
Abstract
Nitric oxide (NO) effects in airways are influenced by the activity of NO-synthase isoforms and NO metabolism. Inducible NO-synthase (iNOS), which produces large amounts of NO, is active during the inflammatory process. NO quickly reacts, producing reactive oxygen species (ROS). In this study we attempted to detect the expression of iNOS and markers of ROS in the airway hyperreactivity (AHR) condition. The study was performed in guinea pigs, divided into four groups. Two groups were treated with the non-selective inhibitor of NO-synthase L-NAME. The other two groups were used as controls. Exhaled NO was monitored in vivo, AHR was assessed both in vivo and in vitro, and the expression of iNOS in lung homogenate, and oxidative stress markers were measured in the venous blood. L-NAME significantly affected the AHR only in in vitro condition, blocked the expression of iNOS in control but not in sensitized animals, and decreased the level of exhaled NO. The results concerning the oxidative stress markers are equivocal. The study confirmed that NO is involved in the regulation of AHR; the effects being mediated via iNOS and ROS activity.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
One of the most important aspects of human physiology is clarifying the status of nitric oxide (NO) in a wide range of different systems and organs, including the airways. NO acts as a neurotransmitter of inhibitory non-adrenergic non-cholinergic neurotransmission. Its sources are different respiratory cells – neural, endothelial, epithelial, vascular, bronchial smooth muscle, inflammatory, and other cells (Ricciardolo 2003). NO is synthesized from L-arginine by three isoforms of NO synthase (NOS). The constitutive isoforms – neuronal (nNOS) and endothelial (eNOS) – produce NO in relatively small amounts, which are mainly involved in the regulation of physiological functions, e.g., bronchodilation, mucous secretion, mucociliary transport, gas exchange, or non-specific defense mechanisms. On the other hand, increased production of NO associated with high activity of inducible isoform (iNOS) is considered to underlie the development of pathological processes. The expression and activity of iNOS is increased in such conditions as exposure to exogenous irritants (smoking, allergens, etc.), bacterial products, or proinflammatory cytokines (Barnes and Belvisi 1993). Symptoms accompanying large NO amounts are proinflammatory changes, vasodilatation, plasma exudation, mucus hypersecretion, free radical productions, and airways hyperreactivity. A disordered balance of NO level in airways contributes to changes in airway smooth muscle tone. Activation of the iNOS is mainly responsible for changes in the body NO level including that in exhaled air.
iNOS is a cytoplasmic enzyme that particularly increases in response to proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), interleukin 2 and 10 (IL-2, IL-10), interferon gamma (IFN-γ), or lipopolysaccharides. NO generated by iNOS has antimicrobial, antitumor, cytostatic, or cytotoxic effects associated with free radicals production. Although NO has a protective role in a variety of infectious and inflammatory conditions (Kröncke et al. 1998), it also may play a major role, along with free radicals, in the pathogenesis of airway inflammation. A hallmark of inflammation is airway hyperreactivity, the pathogenesis of which is still unclear.
In pathology, iNOS can produce up to 1,000-fold greater amounts of NO, whose role shifts from regulatory and protective to cytotoxic. NO can be characterized as a free radical with a very short biological half-life. This property gives NO an opportunity to interact with a number of molecules, such as proteins containing thiol groups or superoxide (.O2 −). High amounts of NO produce, through a reaction with superoxide anion, cytotoxic peroxynitrite (.ONOO−), from which the biologically destructive hydroxyl radical originates (Anderson et al. 2011). ROS formed during airway inflammation accelerate the signaling processes of inflammatory mediators and as a consequence underlie the pathogenesis of airway hyperreactivity (Ghosh and Erzurum 2011; De Boer et al. 2001).
To clarify the involvement of NO and ROS pathways in airway hyperreactivity, in the present study we set out to determine the influence of pre-treatment with L-NAME, a non-selective inhibitor of NO-synthase, on the expression of iNOS and markers of oxidative stress (3-nitrotyrosine and thiobarbituric acid reactive substances – TBARS) in experimental allergic inflammation.
2 Methods
The study design was approved by the Ethical Committee of Jessenius Faculty of Medicine in Martin, Slovakia. All experiments were realized in accordance with the recommendations of Helsinki Declaration of the World Medical Association, Directive of European Commission on the protection of animals used for experimental and other scientific purposes adopted in 1986 (86/609/EEC) and the regulations of the Slovak Republic (Law No. 289/2003 Statute-book Regulation of Slovak Republic).
Thirty two pathogen-free, adult male TRIK strain guinea pigs weighing 180–250 g were used in the experiments. The animals were obtained from the Department of Toxicology and Breeding of Experimental Animals of the Institute of Experimental Pharmacology and Toxicology of the Slovak Academy of Sciences, Dobrá Voda, Slovak Republic, a certified breeding facility. They were group-housed in individual cages in the climate-controlled commercial cages, with a 12/12 h light/dark cycle in place, and had access to water and chow ad libitum. Room temperature was maintained at 21 ± 1 °C.
2.1 Study Design
Guinea pigs were divided into four groups, each consisting of eight animals: two control and two experimental groups:
-
healthy animals which received saline in a dose of 1 ml/kg;
-
animals which were sensitized with ovalbumin (OVA, Sigma Aldrich, St. Louis, MO) in accordance with the sensitizing scheme described below;
-
animals which received L-NAME (Nω-nitro-L-arginine methylester) in a dose of 40 mg/kg daily for 14 days, without sensitization;
-
animals which received L-NAME in a dose of 40 mg/kg daily throughout a 14-day sensitization time.
All injections were intraperitoneal unless otherwise indicated.
2.2 Airway Hyperreactivity Provocation
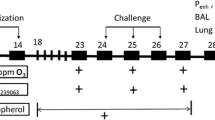
The animals were sensitized with the allergen ovalbumin. The sensitizing scheme was the following. A hundred micrograms of ovalbumin, dissolved in 1 ml saline, were injected on Day 1 (0.5 ml – subcutaneously in the neck and the other 0.5 ml intraperitoneally). On Day 3, the animals received only the intraperitoneal dose of OVA. Then, on Day 14, the animals inhaled 0.1 % OVA solution for 5 min. The inhalation of OVA was realized in a rodent whole-body plethysmograph consisting of glass thoracic and nasal chambers (type 885, Hugo Sachs Electronik; March-Hugstetten, Germany).
2.3 Measurement of Specific Airway Resistance
Specific airway resistance (RxV) was measured plethysmographically 2 min after inhalation of saline solution and subsequently 2 min after inhalation of the bronchoconstrictive mediator histamine in a concentration of 10−6 mol/l (Sigma-Aldrich, St. Louis, MO). The measurement was performed a day before OVA sensitization and on the last day of sensitization, 5 h after ovalbumin administration.
2.4 Airways Smooth Muscle Reactivity
Airway smooth muscle reactivity was recorded in vitro. The animals were euthanized with an overdose of an anesthetic 24 h after the last OVA administration. The trachea and lungs were removed and small tissue strips were prepared and placed into an organ bath consisting of the Krebs-Henseleit solution (110.0 mmol/l NaCl, 4.8 mmol/l KCl, 2.35 mmol/l CaCl2, 1.2 mmol/l MgSO4, 1.2 mmol/l KH2PO4, 25.0 mmol/l NaHCO3, and 10.0 mmol/l glucose in glass-distilled water), which was exchanged every 10 min. The solution was continuously aerated with a mixture of 95 % O2 and 5 % CO2 at pH 7.5 ± 0.1 and temperature 36 ± 0.5 °C. The measurement of smooth muscle reactivity was conducted in the isolated tissue bath system (Experimetria, Budapest, Hungary). Tissue strips were initially exposed to the tension of 4 g for 30 min (loading phase). Thereafter, tension was reduced to the baseline level of 2 g for 30 min (adaptation phase). Tension changes induced by contracted tracheal and lung tissue strips in response to cumulative doses of histamine and acetylcholine (10−8–10−3 mol/l; Sigma-Aldrich, St. Louis, MO) were recorded by computer after an hour’s incubation time. A cumulative concentration-response curve was determined for every strip.
2.5 Exhaled Nitric Oxide Detection
Exhaled NO (eNO) was detected using a chemiluminescence method. The method consists of detection of the luminescence arising during a chemical reaction of NO and ozone, which progresses in the analyzer (NIOX, Aerocrine; Solna, Sweden), which was adapted for small laboratory animals with a hermetic plexiglass chamber (MR Diagnostic; Prague, Czech Republic). The resulting output signal corresponds to the concentration of NO in the input sample (exhaled air). Each guinea pig was placed in the chamber individually. After 5 min of spontaneous breathing, the air from the chamber was discharged through a two-way valve into the analyzer. The off-line mode was used for analysis of all air samples. eNO analysis corresponded with the sensitization scheme. It was performed on Day 1, Day 3, and Day 14 in all groups of animals; in the sensitized ones 30 min before OVA administration.
2.6 Inducible NO-Synthase Expression
Distal parts of the lungs were taken for RT-PCR analysis. Lung tissue was homogenized in a solution consisting of ß-mercaptoethanol and RLT buffer for 20–40 s. Total RNA was isolated using RNeasy Micro Kit (QIAGEN Group; Hilden, Germany) and was then eluted in the RNase-free water. We used 1 μg of total isolated RNA for reverse transcription. cDNA synthesis was conducted using a QuantiTect® Reverse Transcription Kit (QIAGEN Group; Hilden, Germany). The transcription lasted at 42 °C and the reaction time was 20 min. The concentration of total DNA and cDNA was determined using a NanoPhotometer (Implen; Munich, Germany). Isolation of RNA and reverse transcription were performed according to the manufacturer’s instruction.
iNOS primer sequences (forward: GCAGCAGCGGCTTCACA; reverse: ACATCCAAACAGGAGCGTCAT) used for the guinea pig were previously published (Yamada et al. 2005) and checked for a sequence homology against the known sequence of Cavia porcellus nitric oxide synthase-2, inducible (NOS2) mRNA (NCBI Reference Sequence: NM_001172984.1). Hypoxanthine phosphoribosyltransferase (HPRT) was used as a reference gene. The primer sequences were previously published as well (Cho et al. 2005). All data were normalized to HPRT mRNA expression in the same sample.
RT-PCR was performed with QuantiTect® SYBR® Green PCR Kit (QIAGEN Group; Hilden, Germany) according to the manufacturer’s instruction, using 1 μl of cDNA (from RT) in a final volume of 25 μl containing 0.3 μM final F, R primer concentration. Quantitative PCR was performed using iCycler iQ®5 (Bio-Rad Laboratories; Hercules, CA) for 45 cycles at 95 °C for 15 s, primer-specific annealing temperature of 60 °C for 1 min, and 72 °C for 30 s. The crossing point, or the cycle number at which the fluorescence of the sample exceeded that of the background, was determined by the Bio-Rad iQ5–Standard Edition Optical System Software 2.0 using the second derivative method. RT-PCR assays of cDNA samples were performed in triplicate. A relative quantification method was used to assess the differences between tissue samples, employing the cycle number at which the fluorescence signal associated with a particular amplicon accumulation crosses the threshold, referred to as the (ΔCt).
2.7 Detection of Oxidative Stress Markers
We detected 3-nitrotyrosine and thiobarbituric acid reactive substances (TBARS) in the plasma. The presence of the former points to protein damage evoked by modification of tyrosine with peroxynitrite or other oxidative stress products, and the latter is indicative mostly of the level of lipid peroxidation. The blood was drawn from the heart to EDTA tubes immediately after animals’ death and was then centrifuged for 15 min at 1,000 × g. Plasma was stored at −80 °C. The markers were detected with an enzyme-linked immuno-sorbent (ELISA) method using Oxi Select Nitrotyrosine and Oxi Select TBARS Assay kits (Cell Biolabs; San Diego, CA), respectively, according to the manufacturer’s instructions. Plasma 3-nitrotyrosine was expressed in nM and TBARS was expressed as quantitative values of malonedialdehyde (MDH) in μM.
2.8 Statistical Elaboration
All results are expressed as means ± SE. Statistical analysis was performed using one-way analysis of variance (ANOVA). Comparisons of baseline values between groups were performed with a t-test. Differences were considered statistically significant when p-value was below 0.05. A commercial statistical package was used for all calculations (Microsoft Excel and IBM SPSS Statistic Standard; Czech Republic).
3 Results
3.1 Specific Airway Resistance
Specific airway resistance (RxV) was significantly higher in the OVA-sensitized animals than in the control group of healthy non-sensitized L-NAME untreated animals (p < 0.05). L-NAME pretreatment had no effect on RxV in the healthy animals, but it decreased the enhanced RxV in the animals with allergic inflammation, although the effect did not reach statistical significance and the RxV remained above that present in the healthy animals (Fig. 1).

Effects of L-NAME on specific airway resistance (RxV) (reactivity in vivo ). p < 0.05 vs. healthy and healthy + L-NAME groups
3.2 In Vitro Airway Reactivity
Changes in in vitro airway reactivity caused by L-NAME pretreatment in the non-sensitized and OVA-sensitized animals are exemplified by the presentation of the tracheal smooth muscle response to histamine only (Fig. 2). Tracheal smooth muscle contractile reactivity significantly increased in response to 10−6–10−3 mol/l histamine in the healthy non-sensitized L-NAME pretreated animals compared with L-NAME untreated animals (Fig. 2a; p < 0.05*). Likewise, tracheal smooth muscle reactivity strongly increased in the OVA-sensitized L-NAME pretreated compared with L-NAME untreated animals (Fig. 2b; p < 0.05*, p < 0.01**). Thus, L-NAME pretreatment markedly enhanced tracheal smooth muscle reactivity in both non-sensitized and OVA-sensitized animals. Similar tracheal changes were observed in response to acetylcholine; however, lung tissue showed no reaction to L-NAME pretreatment in the OVA-sensitized condition (data not shown).

Effects of L-NAME on tracheal smooth muscle reactivity (reactivity in vitro ). (a) Healthy non-sensitized animals; p < 0.05* for L-NAME pretreated vs. L-NAME untreated and (b) OVA-sensitized animals; p < 0.05*, p < 0.01** for L-NAME pretreated vs. L-NAME untreated
3.3 Exhaled Nitric Oxide
The level of eNO in healthy non-sensitized guinea pigs amounted to 3.4 ± 1.0 ppb. OVA sensitization had no appreciable effect on eNO which remained at 3.6 ± 0.4 ppb. Pretreatment with L-NAME in non-sensitized guinea pigs exerted a dual effect on eNO; it first significantly increased to 4.8 ± 0.6 ppb on Day 1 of sensitization and then decreased to 1.7 ± 0.8 ppb on Day 14 (Fig. 3a; p < 0.05‡), the level lower than that in the control untreated guinea pigs. In the OVA sensitized animals, L-NAME pretreatment caused a decrease in eNO throughout the experiment, to 1.9 ± 0.4 and 1.5 ± 0.6 ppb on Day 3 and Day 14, respectively. The decreases were significant compared with the OVA L-NAME untreated group (p < 0.05*) (Fig. 3a).

Exhaled NO (a) and expression of iNOS in lung homogenate (b) before and after L-NAME pretreatment in non-sensitized and OVA-sensitized guinea pigs. The levels of eNO decreased gradually with therapy duration. OVA-sensitization markedly enhanced iNOS expression in lung tissue. See text for details
3.4 Inducible NO-Synthase
The expression of iNOS in lung tissue of healthy non-sensitized L-NAME pretreated animals was very low. It was strongly enhanced in OVA-induced hyperreactivity and stayed at the enhanced level despite L-NAME pretreatment in this group (Fig. 3b, p < 0.01**). In general, expression of iNOS in lung tissue was rather variable, which likely reflected local differences in inflammatory changes in various parts of lungs, such as epithelium, bronchi smooth muscle cells, blood vessels, etc.
3.5 Oxidative Stress Markers
Both markers of oxidative stress, 3-nitrotyrosine and malondialdehyde (MDA; one of the end products of lipids peroxidation in the TBARS assay), changed in like manner in response to the pharmacological procedures used (Fig. 4a, b). Their levels were significantly higher in the OVA-sensitized animals compared with the healthy non-sensitized ones (p < 0.05*). Pretreatment with L-NAME resulted in a dual effect; it enhanced the markers in the non-sensitized animals (p < 0.05*), but reversed the enhancement due to sensitization (Fig. 4a, p < 0.01‡).

Markers of oxidative stress in the experimental conditions used: (a) 3-nitrotyrosine and (b) malondialdehyde. Both markers were significantly increased by OVA-sensitization (p < 0.05*). L-NAME pretreatment enhanced the markers in non-sensitized (p < 0.05*) but decreased them in OVA-sensitized animals; the decrease was significant in case of 3-nitrotyrosine (p < 0.05‡)
4 Discussion
Airway hyperreactivity (AHR) is characterized as uncontrolled bronchoconstriction in response to various endogenous and exogenous stimuli (Grootendorst and Rabe 2004). It is a hallmark of asthma and chronic obstructive pulmonary disease (COPD), but can also be present in upper respiratory tract infections, rhinitis, or gastroesophageal reflux. Bronchoconstriction may be caused by allergens, chemical irritants, cold air, hypoxia, etc. The genesis of airway hyperreactivity after exposure to such stimuli involves various types of cell which release mediators. The most important of these mediators is nitric oxide.
The literature shows that the role of NO in airways is still largely uncertain. Therefore, we deemed it worthwhile to investigate NO changes in airway hyperreactivity. To invoke AHR we used OVA sensitization, a model of allergic inflammation (Antošová and Strapková 2008; Mokrý et al. 2013), which was confirmed in the present study by increases in specific airway resistance. The NO homeostasis was modulated with L-NAME, an inhibitor of NO-synthase, in both healthy and OVA-sensitized, and thus suffering from airway inflammatory hyperreactivity, animals. Pretreatment with L-NAME did not appreciably affect airway resistance in healthy non-sensitized animals. This finding is at variance with that of Rubini (2011) who found an increase in airway resistance after L-NAME in healthy rats. On the premise that NO causes bronchodilation, the inhibition of NO-synthesis by L-NAME could indeed have been expected to increase airway resistance. That was not the case in the present study. We surmise that there is no tonic NO action at the physiological level of airway resistance, in other words no need to bronchodilate in healthy animals, and thus the lack of appreciable action of L-NAME. In contrast, in allergic inflammation, where airway resistance is increased, which is accompanied by overproduction of NO, L-NAME clearly acts to decrease airway resistance. Jiang et al. (2008) did not report an inhibitory action of L-NAME in lipopolysaccharide-induced airway hyperresponsiveness in guinea pigs, but recorded a significant decrease in airway resistance after aminoguanidine (a selective iNOS inhibitor) pretreatment. In the present study, non-selective inhibition of NO-synthase sufficed to demonstrate a drop in the allergy-enhanced airway resistance.
In the present study L-NAME increased reactivity of tracheal and lung tissue smooth muscles on the background of bronchoconstrictive action of histamine and acetylcholine in non-sensitized animals. In OVA-sensitized animals we observed an increase only in tracheal smooth muscle reactivity; lung tissue did not react. The reason for OVA-sensitized lung tissue not responding with increased reactivity on the background of bronchoconstriction is not readily explicable, but might have to do with a differential action of NO-synthase isoforms in response to airway allergy, depending on the level of the respiratory tract (trachea or lung tissue), and therefore a differential expression of the blocking effect of L-NAME. Plausibly, in OVA-sensitized animals L-NAME blocked predominantly cNO-synthase in lung tissue, but not iNOS through which NO could still be formed. These results correspond with the previously reported data on selective and non-selective inhibitors of NO-synthase (Antošová and Strapková 2008). It also is possible that in OVA-sensitized animals others mediators, such as cytokines, interleukines, or leukotrienes played a role in airway hyperreactivity which was not controlled for in the present study.
Subsequently we examined eNO, a marker of eosinophilic inflammation, in animals without and with L-NAME pretreatment. Detection of eNO is a specific noninvasive method, predominantly used in humans. In our department we used a standard allergic animal model, in which allergic inflammation was extensively confirmed by histology in our previous studies (Antošová 2007). We assumed that the values of eNO in allergic guinea pigs would be increased. Our results showed that after 14 days of OVA sensitization eNO was about the same as that in healthy animals. These results did not correspond well with increased airway reactivity found. However, eNO strongly increased the day following administration of L-NAME in healthy animals to decrease in later days in both healthy and OVA-sensitized animals. This dichotomous response of eNO to L-NAME pretreatment is not readily explainable. The literature data on the topic are scarce and controversial. Samb et al. (2001) described similar results; there was a reduction in eNO and an increase in hyperresponsiveness of tracheal smooth muscle in guinea pigs sensitized with ovalbumin. In the control group, these authors found an average value of eNO of 3.53 ppb, close the 3.37 ppb of the present study, and 2.52 ppb in sensitized animals. The findings were explained by a reduction in nNOS expression and activity in ovalbumin-sensitized animals, as no changes were observed in the other isoforms – eNOS and iNOS. A change in the activity of arginase, an enzyme that competes with the NO synthases for the common substrate – L-arginine, could contribute to a deficit of NO production. In contrast, Sethi et al. (2008) found in healthy mice that the baseline eNO of 7 ppb doubled in response to ovalbumin sensitization; the increase abated down to 10 ppb after 72 h. Likewise, Ahmad et al. (2009) showed a significant increase in eNO that correlated with allergic inflammation. After weeks of ovalbumin sensitization, eNO increased from 10.6 to 18 ppb in mice. The initial increase of eNO after L-NAME pretreatment in healthy animals in the present study could be evoked by pharmacokinetics properties and delayed onset of action of the inhibitor. The following eNO decrease confirms that the inhibition of NO was effective in both healthy and sensitized animals. To this end, our results are similar to those of Hori et al. (2011) who described a suppressive effect of L-NAME in both sensitized and non-sensitized animals. The question, however, remains, which NO synthase isoform is affected in relation to eNO changes.
The literature shows that asthmatic airway hyperreactivity has to do with increased expression of iNOS (Koarai et al. 2002; Schuiling et al. 1998), which suggests that this isoform is involved in the pathogenesis AHR and asthma. We confirmed the role of iNOS in the present study, finding that after L-NAME pretreatment in healthy animals its expression was very low, but it dramatically increased after allergen exposition. It seems that iNOS is the predominantly active NO isoform in airway hyperactivity and L-NAME is unable to fully inhibit it, which would be in line with a decrease in airway resistance in L-NAME-pretreated OVA-sensitized animals. The lack of the assessment of iNOS expression in the healthy untreated animals in the present study makes, however, the final judgment on the role of iNOS difficult.
ROS and reactive nitrogen species are increased in airway hyperreactivity and asthma in bronchoalveolar lavage, blood, and lung tissue, and ROS production is connected to a higher expression of iNOS in airways (Andreadis et al. 2003). In the present study we assessed two markers – 3-nitrotyrosine, a marker of protein damage, and malondialdehyde, a marker of lipid peroxidation. Both markers were enhanced in OVA-sensitized animals. In these animals, expectedly, NO inhibition due to L-NAME pretreatment decreased the oxidative markers. However, in healthy untreated animals, L-NAME pretreatment increased both oxidative markers, which is less readily explainable. If, however, we assume that L-NAME is a predominant cNOS inhibitor, then NO would be formed via iNOS pathway and produced free radicals. Our results correspond with those of Sugiura et al. (1999) who detected an enhanced level of 3-nitrotyrosine during late allergic reaction in guinea pigs. Likewise, Capellier et al. (1996) founded that in acute lung injury TBARS substances are elevated in the plasma, but decreased in lung tissue, with no appreciable effect of L-NAME found.
In conclusion, the present study confirmed that NO is involved in the regulation of airway hyperactivity in the allergic model of asthma in guinea pigs; the effects having to do with iNOS and ROS activity. There is a need for alternative study designs, using specific selective NO synthase inhibitors to sort out the exact interactions between ROS and NO in allergic airway hyperactivity.
References
Ahmad T, Mabalirajan U, Joseph DA, Makhija L, Singh VP, Ghosh B, Anurag A (2009) Exhaled nitric oxide estimation by a simple and efficient noninvasive technique and its utility as a marker of airway inflammation in mice. J Appl Physiol 107:295–301
Anderson JT, Zeng M, Li Q, Stapley R, Moore DR 2nd, Chenna B, Fineberg N, Zmijewski J, Eltoum IE, Siegal GP, Gaggar A, Barnes S, Velu SE, Thannickal VJ, Abraham E, Patel RP, Lancaster JR Jr, Chaplin DD, Dransfield MT, Deshane JS (2011) Elevated levels of NO are localized to distal airways in asthma. Free Radic Biol Med 50:1679–1688
Andreadis AA, Hazen SL, Comhair SA, Erzurum SC (2003) Oxidative and nitrosative events in asthma. Free Radic Biol Med 35:213–225
Antošová M (2007) Nitric oxide and bronchial hyperreactivity. Thesis. Jessenius Faculty of Medicine in Martin, Comenius University in Bratislava, 167 pp
Antošová M, Strapková A (2008) Dual effect of L-NAME on airway hyperreactivity. Acta Medica Martiniana 8:8–16
Barnes PJ, Belvisi MG (1993) Nitric oxide and lung disease. Thorax 48:1034–1043
Capellier G, Maupoil V, Boillot A, Kantelip J-P, Rochette L, Regnard J, Barale F (1996) L-NAME aggravates pulmonary oxygen toxicity in rats. Eur Respir J 9:2531–2536
Cho H, Lasco TM, Allen SS, Yoshimura T, McMurray DN (2005) Recombinant guinea pig tumor necrosis factor alpha stimulates the expression of interleukin-12 and the inhibition of Mycobacterium tuberculosis growth in macrophages. Infect Immun 73:1367–1376
de Boer J, Meurs H, Flendrig L, Koopal M, Zaagsma J (2001) Role of nitric oxide and superoxide in allergen-induced airway hyperreactivity after the late asthmatic reaction in guinea-pigs. Br J Pharmacol 133:1235–1242
Ghosh S, Erzurum SC (2011) Nitric oxide metabolism in asthma pathophysiology. Biochim Biophys Acta 1810:1008–1016
Grootendorst DC, Rabe KF (2004) Mechanisms of bronchial hyperreactivity in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 1:77–87
Hori A, Fujimura M, Ohkura N, Tokuda A (2011) Involvement of nitric oxide (NO) in cough reflex sensitivity between non-sensitized and OVA-sensitized guinea pigs. Cough 7:5
Jiang H, Qu J, He L, CHen X, Pan J, Li L, Zhu D, Cao Y, Shen L (2008) Effects of Nω-nitro-L-arginine methyl ester and aminoguanidine on lipopolysaccharide-induced airway hyperresponsiveness in guinea pigs. Chin Med J 121:1693–1697
Koarai A, Ichinose M, Sugiura H, Tomaki M, Watanabe M, Yamagata S, Komaki Y, Shirato K, Hattori T (2002) iNOS depletion completely diminishes reactive nitrogen-species formation after an allergic response. Eur Respir J 20:609–616
Kröncke KD, Fehsel K, Kolb-Bachofen V (1998) Inducible nitric oxide synthase in human diseases. Clin Exp Immunol 113:147–156
Mokrý J, Jošková M, Mokrá D, Christensen I, Nosáľová G (2013) Effects of selective inhibition of PDE4 and PDE7 on airway reactivity and cough in healthy and ovalbumin-sensitized guinea pigs. Adv Exp Med Biol 756:57–63
Ricciardolo FL (2003) Multiple roles of nitric oxide in the airways. Thorax 58:175–182
Rubini A (2011) The effect of N-nitro-L-arginine methyl ester, a nitric oxide synthase inhibitor, on respiratory mechanics in rats. Respiration 82:468–475
Samb A, Pretolani M, Dinh-Xuan AT, Ouksel H, Callebert J, Lisdero C, Aubier M, Boczkowski J (2001) Decreased pulmonary and tracheal smooth muscle expression and activity of type 1 nitric oxide synthase (nNOS) after ovalbumin immunization and multiple aerosol challenge in guinea pigs. Am J Respir Crit Care Med 164:149–154
Schuiling M, Meurs H, Zuidhof AB, Venema N, Zaagsma J (1998) Dual action of iNOS-derived nitric oxide in allergen-induced airway hyperreactivity in conscious, unrestrained guinea pigs. Am J Respir Crit Care Med 58:1442–1449
Sethi JM, Choi AM, Calhoun WJ, Ameredes BT (2008) Non-invasive measurements of exhaled NO and CO associated with methacholine responses in mice. Respir Res 9:45. doi:10.1186/1465-9921-9-45
Sugiura H, Ichinose M, Oyake T, Mashito Y, Ohuchi Y, Endoh N, Miura M, Yamagata S, Koarai A, Akaike T, Maeda H, Shirato K (1999) Role of peroxynitrite in airway microvascular hyperpermeability during late allergic phase in guinea pigs. Am J Respir Crit Care Med 160:663–671
Yamada H, Udagawa T, Mizuno S, Hiramatsu K, Sugawara I (2005) Newly designed primer sets available for evaluating various cytokines and iNOS mRNA expression in guinea pig lung tissues by RT-PCR. Exp Anim 54:163–172
Acknowledgments
This work was supported by the project “The increasing opportunities for career growth in research and development in the medical sciences”, co-financed from the EU sources: Grant MZ SR 2007/46-UK-11 and VEGA 1/0062/13.
Conflicts of Interest
The authors declare no conflicts of interest in relation to this article.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Antošová, M., Strapková, A., Mikolka, P., Mokrý, J., Medveďová, I., Mokrá, D. (2014). The Influence of L-NAME on iNOS Expression and Markers of Oxidative Stress in Allergen-Induced Airway Hyperreactivity. In: Pokorski, M. (eds) Allergens and Airway Hyperreactivity. Advances in Experimental Medicine and Biology(), vol 838. Springer, Cham. https://doi.org/10.1007/5584_2014_62
Download citation
DOI: https://doi.org/10.1007/5584_2014_62
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-10008-1
Online ISBN: 978-3-319-10009-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)