xing Y, Carstens RP (2015) The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. eLife 4:e08954
Bhate A, Parker DJ, Bebee TW, Ahn J, Arif W, Rashan EH, Chorghade S, Chau A, Lee JH, Anakk S, Carstens RP, Xiao X, Kalsotra A (2015) ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat Commun 6:8768
Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ (2013) Dynamic integration of splicing within gene regulatory pathways. Cell 152:1252–1269
Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, Cheng C (2011) CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest 121:1064–1074
Cieply B, Farris J, Denvir J, Ford HL, Frisch SM (2013) Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res 73:6299–6309
Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7:1267–1278
David CJ, Manley JL (2010) Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24:2343–2364
Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, Xing Y, Carstens RP (2012) Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol 32:1468–1482
Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R (2005) DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 24:2375–2385
Eswarakumar VP, Lax I, Schlessinger J (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 16:139–149
Fagoonee S, Bearzi C, Di Cunto F, Clohessy JG, Rizzi R, Reschke M, Tolosano E, Provero P, Pandolfi PP, Silengo L, Altruda F (2013) The RNA binding protein ESRP1 fine-tunes the expression of pluripotency-related factors in mouse embryonic stem cells. PLoS One 8:e72300
Fiegen D, Haeusler LC, Blumenstein L, Herbrand U, Dvorsky R, Vetter IR, Ahmadian MR (2004) Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J Biol Chem 279:4743–4749
Horiguchi K, Sakamoto K, Koinuma D, Semba K, Inoue A, Inoue S, Fujii H, Yamaguchi A, Miyazawa K, Miyazono K, Saitoh M (2012) TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 31:3190–3201
Horvath A, Pakala SB, Mudvari P, Reddy SD, Ohshiro K, Casimiro S, Pires R, Fuqua SA, Toi M, Costa L, Nair SS, Sukumar S, Kumar R (2013) Novel insights into breast cancer genetic variance through RNA sequencing. Sci Rep 3:2256
Hoshi Y, Endo K, Shirakihara T, Fukagawa A, Miyazawa K, Saitoh M (2016) The potential role of regulator of G-protein signaling 16 in cell motility mediated by δEF1 family proteins. FEBS Lett 590:270–278
Ishii H, Saitoh M, Sakamoto K, Kondo T, Katoh R, Tanaka S, Motizuki M, Masuyama K, Miyazawa K (2014) Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J Biol Chem 289:27386–27399
Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, Masuko T, Shimizu T, Ishikawa T, Kai K, Takahashi E, Imamura Y, Baba Y, Ohmura M, Suematsu M, Baba H, Saya H (2011) CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 19:387–400
Jordan P, Brazåo R, Boavida MG, Gespach C, Chastre E (1999) Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 18:6835–6839
Kaida D, Schneider-Poesch T, Yoshida M (2012) Splicing in oncogenesis and tumor suppression. Cancer Sci 103:1611–1616
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420–1428
Leontieva OV, Ionov Y (2009) RNA-binding motif protein 35A is a novel tumor suppressor for colorectal cancer. Cell Cycle 8:490–497
Lu H, Liu J, Liu S, Zeng J, Ding D, Carstens RP, Cong Y, Xu X, Guo W (2013) Exo70 isoform switching upon epithelial-mesenchymal transition mediates cancer cell invasion. Dev Cell 27:560–573
Mizutani A, Koinuma D, Seimiya H, Miyazono K (2015) The Arkadia-ESRP2 axis suppresses tumor progression: analyses in clear-cell renal cell carcinoma. Oncogene. doi:10.1038/onc2015.412
Ponta H, Sherman L, Herrlich PA (2003) CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol 4:33–45
Preca BT, Bajdak K, Mock K, Sundararajan V, Pfannstiel J, Maurer J, Wellner U, Hopt UT, Brummer T, Brabletz S, Brabletz T, Stemmler MP (2015) A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int J Cancer 137:2566–2577
Reinke LM, Xu Y, Cheng C (2012) Snail represses the splicing regulator epithelial splicing regulatory protein 1 to promote epithelial-mesenchymal transition. J Biol Chem 287:36435–36442
Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, Harbeck N, Schmitt M, Lengyel E (2000) Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 19:3013–3020
Shirakihara T, Horiguchi K, Miyazawa K, Ehata S, Shibata T, Morita I, Miyazono K, Saitoh M (2011) TGF-β regulates isoform switching of FGF receptors and epithelial-mesenchymal transition. EMBO J 30:783–795
Ueda J, Matsuda Y, Yamahatsu K, Uchida E, Naito Z, Korc M, Ishiwata T (2014) Epithelial splicing regulatory protein 1 is a favorable prognostic factor in pancreatic cancer that attenuates pancreatic metastases. Oncogene 33:4485–4495
Warzecha CC, Carstens RP (2012) Complex changes in alternatice pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT). Semin Cancer Biol 22:417–427
Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP (2009a) ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33:591–601
Warzecha CC, Shen S, Xing Y, Carstens RP (2009b) The epithelial splicing regulatory factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol 6:546–562
Warzecha CC, Jiang P, Amirikian K, Ditmmar KA, Lu H, Shen S, Guo W, Xing Y, Carstens RP (2010) An ESRP-regualted splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J 29:3286–3300
Werner S, Frey S, Riethdorf S, Schulze C, Alawi M, Kling L, Vafaizadeh V, Sauter G, Terracciano L, Schumacher U, Pantel K, Assmann V (2013) Dual roles of the transcription factor grainyhead-like 2 (GRHL2) in breast cancer. J Biol Chem 288:22993–23008
Xiang X, Deng Z, Zhuang X, Ju S, Mu J, Jiang H, Zhang L, Yan J, Miller D, Zhang HG (2012) Grhl2 determines the epithelial phenotype of breast cancers and promotes tumor progression. PLoS One 7:e50781
Xu Y, Gao XD, Lee JH, Huang H, Tan H, Ahn J, Reinke LM, Peter ME, Feng Y, Gius D, Siziopikou KP, Peng J, Xiao X, Cheng C (2014) Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev 28:1191–1203
Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ, Wada T, Masuko T, Mogushi K, Tanaka H, Osawa T, Kanki Y, Minami T, Aburatani H, Ohmura M, Kubo A, Suematsu M, Takahashi K, Saya H, Nagano O (2012) Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun 3:883
Acknowledgement
This work was supported by the Vehicle Racing Commemorative Foundation and the JSPS Core-to-Core Program ‘Cooperative International Framework in TGF-β Family Signaling’.
Conflict of Interest
None declared
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Hayakawa, A., Saitoh, M., Miyazawa, K. (2016). Dual Roles for Epithelial Splicing Regulatory Proteins 1 (ESRP1) and 2 (ESRP2) in Cancer Progression. In: Atassi, M. (eds) Protein Reviews. Advances in Experimental Medicine and Biology(), vol 925. Springer, Singapore. https://doi.org/10.1007/5584_2016_50
Download citation
DOI: https://doi.org/10.1007/5584_2016_50
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3709-2
Online ISBN: 978-981-10-3710-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)