
Abstract
The only enzyme that is able to fix nitrogen, nitrogenase, reduces inert and abundant dinitrogen (N2) into bioavailable ammonia (NH3) under ambient conditions. The most investigated variant, the MoFe nitrogenase, uses three metallo-cofactors: the [Fe4S4] cluster in the electron-carrier component (Fe protein), as well as the [Fe8S7] (P-cluster) and [MoFe7S9C] (M-cluster) clusters in the catalytic component (MoFe protein). To better understand the physical properties of these cofactors, various methods have been developed for the chemical synthesis of model metal-sulfur clusters. In this review, we address the following topics with emphasis on recent developments: (a) the synthesis of all-ferrous [Fe4S4]0 clusters, which are isoelectronic to the super-reduced state of the cluster in the Fe protein, (b) the reproduction of the unique [Fe8S7] inorganic core of the P-cluster, and (c) the synthesis of metal-sulfur clusters relevant to the M-cluster and their variants that incorporate a light atom. Even though reproduction of the M-cluster remains elusive, some recent advances seem promising toward new classes of metal-sulfur clusters that satisfy the key structural features of the M-cluster.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction: Biological N2 Fixation and Nitrogenase Systems
Nitrogen is an essential element in nucleic and amino acids, which are in turn indispensable to biological activities. Such organic nitrogen compounds are produced through numerous metabolic pathways, where ammonia (NH3) is used as a raw material. Even though most organisms are unable to supply NH3, a biological process is present for the reduction of inert and abundant dinitrogen (N2).
Nitrogenase is the only known enzyme that catalyzes the reduction of N2 into NH3. Three variants, i.e., MoFe, VFe, and Fe-only nitrogenases, have been identified and named after their essential metal content [1]. These enzymes, encoded in nif, vnf, and anf gene clusters, respectively, are co-induced with the corresponding biosynthetic machinery under nitrogen-deficient environments. As a survival strategy of N2-fixing bacteria under varying conditions, the variant to be expressed is regulated by the availability of the metals. Likely following the order of catalytic activity, the bacteria prioritize the production of the MoFe, VFe, or Fe-only variant [1]. All these variants are homologous and consist of two components, i.e., an electron-carrier oxidoreductase and a catalytic component.
The MoFe nitrogenase is the best-studied variant, whose catalytic component, known as the MoFe protein, is encoded by nifD and nifK genes. The resulting α 2 β 2 tetrameric protein receives electrons from the Fe protein, which is the homodimeric oxidoreductase component encoded by nifH. In the Fe protein, two binding sites for adenosine triphosphate (ATP) are present. The ATP-bound form associates with the MoFe protein to form a transient complex that leads to the electron transfer from the Fe protein to the MoFe protein. Hydrolysis of the protein-bound ATP into adenosine diphosphate (ADP) and monophosphate (Pi) has been suggested to trigger the dissociation of the Fe protein from the MoFe protein [2]. By repeating this ATP-dependent process, nitrogenase transfers electrons from the Fe protein to the MoFe protein, and eventually to N2 together with protons, for the formation of NH3. The reduction of one molecule of N2 is presumably accompanied by the obligate production of one molecule of H2 according to the following chemical equation: N2 + 8H+ + 8e− + 16ATP → 2NH3 + H2 + 16ADP + 16Pi [3, 4].
To achieve its extraordinary activity, the MoFe nitrogenase uses three redox-active metallo-cofactors, which are metal-sulfur clusters consisting of multiple metal and sulfur atoms. The metallo-cofactor in the Fe protein is a typical [Fe4S4] cluster, while the other two in the MoFe protein are unique to nitrogenase and designated as the P-cluster and M-cluster, whose compositions have been determined as [Fe8S7] [5] and [(cit)MoFe7S9C] (cit = R-homocitrate) [6, 7], respectively (Fig. 1). Recent protein crystallographic analyses associated with biochemical studies have further elucidated some properties of these metallo-cofactors, such as the predominant involvement of the 1e− redox process of the P-cluster under the turnover conditions [8] and the proposed displacement of one of the bridging sulfides of the M-cluster for the generation of the reactive form [9, 10]. Recently, the possible removal of a bridging sulfide has been revisited based on the protein crystallographic analyses of the VFe nitrogenase [11, 12], where a light atom (theoretically proposed as an OH moiety derived from H2O) [13] replaces one of the bridging sulfides under reducing conditions. Even though enzymatic studies have uncovered some important clues as to how such nitrogenase metallo-cofactors might work, a number of uncertainties remain regarding their structure-function relationships that represent a major issue to be addressed from a chemical perspective. Thus, the chemical synthesis of model compounds and the analysis of their detailed properties and reactivity could provide valuable insight into the metallo-cofactors. While some reviews have been published on model compounds of nitrogenase metallo-cofactors (for representative reviews of the model chemistry of nitrogenase, see [14, 15]), here we revisit this topic with emphasis on the most recent advances.

Schematic illustration of the electron-transfer pathway in the MoFe nitrogenase, highlighting the metallo-cofactors and the protein-bound MgATP molecules. (a) [Fe4S4] cluster of the Fe protein; (b) P-cluster and (c) M-cluster of the MoFe protein. The Fe protein is colored in green, while the MoFe protein is colored in purple and yellow. Only half of the α 2 β 2-heterotetramer of the MoFe protein is shown for clarity. PDB ID: 4WZA (left) and 3U7Q (right). Color legend: C, gray; Fe, orange; N, blue; Mo, teal; O, red; S, yellow
2 Model [Fe4S4] Clusters of the Fe Protein
Cuboidal [Fe4S4] clusters are arguably the most prominent class of biological iron-sulfur clusters, and their oxidation states typically range between [Fe4S4]+ and [Fe4S4]3+ (for representative reviews, see [16,17,18,19]). In contrast to ordinary [Fe4S4] clusters, the cluster in the Fe protein can be reduced to the formal oxidation state [Fe4S4]0 (for a review specifically focusing on Fe protein, see [20]), the so-called super-reduced state, in the presence of reducing agents [21, 22]. The physiological importance of this super-reduced state still remains unclear; however, it demonstrates the exceptional stability of the [Fe4S4] cluster of the Fe protein under reducing conditions. Furthermore, recent studies have revealed that the Fe proteins from some prokaryotes and archaea are able to catalyze the reduction of carbon dioxide to furnish carbon monoxide and short-chain hydrocarbons [23, 24]. Thus, synthetic [Fe4S4] clusters in the reduced states are of interest not only as the models for the cluster in the Fe protein but also as potential catalyst precursors for artificial carbon fixation and small-molecule activation.
2.1 Synthesis of Super-Reduced [Fe4S4] Clusters
In a pioneering study from 1972, Holm et al. reported the first chemical synthesis of an [Fe4S4] cluster bearing four thiolate ligands [25]. Since then, over 80 examples of thiolate-supported [Fe4S4] clusters in [Fe4S4]+/2+/3+ oxidation states have been synthesized, while only a limited number of [Fe4S4]0 clusters are accessible. As the chemistry of [Fe4S4]+/2+/3+ clusters has been summarized elsewhere (for representative reviews, see [26,27,28,29]), this section focuses on synthetic [Fe4S4]0 clusters.
Even though the [Fe4S4] cluster in the Fe protein is supported exclusively by cysteine residues [30], no thiolate-supported [Fe4S4]0 cluster has been synthesized and isolated thus far. As short-lived species, [Fe4S4(SR)4]4− have been generated under certain electrochemical measurement conditions [31,32,33], but their instability has so far prevented their isolation. This instability arises from the dissociation of thiolate(s) from [Fe4S4(SR)4]4−, as the σ-donation of thiolate anions is not suitable for the stabilization of relatively low-valent, electron-rich metal centers. In contrast, π-acceptor ligands stabilize electron-rich metals through back-bonding [34]. As phosphines (PR3) are a representative class of π-acceptor ligands for transition metals, Holm and co-workers have employed phosphines for the attempted stabilization of the super-reduced [Fe4S4]0 cluster in the form [Fe4S4(PR3)4]0 (R = cyclohexyl (Cy), isopropyl (iPr), tert-butyl (tBu)). However, the synthesis of [Fe4S4(PR3)4]0 via the chemical reduction of [Fe4S4(PR3)4]+ using sodium acenaphthalenide was not successful due to the subsequent dissociation of some of the phosphines from the postulated [Fe4S4(PR3)4]0, resulting in the formation of an [Fe4S4] dimer (R = Cy) or tetramers (R = iPr, tBu), in which the [Fe4S4] units are connected via Fe-S edges [35,36,37]. In order to prevent the dissociation of supporting ligands from Fe, Holm and co-workers then employed cyanide as a more π-acidic ligand and successfully isolated the first super-reduced [Fe4S4]0 cluster, [Fe4S4(CN)4]4− (1), where the 4− net charge results in high susceptibility toward oxidation [38]. Moreover, the strong binding properties of N-heterocyclic carbenes toward Fe [39] were able to stabilized another [Fe4S4]0 cluster, [Fe4S4(IiPrMe2)4] (2, IiPrMe2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) (Fig. 2) [40]. These examples indicate that the use of stabilizing ligands that exhibit a combination of π-acidic and strong σ-bonding properties is crucial for the isolation of synthetic [Fe4S4]0 clusters.

Synthesis of [Fe4S4]0 clusters [Fe4S4(CN)4]4− (1) and [Fe4S4(IiPrMe2)4] (2; IiPrMe2 = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene)
2.2 Physical Properties of the Super-Reduced Clusters
The Fe centers of [Fe4S4]0 clusters 1 and 2 are supported by non-native π-acidic ligands. Nevertheless, their structures closely resemble the [Fe4S4]0 cluster in the Fe protein from Azotobacter vinerandii (Av). As summarized in Table 1, the average Fe-Fe/Fe-S bond distances of 1 and 2 are nearly identical to those of the super-reduced Av Fe protein, as determined by X-ray crystallography [42] and extended X-ray absorption fine structure (EXAFS) spectroscopy [43]. A structural comparison of [Fe4S4]0 clusters and the [Fe4S4]+ cluster [Fe4S4(CN)4]3− [41], which is the one-electron oxidized form of 1, allows evaluating the influence of the oxidation state on the [Fe4S4] core structures. A notable difference in [Fe4S4]0,+ clusters lies in the volumes of the S4 tetrahedra, which are larger for [Fe4S4]0 clusters (6.14–6.21 Å3) than for the [Fe4S4]+ cluster [Fe4S4(CN)4]3− (5.64 Å3). Similarly, the volume of the S4 tetrahedron in the thiolate-supported [Fe4S4]2+ and [Fe4S4]+ clusters, [Fe4S4(SR)4]2−/3–, is smaller than 6 Å3 [27], indicating that the volume of the S4 tetrahedron may serve as a diagnostic parameter to identify the super-reduced [Fe4S4]0 state. Some theoretical studies have been conducted in order to understand the physical properties of the [Fe4S4]0 clusters [44, 45], but the postulated relationship between the oxidation state of [Fe4S4] clusters and the volume of the S4 tetrahedron remains unclear.
Similarities between the [Fe4S4]0 clusters of 1 and 2 and the super-reduced Av Fe protein can also be found in their Mössbauer spectra. The spectra of 1 and 2 display two doublets with δ = 0.65/0.65 mm/s and ΔE Q = 1.45/2.00 mm/s (1:1 ratio; 1) as well as δ = 0.54/0.62 mm/s and ΔE Q = 2.92/1.54 mm/s (1:3 ratio; 2) at 77 K [38, 40]. The spectrum for the super-reduced Av Fe protein exhibits two doublets at δ = 0.68/0.68 mm/s with ΔE Q = 3.08/~1.5 mm/s (1:3 ratio). Electron paramagnetic resonance (EPR) and more detailed Mössbauer spectroscopic investigations on 2 revealed an S = 4 ground state for this cluster [45], and the same assignment should be applicable to the super-reduced Fe protein, as the g tensor of 2 (g = 16.08) observed by parallel-mode EPR is very similar to that of the Av Fe protein (g = 16.4) [22].
3 P-Cluster Models
The [Fe8S7] composition common to the MoFe and VFe nitrogenases is referred to as the P-cluster, which has been suggested to mediate electron-transfer processes through its redox activity. In the reduced form, denoted as the PN state, the [Fe8S7] core has been described as a fused form of two cuboidal [Fe4S4] clusters that share one of the sulfides. This inorganic core is supported by two bridging and four terminal thiolate moieties from cysteine residues. The two-electron oxidized form of PN is denoted as the POX state (or the P2+ state), which has the same core composition but a more open configuration due to the cleavage of two Fe-S bonds with the central sulfide and coordination of a serine residue and a backbone amide moiety (Fig. 3) [5]. The one-electron oxidized P1+ state has been detected as a transient species using spectroscopic methods [46, 47], while its structure has recently been determined by X-ray crystallography upon electrochemical generation of such a P1+ state [48]. In comparison with the PN-cluster, the [Fe8S7] core in the P1+ state lacks an Fe-S bond with respect to the central sulfide and instead forms an Fe-O bond with a serine residue. Thus, the P1+ state displays an intermediary structure between the PN and POX states. The redox-dependent dynamic structural rearrangements across the PN, P1+, and POX states should be important to regulate the electron flow from the [Fe4S4] cluster of the Fe protein to the P-cluster and then to the M-cluster, while the redox couple of the PN/P1+ states has been proposed to be predominant under the turnover conditions of nitrogen fixation [8].

Redox-dependent structural rearrangement of the P-cluster in the MoFe protein. PDB ID: 3U7Q. Color legend: C, gray; Fe, orange; N, blue; O, red; S, yellow
Early structural models of the P-cluster were based on dimers of [Fe4S4] cubes, such as the sulfido-bridged [Fe4S4]-(μ 2-S)-[Fe4S4] and edge-bridged [Fe4S4]-[Fe4S4] clusters [14, 15, 26,27,28,29], because the structure of the P-cluster had initially been proposed as two [Fe4S4] clusters bridged by cysteine residues [49] until the precise structure was reported in 1997 [5]. Although these [Fe4S4] dimers are no longer considered to represent P-cluster models, the former [Fe4S4]-(μ 2-S)-[Fe4S4] cluster was coincidentally discovered to be the cofactor of a double-cubane cluster protein from Carboxydothermus hydrogenoformans (DCCPCh) [50]. The [Fe4S4]-(μ 2-S)-[Fe4S4] cluster of DCCPCh catalyzes the reduction of acetylene, which indicates its potential for the reduction of small molecules.
Previous attempts to extract the P-cluster from the protein by addition of excess thiol (HSR, R = p-[dichloro(fluoro)methyl]phenyl) resulted in the degradation of the [Fe8S7] core, furnishing [Fe4S4] clusters in high yield (>90%) [51]. This result indicates the importance of the specific arrangement of six cysteines for the stabilization of the [Fe8S7] core of the P-cluster, which renders the chemical synthesis of the [Fe8S7] cluster challenging. It should also be noted that the μ 6-S atom at the center of the [Fe8S7] core is not only unique to the P-cluster among the biological iron-sulfur clusters but also rare in synthetic metal-sulfur clusters. Thus, synthetic strategies for the P-cluster models have been directed toward how to generate such an unusual μ 6-S center. Here we address three strategies that have been devised to meet this requirement.
3.1 Rearrangement of Edge-Bridged Mo(V)-Fe-S Double-Cubane Clusters
While the P-cluster core contains only Fe and S atoms, the first structurally identified molecule with μ 6-S atoms was a heterometallic Mo-Fe-S cluster. An edge-bridged [MoFe3S4] double-cubane precursor, [(Cl4-cat)MoFe3S4(PEt3)3]2 (3, Cl4-cat = tetrachlorocatecholate) [52], was treated with 2 equiv. of [NEt4][SH], leading to the rearrangement of the cluster core to give a complicated mixture. From this mixture, crystals of the giant [Mo2Fe6S9]-[Mo2Fe8S12]-[Mo2Fe6S9] cluster, which consists of two P-cluster-like [Mo2Fe6S9] units and a bridging [Mo2Fe8S12] units, were obtained [53]. From a similar reaction of 3 with [NEt4][SH] and KC14H10 (potassium anthracenide), a dimer of P-cluster-like [Mo2Fe6S9] clusters bridged by potassium atoms and sulfides was obtained. This synthetic method was further modified to employ [MFe3S4]-[MFe3S4] (M = Mo (4a), V (4b)) clusters bearing tris(pyrazolyl)hydroborate (Tp) ligands on the heterometals (M), and their structural rearrangement in the presence of [NEt4][SH] proceeds in a more controlled manner to provide the P-cluster models [(Tp)2Mo2Fe6S9(SH)2]3− (5a) and [(Tp)2V2Fe6S9(SH)2]4− (5b) (Fig. 4) [54, 55]. In this case, the protection of M by the tridentate Tp ligand may extend the lifetime of intermediary species generated from the precursor, facilitating the formation of 5a and 5b. A possible intermediate is a sulfur-voided [MFe3S3]-[MFe3S4] cluster that contains an incomplete cubane-type [MFe3S3] fragment (cf. Sect. 4.1.2), in which the sulfur-voided corner can accommodate a sulfur atom of the neighboring [MFe3S4] cube to furnish a central μ 6-S atom.

Core rearrangement of the edge-bridged double-cubane clusters into [(Tp)2Mo2Fe6S9(SH)2]3− (5a) and [(Tp)2V2Fe6S9(SH)2]4− (5b)
The structural rearrangement of the [MFe3S3]-[MFe3S4] double-cubane into PN-type [M2Fe6S9] is triggered by hydrosulfide (HS−), hydroselenide (HSe−), methoxide (MeO−), or ethane thiolate (EtS−). Attempts to introduce further structural modifications on the [M2Fe6S9] clusters have had limited success so far. For example, terminally bound HS− ligands or μ 2-bridging sulfides have been replaced with cyanides [56] and MeO− [57], respectively, while substitution of μ 2-sulfides with thiolates has not been achieved. Recovery of the double-cubane structure from the PN-type cluster has been demonstrated by the reaction of [(Tp)2Mo2Fe6S8(OMe)3]3− with Me3SiX (X = Cl, Br), where MeO− is replaced by X−. Such core convertibility indicates a comparable thermodynamic stability for the [MoFe3S3]-[MoFe3S4] and [Mo2Fe6S9] cores.
A number of M-Fe-S (M = Mo or V) clusters in this section feature a [M2Fe6S9] core, which is a fused form of two cubes with a central μ 6-S atom, two inter-cubane μ 2-sulfides, and peripheral M atoms, that exhibits a similar arrangement to that of the metal and sulfur atoms in the PN-cluster. Their structural similarity is further supported by the superposition of the [M2Fe6S9] cores of 5a (M = Mo) and 5b (M = V) with the [Fe8S7(μ 2-S-Cys)2] core of the PN-cluster and the obtained weighted root mean square deviations (RMSDs) of 0.38 Å (5a vs. PN) and 0.33 Å (5b vs. PN) [54]. In the Mössbauer spectra of clusters 5a and 5b at 4.2 K, a broad doublet is observed for 5a at δ = 0.55 mm/s with ΔE Q = 0.62 mm/s, while two overlapping doublets are found for 5b at δ = 0.52/0.59 mm/s with ΔE Q = 1.23/0.65 mm/s (major/minor = 3:1). These δ values indicate a relatively reduced Fe(II) state, in agreement with an all-ferrous state of the PN-cluster [58]. The relatively low Fe(II) state in [M2Fe6S9] clusters indicates the retention of the oxidation state of Fe in edge-bridged double-cubane precursors prepared by chemical reduction of single cubanes. For instance, cluster 3 was prepared by reduction of the chloride-bound [MoFe3S4] cube in the presence of PEt3 [52]. Similarly, the [MoFe3S3]-[MoFe3S4] precursor for 5a was prepared from [(Tp)MoFe3S4Cl3]− through substitution of the iron-bound chlorides with PEt3 and subsequent reduction with [NBu4][BH4].
3.2 Self-Assembly in a Nonpolar Media
A successful approach to reproduce the [Fe8S7] core of the P-cluster is the self-assembly reaction shown in Fig. 5, where an iron(II) amide complex Fe{N(SiMe3)2}2 is treated with HSTip (Tip = 2,4,6-tri(isopropyl)phenyl), tetramethylthiourea [SC(NMe2)2], and elemental sulfur (S8) in toluene. This reaction selectively furnishes the crystalline [Fe8S7] cluster [Fe4S3{N(SiMe3)2}(SC(NMe2)2)]2(μ 6-S){μ 2-N(SiMe3)2}2 (6) in up to 82% yield [59, 60]. For this assembly reaction, some elementary steps can be postulated: (1) the –N(SiMe3)2 group on iron should serve as a Brønsted base to abstract a proton from HSTip, which leads to a ligand exchange between –N(SiMe3)2 and –STip; (2) a subsequent treatment with S8 results in the oxidation of the Fe centers via the formation of Fe-S bonds; (3) the oxidation reaction in (2) should be followed by a reduction process to retain the average oxidation state of Fe between Fe(II) and Fe(III) through the reductive elimination of disulfide TipS-STip; (4) upon dissociation of some –STip ligands as TipS-STip, vacant coordination sites are generated on the Fe centers, which facilitate the aggregation of small iron-sulfur intermediates into high-nuclearity species. Steps (2)–(4) are repeated until (a) the depletion of S8 and (b) the product becomes thermodynamically and/or kinetically stable enough for isolation. It is interesting to note that once isolated, 6 is stable for a few hours in solution at 50°C, suggesting that the core structure of the P-cluster is one of the thermodynamically stable forms of such iron-sulfur clusters.

Synthesis of the [Fe8S7] clusters [Fe4S3{N(SiMe3)2}(SC(NMe2)2)]2(μ 6-S){μ 2-N(SiMe3)2}2 (6) and [(SAr){CpFe(C6H5S)}Fe4S3]2(μ 6-S){μ-N(SiMe3)2}2 (7; Ar = 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl), which reproduce the core of the P-cluster
The [Fe8S7] core of cluster 6 reproduces well that of the PN-cluster (Fig. 6), and in fact, a structural comparison between 6 and the PN-cluster from the Protein Data Bank (ID: 3U7Q) [6] provided a low RMSD value (0.34 Å) [61]. The Mössbauer spectrum of 6 exhibits two doublets in an approximate ratio of 3:1 at δ = 0.61/0.37 mm/s with ΔE Q = 0.54/1.28 mm/s (major/minor), which indicates a formal Fe(II)6Fe(III)2 oxidation state. This oxidation state corresponds to the POX state, which is the two-electron oxidized form of the all-ferrous PN state [62], while cluster 6 adopts a PN-type structure. The discrepancy between the oxidation states of 6 and the PN-cluster may be partly attributed to tentative hydrogen bonding between the PN-cluster and adventitious water and/or the peptide backbone. The dependence of the redox potentials of [Fe4S4] clusters on the number of hydrogen bonds between the clusters and the water/peptide backbone has been discussed elsewhere [29, 63, 64]. In contrast to the PN-cluster embedded in the protein, cluster 6 is in a completely hydrophobic environment, facilitating a higher oxidation state. An additional factor speculated for the relatively high oxidation state of 6 is the strong electron-donating property of the –N(SiMe3)2 ligands.

Structural comparison of 6 and the PN-cluster (transparent) via an overlay. The PN-cluster is obtained from a crystal structure of MoFe nitrogenase (PDB ID: 3U7Q). Color legend: C, gray; Fe, orange; N, blue; S, yellow; Si, wheat
The [Fe8S7] core of 6 is supported by amide and thiourea ligands, which have less relevance to the native P-cluster. Thus, replacement of these ligands with cysteine analogues was attempted to provide improved models. The –N(SiMe3)2 and thiourea ligands in 6 could be replaced by –SR via reactions with HSR and –SR, respectively; however, such ligand exchange reactions require careful optimization of the conditions due to the facile degradation of the [Fe8S7] core. Thus, the reaction of 6 with 2 equiv. of CpFe(C6H5S) (Cp = cyclopentadienyl) [65] and HSAr (Ar = 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl) at −40°C in fluorobenzene enabled the substitution of the terminal amide and thiourea ligands with thiolates to afford the [Fe8S7] cluster [(SAr){CpFe(C6H5S)}Fe4S3]2(μ 6-S){μ-N(SiMe3)2}2 (7), which bears four terminal thiolate ligands (Fig. 5) [60]. In agreement with the facile degradation of the native P-cluster in the presence of excess thiol [51], the [Fe8S7] core of 6 readily decomposes into [Fe4S4] clusters in the presence of proton sources and nucleophiles, possibly because cleavage of the μ 2-bridging ligand in the middle of the cluster triggers such irreversible degradation. This assumption may also explain why the replacement of the μ 2-N(SiMe3)2 ligands in 6 has not been successful so far.
3.3 Reductive Desulfurization of a High-Valent [Fe4S4] Cluster
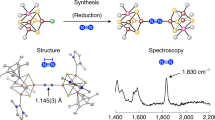
Another “nonpolar” approach for the formation of the PN-type [Fe8S7] cluster is the reductive desulfurization of a highly oxidized [Fe4S4] cluster [66]. [Fe4S3{N(SiMe3)2}(SPR3)]2(μ 6-S){μ 2-N(SiMe3)2}2 (R = Me (8a), Et (8b)), i.e., analogues of 6 that bear phosphine sulfides SPR3 (R = Me, Et) instead of tetramethylthiourea, have been obtained from the reaction of an all-ferric [Fe4S4]4+ cluster [Fe4S4{N(SiMe3)2}4] [67] with phosphines (Fig. 7a). In this reaction, the phosphine abstracts one of the sulfur atoms of the [Fe4S4] cube to produce SPR3 and a transient sulfur-voided [Fe4S3] cluster. The vacant Fe sites of this tentative [Fe4S3] intermediate have been proposed to capture a sulfur atom of the [Fe4S4] cube to furnish the central μ 6-S atom of the resulting [Fe8S7] core (Fig. 7b).

(a) Synthesis of [Fe4S3{N(SiMe3)2}(SPR3)]2(μ 6-S){μ 2-N(SiMe3)2}2 (R = Me (8a), Et (8b)) from an all-ferric [Fe4S4] cluster. (b) Proposed reaction pathway toward the [Fe8S7] core via the formation of an [Fe4S3] intermediate
The reaction pathway proposed for the formation of 8a and 8b has relevance to the biosynthesis of the P-cluster. The maturation of the P-cluster has been postulated as the coupling of two [Fe4S4] clusters under reducing conditions (for recent reviews, see [68, 69]). Gene knockouts and subsequent isolation of the MoFe protein from the resulting strain revealed that there is a precursor state of the P-cluster (P*-cluster) with an S = 1/2 EPR feature in the dithionite-reduced form, which is characteristic for [Fe4S4]+ clusters [70]. The assignment of the P*-cluster as a pair of [Fe4S4] cubes was further supported by an Fe K-edge EXAFS analysis [71]. The P*-cluster can be converted into the P-cluster in the presence of the Fe protein with ATP and a chaperone-like supporting protein (NifZ), as evident from the appearance of the characteristic EPR signal of the P-cluster at g = 11.8 in the parallel-mode spectrum [71, 72]. Thus, the P*-cluster, a pair of [Fe4S4] clusters, is likely converted into the [Fe8S7] core of the P-cluster via removal of a sulfur atom and generation of an [Fe4S3]-type intermediate [73].
4 M-Cluster Models
The catalytic site of MoFe nitrogenase, denoted as the M-cluster, is arguably the most complex and enigmatic metallo-cofactor in nature. The M-cluster core in the resting state consists of one Mo, seven Fe, nine S, and one C atoms. This [MoFe7S9C] core can be viewed as a fused form of [MoFe3S3C] and [Fe4S3C] cubes that share the central C atom and that is supported by three μ 2-S atoms in the middle [6, 7]. As one of the μ 2-S atoms can be exchanged with an inhibitor carbon monoxide molecule or a Se atom [9, 10], the displacement of such “belt” S atoms represents a plausible explanation for the generation of the catalytically active M-cluster.
In an early stage of the biosynthetic pathway of the M-cluster, the coupling of two [Fe4S4] clusters occurs via incorporation of a carbon atom derived from S-adenosylmethionine to give an [Fe8S9C] species, denoted as the L-cluster (Fig. 8) [74]. Subsequent replacement of one of the peripheral Fe atoms of the [Fe8S9C] core with Mo and incorporation of a homocitrate moiety, followed by inter-protein transfer of the cluster, eventually furnishes the M-cluster [68, 69, 75, 76]. Given that most of the details of the biosynthesis of the M-cluster have been uncovered, imitation of the biosynthetic processes seems to be a promising approach for the chemical synthesis of M-cluster models. However, two major obstacles are easily identified when attempting to mimic the biosynthetic pathway based on the current synthetic methods of metal-sulfur clusters: (a) the incorporation of a carbon atom derived from the CH3 moiety of S-adenosylmethionine and (b) the selective and asymmetric substitution of an Fe atom with Mo. Methods to carry out these difficult reactions remain challenging.

Overview of the M-cluster biosynthesis. The precursors, a pair of [Fe4S4] clusters (K-cluster) in the NifB protein, are transformed into the [Fe8S9C] cluster (L-cluster) and then into the M-cluster that is accompanied by inter-protein transfer of the clusters to NifEN and then to NifDK. Color legend: C, gray; Fe, orange; N, blue; Mo, teal; O, red; S, yellow
One of the intriguing properties of the M-cluster is its stability, even in the absence of a protein scaffold. Unlike the P-cluster, which is supported by six cysteine residues, the M-cluster is bound to the MoFe protein only by one cysteine and one histidine residues, and the Mo site carries a bidentate R-homocitrate ligand as a nonprotein ligand. Probably owing to the loose binding from only two protein residues, the M-cluster can be extracted from the protein into organic solvents such as N-methylformamide, N,N-dimethylformamide, or acetonitrile without significant degradation, where the catalytic activity is recovered after reintroduction into the original protein-binding site [77, 78]. The robustness of the M-cluster as a discrete molecule in solution has stimulated the interest of synthetic chemists toward its reproduction. While the significant amount of work related to M-cluster models is summarized elsewhere ([14, 15]; for representative reviews on the functional models of nitrogenase, see [79,80,81]), we will herein focus on some recent advances in synthetic models and potential approaches toward the reproduction of the M-cluster core.
4.1 [MS3] (M = Mo, W) Complexes as Building Blocks
Prior to the structural identification of the M-cluster, the available information was limited to, e.g., the proposed MoFe8S6 composition of the extracted cofactor [77]. Soon after, Holm and co-workers reported the synthesis of a double-cubane cluster with thiolate/sulfide inter-cubane bridges, [MoFe3S4(SEt)3](μ 2-S)2(μ 2-SEt), through the assembly reaction of [MoS4]2−, FeCl3, and EtS− [82]. The [MoFe3S4] cluster was intensively studied thereafter, together with other heterometallic cubanes such as the [VFe3S4] and [WFe3S4] clusters. One of the most significant results from these studies is arguably the synthesis of the PN-type model clusters, which is described in Sect. 3.1 [14]. Although the chemistry of these cubanes in the field of heterometallic cofactor models has been well developed, we herein approach the utility of metal trisulfide [MS3] (M = Mo, W) complexes, which serve as building blocks for heterometallic clusters.
4.1.1 [M6S9]-Type Clusters Derived from [MS3] Precursors
After the synthesis of organometallic trisulfide complexes of the type [Cp*MS3] (M = Mo, W; Cp* = pentamethylcyclopentadienyl) [83], the reactivity of the sulfide moiety was examined through the synthesis of various heterometallic clusters with noble metals such as Cu, Ag, and Au [84]. The successful isolation of discrete heterometallic clusters demonstrated the synthetic potential of [MS3] complexes as building blocks for biomimetic Mo-Fe-S and W-Fe-S clusters. In this context, [(Tp*)WS3]− (9, Tp* = tris(3,5-dimethylpyrazolyl)hydroborate) [85] was the first trisulfide complex in the field of nitrogenase cofactor models. While the initial study [86] reported analogues of relevant Tp-M systems (Tp = tris(pyrazolyl)hydroborate; M = Mo, V) (cf. Sect. 3.1 as well as [54,55,56,57, 87]), later this approach proved the utility of the [WS3] precursor in the synthesis of high-nuclearity clusters.
The reaction of 9 with FeCl2 (2 equiv.) and HS− (2 equiv.) generated [(Tp*)2W2Fe4S9]− (10), which can be further reduced to [(Tp*)2W2Fe4S9]2− (11) by treatment with [BH4]− (Fig. 9) [88]. More importantly, a slightly modified reaction using Se2− instead of HS− led to the formation of [(Tp*)2W2Fe4S6Se3]2− (12), which confirmed the retention of the [(Tp*)WS3] platform even after the assembly reaction with Fe and Se sources (Fig. 9). Retention of three sulfides on M (Mo or W) is a common feature in cluster synthesis employing [MS3] precursors. Analogous reactions of [(tBu3tach)MS3] (M = Mo (13), W (14); tBu3tach = 1,3,5-tri-(ter-butyl)-1,3,5-triazacyclohexane) with FeCl2, RS−, and Se2− provided heterochalcogenide-incorporated [MFe3S3Se] cubes (15–18), in which three sulfides are bound to M and thus the selenide is located at the position opposite to M (Fig. 10) [89].

Synthesis of [(Tp*)2W2Fe4S9]n− (n = 1 (10) or 2 (11); Tp* = tris(3,5-dimethylpyrazolyl)hydroborate) and Se-containing [(Tp*)2W2Fe4S6Se3]2− (12) from a template [WS3] complex

Heterochalcogenide incorporation into cubane clusters by using [(tBu3tach)MS3] (M = Mo (13), W (14); tBu3tach = 1,3,5-tri-(tertiary-Butyl)-1,3,5-triazacyclohexane) as a structural template
The zero-field 57Fe Mössbauer spectra of 10 and 11 show signals at δ = 0.37 (ΔE Q = 1.21) and δ = 0.42 (ΔE Q = 0.98), respectively, suggesting W(IV)2Fe(III)3Fe(II) (10) and W(IV)2Fe(III)2Fe(II)2 (11) states. Cyclic voltammetry (CV) measurements on 11 and 12 revealed that the incorporation of Se stabilizes the reduced states of the [W2Fe4S6Q3] (Q = S, Se) core, which is reflected in the positive shift of the [2–/3–] redox couple (E 1/2 = −1.91 V (12) and −1.97 V (11) in DMF vs. saturated calomel electrode (SCE)) as well as in the appearance of an irreversible [3–/4–] couple for 12. It should be noted that 10–12 are not the only [M6S9]-type clusters, i.e., other precedents of this class exist, e.g., [Fe6S9(SR)2]4− [90,91,92,93], [Fe6Se9(SR)2]4− [94], and [(edt)2Mo2Fe4S9]3−/4– (edt = ethane-1,2-dithiolate) [95], while that their synthesis involves typical assembly reactions employing Fe (and Mo) precursors, thiolates, and sulfide (selenide) sources.
Even though the trisulfide [MS3] (M = Mo, W) complexes are useful precursors for M-Fe-S(Se) clusters, reproduction of the asymmetric arrangement of metals in the M-cluster, in particular the location of Fe and Mo atoms at the opposite ends, has remained a significant challenge. We have recently revisited [Cp*MoS3]− (19) as a precursor of the Mo-Fe-S cluster and found a way to replicate the asymmetric arrangement of metals in the M-cluster. Surprisingly, a simple assembly reaction of 19 with FeCl2 (5 equiv.) and HS− (20 equiv.) resulted in the formation of [Cp*MoFe5S9(SH)]3− (20) in 54% yield (Fig. 11a) [96]. Similarly to other clusters, 20 also exhibits reversible redox properties at E 1/2 = −0.91 V ([2–]/[3–] couple) and −2.06 V ([3–]/[4–] couple) vs. Ag/AgNO3 in acetonitrile. It is interesting to note that 20 and [Fe6S9(SEt)2]4− catalyze the reduction of C1 substrates such as CN−, CO, and CO2 into short-chain hydrocarbons in the presence of reducing agents and proton sources [97].

(a) Synthesis of [Cp*MoFe5S9(SH)]3− (20). Overlay of 20 and the M-cluster (transparent): (b) top view and (c) side view. PDB ID: 3U7Q
A single-crystal X-ray diffraction analysis confirmed the asymmetric [MoFe5S9] core of 20. As shown in Fig. 11b, the peripheral positions of the [M6S9]-type inorganic core are occupied by Mo and Fe atoms. In comparison with the M-cluster [6], 20 lacks one of the Fe-(μ 2-S)-Fe moieties and possesses a central μ 4-S atom instead of the μ 6-C atom of the M-cluster. As a result, cluster 20 adopts a more open conformation than the M-cluster, which is indicated by the longer Mo⋯Fe distance between the opposite ends of 20 (7.473(1) Å) compared to the corresponding distance in the M-cluster (7.00 Å) (Fig. 11b) and the smaller dihedral angles between two Fe-(μ 3-S)-Fe planes opposing the μ 4-S atom in 20 (58.87(5)°) relative to the corresponding angle in the M-cluster with respect to the μ 6-C atom (83.1–86.6°) (Fig. 11c).
4.1.2 Conversion of [MS3] Complexes into Cuboidal Clusters as Potential Precursors for M-Cluster Models
Until the turn of the millennium, the central μ 6-atom of the M-cluster had not been identified, and its core structure had been considered as a pair of sulfur-deficient [M4S3]-type incomplete cubanes linked by three μ 2-S atoms. Thus, sulfur-voided [M4S3]-type clusters drew attention as suitable precursors for M-cluster models, and these are summarized elsewhere [15, 26]. Even after the precise structure of the M-cluster had been determined, the [M4S3]-type clusters or their equivalents remained potential and attractive precursors, given that a carbon atom can be accommodated at the sulfur-voided corner of the [M4S3] core to possibly link two [M4S3] fragments with a central μ 6-C atom. Even though the incorporation of a carbide ligand in a metal-sulfur cluster remains unprecedented, this section provides some examples of cubic and trinuclear clusters with a bridging light atom (N or O). The methods described herein may serve as a guide to devise further strategies to furnish metal-sulfur clusters that contain a μ 6-C atom.
Recently, cubic clusters of the type [WFe3S3Q] (Q = Cl, Br), in which Q is expected to be exchangeable, have been synthesized. For example, [(Tp*)WFe3S3(μ 3-Q)Q3]2− (Q = Cl (21), Br(22)) have been obtained from the reaction of the trisulfide complex [(Tp*)WS3]− (9) with FeQ2 (3 equiv.) in the presence of sodium benzophenone ketyl as the reducing agent (Fig. 12) [98]. As in the cases of other clusters prepared from such trisulfide complexes, the three sulfur atoms in the [WFe3S3Q] core remain attached to the W atom, and thus halide Q in clusters 21 and 22 occupies the corner opposite to W. An analogous reaction in the presence of FeQ2 (2 equiv.) led to the formation of trinuclear clusters [(Tp*)WFe2S3(μ 2-Q)Q2]− (Q = Cl (23), Br (24); Fig. 12). It has been proposed that in these cases, the presence of a reducing agent is important for the successful incorporation of halides in the cubic [WFe3S3(μ 3-Q)]2+ or the trinuclear [WFe2S3(μ 2-Q)]2+ cores. Other notable examples of cubic metal-sulfur clusters with μ 3-RN2− ligands are [Fe4(NtBu) nS4−nCl4]z− (n = 0–3, z = 0–2) [99,100,101], which were synthesized via stepwise assembly reactions using intermediary dinuclear iron-imide or iron-imide-sulfide complexes.

Synthesis of halide-containing clusters [(Tp*)WFe3S3(μ 3-Q)Q3]2− (Q = Cl (21), Br(22)) and [(Tp*)WFe2S3(μ 2-Q)Q2]− (Q = Cl (23), Br (24)) from a template [WS3] complex and their ligand substitution reactions
The structure of trinuclear [WFe2S3] cluster 23, which was determined by a single-crystal X-ray diffraction analysis, revealed that the mean Fe-(μ 3-Cl) distance (2.495(3) Å) is longer than the Fe-Clterminal distance (2.284(4) Å), suggesting a possible substitution of Cl. In fact, the core μ 2-Cl of 23 was replaced through salt metathesis reactions to furnish [WFe2S3] clusters with μ 2-N3 (25) and μ 2-OMe (26) ligands. On the other hand, an analogous approach for the substitution of μ 3-Cl in the cubic [WFe3S3Cl] cluster 21 remained unsuccessful, which indicates that the μ 3-Cl ligand is less labile relative to the μ 2-Cl ligand in 23. Successful examples for the replacement of μ 3-Cl in 21 include reactions with oxidative reactants such as Me3SiN3 − and S8, from which cubic clusters [(Tp*)WFe3S3(μ 3-X)Cl3]− (X = Me3SiN2− (27), S2− (28)) were obtained.
4.2 Nonpolar Approach and the Incorporation of Light Atoms
Following the successful synthesis of [Fe8S7] clusters modeling the PN-cluster (c.f. Sect. 3.2), the nonpolar approach was further extended to the synthesis of relevant iron-sulfur clusters, which are structurally analogous to the M-cluster [102, 103]. The precursors, i.e., an iron-thiolate complex [Fe(STip)(μ-SDmp)]2 (Tip = 2,4,6-tri(isopropyl)phenyl, Dmp = 2,6-di(mesityl)phenyl) and an iron-thiolate-mesityl complex (DME)Fe(SDmp)(mesityl) (DME = 1,2-dimethoxyethane), react with elemental sulfur in toluene at ambient temperature to afford [Fe8S7] clusters [(DmpS)Fe4S3]2(μ-SDmp)2(μ-SR)(μ 6-S) (R = Tip (29a), mesityl (29b)), which feature a central μ 6-S atom (Fig. 13). In these assembly reactions, the use of bulky thiolate ligands appears to be important to dissolve the precursors in toluene and to stabilize the products at an appropriate size with eight Fe atoms. By encapsulating the Fe-S cores, bulky substituents may provide kinetic stabilization that prevents further assembly beyond the target size, while sufficient thermodynamic stability is a general prerequisite for the synthesis of metal-sulfur clusters.

(a) Synthesis of [(DmpS)Fe4S3]2(μ-SDmp)2(μ-SR)(μ 6-S) (R = Tip (29a), mesityl (29b)). (b) Crystal structure of 29a (side and top view). Color legend: C, gray; Fe, orange; S, yellow
The molecular structures of 29a and 29b revealed that their common inorganic core is a fused form of two [Fe4S4] cubes that share the central μ 6-S atom, which is additionally supported by two μ 2-SDmp and one μ 2-SR (R = Tip or mesityl) ligands. The six inner Fe atoms around the μ 6-S atom are arranged in a slightly distorted trigonal prism. The sulfur-centered trigonal prismatic structure of 29a–b resembles that of the M-cluster. Due to the large size of the μ 6-S atom of 29a–b relative to the μ 6-C atom of the M-cluster, the edge Fe-Fe distances of the trigonal prisms of 29a (2.9103(10)–3.7050(10) Å) and 29b (2.9212(7)–3.6506(6) Å) are significantly longer than the corresponding distances of the M-cluster (2.58–2.62 Å). Given their homometallic nature, 29a–b can also be considered as structural models of the L-cluster (Fig. 8), which is a recently identified [Fe8S9C] precursor of the M-cluster [68, 69, 104, 105]. Antiferromagnetic interactions across the eight Fe atoms are a common feature of the L-cluster and 29a–b. In the EPR spectrum, the oxidized form of the L-cluster exhibits an isotropic S = 1/2 signal at g = 1.92 [105, 106], while clusters 29a–b in the [Fe8S7]5+ state display rhombic S = 1/2 signals at g = 2.19, 2.07, and 1.96 (29a) and g = 2.21, 2.07, and 1.95 (29b). These EPR signals are different from the S = 3/2 feature that appears at g = 4.31, 3.67, and 2.01 for the M-cluster in the resting state.
An oxygen atom can be encapsulated within an Fe-S cluster by the reaction of [Fe(OCPh3)(μ 2-SDmp)]2 with H2O and S8 (Fig. 14) [107]. The major product from this reaction, an [Fe8S6O] cluster (30), often co-crystallizes with the [Fe8S7] by-product (31). Therefore, their occupancy ratio within single crystals varies from 100/0 to 75/25 (30/31). In the crystal structure of 30, the central O atom displays a μ 4-binding mode, which stands in sharp contrast to the μ 6-mode of the central atoms of 29a–b (μ 6-S) and the M-cluster (μ 6-C). As a result, two inner Fe atoms of 30 deviate from the oxygen atom and interact with the mesityl groups of the μ 2-SDmp ligands (2.505(2) Å for the shortest Fe-C distance) surrounding the inner Fe atoms. Given the absence of some Fe-Ocentral bonds and the presence of compensating Fe-mesityl interactions, it seems possible to assume an analogous mode of substrate binding to the inner Fe atoms of the M-cluster. This speculation is consistent with a proposal for the M-cluster, where the N2-binding site is generated through the reversible cleavage or weakening of an Fe-(μ 6-C) bond [108, 109].

Synthesis and crystal structure of [(DmpS)Fe4S3O][(DmpS)Fe4S3](μ-SDmp)2(μ-OCPh3) (30)
5 Concluding Remarks and Future Directions for Nitrogenase Model Studies
In this review, we have summarized recent advances in synthetic metal-sulfur clusters that serve as models for the nitrogenase metallo-cofactors as well as some selected notable achievements of older studies. Representative recent developments include (a) the synthesis of all-ferrous [Fe4S4]0 clusters as models for the super-reduced [Fe4S4] cluster of the Fe protein; (b) the synthesis of [Mo2Fe6S9], [V2Fe6S9], and [Fe8S7] clusters, which model or reproduce the P-cluster; and (c) the synthesis of Mo-Fe-S, W-Fe-S, Fe-S, and Fe-S-O clusters, which are structurally relevant to the M-cluster. The chemical synthesis of such model clusters and their structural modifications remain attractive research topics, especially with respect to a better understanding of the properties of metallo-cofactors, given that spectroscopic studies on nitrogenases are often hampered by the presence of nontarget clusters. Since the (potential) models for the M-cluster remain insufficient as they lack key structural features, one of the most important issues to be addressed in future studies is the synthesis of more reliable M-cluster models, e.g., carbon-centered Mo-Fe-S clusters with eight metal atoms.
Another remaining major issue in nitrogenase studies is the relationship between the structure of the M-cluster and its N2-reducing function. Recent protein crystallographic studies on MoFe and VFe nitrogenases have disclosed some important details in this respect [9,10,11,12], implying the displacement of one of the belt μ 2-S atoms may be necessary for the generation of the reactive species. Thus, synthetic metal-sulfur clusters that feature such belt μ 2-S and central μ 6-C atoms are required. Furthermore, the N2 chemistry of metal-sulfur clusters is in a very early stage, and synthetic developments are needed to uncover the requirements for the reduction of N2 on metal-sulfur clusters. In this regard, it should be noted that a cubic Mo-Ti-S cluster is able to activate N2 at the Ti site under reducing conditions. The N2 moiety bridging two [MoS4Ti] cubes was converted into sub-stoichiometric amounts of NH3 and N2H4, demonstrating the molecular basis for the reduction of N2 on metal-sulfur clusters [110].
The application of synthetic metal-sulfur clusters in biochemical studies can offer a relatively new avenue of research. For instance, we have achieved the incorporation of [Fe6S9(SEt)2]4− into the M-cluster-binding site of the apo-MoFe protein and demonstrated the catalytic reduction of acetylene and CN− with this protein [111]. Furthermore, we have recently employed a synthetic [Fe4S4] cluster to elucidate the source of an additional sulfur atom required for the biosynthesis of the M-cluster [112]. Since analogous strategies should be applicable to various iron-sulfur proteins, the combination of synthetic chemistry and biochemistry represents one of the future directions for metal-sulfur chemistry.
References
Eady RR (1996) Structure−function relationships of alternative nitrogenases. Chem Rev 96:3013–3030. https://doi.org/10.1021/cr950057h
Danyal K, Dean DR, Hoffman BM, Seefeldt LC (2011) Electron transfer within nitrogenase: evidence for a deficit-spending mechanism. Biochemistry 50:9255–9263. https://doi.org/10.1021/bi201003a
Burgess BK, Lowe DJ (1996) Mechanism of molybdenum nitrogenase. Chem Rev 96:2983–3012. https://doi.org/10.1021/cr950055x
Hoffman BM, Lukoyanov D, Yang Z et al (2014) Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem Rev 114:4041–4062. https://doi.org/10.1021/cr400641x
Peters JW, Stowell MHB, Soltis SM, Finnegan MG, Johnson MK, Rees DC (1997) Redox-dependent structural changes in the nitrogenase P-cluster. Biochemistry 36:1181–1187. https://doi.org/10.1021/bi9626665
Spatzal T, Aksoyoglu M, Zhang L et al (2011) Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science 334:940–940. https://doi.org/10.1126/science.1214025
Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergmann U, DeBeer S (2011) X-ray emission spectroscopy evidences a central carbon in the nitrogenase iron-molybdenum cofactor. Science 334:974–977. https://doi.org/10.5061/dryad.6m0f6870
Seefeldt LC, Hoffman BM, Dean DR (2009) Mechanism of Mo-dependent nitrogenase. Annu Rev Biochem 78:701–722. https://doi.org/10.1007/978-1-61779-194-9_2
Spatzal T, Perez KA, Einsle O, Howard JB, Rees DC (2014) Ligand binding to the FeMo-cofactor: structures of CO-bound and reactivated nitrogenase. Science 345:1620–1623. https://doi.org/10.1126/science.1256679
Spatzal T, Perez KA, Howard JB, Rees DC (2015) Catalysis-dependent selenium incorporation and migration in the nitrogenase active site iron-molybdenum cofactor. elife 4:e11620. https://doi.org/10.7554/eLife.11620
Sippel D, Einsle O (2017) The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat Chem Biol 13:956–960. https://doi.org/10.1038/nchembio.2428
Sippel D, Rohde M, Netzer J, Trncik C, Gies J, Grunau K, Djurdjevic I, Decamps L, Andrade SLA, Einsle O (2018) A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 359:1484–1489. https://doi.org/10.1126/science.aar2765
Benediktsson B, Thorhallsson AT, Bjornsson R (2018) QM/MM calculations reveal a bridging hydroxo group in a vanadium nitrogenase crystal structure. Chem Commun 54:7310–7313. https://doi.org/10.1039/C8CC03793K
Lee SC, Holm RH (2004) The clusters of nitrogenase: synthetic methodology in the construction of weak-field clusters. Chem Rev 104:1135–1157. https://doi.org/10.1021/cr0206216
Ohki Y, Tatsumi K (2013) New synthetic routes to metal-sulfur clusters relevant to the nitrogenase metallo-clusters. Z Anorg Allg Chem 639:1340–1349. https://doi.org/10.1002/zaac.201300081
Holm RH, Kennepohl P, Solomon EI (1996) Structural and functional aspects of metal sites in biology. Chem Rev 96:2239–2314. https://doi.org/10.1021/cr9500390
Beinert H, Holm RH, Münck E (1997) Iron-sulfur clusters: nature’s modular, multipurpose structures. Science 277:653–659. https://doi.org/10.1126/science.277.5326.653
Beinert H (2000) Iron-sulfur proteins: ancient structures, still full of surprises. J Biol Inorg Chem 5:2–15. https://doi.org/10.1007/s007750050002
Johnson DC, Dean DR, Smith AD, Johnson MK (2005) Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem 74:247–281. https://doi.org/10.1146/annurev.biochem.74.082803.133518
Jasniewski AJ, Sickerman NS, Hu Y, Ribbe MW (2018) The Fe protein: an unsung hero of nitrogenase. Inorganics 6:25. https://doi.org/10.3390/inorganics6010025
Watt GD, Reddy KRN (1994) Formation of an all ferrous Fe4S4 cluster in the iron protein component of Azotobacter vinelandii nitrogenase. J Inorg Biochem 53:281–294. https://doi.org/10.1016/0162-0134(94)85115-8
Angove HC, Yoo SJ, Burgess BK, Münck E (1997) Mössbauer and EPR evidence for an all-ferrous Fe4S4 cluster with S = 4 in the Fe protein of nitrogenase. J Am Chem Soc 119:8730–8731. https://doi.org/10.1021/ja9712837
Rebelein JG, Stiebritz MT, Lee CC, Hu Y (2016) Activation and reduction of carbon dioxide by nitrogenase iron proteins. Nat Chem Biol 13:147–149. https://doi.org/10.1038/nchembio.2245
Stiebritz MT, Hiller CJ, Sickerman NS, Lee CC, Tanifuji K, Ohki Y, Hu Y (2018) Ambient conversion of CO2 to hydrocarbons by biogenic and synthetic [Fe4S4] clusters. Nat Catal 1:444–451. https://doi.org/10.1038/s41929-018-0079-4
Herskovitz T, Averill BA, Holm RH, Ibers JA, Phillips WD, Weiher JF (1972) Structure and properties of a synthetic analogue of bacterial iron-sulfur proteins. Proc Natl Acad Sci U S A 69:2437–2441. https://doi.org/10.1073/pnas.69.9.2437
Rao PV, Holm RH (2004) Synthetic analogues of the active sites of iron − sulfur proteins. Chem Rev 104:527–560. https://doi.org/10.1021/cr020615
Tan LL, Holm RH, Lee SC (2013) Structural analysis of cubane-type iron clusters. Polyhedron 58:206–217. https://doi.org/10.1016/j.poly.2013.02.031
Holm RH, Lo W (2016) Structural conversions of synthetic and protein-bound iron − sulfur clusters. Chem Rev 116:13685–13713. https://doi.org/10.1021/acs.chemrev.6b00276
Ohta S, Ohki Y (2017) Impact of ligands and media on the structure and properties of biological and biomimetic iron-sulfur clusters. Coord Chem Rev 338:207–225. https://doi.org/10.1016/j.ccr.2017.02.018
Georgiadis MM, Komiya H, Chakrabarti P et al (1992) Crystallographic structure of the nitrogenase iron protein from Azotobacter vinelandii. Science 257:1653–1659. https://doi.org/10.1126/science.1529353
DePamphilis BV, Averill BA, Herskovitz T, Que L Jr, Holm RH (1974) Synthetic analogs of the active sites of iron-sulfur proteins. VI. Spectral and redox characteristics of the tetranuclear clusters [Fe4S4(SR)4]2−. J Am Chem Soc 96:4159–4167. https://doi.org/10.1021/ja00820a018
Cambray J, Lane RW, Wedd AG, Johnson RW, Holm RH (1977) Chemical and electrochemical interrelationships of the 1-Fe, 2-Fe, and 4-Fe analogues of the active sites of iron-sulfur proteins. Inorg Chem 16:2565–2571. https://doi.org/10.1021/ic50176a030
Zhou C, Raebiger JW, Segal BM, Holm RH (2000) The influence of net charge on the redox potentials of Fe4S4 cubane-type clusters in aprotic solvents. Inorg Chim Acta 300–302:892–902. https://doi.org/10.1016/S0020-1693(99)00593-9
Crabtree RH (2014) The organometallic chemistry of the transition metals, 6th edn. Wiley, Hoboken
Goh C, Segal BM, Huang J et al (1996) Polycubane clusters: synthesis of [Fe4S4(PR3)4]1+,0 (R = But, Cy, Pri) and [Fe4S4]0 core aggregation upon loss of phosphine. J Am Chem Soc 118:11844–11853. https://doi.org/10.1021/ja9620200
Zhou H-C, Holm RH (2003) Synthesis and reactions of cubane-type iron-sulfur-phosphine clusters, including soluble clusters of nuclearities 8 and 16. Inorg Chem 42:11–21. https://doi.org/10.1021/ic020464t
Deng L, Majumdar A, Lo W, Holm RH (2010) Stabilization of 3:1 site-differentiated cubane-type clusters in the [Fe4S4]1+ core oxidation state by tertiary phosphine ligation: synthesis, core structural diversity, and S = 1/2 ground states. Inorg Chem 49:11118–11126. https://doi.org/10.1021/ic101702b
Scott TA, Berlinguette CP, Holm RH, Zhou H-C (2005) Initial synthesis and structure of an all-ferrous analogue of the fully reduced [Fe4S4]0 cluster of the nitrogenase iron protein. Proc Natl Acad Sci U S A 102:9741–9744. https://doi.org/10.1073/pnas.0504258102
Ingleson MJ, Layfield RA (2012) N-heterocyclic carbene chemistry of iron: fundamentals and applications. Chem Commun 48:3579–3589. https://doi.org/10.1039/c2cc18021a
Deng L, Holm RH (2008) Stabilization of fully reduced iron-sulfur clusters by carbene ligation: the [FenSn]0 oxidation levels (n = 4, 8). J Am Chem Soc 130:9878–9886. https://doi.org/10.1021/ja802111w
Scott TA, Zhou H-C (2004) The first all-cyanide Fe4S4 cluster: [Fe4S4(CN)4]3−. Angew Chem Int Ed 43:5628–5631. https://doi.org/10.1002/anie.200460879
Strop P, Takahara PM, Chiu HJ, Angove HC, Burgess BK, Rees DC (2001) Crystal structure of the all-ferrous [4Fe-4S]0 form of the nitrogenase iron protein from Azotobacter vinelandii. Biochemistry 40:651–656. https://doi.org/10.1021/bi0016467
Musgrave KB, Angove HC, Burgess BK, Hedman B, Hodgson KO (1998) All-ferrous titanium (III) citrate reduced Fe protein of nitrogenase: an XAS study of electronic and metrical structure. J Am Chem Soc 120:5325–5326
Torres RA, Lovell T, Noodleman L, Case DA (2003) Density functional and reduction potential calculations of Fe4S4 clusters. J Am Chem Soc 125:1923–1936. https://doi.org/10.1021/ja0211104
Chakrabarti M, Deng L, Holm RH, Münck E, Bominaar EL (2009) Mössbauer, Electron paramagnetic resonance, and theoretical study of a carbene-based all-ferrous Fe4S4 cluster: electronic origin and structural identification of the unique spectroscopic site. Inorg Chem 48:2735–2747
Tittsworth RC, Hales BJ (1993) Detection of EPR signals assigned to the 1-equiv-oxidized P-clusters of the nitrogenase MoFe-protein from Azotobacter vinelandii. J Am Chem Soc 115:9763–9767. https://doi.org/10.1021/ja00074a050
Chan JM, Christiansen J, Dean DR, Seefeldt LC (1999) Spectroscopic evidence for changes in the redox state of the nitrogenase P-cluster during turnover. Biochemistry 38:5779–5785. https://doi.org/10.1021/bi982866b
Keable SM, Zadvornyy OA, Johnson LE, Ginovska B, Rasmussen AJ, Danyal K, Eilers BJ, Prussia GA, LeVan AX, Raugei S, Seefeldt LC, Peters JW (2018) Structural characterization of the P1+ intermediate state of the P-cluster of nitrogenase. J Biol Chem 293:9629–96354. https://doi.org/10.1074/jbc.RA118.002435
Kim J, Rees DC (1992) Structural models for the metal centers in the nitrogenase molybdenum-iron protein. Science 257:1677–1682
Jeoung J-H, Dobbek H (2018) ATP-dependent substrate reduction at an [Fe8S9] double-cubane cluster. Proc Natl Acad Sci U S A 115:2994–2999. https://doi.org/10.1073/pnas.1720489115
Kurtz DM Jr, McMillan RS, Burgess BK, Mortenson LE, Holm RH (1979) Identification of iron-sulfur centers in the iron-molybdenum proteins of nitrogenase. Proc Natl Acad Sci U S A 76:4986–4989. https://doi.org/10.1073/pnas.76.10.4986
Demadis KD, Campana CF, Coucouvanis D (1995) Synthesis and structural characterization of the new Mo2Fe6S8(PR3)6(Cl4-cat)2 clusters. Double cubanes containing two edge-linked [MoFe3S4]2+ reduced cores. J Am Chem Soc 117:7832–7833
Osterloh F, Sanakis Y, Staples RJ, Münck E, Holm RH (1999) A molybdenum–iron–sulfur cluster containing structural elements relevant to the P-cluster of nitrogenase. Angew Chem Int Ed 38:2066–2070
Zhang Y, Zuo JL, Zhou H-C, Holm RH (2002) Rearrangement of symmetrical dicubane clusters into topological analogues of the P-cluster of nitrogenase: nature’s choice? J Am Chem Soc 124:14292–14293. https://doi.org/10.1021/ja0279702
Zhang Y, Holm RH (2003) Synthesis of a molecular Mo2Fe6S9 cluster with the topology of the P N cluster of nitrogenase by rearrangement of an edge-bridged Mo 2 Fe6S8 double cubane. J Am Chem Soc 125:3910–3920. https://doi.org/10.1021/ja0214633
Pesavento RP, Berlinguette CP, Holm RH (2007) Stabilization of reduced molybdenum-iron-sulfur single- and double-cubane clusters by cyanide ligation. Inorg Chem 46:510–516. https://doi.org/10.1021/ic061704y
Zhang Y, Holm RH (2004) Structural conversions of molybdenum-iron-sulfur edge-bridged double cubanes and PN-type clusters topologically related to the nitrogenase P-cluster. Inorg Chem 43:674–682. https://doi.org/10.1021/ic030259t
McLean PA, Papaefthymiouv V, Orme-Johnson WH, Münck E (1987) Isotopic hybrids of nitrogenase. J Biol Chem 262:12900–12903
Ohki Y, Sunada Y, Honda M, Katada M (2003) Synthesis of the P-cluster core of nitrogenases. J Am Chem Soc 125:4052–4053
Ohki Y, Imada M, Murata A et al (2009) Synthesis, structures, and electronic properties of [8Fe-7S] cluster complexes modeling the nitrogenase P-cluster. J Am Chem Soc 131:13168–13178
The RMSD value was calculated using the PyMOL software package (ver. 2.0.6). PyMol is an open-source software, released under https://pymol.org/2/
Pierik AJ, Wassink H, Haaker H, Hagen WR (1993) Redox properties and EPR spectroscopy of the P-clusters of Azotobacter vinelandii molybdenum-iron protein. Eur J Biochem 212:51–61. https://doi.org/10.1111/j.1432-1033.1993.tb17632.x
Dey A, Jenney FE, Adams MWW et al (2007) Solvent tuning of electrochemical potentials in the active sites of HiPIP versus ferredoxin. Science 318:1464–1468. https://doi.org/10.1126/science.1147753
Cowan JA, Lui SM (1998) Structure-function correlations in high-potential IRON proteins. Adv Inorg Chem 45:313–350. https://doi.org/10.1016/S0898-8838(08)60028-8
Helling JF, Hendrickson WA (1979) Synthesis and deprotonation of η 6-Arene-η 5-cyclopentadienyliron(II) compexes bearing NH2, OH or SH substituents. J Organomet Chem 168:87–95. https://doi.org/10.1016/S0022-328X(00)91996-X
Ohki Y, Tanifuji K, Yamada N, Cramer RE, Tatsumi K (2012) Formation of a nitrogenase P-cluster [Fe8S7] core via reductive fusion of two all-ferric [Fe4S4] clusters. Chem Asian J 7:2222–2224. https://doi.org/10.1002/asia.201200568
Ohki Y, Sunada Y, Tatsumi K (2005) Synthesis of [2Fe–2S] and [4Fe–4S] clusters having terminal amide ligands from an iron(II) amide complex. Chem Lett 34:172–173. https://doi.org/10.1246/cl.2005.172
Hu Y, Ribbe MW (2013) Nitrogenase assembly. Biochim Biophys Acta Bioenerg 1827:1112–1122. https://doi.org/10.1016/j.bbabio.2012.12.001
Ribbe MW, Hu Y, Hodgson KO, Hedman B (2014) Biosynthesis of nitrogenase metalloclusters. Chem Rev 114:4063–4080. https://doi.org/10.1021/cr400463x
Ribbe MW, Hu Y, Guo M et al (2002) The Femoco-deficient MoFe protein produced by a nifH deletion strain of Azotobacter vinelandii shows unusual P-cluster features. J Biol Chem 277:23469–23476. https://doi.org/10.1074/jbc.M202061200
Lee CC, Blank MA, Fay AW et al (2009) Stepwise formation of P-cluster in nitrogenase MoFe protein. Proc Natl Acad Sci U S A 106:18474–18478. https://doi.org/10.1073/pnas.0909149106
Hu Y, Fay AW, Lee CC, Ribbe MW (2007) P-cluster maturation on nitrogenase MoFe protein. Proc Natl Acad Sci U S A 104:10424–10429. https://doi.org/10.1073/pnas.0704297104
Rupnik K, Lee CC, Hu Y et al (2018) A VTVH MCD and EPR spectroscopic study of the maturation of the “second” nitrogenase P-cluster. Inorg Chem 57:4719–4725. https://doi.org/10.1021/acs.inorgchem.8b00428
Wiig JA, Hu Y, Lee CC, Ribbe MW (2012) Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science 337:1672–1675. https://doi.org/10.1126/science.1224603
Hu Y, Ribbe MW (2016) Biosynthesis of the metalloclusters of nitrogenases. Annu Rev Biochem 85:455–483. https://doi.org/10.1146/annurev-biochem-060614-034108
Sickerman NS, Ribbe MW, Hu Y (2017) Nitrogenase cofactor assembly: an elemental inventory. Acc Chem Res 50:2834–2841. https://doi.org/10.1021/acs.accounts.7b00417
Shah VK, Brill WJ (1977) Isolation of an iron-molybdenum cofactor from nitrogenase. Proc Natl Acad Sci U S A 74:3249–3253. https://doi.org/10.1073/pnas.74.8.3249
Burgess BK (1990) The iron-molybdenum cofactor of nitrogenase. Chem Rev 90:1377–1406. https://doi.org/10.1021/cr00106a002
McWilliams SF, Holland PL (2015) Dinitrogen binding and cleavage by multinuclear iron complexes. Acc Chem Res 48:2059–2065. https://doi.org/10.1021/acs.accounts.5b00213
Nishibayashi Y (2015) Recent progress in transition-metal-catalyzed reduction of molecular dinitrogen under ambient reaction conditions. Inorg Chem 54:9234–9247. https://doi.org/10.1021/acs.inorgchem.5b00881
Burford RJ, Fryzuk MD (2017) Examining the relationship between coordination mode and reactivity of dinitrogen. Nat Rev Chem 1:0026. https://doi.org/10.1038/s41570-017-0026
Wolff TE, Berg JM, Warrick C, Hodgson KO, Holm RH, Frankel RB (1978) The molybdenum-iron-sulfur complex [Mo2Fe6S9(SC2H5)8]3−. A synthetic approach to the molybdenum site in nitrogenase. J Am Chem Soc 100:4630–4632. https://doi.org/10.1021/ja00482a070
Kawaguchi H, Yamada K, Lang J, Tatsumi K (1997) A new entry into molybdenum/tungsten sulfur chemistry: synthesis and reactions of mononuclear sulfido complexes of pentamethylcyclopentadienyl–molybdenum(VI) and -tungsten(VI). J Am Chem Soc 119:10346–10358. https://doi.org/10.1021/ja971725e
Lang J, Ji S, Xu Q et al (2003) Structural aspects of copper(I) and silver(I) sulfide clusters of pentamethylcyclopentadienyl trisulfido tungsten(VI) and molybdenum(VI). Coord Chem Rev 241:47–60. https://doi.org/10.1016/S0010-8545(02)00309-0
Seino H, Arai Y, Iwata N et al (2001) Preparation of mononuclear tungsten Tris(sulfido) and molybdenum sulfido-tetrasulfido complexes with hydridotris(pyrazolyl)borate coligand and conversion of the former into sulfido-bridged bimetallic complex having Pt(μ-S)2WS core. Inorg Chem 40:1677–1682. https://doi.org/10.1021/ic0008823
Hong D, Zhang Y, Holm RH (2005) Heterometal cubane-type WFe3S4 and related clusters trigonally symmetrized with hydrotris(3,5-dimethylpyrazolyl)borate. Inorg Chim Acta 358:2303–2311. https://doi.org/10.1016/j.ica.2004.11.051
Fomitchev DV, McLauchlan CC, Holm RH (2002) Heterometal cubane-type MFe3S4 clusters (M = Mo, V) trigonally symmetrized with hydrotris(pyrazolyl)borate(1–) and Tris(pyrazolyl)methanesulfonate(1–) capping ligands. Inorg Chem 41:958–966. https://doi.org/10.1021/ic011106d
Zheng B, Chen XD, Zheng SL, Holm RH (2012) Selenium as a structural surrogate of sulfur: template-assisted assembly of five types of tungsten-iron-sulfur/selenium clusters and the structural fate of chalcogenide reactants. J Am Chem Soc 134:6479–6490. https://doi.org/10.1021/ja3010539
Majumdar A, Holm RH (2011) Specific incorporation of chalcogenide bridge atoms in molybdenum/tungsten-iron-sulfur single cubane clusters. Inorg Chem 50:11242–11251. https://doi.org/10.1021/ic2018117
Christou G, Holm RH, Sabat M, Ibers JA (1981) A hexanuclear iron-sulfide-thiolate cluster: assembly and properties of [Fe6S9(S-t-C4H9)2]4− containing three types of bridging sulfur atoms. J Am Chem Soc 103:6269–6271. https://doi.org/10.1021/ja00410a071
Christou G, Sabat M, Ibers JA, Holm RH (1982) A new structural type in iron-sulfide-thiolate chemistry: preparation, properties, and structure of the hexanuclear cluster [Fe6S9(S-t-C4H9)2]4−. Inorg Chem 21:3518–3526. https://doi.org/10.1021/ic00139a048
Henkel G, Strasdeit H, Krebs B (1982) [Fe6S9(SCH2C6H5)2]4−: a hexanuclear iron–sulfur cluster anion containing the square–pyramidal [(μ 4–S)Fe4] unit. Angew Chem Int Ed 21:201–202. https://doi.org/10.1002/anie.198202011
Strasdeit H, Krebs B, Henkel G (1984) Synthetic route to [Fe6S9(SR)2]4− clusters (R = alkyl). Their spectroscopic and magnetic properties and the solid-state structures of [Fe6S9(SCH2Ph)2]4− and [(Fe6S9(SMe)2)2Na2]6−. Inorg Chem 9:1816–1825. https://doi.org/10.1021/ic00181a008
Strasdeit H, Krebs B, Henkel G (1987) Synthesis and characterization, and the X-ray structure of (PhCH2NEt3)4[Fe6S9(SMe)2]. Z Naturforsch 42b:565–572
Zhou H, Su W, Achim C, Rao PV, Holm RH (2002) High-nuclearity sulfide-rich molybdenum-iron-sulfur clusters: reevaluation and extension. Inorg Chem 41:3191–3201. https://doi.org/10.1021/ic0201250
Tanifuji K, Sickerman N, Lee CC, Nagasawa T, Miyazaki K, Ohki Y, Tatsumi K, Hu Y, Ribbe MW (2016) Structure and reactivity of an asymmetric synthetic mimic of nitrogenase cofactor. Angew Chem Int Ed 55:15633–15636. https://doi.org/10.1002/anie.201608806
Sickerman NS, Tanifuji K, Lee CC, Ohki Y, Tatsumi K, Ribbe MW, Hu Y (2017) Reduction of C1 substrates to hydrocarbons by the homometallic precursor and synthetic mimic of the nitrogenase cofactor. J Am Chem Soc 139:603–606. https://doi.org/10.1021/jacs.6b11633
Xu G, Wang Z, Ling R, Zhou J, Chen X-S, Holm RH (2018) Ligand metathesis as rational strategy for the synthesis of cubane-type heteroleptic iron-sulfur clusters relevant to the FeMo cofactor. Proc Natl Acad Sci U S A 115:5089–5092. https://doi.org/10.1073/pnas.1801025115
Verma AK, Nazif TN, Achim C, Lee SC (2000) A stable terminal imide on iron. J Am Chem Soc 122:11013–11014. https://doi.org/10.1021/ja001147t
Chen X-D, Duncan JS, Verma AK, Lee SC (2010) Selective syntheses of iron-imide-sulfide cubanes, including a partial representation of the Fe-S-X environment in the FeMo cofactor. J Am Chem Soc 132:15884–15886. https://doi.org/10.1021/ja106478k
Chen XD, Zhang W, Duncan JS, Lee SC (2012) Iron–amide–sulfide and iron–imide–sulfide clusters: heteroligated core environments relevant to the nitrogenase FeMo cofactor. Inorg Chem 51:12891–12904. https://doi.org/10.1021/ic301868m
Ohki Y, Ikagawa Y, Tatsumi K (2007) Synthesis of new [8Fe-7S] clusters: a topological link between the core structures of P-cluster, FeMo-co, and FeFe-co of nitrogenases. J Am Chem Soc 129:10457–10465. https://doi.org/10.1021/ja072256b
Hashimoto T, Ohki Y, Tatsumi K (2010) Synthesis of coordinatively unsaturated mesityliron thiolate complexes and their reactions with elemental sulfur. Inorg Chem 49:6102–6109. https://doi.org/10.1021/ic100692v
Kaiser JT, Hu Y, Wiig JA et al (2011) Structure of precursor-bound NifEN: a nitrogenase FeMo cofactor maturase/insertase. Science 331:91–94. https://doi.org/10.1126/science.1196954
Fay AW, Blank MA, Lee CC et al (2011) Spectroscopic characterization of the isolated iron-molybdenum cofactor (FeMoco) precursor from the protein NifEN. Angew Chem Int Ed 50:7787–7790. https://doi.org/10.1002/anie.201102724
Hu Y, Fay AW, Ribbe MW (2005) Identification of a nitrogenase FeMo cofactor precursor on NifEN complex. Proc Natl Acad Sci U S A 102:3236–3241. https://doi.org/10.1073/pnas.0409201102
Ohta S, Ohki Y, Hashimoto T et al (2012) A nitrogenase cluster model [Fe8S6O] with an oxygen unsymmetrically bridging two proto-Fe4S3 cubes: relevancy to the substrate binding mode of the FeMo cofactor. Inorg Chem 51:11217–11219. https://doi.org/10.1021/ic301348f
Rittle J, Peters JC (2013) Fe-N2/CO complexes that model a possible role for the interstitial C atom of FeMo-cofactor (FeMoco). Proc Natl Acad Sci U S A 110:15898–15903. https://doi.org/10.1073/pnas.1310153110
Creutz SE, Peters JC (2014) Catalytic reduction of N2 to NH3 by an Fe–N2 complex featuring a C-atom anchor. J Am Chem Soc 136:1105–1115. https://doi.org/10.1021/ja4114962
Ohki Y, Uchida K, Tada M, Cramer RE, Ogura T, Ohta T (2018) N2 activation on a molybdenum-titanium-sulfur cluster. Nat Commun 9:3200. https://doi.org/10.1038/s41467-018-05630-6
Tanifuji K, Lee CC, Ohki Y, Tatsumi K, Hu Y, Ribbe MW (2015) Combining a nitrogenase scaffold and a synthetic compound into an artificial enzyme. Angew Chem Int Ed 54:14022–14025. https://doi.org/10.1002/anie.201507646
Tanifuji K, Lee CC, Sickerman NS, Tatsumi K, Ohki Y, Hu Y, Ribbe MW (2018) Tracing the ‘ninth sulfur’ of the nitrogenase cofactor via a semi-synthetic approach. Nat Chem 10:568–572. https://doi.org/10.1038/s41557-018-0029-4
Acknowledgment
Y. O. thanks the Japanese Ministry of Education, Culture, Sports, Science and Technology (16H04116 and 18H04246) and the Takeda Science Foundation for funding.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tanifuji, K., Ohki, Y. (2018). Recent Advances in the Chemical Synthesis of Nitrogenase Model Clusters. In: Ribbe, M. (eds) Metallocofactors that Activate Small Molecules. Structure and Bonding, vol 179. Springer, Cham. https://doi.org/10.1007/430_2018_26
Download citation
DOI: https://doi.org/10.1007/430_2018_26
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-25896-2
Online ISBN: 978-3-030-25897-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)