
Abstract
Clusters of group 14 elements show plenty of similarities with borane clusters. As such, chemists often try to understand their structure and bonding on the basis of Wade’s rules to predict and classify various clusters. Such practice, albeit very common, often faces challenges and difficulties due to significant differences in the bonding abilities between group 13 and 14 atoms, as well as the changes in the ionization energies and radial characteristics of atomic orbitals as the groups are descended. In this chapter, we have extensively discussed the structure and bonding of a wide variety of group 14 clusters, including bare clusters, substituent-decorated clusters, endohedral clusters, transition metal doped clusters, and their combinations. By thoroughly analyzing their electronic structures within the framework of molecular orbital theory, we have summarized their bonding patterns and explored the factors that affect the applicability of Wade’s rules in various group 14 clusters.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Group 14 of the periodic table is arguably the most important group of elements in chemistry. Lead and tin are well known for their use in the production of alloys and devices, silicon and germanium are important semi-conductors that established the foundation of modern technology, and needless to say carbon constitutes the very basis of the living world. Because group 14 crosses the dividing line between metals and non-metals, elements in this group feature a wide variety of physical and chemical properties, bringing a rich context into material science.
Being main group elements with 4 valence electrons, group 14 elements are predominantly tetravalent as a straightforward implication of the octet rule, and thus they are often termed “tetrel elements.” Indeed, the dominance of four-connected diamond structure in the bulk state allotropes of tetrel elements, and the wide existence of tetrel dioxides and tetrahalides, both demonstrate the predominant tetravalency of tetrel elements, especially for lighter ones. For the heavier tetrel elements, inert pair effect gives rise to the possibility of divalent tetrel atoms, mostly seen in lead but sometimes also tin, as exemplified by a wide range of ionic compounds and coordination complexes with the metals formally in the +2 oxidation state.
In addition to the above-mentioned classic main group chemistry, however, tetrel elements can also exist in completely different forms. A clear counterexample is the Zintl compound, which is a class of simple binary compounds formed between alkali metals and heavy tetrel elements. The earliest synthesized Zintl compound is Na4Pb9, which greatly mystified chemists because it did not conform to simple valency rules. If we assume the sodium is present as the monovalent Na(I), we might be forced to assign the lead atoms with fractional negative charges or mixed oxidation states. In-depth examination of its crystal structure (Fig. 1), however, reveals that the lead atoms exist in the form of Pb94− clusters, one of the earliest examples of what are later termed Zintl ion clusters.

Structures of diamond and the Zintl ion cluster [Pb9]4− [1]
The presence of clustered units in bulk phase indicates a new chemistry of tetrel elements, and understanding the electronic structures of these clusters would build an important bridge between molecules and bulk materials. The failure of tetrel elements to form four bonds and conform to octet rule hints a completely different bonding behavior in Zintl clusters. This raises the very fundamental question of this book: how to describe the structure and bonding of a cluster compound?
Encouraged by the fundamental importance and interesting chemistry of cluster compounds, numerous experimental and theoretical efforts have been made to understand the geometric and electronic structures of these nanoclusters. Experimentally, a great number of group 14 cluster compounds have been synthesized and characterized, together with their derivatives with different compositions and properties. A straightforward and extensively studied approach is to start from bulk materials and break the bulk into pieces of cluster units by reducing reagents. On the other hand, the bottom-up production of nanoclusters from molecular substances has also received lots of recent attentions as an alternative to the traditional top-down approach, as it allows more precise control of geometric and electronic structures of cluster compounds for further experimental analyses and development. Through this bottom-up approach, syntheses of group 14 clusters are no longer limited to Zintl phase compounds, but enter into the realm of molecular chemistry, allowing chemists to understand the structure and bonding of cluster compounds from a molecular perspective.
While individual atoms in cluster compounds do not necessarily follow the octet rule, bonding among these atoms is far from arbitrary. When we pay attention to cluster units instead of individual atoms, each cluster would exhibit certain patterns in electron counts, as first observed in borane clusters by Wade [2]. Wade’s electron-counting rules were later extended into other main group and transition metal clusters by Mingos, now known as the polyhedral skeletal electron pair theory (PSEPT) [3, 4]. These electron-counting rules can relate the geometries of clusters with their electron counts, thus serve as versatile rules of thumb and play the central role in cluster chemistry similar to that of the octet rule in traditional inorganic chemistry.
In addition to electron-counting approaches, detailed analysis on electronic structures in terms of orbitals is essential for chemists to establish an in-depth yet generalizable understanding of clusters, especially when simple electron-counting rules do not apply. Stone’s tensor surface harmonics (TSH) theory [5], for example, describes the underlying model that relates the geometric structures and electron counts within the framework of orbital interactions (vide infra) and rationalizes apparent “exceptions” to electron-counting rules.
This chapter is dedicated to revealing the structure and bonding of group 14 clusters that may or may not follow the prediction of Wade’s electron-counting rules. We will go through representative examples, understand how the electron-counting rules work and how they would be affected under different cases, and show how new bonding patterns could arise from orbital interactions among chemical fragments.
Because of the importance of Wade’s rules and PSEPT in cluster chemistry and their relevance in group 14 clusters, in Sect. 2, we will first give a brief review on Wade’s rules and why they work in borane clusters. By making use of the isolobal analogy, we will then show by examples how Wade’s rules play a guiding role in helping us understand group 14 clusters despite important differences between tetrel atoms and BH units (Sect. 3). With these established rules and patterns, we will present a wide variety of endohedral clusters that can be understood within the framework of Wade’s rules, despite the existence of quite a number of exceptions (Sect. 4). More complicated clusters will be introduced in Sect. 5, which may fall out of the scope of Wade’s rules at first glance, but can still be understood with the established knowledge through an in-depth analysis. Nevertheless, cluster chemistry is after all a rapidly developing field, and there are still many clusters out there that do not fit into any of the aforementioned classes, as will be introduced in Sect. 6. Via such an organization, we hope that readers can not only get familiar with the discovered group 14 clusters to date and the applicability of electron-counting rules, but more importantly, also understand the underlying concepts that bridge one with another. A brief conclusion and perspective discussion will be given in Sect. 7 to close this chapter.
2 A Brief Review of Wade’s Rules and the Underlying Principles
While Wade’s rules are convenient rules of thumb that can be easily applied in practice in determining overall charge or electron counts of clusters from their geometries without going into detailed electronic structures, there are more rigorous theoretical models that can better explain the electron counts of borane clusters predicted by Wade’s rules [6]. One representative example of these mathematical models is the Stone’s tensor surface harmonics (TSH) theory based on the framework of orbital interactions [5]. To be specific, let us consider one of the [BH] units distributed on a spherical surface and take a local coordinate system to orient its orbitals. Each [BH] unit has a localized B-H bond between the hydrogen 1s orbital and a boron s-p hybridized orbital, leaving three boron orbitals available for skeletal bonding with other [BH] units, one with σ symmetry and two with π symmetry (with respect to the center of the sphere). For a deltahedral borane, orbital interactions among these 3n orbitals contribute (n + 1) skeletal electron pairs (SEPs) in total (Fig. 2), and hence Wade’s rule for closo clusters is sometimes also referred to as the (n + 1) rule [4]. Based on the same framework, we can also derive the (n + 2) rule for nido clusters and (n + 3) rule for arachno clusters.

Schematic orbital interaction diagram for general Wadean clusters showing the origin of the (n + 1) rule
Understanding the (n + 1) rule from the orbital interaction perspective would help us understand some apparent “exceptions” to Wade’s rules in a unified framework. A famous example is the [B4Cl4] cluster, which has a tetrahedral geometry and hence appears to be closo, but has a SEP count of n instead of (n + 1). As mentioned before, each boron vertex contributes three valence orbitals (1 σ and 2 π) to skeletal bonding. The symmetry-adapted linear combinations of the four σ-type orbitals span A1 + T2 irreducible representations while the eight π-type orbitals span E + T1 + T2. Because there are two sets of orbitals with the same T2 irreducible representations, they would further interact with each other to give rise to the skeletal bonding orbitals as in Fig. 3, in addition to orbitals in other irreducible representations. This orbital interaction diagram immediately eliminates the possibility of applying (n + 1) rule in the B4Cl4 cluster (n = 4) since the (n + 1)-th orbital is degenerate with the (n + 2)-th orbital.

Schematic orbital interaction diagram showing the skeletal molecular orbitals for tetrahedral clusters
It should however be noted that, in molecular orbital calculations, the skeletal bonding orbitals and the external B-H σ bonding orbitals are always extensively mixed with each other, so the canonical molecular orbitals will not be able to clearly exhibit pure skeletal nature, as illustrated by calculation on the [B4H4] molecule shown in Fig. 4. The two sets of A1 + T2 orbitals are actually in-phase and out-of-phase combinations of the skeletal bonding orbitals and the symmetry-adapted linear combinations of the external B-H σ bonding orbitals.

Molecular orbital diagram of the tetrahedral [B4H4] molecule computed using PBE0/def2TZVP model chemistry based on geometries optimized at the same level
While electrons only fill up to the n-th orbital in the boron cluster [B4Cl4] which contains relatively electropositive boron atoms, the tetrahedrane molecule [C4H4] actually follows the (n + 2) rule with six SEPs (each vertex having 3 electrons involved in bonding with the other three vertices, totaling 12 skeletal electrons). Such a delocalized view that correlates well to the [B4Cl4] case is in fact equivalent with the usual localized view for tetrahedrane, which describes their skeletal bonding as having six localized C-C bonds, for which reason tetrahedrane is sometimes called an “electron-precise” cluster.
The inadequacy of Wade’s rules for tetrahedral clusters is not a single exception. In fact, it has been found that in clusters which have a threefold axis and a single vertex on this axis (e.g. tetrahedron), the (n + 1)-th and the (n + 2)-th skeletal bonding orbital will be degenerate as discussed above [7, 8]. In such case, a cluster with (n + 1) SEPs will distort from a highly symmetric structure due to (first-order) Jahn-Teller effect, arising from the incomplete occupation of degenerate orbitals. Hence deviation from (n + 1) rule is expected for such clusters. Apart from the aforementioned tetrahedral cluster [B4Cl4], the hypothetical [B10H10] cluster is also said to violate Wade’s rules due to such a degeneracy. [B10H10] might appear to have a closed deltahedral structure (Fig. 5), but is very different from the bicapped square antiprismatic geometry for typical closo 10-vertex clusters. At the same time, this neutral cluster has only n SEPs, instead of (n + 1) SEPs as seen in the bicapped square antiprismatic borane cluster [B10H10]2−. Relevant examples with this kind of unusual electron count and geometry include some metallaboranes as well as some other cases (vide infra) that are sometimes termed “hyper-closo” clusters [7, 8]. These apparent exceptions to Wade’s rules have been extensively studied and become a part of the more generalized Wade-Mingos rules, or the PSEPT, which considers not only removal of vertices from closo clusters to generate nido, arachno, or hypho structures, but also vertex capping and other extensions [4].

Structure of the hypothetical cluster [B10H10] [7]
3 Group 14 Clusters Which Conform to Wade’s Rules
As mentioned in the Introduction section, Wade’s rules are handy empirical rules that bridge the electron count and the geometry of borane clusters. The most typical boranes that fulfill Wade’s rules are closo clusters in the form of [BnHn]2− with deltahedral geometries. The valence electron count associated with this kind of clusters is 4n + 2 (3n from n B atoms, n from n H atoms, and 2 from the negative charge). Although Wade’s rules are originally proposed for borane clusters, this (4n + 2) rule has been extended to (14n + 2) rule in PSEPT for transition metal clusters by taking into consideration the d-shell of each vertex atom, and can also serve as an important guiding principle for group 14 clusters [9,10,11,12]. Despite the general applicability of Wade’s rules, there are still striking differences between boranes and group 14 clusters that are worth careful examination. In this section, we will discuss the applicability of Wade’s rules in group 14 clusters with a number of examples, especially those that are synthesized and characterized in solution or solid phases.
3.1 Bare Clusters [En]q− and Their Derivatives
Let us begin with the simplest group 14 cluster compounds in the formula of [En]q−, E being a tetrel element hereafter, to demonstrate the general applicability of Wade’s rules on group 14 clusters. In such a bare cluster [En]q−, the orbital interaction among tetrel atoms is similar to that among [BH] units in boranes, by noting that a tetrel atom is not only isoelectronic (having the same number of valence electrons), but also isolobal (having the same number of available valence orbitals with similar symmetries and shapes) to a [BH] unit in borane clusters. Hence the same scenario as introduced in the previous section for borane clusters also holds in general for bare group 14 clusters.
Specifically, if we consider each atom having a lone pair pointing outwards, in analogy with an external B-H σ bond in a borane cluster, each tetrel atom is left with two valence electrons and three valence orbitals available for skeletal bonding, making itself isolobal with a [BH] unit. Hence a similar orbital interaction pattern is expected, giving rise to the same electron-counting rules.
Because of the aforementioned isolobal analogy, Wade’s rules would predict that group 14 clusters [En]2− will also adopt a closo geometry, similar to their analogous borane clusters with the same number of vertices. Indeed, there are many clusters following this prediction. Examples include [E5]2− [13,14,15,16,17,18], [E9]2− [19, 20], and [E10]2− [21] (Fig. 6), all of which have similar shapes with the corresponding borane clusters [B5H5]2−, [B9H9]2−, and [B10H10]2−. [E12]2− clusters have also been found to be particularly stable by theoretical studies, although they have only been detected in gas phase [22,23,24].

Group 14 clusters in nido geometries also exist. A notable example is the [E9]4− (E = Ge, Sn, Pb) unit which exists in monocapped square antiprismatic geometry in A4E9 (A = alkali metals) compounds [17, 25,26,27,28,29] (Fig. 7). The Na4Pb9 Zintl phase introduced earlier is one of the earliest examples that features this structure.

Similar to boranes, exceptions to Wade’s rules could also be found in group 14 clusters. For example, [E4]4− clusters have been reported [27, 30] (Fig. 7), which have a similar orbital interaction diagram as [B4Cl4] and thus clearly cannot fulfill Wade’s rules. Instead, there are two more pairs of electrons filling into the non-bonding E-type orbitals, giving rise to n + 2 SEPs. Note that this description is equivalent to a localized picture in which there is a lone pair on each tetrel atom and a two-center-two-electron (2c-2e) bond associated with each neighboring atom pair. This equivalence can be immediately revealed by introducing a proton on each vertex to form an E-H bond and noting that the resulting species [E4H4] is isoelectronic with tetrahedrane C4H4. Alternatively, one can also consider the isoelectronic relationship between an anionic tetrel atom [E−] and a pnictogen atom, say, P, and the cluster will simply be isoelectronic with the white phosphorous molecule P4. This slightly higher tendency for group 14 clusters to fulfill octet, when compared to group 13 counterparts, gives rises to some difference in bonding behaviors, which will be elaborated later in Sect. 6.
Apart from comparisons across groups, we may also make intra-group comparison among different periods. For heavy tetrel atoms like germanium, tin and lead, their atomic ns-np energy gaps are large due to strong shielding effect. Thus, the s-p hybridization is not that significant when compared to carbon. Hence, we would expect that the lone pairs of tetrel atoms in a group 14 cluster are largely contributed by electrons in their ns orbitals, especially when the tetrel element goes down the group. To illustrate such difference, we can perform a principal interacting orbital (PIO) analysis [31, 32] on the [Si12]2− cluster, to investigate the most important orbitals that each vertex utilizes to interact with other vertices. Indeed, the PIO analysis identifies three pairs of orbital interactions, consistent with our previous descriptions based on Wade’s rules and TSH theory (Fig. 8). The PIO analysis on the [Pb12]2− cluster reveals a similar picture, except that the σ-type PIO of the Pb vertex is obviously of more p character compared to the Si analog, leaving the Pb 6s orbital for holding the lone pair. The unusual stability of ns orbitals (especially for lead because of the inert pair effect) is essential for a heavy tetrel atom to hold a lone pair of electrons, so that it does not significantly participate in skeletal bonding.

Principal interacting orbital analysis on [Si12]2− and [Pb12]2− clusters, with one of the vertices as one fragment and the rest of cluster as the other fragment
Compared to heavy tetrel elements, carbon has a much smaller s-p gap than heavier tetrel elements, and thus disfavors a localized lone pair that resembles a B-H σ bond in borane. This explains the fact that while there exist numerous examples of silicon, germanium, tin, and lead clusters that have close resemblance to boranes, we cannot find any similar electron-deficient clusters that consist of only carbon atoms. Instead, carbon has a bonding behavior very different from heavy tetrels. The out-of-plane π-type interactions between carbon 2p orbitals are much stronger than those between p orbitals of heavier tetrel atoms. Hence the commonly seen carbon “clusters” are fullerenes, which have completely different geometries from deltahedral borane clusters.
Other than the aforementioned bare clusters, a larger number of their derivatives have been synthesized by introducing various substituents on some of the vertices of a cluster, ranging from monosubstituted, disubstituted, trisubstituted to tetrasubstituted clusters [33,34,35,36,37,38,39,40,41,42]. These clusters can be easily understood in a similar way, noting the isoelectronic/isolobal relationship among [BH], [E], and [ER]+ units.
For example, the cluster [Ge9R2]2− (R = aliphatic groups) has 40 valence electrons (4 × 9 from Ge atoms, 1 × 2 from R substituents, and 2 from the negative charge), which is isoelectronic with the bare cluster [Ge9]4− and the borane cluster [B9H9]4−. Hence the skeletal bonding of the cluster [Ge9R2]2− should resemble its analogs. Indeed, the [Ge9R2]2− cluster adopts a monocapped square antiprismatic geometry (Fig. 9), falling into the class of nido clusters as predicted by Wade’s rules.

In fact, with an R-group substitution turning the “lone pair” of electrons on carbon into a σ bonding pair, electron-deficient carbon clusters become possible in some very rare cases. A worth-noting example is the dicationic “benzene,” [C6Me6]2+, which has a skeletal electron count of 16 (3 × 6 from carbon atoms minus 2 from positive charge), isoelectronic with [B6H6]4− in terms of skeletal bonding [43,44,45]. Unlike the neutral benzene in which all six carbon atoms lie on a plane, this molecule is electron-deficient and forms a three-dimensional cluster. As there are 6 vertices with a SEP count of 8 (= n + 2), Wade’s rules predict a nido geometry similar to [B6H6]4−, i.e. a pentagonal pyramid, which is indeed the case (Fig. 10).

3.2 Electronic Structure of [E9] Clusters and Its Deviation from Wade’s Rules
Although many group 14 clusters could be easily understood with Wade’s rules, they also show significant differences from borane clusters in terms of structure and bonding. One notable discrepancy can be seen from the observation that although [B6H6]2− and [B12H12]2− are representative structures of borane clusters, they are rarely observed in group 14 clusters. Instead, the most commonly seen group 14 cluster consists of 9 tetrel atoms, with two commonly associated electron counts, [E9]2− and [E9]4−, which are seldom seen for borane compounds. Moreover, open-shell [E9]3− species have also been reported to be stable [15, 28, 46, 47], which clearly falls out of the prediction of Wade’s rules. All of these indicate that, apart from a simple analogy, there are still fundamental differences between group 14 clusters and boranes.
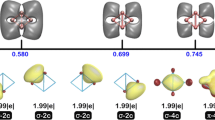
Some theoretical studies have been devoted to understand the origin of the extraordinary stability of the above-mentioned [E9] clusters. Adaptive natural density partitioning (AdNDP) analysis [48] has been performed to decipher the bonding by partitioning the electron density into 1c-2e bonds (lone pairs), 2c-2e bonds, and nc-2e (multi-centered) bonds [49] (Fig. 11). The results show that the analyzed nine-vertex clusters seem to have special σ-aromaticity that stabilizes the skeletal bonding. As there are not yet systematic studies that perform parallel comparisons, how this aromatic behavior translates to the overall stability is still not so clear.

Another interesting attempt should be attributed to King and coworkers who viewed this problem from a different angle [50]. Note that in the aforementioned models, group 14 clusters are described as having out-pointing lone pairs localized at each vertex separated from skeletal bonding. But unlike borane clusters in which the B-H bonds are pointing away from each other, in group 14 clusters, the out-pointing lone pairs have stronger s characters and thus they can overlap with each other as well as with skeletal bonding orbitals. Hence an alternative model that could also be applied to understand the electronic structure of group 14 clusters is the Jellium model [50], which was originally proposed for alkali metal clusters and later extended to group 11 clusters [51,52,53]. Making use of the fact that a large class of clusters are approximately spherical, the Jellium model considers a cluster made up of many atoms as a “superatom,” and the molecular orbitals of a cluster will resemble “superatomic orbitals” that can be well described with angular quantum numbers and hence can be labelled as S, P, D, F, etc. The 2-electron rule, 8-electron rule, and 18-electron rule become applicable, and the related magic numbers continue with 20, 34, 40, etc. Note that in this model, all of the valence electrons of a group 14 cluster are included in the electron counting. For example, the occupied orbitals for the [E12]2− cluster are considered as a 50-electron species having its superatomic 1S, 1P, 1D, 2S, 1F, 2P, and 2D orbitals occupied.
Note that a [E9]4− cluster has 40 (4 × 9 from tetrel atoms and 4 from the negative charge) valence electrons and thus conforms to the magic number (40). Without going into details, its corresponding occupied superatomic orbitals are 1S, 1P, 1D, 2S, 1F, and 2P (Fig. 12). Such “closed-shell” superatomic configuratiion described in the Jellium model might explain why [E9]4− clusters (with or without substituents) are so common, even more common than closo [E9]2− clusters, as well as the extraordinary stability of the open-shell species [E9]3−.

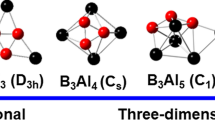
The [E9]4− cluster is particularly interesting not only due to its surprising stability, but also because of its structural flexibility. If we follow the prediction of Wade’s rules, [E9]4− have n + 2 pairs of skeletal electrons and hence should adopt a nido structure, that is, a monocapped square antiprism (MSA).
However, there exist a number of examples showing that substituent-decorated [E9]4− clusters can also be stable in a closo structure, that is, tricapped trigonal prism (TTP), as exemplified by the threefold-symmetric trisubstituted cluster [Ge9(Hyp)3]− (Hyp = hypersilyl, Si(SiMe3)3) [54] (Fig. 13). To understand this observation, one has to first note that the MSA and TTP structures do not differ much in geometry. In fact, the MSA structure can be viewed as elongating one of the prismatic edges of the TTP geometry and opening up a square face (Fig. 14). While the TTP structure of [E9]4− does have an imaginary frequency according to gas phase calculation [12], this distortion is so small that the energy gain has been found to be very little. Therefore, the most stable geometry of [E9]4− could be regulated by solvent, counterions, and substituents [55].


Comparison between D3h (left) and C4v (right) structures of [E9] clusters in which shortening of the distance for a diagonal pair in the basal square face of the C4v structure (top part of the shown structure) leads to the D3h structure while an elongation leads back to the C4v structure
From the electronic structure viewpoint, we may again see the close relationship between [E9]2− and [E9]4− at TTP and MSA geometries. Based on the aforementioned Jellium model, we can easily predict the shapes of the molecular orbitals of [E9]2− at a TTP geometry. According to the Jellium model, the lowest occupied valence orbitals of [Ge9]2− are expected to be 1S, 1P, 1D, and 2S orbitals, respectively (Fig. 12). The subsequent 1F and 2P orbitals are higher in the valence region and are interlaced, with the 2Pz orbital being the HOMO and 1Fz3 orbital being the LUMO (Fig. 15). Note that this LUMO has slight bonding character among capping atoms and anti-bonding character among those base atoms across the vertical edges. Hence in an [E9]4− cluster, occupation of this 1Fz3 orbital induces a second-order Jahn-Teller instability that could bring two of the capping atoms together while elongating a lateral edge, causing the cluster to deform from TTP to MSA geometry.

Frontier molecular orbitals of [Ge9]2− at a TTP geometry with corresponding superatomic orbital labels
It should however be noted that, while Jellium model offers a distinct viewpoint for group 14 clusters, it does not predict different electronic structures but rather provides a different interpretation of the occupied orbitals of the clusters. In particular, for a [E9] cluster, Jellium model describes its molecular orbitals as superatomic orbitals and thus predicts a “closed-shell” configuration with 40 electrons. Wade’s rules and Stone’s TSH theory, on the other hand, separate skeletal bonding from external lone pairs and predict a closo configuration with 38 electrons. These two models should not be taken as contradictory to each other, but rather demonstrate the flexibility of the [E9] moiety in its electron count. The preferred configuration might vary with chemical environment, and the open-shell species [E9]3−, which lies between the two “closed-shell” electronic structures with different electron counts, can also exist. In fact, it has been found that this cluster can undergo electron exchange in solvent and reach a redox equilibrium among [E9]2− and [E9]4−, as well as the intermediate state [E9]3− [56].
Apart from the different possible electron counts shown above in a nine-vertex group 14 cluster, Wade’s rules also have discrepancies in predicting the preferred geometries of 8, 11, and 14-vertex boranes and germanium clusters (Fig. 16). While borane clusters tend to adopt deltahedral structures in which most if not all of the vertices have 4–5 neighboring vertices, germanium clusters prefer more an omnicapped polyhedral structure in which all the faces of a kernel polyhedron are capped with an extra atom. Such omnicapped polyhedral clusters show bispherical feature in geometry and have been systematically studied by Johnston and Mingos based on group theory [57].

4 Endohedral Clusters
As mentioned above, group 14 clusters are mostly formed by heavy tetrel elements. Heavy element implies large atomic radius and thus the cavity inside polyhedral clusters could be large enough to encapsulate another atom, forming the so-called endohedral clusters [58,59,60,61]. Still, the electronic structure of endohedral clusters can be understood under the framework of Wade’s rules in many cases.
4.1 Wadean Endohedral Clusters
Let us take [PtPb12]2− as our first example. Compared to the bare cluster [Pb12]2−, this cluster has an additional Pt center in the cavity of the [Pb12] skeleton (Fig. 17), and is often denoted as [Pt@Pb12]2− to show the endohedral relationship. The effect of the central metal atom on the electronic structure can be revealed via an orbital interaction diagram between the endohedral center and the peripheral atoms as shown in Fig. 18.


Orbital interaction diagram for [Pt@Pb12]2−. For the [Pb12]2− fragment, only the 13 skeletal bonding orbitals are shown for clarity [62]
Let us consider the orbital interactions between the valence orbitals of Pt, namely 5d, 6s, and 6p, and the skeletal orbitals of [Pb12]2− with the Hg, Ag, and T1u irreducible representations, respectively. The consequence of the orbital interactions is that the Ag and T1u skeletal bonding orbitals are stabilized by the empty metal s and p orbitals, while the Hg orbitals interact with metal d orbitals to form bonding and anti-bonding orbitals as shown in Fig. 18. For late transition metals, the metal d orbitals are low-lying in energy and relatively contracted in size, in which case the destabilization of the skeletal Hg set will be insignificant. Hence the existence of an endohedral center often will not affect the electron count: the peripheral atoms will still follow the established rules for bare clusters while the endohedral center has a closed-shell d10 configuration. In such a scenario, if we take into account both the 4n + 2 valence electrons of the Wadean cluster and the 10 d electrons of the interstitial transition metal center, one would come up with the (4n + 12) rule for transition metal doped endohedral cluster compounds.
This observation applies to a variety of endohedral clusters, including structurally characterized clusters [M@Pb12]2− (M = Ni, Pd, Pt), [Rh@Pb12]3−, and [Ir@Sn12]3− [62,63,64,65], as well as spectroscopically detected [M@Pb12]− (M = Cu, Ag, Au), [M@Sn12]− (M = Cu, Au) and [Zn@E12] (E = Sn, Pb) in gas phase [66,67,68]. Note that these examples all have a d10 endohedral center fit into an external icosahedral skeleton following Wade’s (n + 1) rule for closo cluster.
It is not surprising that not all clusters can have a central atom. The cavity size must match the size of the endohedral atom to avoid significant destabilization from the occupied Hg orbitals. For an icosahedral cluster, the distance between central atom and a peripheral atom is not much different from that between two peripheral atoms, suggesting that the central atom and peripheral atoms should have comparable atomic radius for the endohedral cluster to be stable.
This argument is clearly evidenced by the [Co@Ge12]3− cluster [69] (Fig. 19). A simple electron counting shows that this cluster has 60 valence electrons (4 × 12 + 9 + 3), isoelectronic with the [Pt@Pb12]2− cluster. However, this Co-centered cluster does not have the expected perfect icosahedral geometry. Instead, both the crystal structure and computational optimization suggest a distorted icosahedral structure in D5d point group. Such an observation can be explained by the size mismatch between the central atom and the cage cavity. Ge has a much smaller radius than Pb, giving rise to a smaller cavity that is incompatible with the Co center. Thus the cluster has to undergo an expansion that reduces its symmetry through second-order Jahn-Teller distortion.

Endohedral clusters with fewer peripheral atoms have a similar bonding pattern as the [PtPb12]2− cluster, but with a smaller cavity. Hence the possible number of peripheral atoms is usually limited. In addition to aforementioned 12-vertex endohedral clusters, a representative 10-vertex endohedral cluster is [Ni@Pb10]2− (Fig. 20), which also features a d10 center enclosed by a Wadean cluster, albeit its isoelectronic analogs [M@Pb10]2− (M = Pd, Pt) have only been detected in gas phase [63, 70].

Nine-vertex endohedral clusters are also known, including [Cu@E9]3− (E = Sn, Pb), [Ni@Sn9]4−, [Co@E9]5− (E = Ge, Sn) and [Ru@Sn9]6− [36, 71,72,73,74,75,76,77]. Electron counting reveals that these compounds all have 50 valence electrons, featuring a d10 center enclosed by an [E9]4− cage. However, both the tricapped trigonal prismatic and monocapped square antiprismatic structures have been reported, demonstrating the previously discussed geometric flexibility of the [E9]4− cage. The electronic flexibility of [E9] cages is also reflected by the existence of the 49-electron endohedral clusters [Ni@E9]3− (E = Ge, Sn) (Fig. 21) and [Co@Sn9]4− [36, 78,79,80].

4.2 Icosahedral Clusters That Do Not Conform to Wade’s Rules
Endohedral clusters discussed in the previous subsection all followed a simple electron-counting rule: a d10 endohedral center enclosed by a Wadean shell. However, there are cases that the electron counts of the core and the shell are not so easily separated, and more detailed examination is necessary.
For example, the outer cage of the cluster [Au@Pb12]3− also has an icosahedral shape but only the symmetry of a D3d point group (Fig. 22) [81]. At first glance, the icosahedral cage resembles a typical Wadean cluster and is supposed to follow Wade’s rules. But if one tries to perform electron counting, one would find that the total number of valence electrons in this cluster is 62 (4 × 12 + 11 + 3). When compared to the typical endohedral cluster [Pt@Pb12]2− with 60 valence electrons, one might suggest a [(Au−)@(Pb122−)] assignment which formally retains the high symmetry in its electronic structure by putting the two extra valence electrons to the Au 6s orbital. However, computational study indicates that the HOMO of this cluster is completely of skeletal nature (with a2g symmetry in D3d point group and its nodal characteristics resembling the superatomic Gx3z orbital, see ref. [81] for its exact shape) and that the HOMO-LUMO gap of [Au@Pb12]3− (2.10 eV) is also close to that of the hypothetical bare cluster [Pb12]4− (1.90 eV) while the closo [Pb12]2− cluster has a much larger HOMO-LUMO gap (3.26 eV) [81]. This scenario is not completely unexpected based on the observed distorted icosahedral geometry. One should note that the LUMOs of the (hypothetical) Wadean cluster [Au@Pb12]− or [Pb12]2− are expected to be degenerate. Therefore, addition of two extra electrons would lead to the observed symmetry-breaking. These results hint that the assignment [(Au+)@(Pb12)4−] is more appropriate for describing the electronic structure of the whole cluster. In this case, the Au 6s orbital is too high to be occupied with another pair of electrons because it interacts strongly with the skeletal bonding orbital and becomes high-lying in energy.

The [Mn@Pb12]3− cluster shown in Fig. 23 provides another example of icosahedral clusters that do not exactly conform to Wade’s rules. Again, this cluster has 58 (4 × 12 + 7 + 3) valence electrons in total and is distorted in a D2h manner. If this cluster follows the bonding pattern of typical endohedral clusters and has a d10 endohedral center, the icosahedral shell would be left with 48 = 4n electrons as opposed to the 4n + 2 electron count expected for closo clusters. Alternatively, if we consider Mn as an early transition metal adopting its highest oxidation state +7 with d0 configuration, or a half-filled d shell (d5), the skeletal bonding would involve 58 = 4n + 10 or 53 = 4n + 5 electrons, respectively, none of which follows Wade’s rules.

On the other hand, if we force the icosahedral cage to have 4n + 2 electrons, fulfilling Wade’s rules, the Mn center will have an unusual d8 configuration. Computational study suggests that this cluster has a triplet ground state, consistent with the surprising d8 configuration for Mn (5α3β) [82]. However, it should be noted that spin polarization can be significant in such species. The strong Coulomb repulsion between Mn d electrons leads to contracted α d orbitals, while the β unpaired electrons would be pushed away from the metal center. Hence it has been proposed that this cluster is better described as [Mn2+@(Pb12)5−]. The Mn2+ center adopts a rather stable half-filled d5 configuration, while the outer cage has three electrons more than the one predicted by Wade’s rules, leading to an antiferromagnetic coupling between the endohedral center and peripheral cage, as well as the observed (first-order) Jahn-Teller distortion from the perfect Ih symmetry.
It should however be noted that a symmetry-lowering distortion is not always a result of incompletely occupied degenerate orbitals, as exemplified by the [Co@Ge12]3− cluster discussed in the previous subsection [69]. The distorted structure of the latter originates from the size mismatch between the endohedral center and the cage cavity, which leads to a second-order Jahn-Teller distortion. Such possibility suggests that our empirical rules for both “structure” and “bonding” could potentially be violated, complicating the analysis of cluster compounds.
4.3 Competing Isomers of Endohedral Clusters
Deviation from the empirical structural rule can occur way beyond mere symmetry-lowering Jahn-Teller distortion as introduced in the last subsection. The potential energy surface of a cluster compound can be so complicated that, under certain circumstances, the structure could undergo an extensive rearrangement into a completely different shape, challenging our established understanding.
4.3.1 10-Vertex Clusters
A representative case for such a rearrangement could be seen even in very simple endohedral 10-vertex group 14 clusters. Wade’s rules predict a 10-vertex closo [E10]2− cluster would adopt a bicapped square antiprismatic geometry in a D4d point group. According to our discussion in Sect. 4.1, we might expect that a similar situation would occur in endohedral cluster, and indeed [Ni@Pb10]2− does exhibit a typical pattern of d10 center enclosed by a cluster following Wade’s rules.
However, the [Co@Ge10]3− cluster (Fig. 24), which could be seen as an isoelectronic analog of [Ni@Pb10]2−, was found to possess a completely different shape [83]. The 10 Ge atoms now arrange in a pentagonal prism, and the cluster seems to conform to octet rule at first glance, each having an external lone pair and three bonds with neighboring atoms. Such bonding scheme would require 50 valence electrons in total (10 lone pairs and 15 Ge-Ge bonds). Electron counting, however, does not support such a bonding scheme, since its total number of valence electrons is 52 (4 × 10 + 9 + 3), different from that required by the octet configuration, not to mention it requires the central metal to be in a very high oxidation state (+7) if 50 electrons are assigned to the outer cage.

Another closely related cluster, [Fe@Ge10]3−, also has a pentagonal prismatic structure (Fig. 25), despite that it has 1 electron less than the Co-centered analog. Computational study suggests that its single electron is predominantly localized on the dz2 orbital of the Fe center, assuming the z axis is coincident with the fivefold axis [84].

What further complicates the situation is the characterization of a [Fe@Sn10]3− cluster (Fig. 26), which is isoelectronic with [Fe@Ge10]3− in composition, but has a geometry that differs from all of the other 10-vertex clusters introduced above. It has an unusual C2v geometry, which has been described as an intermediate structure between the two aforementioned D4d and D5h geometries (Fig. 27) [85].


A systematic computational study suggests that there is a universal trend in the energy differences among D5h, D4d, and C2v structures of 51-electron or 52-electron [M@E10] species (Fig. 28) [85]. With respect to the D4d structure as a reference, late transition metals always prefer the D5h structure to a greater extent, while the C2v structure in most cases lies in between. Compared to the [M@Ge10] series, the tin variants also have a higher tendency to deform toward the D4d structure, although the C2v structure can sometimes be even more stable than both, giving rise to the unexpected C2v [Fe@Sn10]3− species.

Geometric preference of [M@E10]q- clusters where each panel showing the energies of various geometric isomers relative to the D4d structure across the periodic table for different tetrel elements and electron counts [85] (Reproduced from Ref. [85] with permission from The Royal Society of Chemistry)
The competition among multiple low-lying isomers has also been found in related 10-vertex group 13 cluster compounds with 50 electrons such as [Zn@In10]8− [86] and [Ni@In10]10− [87], where the candidate structures are, respectively, bicapped square antiprism (D4d) and tetracapped trigonal prism (C3v, c.f. [B10H10]) [85].
4.3.2 12-Vertex Clusters
A somewhat similar scenario occurs in some 12-vertex group 14 clusters. Typical 12-vertex clusters are certainly of icosahedral shape with 4n + 2 = 50 valence electrons, or 60 if there is an endohedral d10 center. Variants with 62 electrons ([Au@Pb12]3−) or 58 electrons ([Mn@Pb12]3−, albeit a high spin species) are also known as discussed in Sect. 4.2, which, though distorted, still roughly retain icosahedral shapes.
Other isomers are possible, however, including hexagonal prismatic (D6h) and bicapped pentagonal prismatic (D2d) structures. One cluster with the latter geometry, [Ru@Ge12]3− (Fig. 29), has been synthesized and structurally characterized. Similar to the [Co@Ge10]3− case (Fig. 24), the Ge atoms in the outer cage of [Ru@Ge12]3− seem to retain octet configurations at first glance, each having a lone pair and forming three covalent bonds. But electron counting shows this cluster has 59 (4 × 12 + 8 + 3) valence electrons, one less than the electron count 60 (12 lone pairs and 18 Ge-Ge bonds) required by octet assignment.

A representative of hexagonal prismatic group 14 clusters would be the [CrSi12] cluster, which has 54 (4 × 12 + 6) valence electrons. This cluster has only been detected in mass spectroscopy [89, 90], but has attracted special interest because it seems that the cluster can be viewed as an analog of the coordination complex [Cr(C6H6)2] [91], if one considers all carbon atoms to be replaced with isoelectronic silicon atoms and all equatorial C-H bonds to be replaced by vertical Si-Si bonds. For this reason, this complex has once been proposed to follow the 18-electron rule for coordination complexes, since it does not follow any known electron-counting rules for clusters [92,93,94]. But detailed orbital analysis revealed that this cluster does not follow the 18-electron rule in an orbital sense, as demonstrated by the fact that the Cr dz2 orbital is found to be the LUMO of the cluster (Fig. 30) [95].

Computational studies have been performed to understand the geometric preference of various 12-vertex group 14 clusters among icosahedron (Ih), hexagonal prism (HP), hexagonal antiprism (HAP), puckered hexagonal prism (PHP), and bicapped pentagonal prism (BPP) (Fig. 31) [96]. Although a general understanding is hard to achieve from the computational results, it is yet interesting to notice that, when compared to Si counterparts, Ge-containing clusters in general favor more the icosahedral geometry as predicted by Wade’s rules. This is in accordance with the finding in the case of 10-vertex clusters in which heavier elements also prefer traditional Wadean geometries.

While in these competing cases we do not yet have a systematic rule for correlating their electron counts and geometries, we are still fortunate in the sense that these clusters still possess relatively high symmetry and nearly spherical shapes, which offers great help to our orbital analyses. Even though there are clusters with intermediate symmetries (e.g., the 10-vertex C2v structure and the 12-vertex D2d structure) that their superatomic electronic configurations might be difficult to assign, comparison against more symmetric structures still offers insight to the governing rules of structure and bonding.
5 Cluster Assemblies
Most clusters discussed up to this point are largely based on one single (quasi-)spherical unit. But these units can be further extended into more complicated clusters, sometimes in an unapparent way. Even so, a modular picture connecting the bonding of fragments to that of large systems is often a very powerful aid in chemical understanding. Hence in this section, we are going to discuss a number of more complicated cluster compounds, and decipher their bonding with the established bonding rules and models discussed in previous sections to show their transferability. This kind of understanding on how clusters can interact with each other or with other chemical moieties would then serve as an important step toward modelling of larger clusters.
5.1 Clusters with Transition Metal Vertex
One simple extension to (quasi-)spherical clusters is to take into account of their interaction with transition metal centers. Apart from endohedral clusters discussed in the previous section, there are also many metal doped group 14 clusters in which the metal center lies on the same surface as the cluster’s (quasi-) spherical skeleton, thus itself could also be considered as one vertex on the polyhedron.
In Mingos’ PSEPT, it has already been stated how we could view an electron-deficient cluster that combines main group and transition metal vertices. As an illustrative example, let us first take a look at the clusters [E9ZnPh]3− (E = Si, Ge, Sn or Pb) (Fig. 32) [97,98,99]. These clusters adopt a perfect bicapped square antiprismatic geometry, with the transition metal moiety being one of the vertices. The cluster can clearly be understood by Wade’s rules, by considering the fact that a [ZnPh]− moiety has 4 valence electrons (if Zn is considered as a main group, or 14 as a transition metal; either way leads to the same analysis), isoelectronic with a tetrel element, making the whole cluster isoelectronic with a closo [E10]2− cluster with 42 valence electrons.

The same analysis would apply to the cluster [Ge9R3EtPd(PPh3)] (Fig. 33) [100], which has 52 valence electrons (4 × 9 + 1 × 3 + 1 + 10 + 2) and is also isoelectronic with [E10]2− clusters noting that the additional 10 electrons arise from the d10 shell of the transition metal Pd center. However, this cluster differs from the [E9ZnPh]3− cluster in the sense that the transition metal center occupies the antiprismatic vertex, instead of a capping position as in [E9ZnPh]3−.

The reason that the transition metal atoms Zn and Pd occupy different sites in the above two cases is actually not yet well understood. In fact, the preferred occupation site of transition metal can even be altered by the ligand exchange on the metal center in the case of [Ge9R3RhL2] [101]. It has been proposed that different structural isomers of a bicapped square antiprismatic cluster can rapidly interconvert to each other under certain circumstances [102], based on the existence of both isomers of [E9M(CO)3]4− (E = Sn, Pb, M = Cr, Mo, W) species [102,103,104,105]. Early studies based on perturbative approach suggest that site preference might be related to factors like electronegativities of the heteroatom, interaction strengths, and connectivities of different sites [106].
Apart from variants of the bicapped square antiprismatic structure, there exists another competing structure for [E9M]-based 10-vertex clusters. The cluster [Ge9(SiiBu3)3CuNHCDipp] (Fig. 34) adopts a tetracapped trigonal prismatic structure that may seem to resemble the hyper-closo [B10H10]. However, its valence electron count sums up to 52 (4 × 9 + 3 + 11 + 2), which is isoelectronic with the closo cluster [B10H10]2− but not the hyper-closo cluster [B10H10]. To understand this anomaly, one should notice that the [Ge9(SiiBu3)3] moiety itself has a flexible electron count for [E9] clusters and may exist in tricapped trigonal prismatic structure as discussed in Sect. 3.2. Hence the copper doped cluster could be understood simply as a coordination complex in which the [Ge9R3]− moiety acts as a ligand, donating to the [CuL]+ fragment.

Such a fragment-based view of transition metal doped clusters is convenient in explaining the various geometries adopted by the [E9] moiety when interacting with a transition metal fragment. Just as what we have elaborated in Sect. 3.2, [E9] fragment is very flexible in its geometry and electron count, which could explain its versatility in interacting with transition metal centers in different clusters. The [E9M]-based 10-vertex clusters could be considered as a closo bicapped square antiprismatic cluster in which the transition metal center interacts with [E9] in η4 or η5 modes. Alternatively, it could adopt a structure that can be described as a transition metal center interacting with a tricapped trigonal prismatic [E9] in an η3 mode. In addition, as each vertex of the [E9]4− fragment has an out-pointing lone pair, it is also possible for the fragment to coordinate to the transition metal center in an η1 manner via one of its lone pairs, for example, in the [Ge9(Hyp)3FeCp(CO)2] cluster (Fig. 35) [108].

To better understand the role of the [E9]4− cage as a ligand, let us take a look at the [(η1-Ge9R3)Cr(CO)5]− and [(η5-Ge9R3)Cr(CO)3]− compounds [109, 110] (Fig. 36). In the former, the Cr center is coordinated by one lone pair of the [Ge9R3]− cage as well as 5 carbonyl ligands, hence conforming to the 18-electron rule. Similarly, in the latter species the Cr center also conforms to the 18-electron rule because the [η5-Ge9R3]− ligand donates 6 electrons to the metal center (in analogy with a Cp− ligand in coordination chemistry). Similar analysis applies to all of the mentioned clusters in this section, where the doping transition metal atoms all conform to the 18-electron rule and follow our traditional understanding in coordination chemistry, despite the presence of cluster moiety as a mono- or multi-dentate “ligand”.

As discussed before, deviations from the (n + 1) rule are not uncommon. The cluster [Ni@Ge9Ni(CO)]2− (Fig. 37) is another example of this, which can actually be viewed as an endohedral Ni center inside a [Ge9Ni(CO)]2− cage. If we make our first guess by assigning the endohedral Ni center as Ni(0) with a d10 configuration, the outer cage would be left with 50 valence electrons (4 × 9 + 10 + 2 + 2), different from what we have introduced above. On the other hand, one should note that although this cluster also adopts a tetracapped trigonal prismatic structure that seems to resemble the [Ge9(SiiBu3)3CuNHCDipp] structure shown in Fig. 34, it is actually isoelectronic with the hyper-closo cluster [B10H10] in Fig. 5, which is different from the previously discussed examples in electron count.

To understand this, we note that there are two different ways to describe a tricapped trigonal prismatic [E9] cage. If one takes it as a whole and considers it as a nine-vertex closo cluster, Wade’s rules predict it to be [E9]2−, although we have discussed in Sect. 3.2 that its low-lying LUMO makes it possible to adopt [E9]4− configuration within D3h point group under certain circumstances. On the other hand, one can view it as a “hyper-nido” structure derived from a hypothetical hyper-closo [E10] cluster (analogous with the hyper-closo [B10H10]) by removing its top vertex on its threefold axis. Compared to the ordinary closo [E9]2− cluster, this hypothetical “hyper-nido” cluster has one of its trigonal bases slightly open when interacting with the 10th vertex, but its electronic structure is essentially the same as the ordinary one except that its frontier orbitals could be rearranged a bit due to slight geometric distortion.
Similarly, the [Ni@Ge9Ni(CO)]2− cluster can also be understood in two different ways. Taking the cluster as a whole would result in a 50-electron hyper-closo 10-vertex cluster, isoelectronic with the borane cluster [B10H10], while considering it as a coordination complex would lead to a closo nine-vertex [Ni@Ge9]2− cluster conforming to Wade’s rules coordinated to the transition metal moiety [Ni(CO)].
Relationship among different geometries and electron counts could be better understood by taking a closer look at their frontier orbitals. The right-hand side of Fig. 38 shows the arrangement of the frontier orbitals of nido [E9]4− in the monocapped square antiprism (MSA) geometry. As discussed in Sect. 3.2, MSA can also rearrange into tricapped trigonal prism (TTP) without significant energy penalty. One can see that at TTP geometry, the HOMO of [E9]4− is singly degenerate, consistent with our prediction of the closo TTP [E9]2− (with the topmost electron pair removed) based on Wade’s rules. When it comes to the “hyper-nido” structure which has a triangular base face opened up compared to the ordinary TTP structure, such a distortion will lead to a rearrangement of the LUMOs but the occupied orbitals still show clear correspondence in terms of symmetry as shown in the leftmost column of Fig. 38. It turns out that such “open-TTP” structure would serve as an important fragment for understanding many tetrel clusters, which will be further discussed in later analyses.

Frontier molecular orbital diagram of the [Ge9] cluster with different charges at different geometries
While the above two approaches to understand [Ge9Ni2L]2− make no essential difference in its bonding, the fragment-based approach, which considers a cluster fragment as a ligand, can be more flexible when extended to clusters that do not follow Wade’s rules. Because of the electronic flexibility of the [E9] cage, not only the Wadean 50-electron η3-coordinated clusters and the 52-electron η4-coordinated clusters can be formed, but we are also able to obtain 52-electron η3-coordinated clusters such as the aforementioned [(η3-Ge9R3)CuL] (shown in Fig. 34), or even 50-electron η4-coordinated clusters such as [Co@Ge9NiL]3− (E = Ge, L = CO; E = Sn, L = C2H4) [80]. Open-shell species can also exist, including [{η4-Ge9}Ni(CO)]3− (Fig. 39), [{η4-Ge9}PdPPh3]3−, [{η1-Ge9(Si(SiMe3)3)3}TiCp2(NCMe)], [{η3-Ge9(Hyp)3}Ni(dppe)], [{η3-Ni@Ge9}Ni(en)]3−, and [{η4-Ni@Sn9}Ni(CO)]3− [79, 111,112,113,114]. However, given that both an [E9] cluster and a transition metal can have the potential to form open-shell species, whether the unpaired electron(s), when present, actually locate on the cage or on the transition metal center has to be determined on a case-by-case basis, and no general rule of thumb is currently available.

Finally, a rather surprising case is associated with the cluster [Pd@Sn9SnCy3]3− (Fig. 40) in which there is an SnCy3 group attached to a [Pd@Sn9] cluster in an unprecedented η2 manner [115]. Despite the substituent SnCy3 is not a transition metal here, the cluster can still be understood as a coordination adduct of [Pd@Sn9]4− and [SnCy3]+. While it has been shown that the stannyl group, which is isolobal to H, Me, or AuL, can serve as a flexible bridging group bonded to the cluster in multiple ways (Fig. 41) [33], the preference among different coordination modes under different situations is still not clear.


To summarize, a list of transition metal doped group 14 clusters is given in Table 1, in which the [E9] moiety may adopt η3, η4, or η5 coordination modes. The structural flexibility of the [E9]-based clusters results in a very rich chemistry of transition metal doped group 14 clusters. One will see in later parts that the fragment-based understanding of cluster compounds introduced in this section can actually serve as the basis for a more general bonding picture to understand the electronic structure of more complicated clusters.
5.2 Metal-Bridged Cluster Assemblies
With the interaction between transition metal and group 14 clusters being well elucidated above, we can proceed to discuss clusters having metal center(s) as bridges which link various (quasi-)spherical clusters together. Specifically, the previously introduced nine-vertex group 14 clusters are important building blocks in large cluster compounds, and multiple [E9] units can be connected in a variety of ways.
In the class of [MGe18R6]q- (q = 0, M = Zn, Cd, Hg, Mn; q = 1, M = Cu, Ag, Au; q = 2, M = Pd) clusters, there are two [Ge9R3]− cages bridged by a transition metal center (Fig. 42) [114, 116, 124,125,126,127,128,129]. Note that all metal centers adopt d10 configurations (except Mn which adopts a half-filled d5 configuration) and have 6 neighboring Ge atoms arranged in octahedral geometry. The Ge-Ge distances in the coordinating triangular base face (2.87 Å in the case of Pd) are significantly longer than those in the uncoordinated triangular faces (2.63 Å in the case of Pd), suggesting a D3h-to-C3v distortion within each [E9R3]− unit in the overall cluster, implying a combination of the donation from the HOMOs of the [Ge9R3]− unit to the metal center and the back-donation from the metal center to the LUMOs of the cluster unit. The same scenario also holds for other metal-bridged group 14 clusters including but not limited to [RAu ← Sn9R3 → Au ← Sn9R3 → AuR]−, [Ge9R3 → Cu ← Ge9R3 → CuPPh3] and [RZn ← Ge9R3 → Pt ← Ge9R3 → ZnR] (Fig. 42) [124, 130, 131].

Multiple bare [E9] units can also be bridged via different coordination modes. In the cluster [Ge9 → Hg ← Ge9 → Hg ← Ge9 → Hg ← Ge9]10−, for example, it features a union of multiple Hg2+ and [Ge9]4− moieties in which η1 and η2 coordination modes exist at the same time [132] (Fig. 43). Such a coordination chain can grow in length and result in a polymer as in the [K2HgGe9] compound, though the coordination in the polymeric structure consists of both η1 and η3 modes instead [133]. Besides, other coordination modes can also coexist, such as [(η4-Ge9)-Cu-(η1-Ge9)]7−, [(η4-Sn9)-Ag-(η1-Sn9)]7−, [(η4-Ge9)-Zn-(η3-Ge9)]6−, [(η2-Sn9)-Hg-(η2-Sn9)]6−, [(η4-Ge9)-Zn-{μ2(η4:η1-Ge9)}-Zn-(η4-Ge9)]8−, and polymeric ∞[Zn{μ2(η4:η1-Ge9)}]2− [121, 122, 134,135,136]. The main group metal-bridged cluster [(Ni@Sn9)In(Ni@Sn9)]5− exhibits a similar structural isomerism among η4:η4, η4:η3 and η3:η3 isomers [137].

The cluster [Au3Ge18]5− (Fig. 44) provides another possibility of bridging two [E9] moieties with an [Au3] triangle [138]. While each Au center has a formal charge of +1 and seems to be repulsive with each other, we would also expect the aurophilic interaction commonly discovered in multinuclear gold(I) compounds can hold multiple Au(I) centers together and in turn bridge the two [E9] cages [139]. Additional ligands can also be present at the bridging moiety, for example the [{Ni(CO)2}2(Si9)2]8− cluster is an assembly of two [Si9]4− moieties bridged by two separated Ni(CO)2 fragments (Fig. 44) [117, 119, 140]. Each Ni center is Ni(0) with four ligands arranged in a tetrahedral geometry, perfectly satisfying its own coordination chemistry.

Another example, the [Ni3Ge18]4− cluster, may seem to be three endohedral Ni centers in a [Ge18] cage [78] (Fig. 45). However, the 18 Ge atoms are actually separated into two groups of atoms, and hence this cluster is better described as two [Ni@Ge9]2− cages linked by a Ni(0) center [141].

5.3 Directly Bonded Cluster Assemblies
In addition, multiple [E9] units can also be explicitly bonded together via covalent bonds or other direct interactions. Recall that in the [Ge9R2]2− cluster discussed in Sect. 3.1, the [Ge9] moiety is directly bonded with two substituents with localized covalent bonds. In addition to having a substituent in the traditional sense, a Ge vertex could also be bonded to another [Ge9] moiety. This is the case in the cluster [R-Ge9-Ge9-R]4− (R = alkyls, SbPh2 or ER’3 where E = Ge, Sn and R’ = Me, Ph) (Fig. 46), in which each [Ge9] cage has two covalent bonds, one bonded with substituent and the other bonded with another cage [56, 142, 143]. The skeletal bonding of each cage still resembles that of the [Ge9R2]2− cluster, hence it is totally expected that each cage in [R-Ge9-Ge9-R]4− adopts a nido MSA geometry. The same analysis also applies to the [Ge18]6− (Fig. 46) with two [Ge9] cages bonded via a single bond [144, 145]. Other examples include [Ag(Sn9-Sn9)]5− (Fig. 46) and [InPh(Ge9-Ge9)]4−, each with both a Ge-Ge σ-bond and a metal ion that bridges two Ge9 cages [121, 146, 147]. The cluster polymer ∞[Ge9]2− shows extended inter-cage bonding (Fig. 47), illustrating how these interactions could be utilized in much larger structures [148,149,150].


The [E9] building blocks can also be linked in a more delocalized manner. The clusters [Pd3Ge18R6]2− (R = SniPr3, SiiPr3) provide specific examples for such scenario [151, 152]. Each cluster has two [Ge9R3] cages bridged by a Pd3 triangle in between and may be described as a pair of icosahedral cages linked by a common Pd3 triangular face (Fig. 48). Given our discussion on [R6Ge18Pd]2− in Sect. 5.2, one might follow our previous analysis and assign each [Ge9R3] cage to be [Ge9R3]− and each Pd to be Pd(0). However, it is interesting to notice that the [Ge9R3] cages are different in these two clusters. Compared to [PdGe18R6]2−, the cluster [Pd3Ge18R6]2− has two [Ge9R3] cages each with one base face widely open, a structure that we have often seen in the previously discussed coordinated clusters. As mentioned in Sect. 5.1, a [Ge9R3] cage in an open-TTP conformation has its frontier orbitals reordered, hence [Ge9R3]− is expected to have degenerate HOMO and LUMO in the open-TTP geometry if electrons are forced to be paired up. In this sense, the [Ge9R3]− cage is better described as a triplet fragment in this scenario. Formally, these two triplet cages form a pair of delocalized π-bonds (Fig. 48). Although the two cages lie across the Pd3 triangle and such a delocalized bonding seems remote and unstable, the cage SOMOs are actually quite localized on the three Ge atoms on its open face [151]. This also explains the eclipsed conformation of the [Pd3Ge18R6]2− clusters which bring the two open faces close to each other to stabilize the “remote π-bonds” [151].

Such an atypical delocalized bonding is not unique to [Pd3Ge18R6]2−. The clusters [Pd2E18]4− (E = Ge, Sn) also have two [E9] cages close to each other [154, 155] (Fig. 49). But different from the previous case, the two cages now adopt a staggered conformation. Compared to the Pd3 triangle in [Pd3Ge18R6]2−, the Pd-Pd distance in [Pd2E18]4− is even longer, hence there should not be any Pd-Pd bond. Therefore, each Pd center is again expected to be Pd(0), and the [E9] cage is assigned to be [E9]2−, which is expected to be at singlet state at an open-TTP structure. The interaction between the two [E9] cages is thus different from the case in [Pd3Ge18R6]2−. Specifically, their frontier orbital interactions give rise to three pairs of mutual delocalization, which hold the two cages together although their formal bond order is zero [31, 141] (Fig. 49).

In fact, when we cross-compare different cases, we can understand the role of an [E9] unit in a unified framework. If we examine the “principal interacting orbitals” (PIOs) [31, 32] of the formal [E9] units (together with their substituents or endohedral center) in [Ge9Ni2L]2− (Fig. 37), [Pd2E18]4− (Fig. 49), and [Pd3Ge18R6]2− (Fig. 48), we would find very similar PIOs among different cases (as illustrated by the resemblance of π-type PIOs in Figs. 48 and 49), giving rise to a more generalized picture of the “frontier interactions” of the [E9] unit [31, 153].
Finally, it should be noted that not all [E9]-based clusters have been well studied. The clusters [Ge27]6− and [Ge36]8− are two of such examples (Fig. 50). Our knowledge introduced in this chapter can still be used to understand their bonding patterns despite the absence of computational results. These clusters are apparently made up of three and four [Ge9]2− units, respectively. However, adjacent [Ge9] units are doubly connected, with two close Ge-Ge contacts in the range of 2.546 ~ 2.752 Å. The Ge-Ge distances are longer than typical Ge-Ge single bond lengths of approximately 2.48 Å, hence distinguishing these cases from the aforementioned [Ge18]6− cluster (Fig. 46), which has two nido-Ge93− moieties linked by a Ge-Ge single bond. Even if we ignore the atypical Ge-Ge bond length and assume that there are two localized bonds between adjacent [Ge9] units, the middle [Ge9]2− unit will formally be isoelectronic with [Ge9R4]2−, an arachno cluster according to the total number of valence electrons and Wade’s rules. This is different from the familiar skeletal bonding patterns for nine-vertex group 14 clusters. We thus expect these [Ge9] units are not connected by localized single bonds but are bonded in a rather delocalized manner.

Noting that the structure of each [Ge9] unit in Fig. 50 is close to the MSA geometry, with the open square face vertices being connected with adjacent units, we may speculate their electronic structures based on the established models introduced in Sect. 3.2 and 5.1, in which the degenerate frontier orbitals are localized on the square face. As a starting point, the (almost) degenerate frontier orbitals are only partially filled for each [Ge9]2− unit. Then the linear combination of these two frontier orbitals will span two bands, with their partial occupation finally giving rise to weakly bonded [Ge9]2− units with each individual unit still resembling typical nine-vertex group 14 clusters. Such a delocalized bonding picture is, however, much harder to illustrate through traditional bonding analysis methods, because most of them are based on identifying localized bonds among a couple of adjacent atoms. But with the recent development of computational tools, it is still conceivable that a general bonding picture for these extended clusters could be achieved in the near future.
6 Clusters Beyond Wade’s Rules
In the previous sections, we have introduced Wade’s rules and their application on a wide variety of group 14 clusters. We have shown that many group 14 clusters can be interpreted using Wade’s rules if more specific orbital interactions are introduced. There are still some clusters which require rather different approaches. We will briefly examine some representative examples in this section.
Although in a large part of this chapter we have greatly relied on the “isolobal relationship” among [BH], [E], and [ER]+, we have discussed in Sect. 3 that they do have some differences. In particular, we have briefly mentioned in Sect. 3.1 the role of substituents in stabilizing electron-deficient “carbon cluster.” We have also discussed the relationship of tetrel clusters to Jellium model due to the relatively active lone pair in Sect. 3.2. In fact, the availability of skeletal electron pairs and the total electron count can shift the governing bonding models from one to the other.
Recall the two examples we mentioned in Sect. 2: [B4Cl4] and [C4H4], we considered [B4Cl4] to follow a variant of Wade’s rules, and described C4H4 simply with localized bonding, but what they actually differ is the total electron count.
In fact, similar case also occurs in group 15 clusters where they are often described by a dedicated electron-counting rule for these octet-conforming cases: the 5n rule. Because in the case of pnictogen clusters, this often means each vertex would have 3 bonds, and clusters of this kind are therefore often also called “3-connected clusters” [4].
Tetrel elements have 4 valence electrons, hence in most cases electroneutrality principle forces group 14 clusters to fall into the class of Wadean clusters as opposed to 3-connected clusters. However, more electrons could be present in skeletal bonding if the overall cluster is particularly stable. A typical example, albeit complicated, is the Matryoshka cluster [Sn@Cu12@Sn20]12− (Fig. 51), which has a highly symmetric three-layer geometry [158]. Despite its compositional complexity, its bonding can be simply deciphered via a divide-and-conquer approach like peeling an onion: the innermost tin atom fulfills the octet rule, the sandwiched layer simply consists of 12 Cu centers all with a closed-shell d10 configuration, and the outermost layer is a 3-connected cluster in which each tin atom holds an external lone pair and forms three covalent bonds with neighboring vertices, also fulfilling the octet rule. Such assignment indicates that this cluster can be formally written as [(Sn4−)@(Cu+)12@(Sn−)20], where each individual layer is isoelectronic with that in another famous transition metal doped pnictogen Matryoshka cluster [(As3−)@Ni12@As20] [159, 160].

Crystal structure of [Sn@Cu12@Sn20]12− and its bonding Scheme [159] – Reproduced by permission of The Royal Society of Chemistry
Generally, group 14 clusters in most cases cannot sustain such a high negative charge of an electron-precise cluster and require substituents to balance and stabilize the charge. For example, the substituent-stabilized clusters [EnRm](n-m)- (n = 4, E = Si, Ge; n = 8, E = Sn) follow the desired valence electron count for 3-connected clusters (not counting substituents in the connectivity) [161,162,163,164,165,166] (Fig. 52a). Hence these clusters are isoelectronic to corresponding pnictogen clusters [Pnn] and polyhedranes [CnHn].

In many cases, substituent-decorated group 14 clusters fall into the intermediate regime between Wadean and 3-connected clusters, with intermediate valence electron counts between 4n + 2 for Wadean closo clusters and 5n for 3-connected clusters. Taking 8-vertex clusters as an example, an [E8Rx] cluster has 32 + x valence electrons. The case with x = 2 corresponds to a Wadean closo cluster while the case with x = 8 corresponds to a 3-connected cluster. The cases with x = 4 or x = 6 are predicted to adopt nido or arachno structures, respectively, according to Wade’s rules, which are deltahedral clusters with one or two vertices removed.
On the other hand, experimentally synthesized and characterized [E8R4] and [E8R6] molecules are seen to adopt (distorted) cubic structures, including [Ge8{N(SiMe3)2}6] (Fig. 52b), [Ge8{C6H3(OtBu)2}6], [Sn8(2,6-Mes2C6H3)4], and [Sn8(SitBu3)6] [164, 167, 169, 170]. In terms of electron count, more edges could be added to the cubic structure to increase electron-sharing and formally make all vertices tetravalent, although the undecorated vertices may not maintain the typical tetrahedral tetravalent geometry in main group chemistry.
At the same time, their isoelectronic silicon analog, [Si8(SitBu3)6] (Fig. 52c), has a completely different structure [168]. This cluster has a Si2 moiety sandwiched by two Si3R3 moieties, in which all Si atoms are tetravalent and fulfilling octet rule, showing a rather localized bonding pattern. These compounds clearly show the diversity in structure and bonding of cluster compounds because geometrically similar clusters may have different electron counts while isoelectronic clusters may adopt diverse geometries. It remains a question why these clusters do not follow Wade’s rules but instead adopt a 3-connected structure. We anticipate more clusters in this transition regime synthesized in the future could help us understand their preferences.
When it comes to 10-vertex substituted clusters, the diverse scenario is somewhat similar. The cluster [Ge10{Si(SiMe3)2}(Hyp)4Me]− (Fig. 53) has a [Ge10] skeleton with 7 attached substituents and hence can be formally described as a [Ge10R7]− if we consider that the substituents are singly bonded to the skeletal atoms (note that SiR2 is counted as two separate substituents) [171]. Thus, this cluster has 48 valence electrons (4 × 10 + 7 + 1), two less than that required by a 3-connected bonding scheme, although each vertex has three closest neighboring Ge atoms in around 2.5 Å (except that one of the edges, Ge9-Ge10, has a bond length of 2.74 Å). Based on the electron count, one might assign an additional Ge1-Ge6 bond, despite its relatively long bond length of 2.96 Å. However, this assignment will again lead to non-tetrahedral Ge atoms which are not typical for tetravalent main group elements.

Nevertheless, the locally inverted geometries might not be evident enough to eliminate the possibility of an additional bond between unsubstituted tetrel atoms in above clusters. It is well established that an inverted bond can exist in the [1.1.1]propellane molecule (Fig. 54) with charge-shift bonding character [172,173,174]. We would not judge the charge-shift bonding character in the aforementioned cluster compounds here, but would still like to point out that the inverted nature of local center does not completely exclude the possibility of tetravalency.

The cluster [Sn10R8] (Fig. 55) is an isoelectronic analog with the previously discussed cluster shown in Fig. 53, which again adopts a different geometry [175]. There are 4 apparent trivalent Sn atoms, in addition to 6 tetravalent Sn atoms. Since this cluster is only short by two electrons to fulfill the electron count for a 3-connected cluster, the central four Sn atoms must share three lone pairs. In other words, this cluster exhibits a 4-center-6-electron delocalized bonding pattern in the middle, even though the overall structure largely resembles a 3-connected geometry.

Crystal structure of [Sn10R8] [175] – Published by The Royal Society of Chemistry, used under CC BY-NC 3.0
We may find other examples of intermediate 10-vertex clusters with even fewer valence electrons. The cluster [Ge10(SitBu3)6I]+ (Fig. 56) has 46 valence electrons (4 × 10 + 6 + 1–1), four electrons less than that required by 5n rules, despite its apparent 3-connected structure with 15 close Ge-Ge contacts and 7 external bonds [176]. On the other hand, if one deducts the 22 localized bonding electron pairs from overall valence electrons, one would find that there are two remaining electrons while three Ge atoms are not tetravalent yet. This will lead us to assign a 3-center-2-electron bond among the three trivalent Ge atoms, with each Ge atom contributing its external orbital that is normally associated with a lone pair in 3-connected cluster but is now sharing a common electron pair with the other two Ge atoms.

The above 3-center-2-electron bond is not unique in this single cluster. In fact, 2-center-2-electron localized bond, 3-center-2-electron semi-localized bond, and multi-centered bonding (following Wade’s rules) can be present in a single cluster at the same time. In the very complicated cluster [Au3Ge45]9− (Fig. 57), there are four Ge9 moieties in typical closo or nido geometries, while the remaining 9 Ge atoms appear to arrange in a 3-connected manner [177]. Further inspection shows that the central [Ge9] skeleton has a similar structure with the [Ge9] skeleton in [Ge10(SitBu3)6I]+ discussed above, while the [Ge9] unit IV plays the role of iodine substituted Ge atom, and the [Ge9] units I, II, and III play the role of the silyl substituents [177]. Eventually, there is a 3-center-2-electron bond among Ge54, Ge55, and Ge56, in addition to the delocalized bonding within the four [Ge9] units and the localized 2-center-2-electron bonds within the central [Ge9] skeleton.

In the above seemingly 3-connected clusters, there are vertices that clearly conform to the octet rule and vertices that do not, but this is not always the case. The cluster [Ge14R5]3− (R = Ge(SiMe3)3) (Fig. 58a) has a 3-connected skeletal geometry with 5 out of 14 Ge atoms decorated by substituents [178]. However, this cluster only has 64 (4 × 14 + 5 + 3) valence electrons, much smaller than 5n = 70 as required by a typical 3-connected cluster predicted by PSEPT. Its electron count does not follow either 4n + 2 or 5n rule and the cluster shows non-deltahedral geometry, both suggesting the inapplicability of Wade’s rules on this cluster.

Crystal structure of [Ge14{Ge(SiMe3)3}5]3−, [Eu@Sn6Bi8]4− and [Pd3Sn8Bi6]4− and comparison among their geometries [178,179,180] (Reprinted with permission from [178], Copyright (2008) The Royal Society of Chemistry; from [179], Copyright (2010) John Wiley and Sons; and from [180], Copyright (2011) American Chemical Society)
A closely related cluster is the intermetalloid cluster [Pd3Sn8Bi6]4− (Fig. 58c), in which tetrel and pnictogen atoms are mixed in the 14-vertex peripheral cage [180]. The geometry of this cluster is very similar to that of [Ge14R5]3−, except that there are three endohedral Pd atoms arranged in a triangle. If we follow the usual rule-of-thumb for endohedral centers and assign each Pd to be Pd0 (d10 configuration), then the cage would have 66 valence electrons (4 × 8 + 5 × 6 + 4), still 4 electrons short when compared to that for a regular 3-connected cluster. However, 3-connected clusters with similar composition do exist, as demonstrated by the clusters [Nb@Ge8As6]3− and [Eu@Sn6Bi8]4− (Fig. 58b) [179, 181], both of which have similar geometries with [Ge14R5]3− and [Pd3Sn8Bi6]4−. Nb is an early transition metal with small electronegativity, hence it is hard to imagine Nb to adopt a d10 configuration in [Nb@Ge8As6]3−. Instead, if we consider Nb to be Nb5+, then the outer cage will become [Ge8As6]8−, isoelectronic with [As14] having 70 valence electrons, thus conforming to the electron count of a 3-connected cluster. The f-block element doped endohedral cluster [Eu@Sn6Bi8]4− can be deciphered in a similar way, if we notice that the outer cage should be [Sn6Bi8]6− in order to be 3-connected, hence the Eu center is Eu2+ with a half-filled f7 configuration.
These examples again demonstrate the great flexibility in electron count for group 14 clusters. Despite such flexibility, one can also find patterns among them. From a geometric point of view, the clusters [Nb@Ge8As6]3− and [Eu@Sn6Bi8]4− are fairly spherical, while [Ge14R5]3− and [Pd3Sn8Bi6]4− are oblate (Fig. 58d). Clearly there is a relationship between valence electron count and sphericity, implying that the additional 4/6 electrons would have vertical anti-bonding character.
While the previous examples with intermediate electron counts feature 3-connected geometries to some extent, there are also a number of other examples with intermediate electron counts closely resembling Wadean clusters. For example, the clusters Sn10(Hyp)6 (Fig. 59), [Sn10(Hyp)5]−, [Sn10(Hyp)4]2−, and [Pb10(Hyp)6] all have 46 = 4n + 6 valence electrons, but adopt arachno geometries that do follow the prediction of Wade’s rules [182,183,184,185,186].

Similarly, the 12-vertex [Pb12(Hyp)6] cluster (Fig. 59) has an approximately icosahedral skeleton, with six out of twelve Pb atoms decorated with substituents [183]. This cluster has a valence electron count of 54 (4 × 12 + 6), 4 electrons more than that required by a Wadean cluster (50) but still less than that in 5n rule. While skeletal bonding has been partially broken because of the additional electrons, leading to a kernel in (approximate) D3d symmetry, the unexpected hypervalent interaction between silyl groups with another proximate Pb atom shortens the bridge edge, further reducing the molecular symmetry to a pseudo-C2h point group. Although it has been proposed based on electron counting that the overall cluster should be described as an arachno-type cluster as predicted by Wade’s rules [183], the narrow spread of the bond lengths of the icosahedral edges hints a different electronic structure from its isoelectronic arachno borane cluster [B12H18], which has a macropolyhedral geometry [187].
From these examples, we can see that group 14 clusters made up of heavier tetrel elements may not necessarily share the same geometry as those made up of lighter tetrel elements with the same number of electrons. On the other hand, without going into detailed comparison among individual cases, we can still see a similar trend as previously discussed in Sect. 4.3, that heavier tetrel elements seem to have a greater tendency to form Wadean clusters while lighter tetrel elements are more likely to adopt 3-connected structures. The failure of Wade’s rules on the above examples as well as these subtle features again reveals the difference between tetrel atoms and [BH] units and even between lighter and heavier tetrel atoms, and demonstrates the incompleteness of only applying Wade’s rules to understand the electronic structures of group 14 clusters. Nevertheless, Wade’s rules still serve as a good reference and a guiding principle when we first face the constantly synthesized novel clusters, since detailed analysis on their electronic structures is often given on top of Wade’s bonding description of cluster compounds.
7 Perspective
Cluster chemistry always challenges our perception and interpretation of their structures using the current bonding models, manifesting why this field is particularly fascinating. Although Wade’s rules cannot fully explain the electronic structures of all group 14 clusters, it has become a norm to treat them as a starting point when a new cluster is synthesized. The role of Wade’s rules in cluster chemistry is similar to the role of the octet rule in classical inorganic chemistry: not all atoms conform to the octet rule in all molecules, but it is always a good starting point when understanding the bonding of a molecule. We often draw a Lewis structure for a molecule to see whether a satisfactory bonding picture can be achieved. If not, extra attention will be paid to explore the special bonding mode. Although individual atoms in cluster compounds do not necessarily follow the octet rule, we can similarly draw a “Lewis-like structure” in a modular way for many cluster compounds, on the basis of Wade’s rules and polyhedral skeletal electron pair theory [188]. In such a modular representation, we can identify the Wadean units in a large cluster and treat them as “superatoms” that can form covalent and dative bonds with other moieties (see Sects. 5.2 and 5.3). Such a modular bonding picture allows us to understand the structure and bonding for each constituent unit in a cluster. In this way, cluster compounds are nothing more than an aggregation of multiple units, each of which falls into the scope of an existing bonding model.
Still, there are a couple of cluster compounds out there that do not follow Wade’s rules or their extensions, nor conform to the octet rule as in 3-connected clusters, which encourages us to explore their bonding patterns more carefully and thoroughly, and to develop new bonding models that not only explain the electronic structure of a single cluster, but also have the potential to be transferred to a number of other clusters.
So far, a lot of efforts have been paid to formulate a better understanding for more clusters, and many models arise. For example, aromaticity is a longstanding concept that has been extensively used across various contexts when one intends to describe certain unexpected stability of a compound, and has also been widely applied in group 14 clusters. The Jellium model, which originated from alkali metal clusters and has been widely used for gold clusters, can also be used to understand the electronic structures of some group 14 clusters, as introduced in Sect. 3.2.
While both Wade’s rules and Jellium model were developed to understand electronic structure for species with a near-spherical shape, bonding rules proposed for vertex capping and cluster fusion described in the polyhedral skeletal electron pair theory are important steps toward complicated non-spherical clusters. Following this spirit, new bonding models based on the nine-vertex cluster units have been developed, inspired by their frequent presence in cluster assemblies, to understand a number of group 14 cluster assemblies as introduced in Sect. 5.
Nevertheless, cluster compounds covered in this chapter are still far from comprehensive. There are a lot of clusters for which a general bonding model has not emerged. While we are halfway toward a comprehensive understanding of cluster chemistry, the bonding models introduced in this chapter are powerful tools that we are armed with to explore this terra incognita ahead of us.
In particular, the [E10]-fused clusters [Co2Ge16]4− and [Rh3Sn24]5− [189, 190], though very different in composition and shape, feature the geometries in which identical cluster fragments are fused, which may hint us to adopt a fragment-based approach to understand their relationship, similar to what we have presented in Sect. 5.
The bonding model we discussed for the gold-bridged group 14 cluster [Au3Ge18], which is a coordination bridged cluster with no explicit Au-Au bonding, could potentially be used to understand the gold-bridged clusters [Au8Pb33]6− and [Au12Pb44]8− [191] where Au-Au bonding is apparent and a joint model that bridges group 14 clusters and group 11 clusters becomes obligatory, although the existence of the icosahedral cluster [Au@Pb12]3−, which does not conform to Wade’s rules, hints us there might be a more complicated chemistry ahead.
Nevertheless, new clusters are constantly synthesized, challenging our established chemistry and urging us to develop new bonding models. After all, this is why Wade’s rules were proposed and all the succeeding models were developed. We anticipate novel clusters and innovative models will persistently arise in the future, presenting us with new chemistry.
Abbreviations
- AdNDP:
-
Adaptive natural density partitioning
- HOMO:
-
Highest occupied molecular orbital
- Hyp:
-
Hypersilyl, Si(SiMe3)3
- LUMO:
-
Lowest unoccupied molecular orbital
- MSA:
-
Monocapped square antiprism
- nc-2e:
-
n-center-2-electron
- PIO:
-
Principal interacting orbital
- PSEPT:
-
Polyhedral skeletal electron pair theory
- SEP:
-
Skeletal electron pair
- TSH:
-
Tensor surface harmonics
- TTP:
-
Tricapped trigonal prism
References
Fässler TF, Hoffmann R (1999) Novel synthetic route to soluble polyanions: synthesis and crystal structure of [K(18-crown-6)]4[Pb9]·en·tol†. J Chem Soc Dalton Trans:3339–3340
Wade K (1971) The structural significance of the number of skeletal bonding electron-pairs in carboranes, the higher boranes and borane anions, and various transition-metal carbonyl cluster compounds. J Chem Soc Chem Commun 0:792–793
Mingos DMP (1984) Polyhedral skeletal electron pair approach. Acc Chem Res 17:311–319
Mingos DMP, Wales DJ (1990) Introduction to cluster chemistry. Prentice-Hall
Stone AJ (1981) New approach to bonding in transition-metal clusters and related compounds. Inorg Chem 20:563–571
Welch AJ (2013) The significance and impact of Wade’s rules. Chem Commun 49:3615–3616
Johnston RL, Mingos DMP (1986) Molecular orbital calculations relevant to the hypercloso vs. iso-closo controversy in metallaboranes. Inorg Chem 25:3321–3323
Bould J, Greenwood NN, Kennedy JD, McDonald WS (1982) Quantitative ortho-cycloboronation of P-phenyl groups in metallaborane chemistry and the crystal and molecular structure of the novel iso-closo-ten-vertex metallaborane [1,1,1-H(PPh3)(Ph2P-ortho-C6H4)-iso-closo-(1-IrB9H8-2-)]. J Chem Soc Chem Commun:465–467
King RB (2011) Structure and bonding in zintl ions and related main group element clusters. In: Fässler TF (ed) Zintl ions Princ. Recent dev. Springer, Berlin, pp 1–24
Fässler TF (2011) Relationships between soluble zintl anions, ligand-stabilized cage compounds, and intermetalloid clusters of tetrel (Si–Pb) and pentel (P–bi) elements. In: Fässler TF (ed) Zintl ions Princ. Recent dev. Springer, Berlin, pp 91–131
Scharfe S, Kraus F, Stegmaier S, Schier A, Fässler TF (2011) Zintl ions, cage compounds, and intermetalloid clusters of group 14 and group 15 elements. Angew Chem Int Ed 50:3630–3670
Liu C, Sun Z-M (2019) Recent advances in structural chemistry of group 14 Zintl ions. Coord Chem Rev 382:32–56
Hoch C, Wendorff M, Röhr C (2003) Synthesis and crystal structure of the tetrelides A12M17 (A=Na, K, Rb, Cs; M=Si, Ge, Sn) and A4Pb9 (A=K, Rb). J Alloys Compd 361:206–221
Edwards PA, Corbett JD (1977) Stable homopolyatomic anions. Synthesis and crystal structures of salts containing the pentaplumbide(2-) and pentastannide(2-) anions. Inorg Chem 16:903–907
Goicoechea JM, Sevov SC (2004) Naked deltahedral silicon clusters in solution: synthesis and characterization of Si93− and Si52−. J Am Chem Soc 126:6860–6861
Campbell J, Schrobilgen GJ (1997) The closo-Ge52− anion: synthesis, crystal structure, and raman spectrum of (2,2,2-crypt-K+)2Ge52−·THF. Inorg Chem 36:4078–4081
Corbett JD, Edwards PA (1975) Stable homopolyatomic anions: the crystal structures of salts of the anions pentaplumbide(2–) and enneastannide(4–). J Chem Soc Chem Commun:984–985
Suchentrunk C, Korber N (2006) Ge52− Zintl anions: synthesis and crystal structures of [K([2.2.2]-crypt)]2Ge5·4NH3 and [Rb([2.2.2]-crypt)]2Ge5·4NH3. New J Chem 30:1737–1739
Goicoechea JM, Sevov SC (2005) Ligand-free deltahedral clusters of silicon in solution: synthesis, structure, and electrochemistry of Si92–. Inorg Chem 44:2654–2658
Belin CHE, Corbett JD, Cisar A (1977) Homopolyatomic anions and configurational questions. Synthesis and structure of the nonagermanide(2-) and nonagermanide(4-) ions, Ge92− and Ge94−. J Am Chem Soc 99:7163–7169
Spiekermann A, Hoffmann SD, Fässler TF (2006) The zintl ion [Pb10]2−: a rare example of a homoatomic closo cluster. Angew Chem Int Ed 45:3459–3462
Cui L-F, Huang X, Wang L-M, Zubarev DY, Boldyrev AI, Li J, Wang L-S (2006) Sn122−: stannaspherene. J Am Chem Soc 128:8390–8391
Cui L-F, Huang X, Wang L-M, Li J, Wang L-S (2006) Pb122−: plumbaspherene. J Phys Chem A 110:10169–10172
Cui L-F, Wang L-S (2008) Stable icosahedral hollow cage clusters: stannaspherene and plumbaspherene. Int Rev Phys Chem 27:139–166
Corbett JD, Edwards PA (1977) The nonastannide(4-) anion Sn94−, a novel capped antiprismatic configuration (C4.Upsilon.). J Am Chem Soc 99:3313–3317
Yong L, Hoffmann SD, Fässler TF (2006) ∞2[K4Pb9] – a low-dimensional arrangement of [Pb9]4− clusters in [K(18-crown-6)]2K2Pb9·(en)1.5. Inorg Chim Acta 359:4774–4778
Schnering HGV, Baitinger M, Bolle U et al (1997) Binary alkali metal compounds with the zintl anions [Ge9]4− and [Sn9]4−. Z Für Anorg Allg Chem 623:1037–1039
Campbell J, Dixon DA, Mercier HPA, Schrobilgen GJ (1995) The nido-Pb94− and the Jahn-Teller distorted closo-Pb93− Zintl anions: syntheses, X-ray structures, and theoretical studies. Inorg Chem 34:5798–5809
Joseph S, Suchentrunk C, Kraus F, Korber N (2009) Si94− anions in solution – structures of the solvates Rb4Si9·4.75NH3 and [Rb(18-crown-6)]Rb3Si9·4NH3, and chemical bonding in Si94−. Eur J Inorg Chem 2009:4641–4647
Wiesler K, Brandl K, Fleischmann A, Korber N (2009) Tetrahedral [Tt4]4− zintl anions through solution chemistry: syntheses and crystal structures of the ammoniates Rb4Sn4·2NH3, Cs4Sn4·2NH3, and Rb4Pb4·2NH3. Z Für Anorg Allg Chem 635:508–512
Zhang J-X, Sheong FK, Lin Z (2018) Unravelling chemical interactions with principal interacting orbital analysis. Chem Eur J 24:9639–9650
Zhang J-X, Sheong FK, Lin Z (2020) Principal interacting orbital: a chemically intuitive method for deciphering bonding interaction. WIREs Comput Mol Sci 10:e1469
Li F, Sevov SC (2014) Synthesis, structures, and solution dynamics of tetrasubstituted nine-atom germanium deltahedral clusters. J Am Chem Soc 136:12056–12063
Geitner FS, Dums JV, Fässler TF (2017) Derivatization of phosphine ligands with bulky deltahedral zintl clusters – synthesis of charge neutral zwitterionic tetrel cluster compounds [(Ge9{Si(TMS)3}2)tBu2P]M(NHCDipp) (M: Cu, Ag, Au). J Am Chem Soc 139:11933–11940
Sevov SC, Goicoechea JM (2006) Chemistry of deltahedral zintl ions. Organometallics 25:5678–5692
Rios D, Gillett-Kunnath MM, Taylor JD, Oliver AG, Sevov SC (2011) Addition of a thallium vertex to empty and centered nine-atom deltahedral zintl ions of germanium and tin. Inorg Chem 50:2373–2377
Hull MW, Sevov SC (2007) Organo-zintl clusters soluble in conventional organic solvents: setting the stage for organo-zintl cluster chemistry. Inorg Chem 46:10953–10955
Hull MW, Sevov SC (2009) Functionalization of nine-atom deltahedral zintl ions with organic substituents: detailed studies of the reactions. J Am Chem Soc 131:9026–9037
Schnepf A (2003) [Ge9{Si(SiMe3)3}3]−: a soluble polyhedral Ge9 cluster stabilized by only three silyl ligands. Angew Chem Int Ed 42:2624–2625
Li F, Muñoz-Castro A, Sevov SC (2012) [Ge9{Si(SiMe3)3}3SnPh3]: a tetrasubstituted and neutral deltahedral nine-atom cluster. Angew Chem Int Ed 51:8581–8584
Hull MW, Sevov SC (2007) Addition of alkenes to deltahedral zintl clusters by reaction with alkynes: synthesis and structure of [Fc–CH–CH–Ge9–CH–CH–Fc]2−, an organo-zintl–organometallic anion. Angew Chem Int Ed 46:6695–6698
Ugrinov A, Sevov SC (2002) [Ph2Bi−(Ge9)−BiPh2]2−: a deltahedral zintl ion functionalized by exo-bonded ligands. J Am Chem Soc 124:2442–2443
Hogeveen H, Kwant PW (1974) Chemistry and spectroscopy in strongly acidic solutions. XL. (CCH3)62+, an unusual dication. J Am Chem Soc 96:2208–2214
Jašík J, Gerlich D, Roithová J (2014) Probing isomers of the benzene dication in a low-temperature trap. J Am Chem Soc 136:2960–2962
Malischewski M, Seppelt K (2017) Crystal structure determination of the pentagonal-pyramidal hexamethylbenzene dication C6(CH3)62+. Angew Chem Int Ed 56:368–370
Faessler TF, Hunziker M (1994) Ge93− and Pb93−: two novel, naked, homopolyatomic zintl ions with paramagnetic properties. Inorg Chem 33:5380–5381
Critchlow SC, Corbett JD (1983) Homopolyatomic anions. The synthesis and characterization of the novel paramagnetic nonastannide(3-) anion Sn93−, a D3h cluster with 21 skeletal electrons. J Am Chem Soc 105:5715–5716
Zubarev DY, Boldyrev AI (2008) Developing paradigms of chemical bonding: adaptive natural density partitioning. Phys Chem Chem Phys 10:5207–5217
Tkachenko VN, Boldyrev AI (2019) Multiple local σ-aromaticity of nonagermanide clusters. Chem Sci 10:5761–5765
King BR, Silaghi-Dumitrescu I (2008) The role of “external” lone pairs in the chemical bonding of bare post-transition element clusters: the Wade–Mingos rules versus the jellium model. Dalton Trans 0:6083–6088
Knight WD, De Heer WA, Saunders WA, Clemenger K, Chou MY, Cohen ML (1987) Alkali metal clusters and the jellium model. Chem Phys Lett 134:1–5
Lin Z, Slee T, Mingos DMP (1990) A structural jellium model of cluster electronic structure. Chem Phys 142:321–334
Walter M, Akola J, Lopez-Acevedo O, Jadzinsky PD, Calero G, Ackerson CJ, Whetten RL, Grönbeck H, Häkkinen H (2008) A unified view of ligand-protected gold clusters as superatom complexes. Proc Natl Acad Sci 105:9157–9162
Li F, Sevov SC (2012) Rational synthesis of [Ge9{Si(SiMe3)3}3]− from its parent zintl ion Ge94−. Inorg Chem 51:2706–2708
Downie C, Mao J-G, Guloy AM (2001) Synthesis and structure of [K+-(2,2)diaza-[18]-crown-6][K3Ge9]·2Ethylenediamine: stabilization of the two-dimensional layer ∞2[K3Ge91−]. Inorg Chem 40:4721–4725
Ugrinov A, Sevov SC (2004) Rationally functionalized deltahedral zintl ions: synthesis and characterization of [Ge9–ER3]3−, [R3E–Ge9–ER3]2−, and [R3E–Ge9–Ge9–ER3]4− (E=Ge, Sn; R=Me, Ph). Chem Eur J 10:3727–3733
Johnston RL, Mingos DMP (1987) A group theoretical paradigm for describing the skeletal molecular orbitals of cluster compounds. Part 2. Bispherical clusters. J Chem Soc Dalton Trans:1445–1456
Chen Z, Neukermans S, Wang X, Janssens E, Zhou Z, Silverans RE, King RB, Schleyer P von R, Lievens P (2006) To achieve stable spherical clusters: general principles and experimental confirmations. J Am Chem Soc 128:12829–12834
Fässler TF, Hoffmann SD (2004) Endohedral zintl ions: intermetalloid clusters. Angew Chem Int Ed 43:6242–6247
Zhao J, Du Q, Zhou S, Kumar V (2020) Endohedrally doped cage clusters. Chem Rev 120:9021–9163
Arcisauskaite V, Jin X, Goicoechea JM, McGrady JE (2016) Electronic properties of endohedral clusters of group 14. In: Mingos DMP (ed) Chem bond 100 years old get. Stronger. Springer, Cham, pp 181–197
Esenturk EN, Fettinger J, Lam Y-F, Eichhorn B (2004) [Pt@Pb12]2−. Angew Chem Int Ed 43:2132–2134
Esenturk EN, Fettinger J, Eichhorn B (2006) The Pb122− and Pb102− zintl ions and the M@Pb122− and M@Pb102− cluster series where M = Ni, Pd, Pt. J Am Chem Soc 128:9178–9186
Yi W, Lu-Lu W, Hua-Peng R, Ben-Long L, Rui-Li S, Li X (2015) Synthesis and characterization of the endohedral plumbaspherene[Rh@Pb12]3−. Chin J Struc Chem 34:1253–1258
Wang J-Q, Stegmaier S, Wahl B, Fässler TF (2010) Step-by-step synthesis of the endohedral stannaspherene [Ir@Sn12]3− via the capped cluster anion [Sn9Ir(cod)]3−. Chem Eur J 16:1793–1798
Xing X, Tian Z, Liu H, Tang Z (2003) Magic bimetallic cluster anions of M/Pb (M = Au, Ag and Cu) observed and analyzed by laser ablation and time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 17:1411–1415
Neukermans S, Wang X, Veldeman N, Janssens E, Silverans RE, Lievens P (2006) Mass spectrometric stability study of binary MSn clusters (S=Si, Ge, Sn, Pb, and M=Cr, Mn, Cu, Zn). Int J Mass Spectrom 252:145–150
Cui L-F, Huang X, Wang L-M, Li J, Wang L-S (2007) Endohedral stannaspherenes M@Sn12−: a rich class of stable molecular cage clusters. Angew Chem Int Ed 46:742–745
Liu C, Li L-J, Popov IA, Wilson RJ, Xu C-Q, Li J, Boldyrev AI, Sun Z-M (2018) Symmetry reduction upon size mismatch: the non-icosahedral intermetalloid cluster [Co@Ge12]3−. Chin J Chem 36:1165–1168
Esenturk EN, Fettinger J, Eichhorn B (2005) The closo-Pb102− Zintl ion in the [Ni@Pb10]2− cluster. Chem Commun:247–249
Scharfe S, Fässler TF, Stegmaier S, Hoffmann SD, Ruhland K (2008) [Cu@Sn9]3− and [Cu@Pb9]3−: intermetalloid clusters with endohedral Cu atoms in spherical environments. Chem Eur J 14:4479–4483
Gillett-Kunnath MM, Paik JI, Jensen SM, Taylor JD, Sevov SC (2011) Metal-centered deltahedral zintl ions: synthesis of [Ni@Sn9]4− by direct extraction from intermetallic precursors and of the vertex-fused dimer [{Ni@Sn8(μ-Ge)1/2}2]4−. Inorg Chem 50:11695–11701
Hlukhyy V, He H, Jantke L-A, Fässler TF (2012) The neat ternary solid K5−xCo1−xSn9 with endohedral [Co@Sn9]5− cluster units: a precursor for soluble intermetalloid [Co2@Sn17]5− clusters. Chem Eur J 18:12000–12007
Yue C-Y, Wang M-F, Yuan Z-D, Zhou F-X, Zhang H-P, Lei X-W (2013) K13CoSn17–x (x = 0.1): a new ternary phase containing cobalt centered [Sn9] cluster synthesized via high-temperature reaction. Z Für Anorg Allg Chem 639:911–917
Hlukhyy V, Stegmaier S, van Wüllen L, Fässler TF (2014) Endohedrally filled [Ni@Sn9]4− and [Co@Sn9]5− clusters in the neat solids Na12Ni1−xSn17 and K13−xCo1−xSn17: crystal structure and 119Sn solid-state NMR spectroscopy. Chem Eur J 20:12157–12164
Benda CB, Waibel M, Köchner T, Fässler TF (2014) Reactivity of liquid ammonia solutions of the zintl phase K12Sn17 towards Mesitylcopper(I) and Phosphinegold(I) chloride. Chem Eur J 20:16738–16746
Witzel BJL, Klein W, Dums JV, Boyko M, Fässler TF (2019) Metallocages for metal anions: highly charged [Co@Ge9]5− and [Ru@Sn9]6− clusters featuring spherically encapsulated Co1− and Ru2− anions. Angew Chem Int Ed 58:12908–12913
Goicoechea JM, Sevov SC (2005) [(Ni-Ni-Ni)@(Ge9)2]4−: a linear triatomic Nickel filament enclosed in a dimer of nine-atom germanium clusters. Angew Chem Int Ed 44:4026–4028
Goicoechea JM, Sevov SC (2006) Deltahedral germanium clusters: insertion of transition-metal atoms and addition of organometallic fragments. J Am Chem Soc 128:4155–4161
Liu C, Li L-J, Jin X, McGrady JE, Sun Z-M (2018) Reactivity studies of [Co@Sn9]4− with transition metal reagents: bottom-up synthesis of ternary functionalized zintl clusters. Inorg Chem 57:3025–3034
Li L-J, Pan F-X, Li F-Y, Chen Z-F, Sun Z-M (2017) Synthesis, characterization and electronic properties of an endohedral plumbaspherene [Au@Pb12]3−. Inorg Chem Front 4:1393–1396
Zhou B, Krämer T, Thompson AL, McGrady JE, Goicoechea JM (2011) A highly distorted open-shell endohedral zintl cluster: [Mn@Pb12]3−. Inorg Chem 50:8028–8037
Wang J-Q, Stegmaier S, Fässler TF (2009) [Co@Ge10]3−: an intermetalloid cluster with archimedean pentagonal prismatic structure. Angew Chem Int Ed 48:1998–2002
Zhou B, Denning MS, Kays DL, Goicoechea JM (2009) Synthesis and isolation of [Fe@Ge10]3−: a pentagonal prismatic zintl ion cage encapsulating an interstitial iron atom. J Am Chem Soc 131:2802–2803
Krämer T, Duckworth JCA, Ingram MD, Zhou B, McGrady JE, Goicoechea JM (2013) Structural trends in ten-vertex endohedral clusters, M@E10 and the synthesis of a new member of the family, [Fe@Sn10]3−. Dalton Trans 42:12120–12129
Sevov SC, Corbett JD (1993) Potassium indium zinc compound K8In10Zn: interstitially-stabilized analogs of early-transition-metal halide clusters. Inorg Chem 32:1059–1061
Sevov SC, Corbett JD (1993) K10In10Z (Z = Ni, Pd, Pt): Zintl phases containing isolated decaindium clusters centered by transition elements. J Am Chem Soc 115:9089–9094
Espinoza-Quintero G, Duckworth JCA, Myers WK, McGrady JE, Goicoechea JM (2014) Synthesis and characterization of [Ru@Ge12]3−: an endohedral 3-connected cluster. J Am Chem Soc 136:1210–1213
Beck SM (1987) Studies of silicon cluster–metal atom compound formation in a supersonic molecular beam. J Chem Phys 87:4233–4234
Janssens E, Gruene P, Meijer G, Wöste L, Lievens P, Fielicke A (2007) Argon physisorption as structural probe for endohedrally doped silicon clusters. Phys Rev Lett 99:063401
Pandey R, Rao BK, Jena P, Blanco MA (2001) Electronic structure and properties of transition metal−benzene complexes. J Am Chem Soc 123:3799–3808
Khanna SN, Rao BK, Jena P (2002) Magic numbers in metallo-inorganic clusters: chromium encapsulated in silicon cages. Phys Rev Lett 89:016803
Ulises Reveles J, Khanna SN (2006) Electronic counting rules for the stability of metal-silicon clusters. Phys Rev B 74:035435
Guo L, Zhao G, Gu Y, Liu X, Zeng Z (2008) Density-functional investigation of metal-silicon cage clusters MSin (M=Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn; n=8–16). Phys Rev B 77:195417
Abreu MB, Reber AC, Khanna SN (2014) Does the 18-electron rule apply to CrSi12? J Phys Chem Lett 5:3492–3496
Goicoechea JM, McGrady JE (2015) On the structural landscape in endohedral silicon and germanium clusters, M@Si12 and M@Ge12. Dalton Trans 44:6755–6766
Goicoechea JM, Sevov SC (2006) Organozinc derivatives of deltahedral zintl ions: synthesis and characterization of closo-[E9Zn(C6H5)]3− (E = Si, Ge, Sn, Pb). Organometallics 25:4530–4536
Zhou B, Denning MS, Chapman TAD, Goicoechea JM (2009) Coupling reactions of functionalized zintl ions [E9Cd(C6H5)]3− (E = Sn, Pb) with tributyltinhydride: synthesis and isolation of {Sn9CdSn[(CH2)3CH3]3}3−. Inorg Chem 48:2899–2907
Zhou B, Denning MS, Jones C, Goicoechea JM (2009) Reductive cleavage of Zn–C bonds by group 14 Zintl anions: synthesis and characterisation of [E9ZnR]3− (E = Ge, Sn, Pb; R = Mes, iPr). Dalton Trans 0:1571–1578
Li F, Muñoz-Castro A, Sevov SC (2016) [(Me3Si)Si]3EtGe9Pd(PPh3)], a pentafunctionalized deltahedral zintl cluster: synthesis, structure, and solution dynamics. Angew Chem Int Ed 55:8630–8633
Townrow OPE, Chung C, Macgregor SA, Weller AS, Goicoechea JM (2020) A neutral heteroatomic zintl cluster for the catalytic hydrogenation of cyclic alkenes. J Am Chem Soc 142:18330–18335
Kesanli B, Fettinger J, Eichhorn B (2001) The closo-[Sn9M(CO)3]4− zintl ion clusters where M=Cr, Mo, W: two structural isomers and their dynamic behavior. Chem Eur J 7:5277–5285
Eichhorn BW, Haushalter RC (1990) Closo-[CrPb9(CO)3]4−: a 100 year history of the nonaplumbide tetra-anion. J Chem Soc Chem Commun:937–938
Campbell J, Mercier HPA, Franke H, Santry DP, Dixon DA, Schrobilgen GJ (2002) Syntheses, crystal structures, and density functional theory calculations of the closo-[1-M(CO)3(η4-E9)]4− (E = Sn, Pb; M = Mo, W) cluster anions and solution NMR spectroscopic characterization of [1-M(CO)3(η4-Sn9)]4− (M = Cr, Mo, W). Inorg Chem 41:86–107
Yong L, Hoffmann SD, Fässler TF (2005) Crystal structures of [K(2.2.2-crypt)]4[Pb9Mo(CO)3] – isolation of the novel isomers [(η5-Pb9)Mo(CO)3]4− beside [(η4-Pb9)Mo(CO)3]4−. Eur J Inorg Chem 2005:3663–3669
Mingos DMP, Zhenyang L (1989) Site preference effects in heterometallic clusters. Comments Inorg Chem 9:95–122
Schiegerl LJ, Geitner FS, Fischer C, Klein W, Fässler TF (2016) Functionalization of [Ge9] with small silanes: [Ge9(SiR3)3]− (R = iBu, iPr, Et) and the structures of (CuNHCDipp)[Ge9{Si(iBu)3}3], (K-18c6)Au[Ge9{Si(iBu)3}3]2, and (K-18c6)2[Ge9{Si(iBu)3}2]. Z Für Anorg Allg Chem 642:1419–1426
Michenfelder NC, Gienger C, Schnepf A, Unterreiner A-N (2019) The influence of the FeCp(CO)2+ moiety on the dynamics of the metalloid [Ge9(Si(SiMe3)3)3]− cluster in thf: synthesis and characterization by time-resolved absorption spectroscopy. Dalton Trans 48:15577–15582
Schenk C, Schnepf A (2009) {Ge9R3Cr(CO)5}− and {Ge9R3Cr(CO)3}− : a metalloid cluster (Ge9R3−) as a flexible ligand in coordination chemistry [R = Si(SiMe3)3]. Chem Commun:3208–3210
Henke F, Schenk C, Schnepf A (2011) [Si(SiMe3)3]3Ge9M(CO)3− (M = Cr, Mo, W): coordination chemistry with metalloid clusters. Dalton Trans 40:6704–6710
Sun Z-M, Zhao Y-F, Li J, Wang L-S (2009) Diversity of functionalized germanium zintl clusters: syntheses and theoretical studies of [Ge9PdPPh3]3− and [Ni@(Ge9PdPPh3)]2−. J Clust Sci 20:601–609
Kesanli B, Fettinger J, Gardner DR, Eichhorn B (2002) The [Sn9Pt2(PPh3)]2− and [Sn9Ni2(CO)]3− complexes: two markedly different Sn9M2L transition metal zintl ion clusters and their dynamic behavior. J Am Chem Soc 124:4779–4786
Geitner FS, Klein W, Storcheva O, Tilley TD, Fässler TF (2019) Early-transition-metal complexes of functionalized nonagermanide clusters: synthesis and characterization of [Cp2(MeCN)Ti(η1-Ge9{Si(TMS)3}3)] and K3[Cp2Ti(η1-Ge9{Si(TMS)3}2)2]. Inorg Chem 58:13293–13298
Kysliak O, Schrenk C, Schnepf A (2016) Reactivity of [Ge9{Si(SiMe3)3}3]− towards transition-metal M2+ cations: coordination and redox chemistry. Chem Eur J 22:18787–18793
Kocak FS, Zavalij P, Eichhorn B (2011) Reactions of exo-substituted RSn93− clusters with Pd: endohedral cluster formation and oxidative insertion. Chem Eur J 17:4858–4863
Geitner FS, Fässler TF (2016) Introducing tetrel zintl ions to N-heterocyclic carbenes – synthesis of coinage metal NHC complexes of [Ge9{Si(SiMe3)3}3]−. Eur J Inorg Chem 2016:2688–2691
Mayer K, Schiegerl LJ, Fässler TF (2016) On the reactivity of silylated Ge9 clusters: synthesis and characterization of [ZnCp*(Ge9{Si(SiMe3)3}3)], [CuPiPr3(Ge9{Si(SiMe3)3}3)], and [(CuPiPr3)4{Ge9(SiPh3)2}2]. Chem Eur J 22:18794–18800
Geitner FS, Klein W, Fässler TF (2018) Synthesis and reactivity of multiple phosphine-functionalized nonagermanide clusters. Angew Chem Int Ed 57:14509–14513
Geitner FS, Wallach C, Fässler TF (2018) On the variable reactivity of phosphine-functionalized [Ge9] clusters: zintl cluster-substituted phosphines or phosphine-substituted zintl clusters. Chem Eur J 24:4103–4110
Downing DO, Zavalij P, Eichhorn BW (2010) The closo-[Sn9Ir(cod)]3− and [Pb9Ir(cod)]3− zintl ions: isostructural IrI derivatives of the nido-E94− anions (E = Sn, Pb). Eur J Inorg Chem 2010:890–894
Geitner FS, Klein W, Fässler TF (2017) Formation of the intermetalloid cluster [AgSn18]7− − the reactivity of coinage metal NHC compounds towards [Sn9]4−. Dalton Trans 46:5796–5800
Scharfe S, Fässler TF (2010) Varying bonding modes of the zintl ion [Ge9]4− in CuI complexes: syntheses and structures of [Cu(η4-Ge9)(PR3)]3– (R = iPr, Cy) and [Cu(η4-Ge9)(η1-Ge9)]7−. Eur J Inorg Chem 2010:1207–1213
Esenturk EN, Fettinger J, Eichhorn B (2006) Synthesis and characterization of the [Ni6Ge13(CO)5]4− and [Ge9Ni2(PPh3)]2− Zintl ion clusters. Polyhedron 25:521–529
Li F, Sevov SC (2015) Coordination of tri-substituted Nona–Germanium clusters to Cu(I) and Pd(0). Inorg Chem 54:8121–8125
Schenk C, Schnepf A (2007) [AuGe18{Si(SiMe3)3}6]−: a soluble Au–Ge cluster on the way to a molecular cable? Angew Chem Int Ed 46:5314–5316
Henke F, Schenk C, Schnepf A (2009) [Si(SiMe3)3]6Ge18M (M = Zn, Cd, Hg): neutral metalloid cluster compounds of germanium as highly soluble building blocks for supramolecular chemistry. Dalton Trans:9141–9145
Schenk C, Henke F, Santiso-Quiñones G, Krossing I, Schnepf A (2008) [Si(SiMe3)3]6Ge18M (M = Cu, Ag, Au): metalloid cluster compounds as unusual building blocks for a supramolecular chemistry. Dalton Trans:4436–4441
Kysliak O, Schrenk C, Schnepf A (2015) {Ge9[Si(SiMe3)2(SiPh3)]3}−: ligand modification in metalloid germanium cluster chemistry. Inorg Chem 54:7083–7088
Kysliak O, Kunz T, Schnepf A (2017) Metalloid Ge9R3− clusters with various silyl substituents: from shielded to open cluster cores. Eur J Inorg Chem 2017:805–810
Binder M, Schrenk C, Block T, Pöttgen R, Schnepf A (2017) [Hyp-Au-Sn9(Hyp)3-Au-Sn9(Hyp)3-Au-Hyp]−: the longest intermetalloid chain compound of tin. Chem Commun 53:11314–11317
Kysliak O, Nguyen DD, Clayborne AZ, Schnepf A (2018) [PtZn2Ge18(Hyp)8] (Hyp = Si(SiMe3)3): a neutral polynuclear chain compound with Ge9(Hyp)3 units. Inorg Chem 57:12603–12609
Denning MS, Goicoechea JM (2008) [Hg3(Ge9)4]10− : a nanometric molecular rod precursor to polymeric mercury-linked cluster chains. Dalton Trans:5882–5885
Nienhaus A, Hauptmann R, Fässler TF (2002) ∞1[HgGe9]2− – a polymer with zintl ions as building blocks covalently linked by heteroatoms. Angew Chem Int Ed 41:3213–3215
Bentlohner MM, Jantke L-A, Henneberger T, Fischer C, Mayer K, Klein W, Fässler TF (2016) On the nature of bridging metal atoms in intermetalloid clusters: synthesis and structure of the metal-atom-bridged zintl clusters [Sn(Ge9)2]4− and [Zn(Ge9)2)]6−. Chem Eur J 22:13946–13952
Yong L, Boeddinghaus MB, Fässler TF (2010) [Sn9HgSn9]6−: an intermetalloid zintl ion with two Sn9 connected by heteroatom. Z Für Anorg Allg Chem 636:1293–1296
Mayer K, Jantke L-A, Schulz S, Fässler TF (2017) Retention of the Zn−Zn bond in [Ge9Zn−ZnGe9]6− and formation of [(Ge9Zn)−(Ge9)−(ZnGe9)]8− and polymeric [−(Ge9Zn)2−−]. Angew Chem Int Ed 56:2350–2355
Zhang C, Morgan HWT, Wang Z-C, Liu C, Sun Z-M, McGrady JE (2019) Structural isomerism in the [(Ni@Sn9)In(Ni@Sn9)]5− Zintl ion. Dalton Trans 48:15888–15895
Spiekermann A, Hoffmann SD, Kraus F, Fässler TF (2007) [Au3Ge18]5− – a gold–germanium cluster with remarkable Au–Au interactions. Angew Chem Int Ed 46:1638–1640
Schmidbaur H (2000) The aurophilicity phenomenon: a decade of experimental findings, theoretical concepts and emerging applications. Gold Bull 33:3–10
Joseph S, Hamberger M, Mutzbauer F, Härtl O, Meier M, Korber N (2009) Chemistry with bare silicon clusters in solution: a transition-metal complex of a polysilicide anion. Angew Chem Int Ed 48:8770–8772
Sheong FK, Chen W-J, Zhang J-X, Li Y, Lin Z (2017) Structure and bonding of [Pd2Sn18]4−: an interesting example of the mutual delocalisation phenomenon. Dalton Trans 46:2214–2219
Ugrinov A, Sevov SC (2003) Derivatization of deltahedral zintl ions by nucleophilic addition: [Ph−Ge9−SbPh2]2− and [Ph2Sb−Ge9−Ge9−SbPh2]4−. J Am Chem Soc 125:14059–14064
Hull MW, Ugrinov A, Petrov I, Sevov SC (2007) Alkylation of deltahedral zintl clusters: synthesis of [R−Ge9−Ge9−R]4− (R = tBu, sBu, nBu, tAm) and structure of [tBu−Ge9−Ge9−tBu]4−. Inorg Chem 46:2704–2708
First Synthesis of Group-14 Polyanions by Extraction of a Binary Alloy with dmf and a Novel Conformation of the (Ge9−Ge9)6− Dimer: Crystal Structures of [K6(Ge9−Ge9)](dmf)12, [Rb6(Ge9−Ge9)](dmf)12 and [K2.5Cs3.5(Ge9−Ge9)](dmf)12 (2006) Z Für Anorg Allg Chem 632:1752–1758
Xu L, Sevov SC (1999) Oxidative coupling of deltahedral [Ge9]4− zintl ions. J Am Chem Soc 121:9245–9246
Wang J-Q, Wahl B, Fässler TF (2010) [Ag(Sn9–Sn9)]5−: a homoleptic silver complex of a dimeric Sn9 zintl anion. Angew Chem Int Ed 49:6592–6595
Hansen DF, Zhou B, Goicoechea JM (2012) Further studies into the reactivity and coordination chemistry of [Ge9]4− zintl ions. The indium-containing anions [In(Ge9)2]5−, [(Ge9)2In(C6H5)]4− and [Ge9{In(C6H5)3}2]4−. J Organomet Chem 721–722:53–61
Ugrinov A, Sevov SC (2005) Synthesis of a chain of nine-atom germanium clusters accompanied with dimerization of the sequestering agent. Comptes Rendus Chim 8:1878–1882
Downie C, Tang Z, Guloy AM (2000) An unprecedented ∞1[Ge9]2− polymer: a link between molecular zintl clusters and solid-state phases. Angew Chem 112:346–348
Downie C, Mao J-G, Parmar H, Guloy AM (2004) The role of sequestering agents in the formation and structure of germanium anion cluster polymers. Inorg Chem 43:1992–1997
Perla LG, Sevov SC (2016) A stannyl-decorated zintl ion [Ge18Pd3(SniPr3)6]2−: twinned icosahedron with a common Pd3-face or 18-vertex hypho-deltahedron with a Pd3-triangle inside. J Am Chem Soc 138:9795–9798
Perla LG, Muñoz-Castro A, Sevov SC (2017) Eclipsed- and staggered-[Ge18Pd3{EiPr3}6]2− (E = Si, Sn): positional isomerism in deltahedral zintl clusters. J Am Chem Soc 139:15176–15181
Zhang J-X, Sheong FK, Lin Z (2019) Remote bonding in clusters [Pd3Ge18R6]2−: modular bonding model for large clusters via principal interacting orbital analysis. Inorg Chem 58:3473–3478
Goicoechea JM, Sevov SC (2005) [(Pd−Pd)@Ge18]4−: a palladium dimer inside the largest single-cage deltahedron. J Am Chem Soc 127:7676–7677
Sun Z-M, Xiao H, Li J, Wang L-S (2007) Pd2@Sn184−: fusion of two endohedral stannaspherenes. J Am Chem Soc 129:9560–9561
Ugrinov A, Sevov SC (2002) [Ge9Ge9Ge9]6−: a linear trimer of 27 germanium atoms. J Am Chem Soc 124:10990–10991
Ugrinov A, Sevov SC (2003) [Ge9Ge9Ge9Ge9]8−: a linear tetramer of nine-atom germanium clusters, a nanorod. Inorg Chem 42:5789–5791
Stegmaier S, Fässler TF (2011) A bronze matryoshka: the discrete intermetalloid cluster [Sn@Cu12@Sn20]12− in the ternary phases A12Cu12Sn21 (A = Na, K). J Am Chem Soc 133:19758–19768
Sheong FK, Chen W-J, Kim H, Lin Z (2015) Peeling the onion: a revised model of the electron count for matryoshka clusters. Dalton Trans 44:7251–7257
Moses MJ, Fettinger JC, Eichhorn BW (2003) Interpenetrating As20 fullerene and Ni12 icosahedra in the onion-skin [As@Ni12@As20]3− ion. Science 300:778–780
Bashkurov R, Kratish Y, Fridman N, Bravo-Zhivotovskii D, Apeloig Y (2021) A high yield synthesis of an octastannacubane and a Bis(silyl) stannylene via reductive elimination of a silane. Angew Chem Int Ed 60:2898–2902
Wiberg N, Finger CMM, Polborn K (1993) Tetrakis(tri-tert-butylsilyl)-tetrahedro-tetrasilane (tBu3Si)4Si4: the first molecular silicon compound with a Si4 tetrahedron. Angew Chem Int Ed Engl 32:1054–1056
Wiberg N, Hochmuth W, Nöth H, Appel A, Schmidt-Amelunxen M (1996) Tetrakis(tri-tert-butylsilyl)-tetrahedro-tetragermane (tBu3Si)4Ge4 – the first molecular germanium compound with a Ge4 tetrahedron. Angew Chem Int Ed Engl 35:1333–1334
Wiberg N, Lemer H-W, Wagner S, Nöth H, Seifert T (1999) Über das Octastannandiid R*6Sn8[Na(THF)2]2 und zur möglichen Existenz des Octastannans R*6Sn8 [1] / on an Octastannanediide R*6Sn8[Na(THF)2]2 and the possible existence of an octastannane R*6Sn8 [1]. Z Für Naturforschung B 54:877–880
Ichinohe M, Toyoshima M, Kinjo R, Sekiguchi A (2003) Tetrasilatetrahedranide: a silicon cage anion. J Am Chem Soc 125:13328–13329
Klapötke TM, Vasisht SK, Fischer G, Mayer P (2010) A reactive Si4 cage: K(SitBu3)3Si4. J Organomet Chem 695:667–672
Schnepf A, Köppe R (2003) [Ge8{N(SiMe3)2}6]: a ligand-stabilized Ge cluster compound with formally zero-valent Ge atoms. Angew Chem Int Ed 42:911–913
Fischer G, Huch V, Mayer P, Vasisht SK, Veith M, Wiberg N (2005) Si8(SitBu3)6: a hitherto unknown cluster structure in silicon chemistry. Angew Chem Int Ed 44:7884–7887
Eichler BE, Power PP (2001) Synthesis and characterization of [Sn8(2,6-Mes2C6H3)4] (Mes=2,4,6-Me3C6H2): a main group metal cluster with a unique structure. Angew Chem 113:818–819
Schnepf A, Drost C (2005) Ge8R6: the ligands define the bonding situation within the cluster core. Dalton Trans:3277–3280
Schnepf A (2006) {Ge10Si[Si(SiMe3)3]4(SiMe3)2Me}−: a Ge10Si framework reveals a structural transition onto elemental germanium. Chem Commun:192–194
Wu W, Gu J, Song J, Shaik S, Hiberty PC (2009) The inverted bond in [1.1.1]propellane is a charge-shift bond. Angew Chem Int Ed 48:1407–1410
Shaik S, Danovich D, Wu W, Hiberty PC (2009) Charge-shift bonding and its manifestations in chemistry. Nat Chem 1:443–449
Shaik S, Danovich D, Galbraith JM, Braïda B, Wu W, Hiberty PC (2019) Charge-shift bonding: a new and unique form of bonding. Angew Chem Int Ed 59:984–1001
Wiederkehr J, Wölper C, Schulz S (2016) Synthesis and solid state structure of a metalloid tin cluster [Sn10(trip8)]. Chem Commun 52:12282–12285
Sekiguchi A, Ishida Y, Kabe Y, Ichinohe M (2002) The cation cluster of heavier group 14 elements: a free germyl cation with trishomoaromaticity. J Am Chem Soc 124:8776–8777
Spiekermann A, Hoffmann SD, Fässler TF, Krossing I, Preiss U (2007) [Au3Ge45]9− – a binary anion containing a Ge45 cluster. Angew Chem Int Ed 46:5310–5313
Schenk C, Schnepf A (2008) Ge14[Ge(SiMe3)3]5Li3(THF)6 : the largest metalloid cluster compound of germanium: on the way to fullerene -like compounds? Chem Commun:4643–4645
Lips F, Clérac R, Dehnen S (2011) [Eu@Sn6Bi8]4−: a mini-fullerane-type zintl anion containing a lanthanide ion. Angew Chem Int Ed 50:960–964
Lips F, Clérac R, Dehnen S (2011) [Pd3Sn8Bi6]4−: a 14-vertex Sn/bi cluster embedding a Pd3 triangle. J Am Chem Soc 133:14168–14171
Mitzinger S, Broeckaert L, Massa W, Weigend F, Dehnen S (2015) [V@Ge8As4]3− and [Nb@Ge8As6]3− : encapsulation of electron-poor transition metal atoms. Chem Commun 51:3866–3869
Schrenk C, Schellenberg I, Pöttgen R, Schnepf A (2010) The formation of a metalloid Sn10[Si(SiMe3)3]6 cluster compound and its relation to the α↔β tin phase transition. Dalton Trans 39:1872–1876
Klinkhammer KW, Xiong Y, Yao S (2004) Molecular lead clusters – from unexpected discovery to rational synthesis. Angew Chem Int Ed 43:6202–6204
Schrenk C, Helmlinger J, Schnepf A (2012) Sn10[Si(SiMe3)3]5−: an anionic metalloid tin cluster from an isolable SnI halide solution. Z Für Anorg Allg Chem 638:589–593
Schrenk C, Gerke B, Pöttgen R, Clayborne A, Schnepf A (2015) Reactions with a metalloid tin cluster Sn10[Si(SiMe3)3]42−: ligand elimination versus coordination chemistry. Chem Eur J 21:8222–8228
Schrenk C, Winter F, Pöttgen R, Schnepf A (2015) Sn10[Si(SiMe3)3]42−: a highly reactive metalloid tin cluster with an open ligand shell. Chem Eur J 21:2992–2997
Kiani FA, Hofmann M (2006) Cluster increments for macropolyhedral boranes. Dalton Trans:5515–5520
Sheong FK, Zhang J-X, Lin Z (2017) Localized bonding model for coordination and cluster compounds. Coord Chem Rev 345:42–53
Liu C, Popov IA, Li L-J, Li N, Boldyrev AI, Sun Z-M (2018) [Co2@Ge16]4−: localized versus delocalized bonding in two isomeric intermetalloid clusters. Chem Eur J 24:699–705
Liu C, Jin X, Li L-J, Xu J, McGrady JE, Sun Z-M (2019) Synthesis and structure of a family of rhodium polystannide clusters [Rh@Sn10]3−, [Rh@Sn12]3−, [Rh2@Sn17]6− and the first triply-fused stannide, [Rh3@Sn24]5−. Chem Sci 10:4394–4401
Shu C-C, Morgan HWT, Qiao L, McGrady JE, Sun Z-M (2020) A family of lead clusters with precious metal cores. Nat Commun 11:3477
Acknowledgments
We would like to dedicate this chapter to the memory of Roy Johnston who made seminal contribution in the application of molecular orbital theory to the understanding of structure and bonding in molecular cluster compounds. We would also like to acknowledge the financial support from the Hong Kong Research Grants Council (HKUST 16305119).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zhang, JX., Sheong, F.K., Lin, Z. (2021). Structure and Bonding of Group 14 Clusters: Wade’s Rules and Beyond. In: Mingos, D. (eds) 50th Anniversary of Electron Counting Paradigms for Polyhedral Molecules . Structure and Bonding, vol 188. Springer, Cham. https://doi.org/10.1007/430_2021_84
Download citation
DOI: https://doi.org/10.1007/430_2021_84
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-84870-5
Online ISBN: 978-3-030-84871-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)