
Abstract
Iron–sulfur clusters are important biological cofactors that are used for electron transfer and also for reactivity. Though the iron atoms in these clusters are typically a mixture of ferrous iron(II) and ferric iron(III), there have been reports of biological iron–sulfur clusters in which all the iron atoms are reduced to the iron(II) oxidation state. These reports have inspired synthetic studies on all-ferrous iron–sulfur clusters. This chapter describes both the biological and synthetic work on all-iron(II) clusters, drawing comparisons and noting promising avenues for future research.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Iron–sulfur clusters have been an important topic of chemical research for several decades [1, 2]. As “clusters”, they contain more than one iron atom held close to other iron atoms, and the number of iron atoms can vary from 2 to 16 or more. Some examples of iron–sulfur cluster types are shown in Fig. 1. The bridges between iron atoms in these clusters are sulfides (S2–), which form strong bonds with Fe2+ and Fe3+ and often bridge multiple metal ions. Though the Fe–S bonds are thermodynamically stable, they are labile by virtue of (a) the weak ligand-field strength of sulfide, which leads to a high-spin electronic configuration of the iron atoms that places electrons in metal–ligand antibonding orbitals, and (b) the spherical charge density on the sulfide, which admits a range of bond angles and lengths and presumably gives low activation barriers for bond rearrangements.

The most common structures and oxidation levels of [2Fe–2S], [3Fe–4S], and [4Fe–4S] clusters. White circles represent Fe3+ (“ferric”), black circles represent Fe2+ (“ferrous”), and grey circles represent Fe2.5+ (delocalized valence)
Iron–sulfur clusters are naturally found in all known bacteria, plants, animals, and archaea [3, 4]. In biological systems, they are always part of metalloproteins, where they are covalently attached to thiolates from deprotonated cysteine amino acid sidechains. Because the cysteinates are anions and the sulfides are dianions, the overall charge on most biological iron–sulfur clusters is negative. This has important consequences for their chemistry, especially that a more polar medium tends to stabilize the more reduced form (i.e., gives a less negative redox potential), because this form bears the greater charge. Often the biological clusters can be extracted intact from the protein environment, through exchange with exogenous thiolate donors [5].
Iron–sulfur clusters play many roles in biological systems [3, 6, 7]. Much recent attention has focused on sensory and transcriptional regulation roles [8, 9]. Nevertheless, one of the most important functions of iron–sulfur clusters in biological systems is to be a one-electron redox shuttle. Many metabolic processes use iron–sulfur enzymes (also denoted as “ferredoxins”) as part of the electron transport chain. Though the proteins are generally air sensitive, they are used in both aerobic and anaerobic organisms. They are well adapted to electron-transfer roles because their structures change little between redox levels. From a coordination chemistry point of view, the structural invariance with redox changes arises from two factors: (a) the tetrahedral geometry at the iron atoms is similar for both Fe2+ and Fe3+, and (b) the accessible states are often mixed valence, with delocalized charge. Because there is little reorganization energy associated with redox changes, rapid electron transfer is possible. A number of proteins have several iron–sulfur clusters of graded potential that act as electron conduits (for example, [10]).
Other iron–sulfur clusters are at the active sites of enzymes. The longest-known iron–sulfur enzyme is aconitase [11], which brings about a non-redox reaction within the tricarboxylic acid (TCA) cycle, but most iron–sulfur cluster enzymes accomplish redox reactions. Iron–sulfur clusters are especially common in enzymes that perform redox transformations of simple small molecules, such as H2 (Fe-only and Fe–Ni hydrogenases), N2 (nitrogenase), CO and CO2 (Ni–Fe CO dehydrogenase) in bacteria and archaea. It is possible that the widespread role of FeS clusters in small-molecule transformations is a reflection of the conditions of early Earth: when these enzymes evolved, Earth’s environment was anaerobic and sulfidic with a significant amount of dissolved Fe2+ [12]. It is notable that iron–sulfur clusters can self-assemble from dissolved iron, hydrosulfide, and thiolates [13], indicating that they could have been formed readily from available raw materials early in the history of life. Both iron–sulfur clusters and iron–sulfur minerals have even been postulated as key catalysts in primordial metabolism that led to the first life forms [14, 15].
The role of iron–sulfur clusters in the biological reduction of small molecules is paralleled by the catalytic ability of solid iron sulfides in small-molecule reactions of CO [15] and CO2 [16]. In addition, solid iron sulfide has been observed to catalyze ammonia formation from N2 [17]. Understanding soluble iron–sulfur clusters (in biological and abiological systems) may give insight into possible mechanisms at the surfaces of these solids.
Because of the redox activity of the clusters, there have been many studies on the redox potentials and spectroscopic features of FeS cluster-containing compounds in different oxidation states. The accumulated data have shown that the most common oxidation levels of iron–sulfur clusters have a mixture of Fe2+ (ferrous) and Fe3+ (ferric). Often iron atoms have delocalized valence, with some iron ions best described as Fe2.5+ because they share an electron equally with another iron site. In general, the Kramers states (those with an odd number of total electrons, corresponding to an odd number of iron(III) centers) are more easily detected and characterized because they show characteristic signals in perpendicular-mode electron paramagnetic resonance (EPR) spectra. Iron-57 Mössbauer spectroscopy is a useful technique, because it shows all iron ions, and the characteristic isomer shift (δ) and quadrupole splitting (ΔE Q) often are used to specify the oxidation state of the iron. Other techniques, such as X-ray absorption, magnetic circular dichroism, and resonance Raman are also useful. The combination of these spectroscopic methods has shown that the common oxidation states are those shown in Fig. 1 [1, 2].
It is interesting that none of the common cluster states in Fig. 1 has all of the iron atoms as Fe2+ (“all-ferrous”). All-ferrous states are uncommon because the buildup of additional negative charge on the cluster is difficult, and could require a reducing agent outside the physiologically possible range. Despite these difficulties, a few teams of biochemists, chemists, and spectroscopists have been able to identify iron–sulfur clusters in biological and synthetic systems that are all ferrous. These rare and interesting systems are the focus of this review. They push the limits of chemistry for several reasons. First, they are usually very sensitive and difficult to isolate, because of the very negative redox potential at which they are formed, and because of oxygen sensitivity. Second, all-ferrous clusters are non-Kramers systems, and thus give little to no EPR signal, especially with the common X-band microwave frequency. Because of the muted EPR signature, it is possible that other all-ferrous clusters in biology have not yet been detected. Finally, they offer interesting spectroscopic challenges, which have been highlighted in a recent minireview [18].
This review describes the biological examples of all-ferrous clusters, followed by synthetic clusters. In order to limit the review to a reasonable size, we do not describe the large literature on nitrosyl, cyclopentadienyl, and carbonyl-containing clusters, which have been comprehensively reviewed [19]. We have also omitted other related clusters that have a low-spin electronic configuration at iron, in order to focus on the high-spin clusters that are most comparable to the biological systems. We do not include heteronuclear clusters that contain iron in addition to other transition metals (most often nickel or molybdenum), which have been included in larger reviews [7, 20–22].
It is our hope that this organized description of highly reduced iron–sulfur clusters will assist biological chemists who may encounter these clusters in new proteins and enzymes. In addition, we hope that this systematic review of the recent exciting developments is of use to synthetic chemists who desire to expand the range of known clusters. In order to facilitate comparisons between the biological and synthetic systems, all redox potentials in this review are given versus the hydrogen electrode (NHE), even though the synthetic clusters have been reported in the primary literature against a variety of different reference potentials.
2 Biological Examples of All-Ferrous Iron–Sulfur Clusters
One of the interesting aspects of the field of highly reduced iron–sulfur clusters is that the examples in biological molecules often preceded the synthetic examples. In several cases, no synthetic analogue exists. This may seem surprising, given that synthetic compounds and non-aqueous solvents offer more options for protecting sensitive, highly reducing metal centers. However, natural iron–sulfur clusters are held in protein environments that stabilize negative charges on the cluster. As a result, the same formal redox couple typically has a much more negative redox potential in synthetic clusters than in the analogous biological system with the same coordinating ligands. The synthetic clusters are accordingly more difficult to isolate in highly reduced forms, and recent progress (described below) has depended on the use of abiological ligand sets.
What makes the protein able to stabilize large negative charges on such highly reduced sites? As will become evident below, moderation of the redox potential in the biological systems often comes from interactions with protons: either the cluster may become protonated, or there may be hydrogen bonds to the rest of the protein. Hydrogen bonds to the coordinating cysteine thiolate residues are a ubiquitous feature of biological iron–sulfur sites. The electron-withdrawing influence of hydrogen bonds diffuses the buildup of charge density on the cluster, and acts to make the redox potential more positive [23, 24].
2.1 Two-Iron Clusters
The simplest type of iron–sulfur cluster in biological systems is the [2Fe–2S] cluster, which typically transfers a single electron to convert between the [2Fe–2S]2+ (oxidized, with two Fe3+) and [2Fe–2S]+ (reduced, with mixed-valence Fe2+Fe3+) oxidation levels. However, a few situations have been identified in which both iron atoms may be reduced to the ferrous oxidation level, giving a [2Fe–2S]0 cluster.
The first example is the reduction of plant [2Fe–2S] ferredoxins using chromium(II) 15-aneN4 (Fig. 2) [25–27]. Interestingly, Cr2+ is the only reductant reported to perform this reaction, and NMR studies show that the reduced protein binds the Cr3+ ion. It is likely that this binding imparts extra energy that enables Cr2+ to achieve the difficult reduction. Because of the influence of chromium binding, a formal redox potential is not defined for this transition. Mössbauer studies have shown that the [2Fe–2S]0 center in the protein is diamagnetic, resulting from strong (|J| > 80 cm−1) anti-ferromagnetic coupling between high-spin (S Fe = 2) sites to give an S total = 0 ground state [28]. This diamagnetic all-ferrous state has also been produced in frozen glasses using radiolytic reduction by gamma rays [28].

Reducing agents that have been used to generate the all-ferrous redox level in biological iron–sulfur clusters
Using electrochemical methods or europium(II) salts, certain Rieske proteins can be reduced beyond the usual mixed-valence [2Fe–2S]+ form to give diamagnetic [2Fe–2S]0 clusters [29, 30]. The iron–sulfur clusters in Rieske proteins are special because one of the iron atoms is coordinated by two histidine residues (Fig. 3). This change in coordination reduces the overall negative charge on the cluster, and makes all of the cluster redox potentials more positive by several tenths of a volt. Therefore, it is not surprising that these clusters are the least difficult to reduce to the unusual all-ferrous state. Despite this influence, the [2Fe–2S]+/0 potentials remain quite cathodic, near −0.8 V. This potential is pH-dependent, which implies that the reduced form is protonated, most likely on a bridging sulfide [30].

There is more negative charge on a normal [2Fe–2S]0 cluster (left) than on a Rieske [2Fe–2S]0 cluster, and so the Rieske cluster is less difficult to reduce to the all-ferrous state
2.2 Three-Iron Clusters
[3Fe–4S] clusters are typically observed in the [3Fe–4S]+ (Fe 3+3 ) and [3Fe–4S]0 (Fe2+Fe 3+2 ) states, but protein film voltammetry at low potentials has shown a two-electron reduction of the [3Fe–4S]0 form to the all-ferrous [3Fe–4S]2– oxidation level (Fig. 4) [31]. This phenomenon was observed in several different three-iron ferredoxins [32]. Bulk electrolysis yielded samples for UV–vis, EPR, and magnetic circular dichroism (MCD) characterization that were consistent with this formulation, although the data did not elucidate the ground spin state of the cluster. Though the standard potential for forming the highly reduced state is ca. −0.7 V, the potential is very pH-dependent. At pH 4, the potentials are close to −0.5 V.

In some [3Fe–4S] clusters, a two-electron reduction gives an all-ferrous state. Though protonation of the reduced form is evident from electrochemical data, the proton locations given here are speculative
This pH dependence also gives insight into the mechanism through which the protein stabilizes the significant negative charge on the cluster. The slope of the plot of pH vs. potential (the “Pourbaix diagram”) gives information on the number of protons transferred in the process. Interestingly, two protons accompany the two electrons during reduction of [3Fe–4S]0 to [3Fe–4S]2–, neutralizing the added positive charge and moderating the reduction potential. The site of protonation is not yet known for the three-iron ferredoxins, but the protonation may be one factor that makes electron transfer relatively slow in this system [33].
2.3 Four-Iron and Larger Clusters
2.3.1 Introduction to Nitrogenase
Considering that nitrogenase reduces N2, one of the most inert enzyme substrates in biological chemistry, it is not surprising that nitrogenase contains several examples of highly reduced iron–sulfur clusters. Because these clusters have been subjected to especially intense study, two sections below deal with the iron–sulfur clusters in nitrogenase. The molybdenum-dependent nitrogenase system has a total of three clusters that lie in two proteins: the Fe protein and the MoFe protein. The MoFe protein contains two eight-metal clusters, one of which is termed the “P cluster” and the other is called the “M cluster” or “FeMoco.” The orientation of the clusters in the Fe protein–MoFe protein complex indicates that electrons flow from the Fe protein to the P cluster, and from the P cluster to the FeMoco. (As discussed below, there is controversy about the order of these events, and about the number of electrons transferred in each event.) Both the P cluster and the Fe protein cluster have been characterized in states that are all-ferrous. The P cluster of the MoFe protein will be described first, and then the four-iron cluster of the Fe protein. The discussion will be selective, focusing only on the highly-reduced (all-ferrous) forms of each cluster.
2.3.2 Nitrogenase P Cluster
The P cluster has eight iron atoms and seven sulfides. The X-ray crystal structures of both Azotobacter vinelandii and Klebsiella pneumoniae proteins have been obtained in the dithionite-reduced form (PN). As indicated in Fig. 5, the reduced cluster is a “double-cubane” structure of two [4Fe–4S] clusters joined at a common six-coordinate sulfide [34, 35]. The cluster lies at the interface of the α and β subunits of the MoFe protein, and is held by three cysteine residues from each subunit. Two cysteine sulfur atoms bridge the two cubes, and each iron atom is four-coordinate (pseudotetrahedral) in the PN state.

The structure of the reduced PN cluster, which has been crystallographically characterized in this likely all-ferrous state
The reduced PN state is EPR silent and shows no magnetic splitting in its Mössbauer spectrum, indicating that it is diamagnetic [36]. Interestingly, it can be oxidized by two one-electron steps with potentials of −0.31 V (P1+/PN) and +0.08 V (P2+/P1+) [37]. Based on the isomer shifts in Mössbauer spectra, the PN form is thought to be all-ferrous [38]. Although it is beyond the scope of this review, it is worth noting that the X-ray crystal structure of the P2+ form displays a structural change where the two cubanes move apart [35]. The coupling of conformational and redox changes suggests a potential gating role for electron flow into the FeMoco [39]. The redox changes also correlate with the number of surface-exposed water molecules [40].
The P cluster is likely to play a role in electron transfer, because it is situated about 15 Å from the site at which the Fe protein binds and about the same distance from the FeMoco. It has been difficult to find direct support for this idea, because the resting-state PN form is EPR silent and thus difficult to follow spectroscopically. However, a Ser188Cys variant of the MoFe protein has a modified P cluster with a paramagnetic resting state, for which the EPR signal disappears during turnover and then reappears. This behavior is as expected for an electron relay [41, 42].
If the P cluster is indeed all ferrous and participates in electron transfer, it raises interesting issues that form the basis for continued research. First, the ferrous oxidation state is generally considered to the lowest possible oxidation state for biological iron (in the absence of strong-field donors like CO and CN– found in hydrogenases). However, if the P cluster in its resting PN (all-ferrous) state is to act as a typical relay by accepting an electron from the Fe protein and then passing it along to the FeMoco, it would need to be reduced to a state P1– that would have at least one iron atom below the ferrous state (Fig. 6a). Alternatively, the P cluster would need to transfer an electron to the FeMoco prior to being reduced by the Fe protein. This model has been termed “deficit spending” [43]. Though the deficit spending model implies that P would become oxidized, EPR spectroscopy shows no sign of an oxidized P cluster under conditions of low electron flux [45]. Thus, the FeMoco would pull an electron out of the P cluster, which would then suck an electron out of the Fe protein rapidly enough that no significant concentration of oxidized P cluster builds up (Fig. 6b). Dean, Hoffman, and Seefeldt have recently shown using a S188C mutant (which substitutes one donor to the P cluster to shift its potential) that electron transfer from the Fe protein to the P cluster is much faster than from the P cluster to FeMoco, supporting the feasibility of the deficit spending model [43].

Three models for electron transfer from the Fe protein (FN in its reduced, resting state) to the FeMoco (MN in its resting state; MR in its reduced state) through the P cluster (PN in its resting state). Mechanism (a) would require an unprecedented subferrous oxidation level. Mechanism (b) is termed the “deficit spending” model, where P → M electron transfer is followed by rapid backfilling by the Fe protein [43]. Mechanism (c) would skip the P cluster entirely, but seems unlikely given the spatial disposition of proteins and the influences of mutations near the P cluster on activity [42, 44]
The deficit spending mechanism requires docking of Fe protein with MoFe protein before the rate-limiting electron transfer from the P cluster to the FeMoco. But how does the FeMoco sense whether the Fe protein is present? Association, dissociation, and movement of the Fe protein on the outside of the MoFe protein cause no obvious conformational change near the FeMoco [46]. It is not clear how this dilemma will be resolved: perhaps there is actually transient formation of P1–; perhaps PN is not actually all-ferrous but instead Fe 2+6 Fe 3+2 ; or perhaps there is a “trigger” for electron flow that has not yet been discovered. One interesting possibility is that that Fe protein binding is coupled to coordination of S188 to the P cluster, lowering its potential and inducing electron transfer to FeMoco and then “backfilling” by the Fe protein [43].
Another issue is raised by the possibility of two-electron redox processes in both the Fe protein (see below) and the P cluster. This naturally raises the possibility of transferring two electrons at a time, which would make the enzyme more efficient. A two-electron transfer would explain why P is a “double cluster” and why all nitrogenase substrates are reduced by an even number of electrons. Two-electron transfers are rare because it would be difficult to balance the large amount of charge displacement; in nitrogenase, this charge balance could come from protonation of substrates at the FeMoco.
2.3.3 Nitrogenase Fe Protein
The Fe protein contains a single [4Fe–4S] cluster at the interface of two identical polypeptide chains. It may be reduced to a [4Fe–4S]+ (Fe 2+3 Fe3+) form by dithionite reduction. This reduced form, which has spectroscopic features typical of reduced [4Fe–4S] clusters, is essential for both the biosynthesis and the catalytic function of the nitrogenase system. Classic studies on the turnover mechanism by Thorneley and Lowe were understood in the context of the reduced Fe protein associating and dissociating from the catalytic MoFe protein once for each electron transferred [47]. This corresponded with the usual one-electron-transfer ability of iron–sulfur clusters. However, in 1994, Watt reported the reduction of the [4Fe–4S]+ (Fe 2+3 Fe3+) form of the Fe protein to a “super-reduced” form using two equivalents of reduced methylviologen (illustrated in Fig. 2b) at −0.46 V [48]. The reduced form was proposed to be all-ferrous [4Fe–4S]0 based on the stoichiometry of reduction and reoxidation. The iron(II) was also extracted by 2,2′-bipyridine, giving >93% of the expected iron(II) concentration.
Subsequent studies have used a variety of strong reducing agents to generate super-reduced clusters in the nitrogenase Fe protein, and evidence exists for two different forms, one with a total spin S = 4 and another with S = 0. The reason why it is possible to generate two different forms at the same redox level is not clear. (The possibility that spent reducing agent binds to the protein has been ruled out in each case described below.) As will be seen, the S = 4 state has been characterized in much greater detail and by multiple research groups, and its formation is well established. The S = 0 state has been reported in only one paper, and is much more difficult to study due to the inability to use magnetic measurements.
The S = 4 state of the all-ferrous cluster can be generated using Ti(III) citrate, deazariboflavin, Eu2+-DTPA [49], or Cr2+(15-aneN4), as well as by radiolytic reduction. Each of these reductants gives a characteristic g eff = 16 signal in EPR spectra [50]. The first definitive spectroscopic characterization of the S = 4 form came from Ti(III) citrate reduced protein that was examined using Mössbauer spectroscopy and parallel-mode X-band EPR spectroscopy at 2 K [51]. Both the Mössbauer isomer shift of δ = 0.68, and XAS studies showing the iron edge energy, indicate that all four sites have the Fe2+ oxidation state [51, 52]. The EPR transition at g eff = 16 is characteristic of an integer-spin system. The EPR and variable-field Mössbauer spectra indicate the S = 4 ground state. A signature visible absorption band is present at λ max = 520 nm, and apparent d–d transitions can be detected using MCD [53].
The Mössbauer signal of the S = 4 cluster may be resolved into a 3:1 ratio of signals with different quadrupole splitting values, suggesting that the “cubane” structure is compressed along a body diagonal (C 3 axis) [50, 51]. EXAFS data suggest that the cluster has a longer (2.77 Å) and a shorter (2.53 Å) Fe–Fe distance [52]. X-ray crystallography shows that the cluster has identical coordination in the all-ferrous form as in the other oxidation states, but the resolution of the X-ray data was not sufficient to clearly distinguish the bond lengths within the cluster [54]. Computational investigations indicate that all of these data can be reconciled within a model in which one of the high-spin iron atoms has its spin aligned opposite to the spin on the three other iron atoms [23].
Potentiometric titration with Cr3+/2+-EDTA indicated that the redox potential of the S = 4 form is −0.79 V at pH 8 [55]. This is roughly 0.48 V more negative than the potential for the reduction of [4Fe–4S]2+ to [4Fe–4S]+. What stabilizes the very low oxidation state enough for its observation? The X-ray crystal structure indicates an unusually large number of NH-thiolate hydrogen bonds [54]. Hydrogen bonds are well known to modulate the redox potentials of iron–sulfur clusters [24]. Even though the super-reduced state is stable enough for isolation and characterization in vitro, the reduction is outside the range of typical physiological electron donors in vivo, raising questions about whether this form is relevant in living organisms.
An apparently different form of the all-ferrous Fe protein has been generated using flavodoxin [56]. A combination of UV–vis, EPR, stoichiometry of reduction and solution magnetic susceptibility indicated that this form has S = 0, because it lacks the EPR signal and visible UV–visible band. Flavodoxin is a physiological electron donor whose potential is −0.52 V. This implies that the [4Fe–4S]+/0 potential is less negative than −0.52 V, and much less negative than the −0.79 V value measured for the S = 4 form. In the future, it would be valuable to pursue CD and/or X-ray diffraction studies to show if the protein conformation is different between these different forms.
If the results on the flavodoxin reduction are correct, it is not clear why dithionite, which has a nominal redox potential of −0.66 V, does not reduce the [4Fe–4S]+ state to this [4Fe–4S]0 state. This discrepancy has been attributed to a reaction between dithionite and the all-ferrous protein [48]. It is worth noting that dithionite does not have a simple reversible redox couple, and that dithionite purity can be problematic [57]. Titanium(III) citrate, the reductant used to form the S = 4 state, also has a complicated speciation in aqueous solution [58]. Further study is needed to understand the reason for the reductant-dependent generation of different all-ferrous species.
If a doubly-reduced Fe protein is involved in nitrogenase catalysis, it could in principle achieve a two-electron reduction of the MoFe protein. This idea found support in single-turnover experiments from two different labs showing that the all-ferrous state could be used productively for reduction of substrates (protons, acetylene, or N2) [48, 53]. Spectroscopic studies also demonstrated nucleotide binding to the [4Fe–4S]0 state, and binding to the MoFe protein. Watt has also shown that use of Ti(III) (which generates the [4Fe–4S]0 state) eliminates the characteristic “lag phase” or induction period before substrate reduction [59]. The most direct evidence for two-electron transfer comes from experiments in which the turnover frequencies of ATP and substrate indicated that 2 ATPs were consumed per 2 electrons when using Ti(III) as the reductant [60]. Since 2 ATP molecules are hydrolyzed in the course of each Fe protein–MoFe protein dissociation cycle, this implies that two electrons are passed each time the all-ferrous Fe protein binds to the MoFe protein. The product distributions were similar with the different reducing agents, suggesting that the FeMoco functions similarly despite the change in electron supply [60]. However, since Ti(III) produces the state of the cluster which apparently has a redox potential of −0.79 V, it is unclear whether this very reduced species is relevant in vivo.
2.3.4 Other Four-Iron Clusters
A second example of a protein that supports a [4Fe–4S]0 cluster was identified more recently in HgdC, the activator protein for 2-hydroxyglutaryl-coenzyme A dehydratase (HgdAB) [61]. Each Hgd protein has a [4Fe–4S] cluster, and reduced cluster in the reduced activator transfers an electron to the iron–sulfur cluster of the enzyme. This electron transfer ultimately activates the enzyme for catalysis. When the reduced [4Fe–4S]+ form of the activator protein is reduced with 10M equivalents of titanium(III) citrate, about 50% of it is converted to a super-reduced form that is spectroscopically similar to the S = 4 all-ferrous state of the nitrogenase Fe protein. Mössbauer and EPR studies were used for comparison and characterization. It is not clear why excess titanium(III) citrate is necessary: it may be that the redox potential is very negative, or that there is a distribution of cluster environments in the protein sample. In addition, the functional role (if any) of a doubly reduced cluster is not clear in this system, because one electron is sufficient to fulfill its activator role.
Both the nitrogenase Fe protein and HgdC have similar quaternary structures with the cluster at the interface of two identical peptides. This suggests that a [4Fe–4S]-bridged homodimer may be unusually adept at stabilizing the large negative charge of the super-reduced clusters. It also suggests that other proteins with clusters at solvent-exposed interfaces (which often couple nucleotide triphosphate hydrolysis to reduction) may provide additional examples of biological [4Fe–4S]0 clusters.
3 Synthetic All-Ferrous Iron–Sulfur Clusters of Nuclearities Two and Three
3.1 Two-Iron Clusters [2Fe–2S]0
In 1973, the first complex having a [2Fe–2S] core was isolated as the diiron(III) compound [Fe2S2(o-(CH2S)2Ph)2]2–, which has the structure shown as 6c in Fig. 7 [62]. Cyclic voltammetry (CV) of this complex in DMF (dimethylformamide) or acetonitrile gave two well-separated one-electron cathodic peaks. This indicates the stepwise two-electron reduction of the Fe 3+2 core to a Fe 2+2 state (Eq. 1) [62, 63]. The second reduction, which leads to the all-ferrous [2Fe–2S]0, has a very negative potential of E 1/2 = −1.81 V and is quasireversible.

Synthetic [2Fe–2S] complexes that can be reduced by two electrons from the [2Fe–2S]2+ state, presumably generating all-ferrous clusters in solution. Charges are not shown for simplicity
In the years since then, numerous ligands have been used to construct complexes with the [2Fe–2S] core. Some of them are shown in Fig. 7 [62, 64–68].
All-thiolate terminated clusters were the first and most widely studied. Table 1 shows that arylthiolate complexes are more easily reduced than alkylthiolate species. Within the arylthiolate series, the reduction potential becomes less negative as the electron-withdrawing power of the para substituent on the aryl group increases [64, 77]. Thus the choice of thiolate substituent can lead to a marked influence on the redox potential. Other complexes with chelating supporting ligands, for example a Rieske model with one NN-coordinated iron and one SS-coordinated iron, show an irreversible cathodic feature at a much more negative potential around −1.8 V [66, 67, 77].
Though these high spin [2Fe–2S]0 clusters have been generated in solution by reducing [2Fe–2S]2+ species in electrochemical experiments, they have not been isolated as solids. The instability can be attributed to two factors. First, most of the synthetic [2Fe–2S]0 clusters are tetraanions, because they are supported by four anionic donors. As a result, the [2Fe–2S]+/0 potentials for the synthetic compounds are usually more cathodic than −1.0 V, which is much more extreme than those for the proteins discussed in Sect. 2. It is likely that this difference is due to the lack of hydrogen bonds that withdraw charge density from the thiolate sulfur atoms. Therefore, it would be an interesting challenge in future research to append groups that can form hydrogen bonds to the coordinated sulfur atoms, to learn the effect on the redox potential. This may lead to isolable all-ferrous [2Fe–2S] compounds.
A second factor contributing to the difficulty in isolating all-ferrous [2Fe–2S] clusters is that the two-electron reduction of [2Fe–2S]2+ clusters takes place through the intermediacy of [2Fe–2S]+ clusters, which are often unstable themselves. Many [2Fe–2S]+ clusters couple with loss of two terminal ligands to form [4Fe–4S]2+ clusters, leaving the iron atoms in the same oxidation states (Eq. 2). This coupling is especially rapid in halide-ligated dinuclear complexes [Fe2S2X4]2−. In these systems, the initial cathodic CV features are associated with the 2−/3− couples of the dinuclear clusters, but the next wave appears at the same potential as the analogous four-iron clusters in the CV [78, 79]. This observation suggests that the second reduction is associated with four-iron clusters rather than two-iron clusters. However, since thiolate ligands are less labile than halides, the conversion to the four-iron cluster is not as fast in sulfur-ligated dinuclear complexes [Fe2S2(SR)4]2−, and thus a quasireversible reduction to [2Fe–2S]0 can still be observed using CV [64].
Singly reduced [2Fe–2S]+ clusters can be stabilized by using chelating capping ligands, thus slowing the ligand loss that leads to dimerization. The combination of this strategy and the use of electron-withdrawing benzimidazole groups is evident in 11a, which was the first reduced [2Fe–2S]+ cluster to be isolable as a solid [69]. A close relative of this compound, 11b, was crystallographically characterized recently [75]. Cyclic voltammetry studies on 11b and another close relative, 11c [76], show reversible reduction to the all-ferrous level at −1.46 V and −1.56 V, respectively. These observations suggest that the all-ferrous form might also have enhanced stability with these capping ligands.
Since the formation of the all-ferrous clusters in synthetic systems has been observed only by CV, there are no spectroscopic data on the putative [2Fe–2S]0 clusters. These data would be very useful to compare to the biological examples in Sect. 2, and future advances are eagerly awaited.
3.2 Two-Iron Clusters [2Fe–1S]2+
Tetradentate ligands, such as acen [80], salen [81, 82] and tmdbtd [83] (H2acacen = N,N′-ethylenebis(acetylacetoneiminate); H2salen = 1,2-bis(salicylideneaminato)ethanediyl; H2tmdbtd = 5H,14H-6,8,15,17-tetramethyldibenzo[b,i][1,4,8,11]tetraazacyclotetradecene) (Fig. 8), support sulfide-bridged diiron cores at the diiron(III) oxidation level ([2Fe–1S]4+). No reduction to all-ferrous [2Fe–1S]2+ clusters appears to be accessible using these ligand systems. For example, the CV of [2Fe–1S]4+ clusters supported by salen ligands showed a reversible reduction to the mixed-valence [2Fe–1S]3+ level, followed by a broad, irreversible reduction around −1.5 V. Fe–S bond cleavage apparently occurs during the electrochemical decomposition, because an iron(III) salen monomer is formed [82].

Ligands used for [2Fe–1S]4+ compounds, which could not be reduced to the all-ferrous [2Fe–1S]2+ state
In 2004, a stable all-ferrous [2Fe–1S]2+ cluster 13 was isolated, and it was stabilized by a very bulky β-diketiminate ligand (LMe = C[C(Me)N(2,6-diisopropylphenyl)]2)–) that made each iron center three-coordinate [84]. Two novel methods have been reported for the synthesis of 13 (Fig. 9). One is the oxidation of a formally diiron(I) N2 complex with elemental sulfur [84], and the other is heating a mixture of a phosphine sulfide and an iron(II) alkyl complex, a reaction that presumably proceeds by β-hydride elimination followed by reductive elimination of H2 [85]. It remains to be seen whether these synthetic methods are general to other series of iron sulfides in which high-spin iron(I) precursors and high-spin iron(II) alkyl/hydride precursors are available.

Formation and reactions of the diiron(II) sulfido complex 13, which is supported by bulky β-diketiminate ligands. It may also be reduced to the first iron(I)-containing iron-sulfido complexes 13 red , which feature alkali metals bound to the sulfur and the arene rings of the β-diketiminates
The spins on the two Fe2+ ions in 13 antiferromagnetically couple to give a diamagnetic ground state, as shown by variable-field Mössbauer spectroscopy. In contrast to the all-ferric [2Fe–1S]4+ species with a diiron(III) core [80, 81, 83], the diiron(II) cluster 13 is quite bent at the bridging sulfur (Fe–S–Fe < 120°). Unfortunately, the exact bond angle is not known with precision because of disorder in X-ray crystal structures of the compound, and computational studies would be helpful in defining this angle and determining whether the lower oxidation state is the cause of the more acute angle (perhaps through a decrease in metal–sulfur π-bonding).
Compound 13 was the first iron–sulfur cluster to be structurally characterized to have three-coordinate iron atoms. This coordinative unsaturation enables 13 to react with donor ligands that give the products pseudotetrahedral iron atoms [84]. An interesting example of cooperative reactivity is the reaction of 13 with phenylhydrazine (PhNHNH2), which gives the mixed-valence iron(II)–iron(III) compound (LMeFe)2(μ-S)(μ-PhNNH2) (Fig. 9a) [84]. The overall reaction is a two-electron reduction of the N–N bond of hydrazine by two of the diiron clusters (each cluster is oxidized by one electron). Other hydrazines were not reduced by 13, with 2:1 adducts formed instead (Fig. 9b); similar adducts were characterized using nitrogen-based donors such as NH3 and MeCN (Fig. 9c, d).
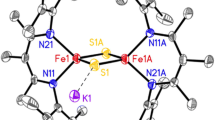
In recent work, addition of Na or K (as potassium graphite) to 13 was shown to give two-electron reduction to the diiron(I) complex 13 red [85]. The iron atoms in both the sodium and potassium versions of 13 red are again three coordinate. They have a high-spin d 7 configuration, as demonstrated by Mössbauer spectroscopy and density-functional calculations, and the iron(I) ions are antiferromagnetically coupled to give a diamagnetic (S total = 0) ground state. The surprising ability to reduce these compounds to the Fe1+ (subferrous) oxidation level was attributed to the stabilizing effect of the main-group cations on the anionic core, as they are not stable without the alkali metals incorporated into the structure. It remains to be seen whether the presence of closely held cations will be more generally useful for the isolation of other highly reduced iron–sulfur clusters.
The open coordination sites on the three-coordinate iron atoms in these iron-sulfide compounds offer significant promise for modifying the coordination sphere. In particular, a second sulfide could be introduced into the ligand sphere to form a [2Fe–2S] cluster, or perhaps other iron–sulfur clusters could be incorporated to form larger structures. The ability to vary the substituents in the β-diketiminate also promises the ability to tune the redox potential and sterics of this unusual type of cluster.
3.3 Three-Iron Clusters
Despite the biological examples of [3Fe–4S]2– clusters (Sect. 2.2), there are not yet reports of generating this redox level in a synthetic cluster, even using electrochemistry. As described in Sect. 2.2, the highly reduced biological clusters are protonated during reduction, and this process of charge balancing is typically not observed in synthetic clusters. Therefore, it seems possible that mimicking the hydrogen-bond donors (or Brønsted acids) that stabilize the biological [3Fe–4S]2– clusters might render this goal a reality.
There are examples of all-ferrous [3Fe–1S]4+ cores with all ferrous sites, in compounds of the type [Fe3S(S2-o-xyl)3]2− [86, 87]. These have a μ3-bridging sulfide, three μ2-bridging thiolates, and three terminal thiolates, as shown in Fig. 10. No electrochemically reversible waves were seen in the CV responses of these compounds.

All-ferrous three-iron sulfide clusters supported by chelating ligands
4 Synthetic All-Ferrous Iron–Sulfur Clusters of Nuclearity Four
4.1 Four-Iron Clusters [4Fe–4S]0 with Thiolate and Phenoxide Ligands
Four-iron clusters at the [4Fe–4S]0 oxidation level were first detected in electrochemical investigations of the thiolate-terminated clusters [Fe4S4(SR)4]2– in aprotic solvents; because the starting material formally has two iron(II) and two iron(III), the second reduction of the cluster around −1.4 V was postulated to give the all-ferrous form [78, 88, 89]. Analogous reductions have been done with dithiolene-terminated clusters, but there are no thorough studies of their voltammetry at negative potentials [90]. Because the redox ambiguity of the dithiolene ligands makes it difficult to assign the oxidation state of the iron atoms in these clusters, the discussion here focuses on the thiolate-terminated clusters. The key reduction of [Fe4S4(SR)4]3– to [Fe4S4(SR)4]4– occurs at potentials more cathodic than −1.0 V (Table 2), and thus the reduced species are highly reducing and very sensitive.
Table 2 shows the potentials for a number of these clusters. In general, these follow the same trend as the 2−/3− potentials, with potentials more cathodic by 0.7 V. The potentials correlate with the Hammett parameter for aromatic substituents, with electron-withdrawing groups giving a less negative potential [78, 89]. The use of positively charged ammonium substituents gave a positive shift in the redox potential, but the amount of shift was consistent with the expected substituent effect, without any extra consideration for the change in charge [89]. The 3−/4− waves are quasireversible, implying that the all-ferrous species are unstable, except for the bulkiest thiolate substituents like R = t-butyl. Thus, they have not been isolated.
The electrochemically generated all-ferrous clusters have been used for reduction of substrates. In research using [Fe4S4(SR)4] systems (R = benzyl, t-butyl or macrocycle), electrocatalytic reduction of CO2 to formate proceeded from the all-ferrous cluster [91–93]. Likewise, the exchange of H2 and D2 to form HD occurs through the action of a [4Fe–4S]0 cluster [94]. In these reports, the intermediacy of [4Fe–4S]0 rather than [4Fe–4S]+ was shown by the observation of cathodic electrocatalytic current only when the potential entered the range when [4Fe–4S]0 is formed. In the case of acetylene reduction to ethylene, [Fe4S4(SPh)4]3– is capable of catalysis, but [Fe4S4(SPh)4]4– reduces the acetylene more quickly [95]. A wider range of electrocatalytic reductions has been achieved with mixed molybdenum/iron systems, which are beyond the scope of this review and have been reviewed [96]. Other reports use extremely strong chemical reducing agents (e.g. sodium sand) to generate the tetraanion, followed by treatment with the substrate. This treatment has given catalytic thiol addition to isocyanides [97]. In combination with a Mo–N2 coordination complex, one of the dithiolene-appended clusters was reported to give small amounts of ammonia [98, 99]. However, given the short lifetimes of the reduced [Fe4S4(SR)4]4– clusters (as demonstrated by CV), it is not clear whether the reactions that involve chemical reduction of the cluster are mediated by the all-ferrous cluster or by its decomposition products.
The halide-terminated clusters with X = Cl and Br give irreversible [Fe4S4X4]3–/4– reductions with E pc of −1.4 to −1.5 V [79]. Thus, the halide species are more easily reduced than the thiolate species, but the reductions give tetraanions that are not stable on the time scale of the CV experiment. On the other hand, the phenoxide-terminated clusters [Fe4S4(OPh)4]2– have two electrochemically reversible reduction features at −1.6 V [100]. These potentials are somewhat more cathodic than the thiolate analogues, suggesting that the phenoxides do not support the buildup of negative charge as effectively as the thiolates (the opposite of the trend expected from the relative electronegativities). Again, the all-ferrous forms have been studied only transiently in solution using CV, and no spectroscopic studies have been reported.
4.2 Four-Iron Clusters [4Fe–4S]0 with Phosphine Ligands
Because phosphines are neutral and have some π-accepting character, they ameliorate the problem of charge buildup in the most reduced forms. Initial attempts to obtain reduced [4Fe–4S] clusters with aliphatic, sterically unencumbered phosphines as terminal ligands were unsuccessful, because addition of small phosphines gave known [Fe6S6(PR3)4Cl2] basket clusters [101, 102]. Larger phosphines with isopropyl, cyclohexyl, or tert-butyl substituents cannot fit into the basket structure, and so the [4Fe–4S] cubane core structure is maintained [103–105]. Interestingly, phosphines can be introduced with concomitant reduction of the cluster: the synthesis begins from the halide-terminated [4Fe–4S]2+ clusters [Fe4S4X4]2–, and addition of phosphine reduces them to the [4Fe–4S]+ level in the neutral product [Fe4S4(PtBu3)3X]. Some of the phosphine is oxidized to the phosphine sulfide [104]. These clusters give an irreversible reduction waves around −1.2 V, indicating that the all-ferrous [Fe4S4(PBut 3)3X]– with mixed ligation are not stable [104]. However, using a slight excess of large tertiary phosphines, it is possible to substitute all four iron atoms to yield [Fe4S4(PR3)4]+ (R = But, Cy, Pri) [103]. The four-phosphine clusters could be reduced to the all-ferrous [Fe4S4(PR3)4]0 level reversibly using electrochemistry; the relevant redox potentials are given in Table 3. Alternatively, [Fe4S4(PR3)4]0 can be prepared by chemical reduction of the monocation with a slight excess of sodium acenaphthalenide. Unlike their positively charged precursors, the neutral black cubane clusters are soluble in benzene and toluene.
These neutral clusters have higher stability than the anionic clusters in Sect. 4.1. They decompose only over days in solution; however, they are still very sensitive to oxidation, and they have not been isolated as pure solids or subjected to X-ray diffraction [103, 105]. Upon standing in solution, the neutral [Fe4S4(PR3)4] clusters aggregate to form insoluble edge-bridged dicubane or tetracubane clusters [Fe8S8(PCy3)6] and [Fe16S16(PBut)8], in which one of the sulfur vertices forms a bond to a fourth iron center (Fig. 11; see Figs. 17 and 18 for structures) [103]. The Lewis basicity of the sulfur in the all-ferrous clusters is greater because of the relatively low oxidation level of the cluster; this apparently makes the sulfur a strong enough nucleophile to displace phosphine from an adjacent cluster. It will be interesting to learn whether the combination of cluster stability and sulfur nucleophilicity will lead to sulfur-based reactivity from these [Fe4S4(PR4)4] clusters with substrates such as alkynes and CO2.

(a) An example of the synthetic route that leads to all-ferrous [Fe4S4(PR3)4] (R = iPr, Cy, tBu) in solution using potassium benzophenone. (b) Over days in solution, these decompose to stable, higher nuclearity edge-bridged clusters
4.3 Four-Iron Clusters [4Fe–4S]0 with Cyanide Ligands
The studies in Sect. 4.2 point toward the use of terminal ligands that maintain the binding ability of an anionic donor but do not place as much electron density on the metal as a halide, aryloxide, or thiolate. Cyanide (CN–) strikes a good balance, and reduction of a phosphine cluster in the presence of cyanide was effective (Fig. 12). This led to the first successful crystallographic characterization of an all-ferrous [4Fe–4S]0 cluster [106]. The [4Fe–4S]+/0 redox couple has E 1/2 = −1.18 V, showing that cyanide falls between thiolate/halide (more electron donating) and phosphine (less electron-donating) in terms of its influence on the cluster potential.

Synthetic routes to the isolable all-ferrous cluster [Fe4S4(CN)4]4−
The isolable [Fe4S4(CN)4]4− species was characterized by visible absorption and Mössbauer spectroscopies as well as X-ray diffraction, which allowed comparison to the biological cubane clusters (see Sect. 2.3.3) [106]. It has a distinct red color in solution and an absorption band around 520 nm that is reminiscent of the protein-bound [4Fe–4S]0 cluster. Like the biological examples, the isomer shifts in the Mössbauer spectrum are indicative of iron(II), but they are not split into the 3:1 ratio as seen in the protein (Table 4). This is probably because the synthetic cluster is more symmetrical, and accordingly the X-ray diffraction analysis reveals that the structure is very close to idealized D 2d symmetry. The mean Fe–Fe and Fe–S distances, as well as the core volumes, are close to those of the [4Fe–4S]0 cluster of the fully reduced Fe–protein (Table 5).
Although [Fe4S4(CN)4]4− was stable enough for solution and crystallographic studies, it could only be crystallized in the presence of excess of reductant [106]. As judged by the redox potentials (see above), the Fe2+ oxidation state is more stabilized by phosphine ligation. This suggests that neutral compounds have greater oxidative stability, and synthesis of neutral [4Fe–4S]0 clusters requires neutral terminating ligands.
4.4 Four-Iron Clusters [4Fe–4S]0 with NHC Ligands
The trends shown in the previous sections suggest that for isolation of highly reduced clusters that do not undergo aggregation, the ideal capping groups would be neutral ligands that bind very tightly to the iron center. The N-heterocyclic carbene (NHC) ligands have this combination of strong electron donating ability and lack of negative charge, which led to success [107]. In this work, the NHC ligand shown in the lower left of Fig. 13 was used. The all-ferrous cubane cluster 15 can be synthesized by displacement of phosphine from pre-formed clusters like 16, or by self-assembly of a double-cubane NHC cluster 17 and subsequent cluster fission with additional NHC (Fig. 13). In contrast with phosphines (which are displaced by sulfides during the formation of cubane aggregates) [103], NHC ligands can break up the double and quadruple cubanes. They have much greater solution stability as well, and the cluster [Fe4S4(NHC)4] (7) can survive in aprotic solvents under anaerobic conditions for at least 3–4 days. The [4Fe–4S]+/0 redox potential of cluster 15 is at −1.06 V, less negative than for clusters ligated by anions like CN– or SR– but more negative than the phosphine-terminated clusters.

Synthetic routes to NHC-based all-ferrous [Fe4S4(NHC)4]
Cluster 15 has been fully characterized by X-ray crystallography, as well as 1H NMR, Mössbauer, and electronic absorption spectroscopies [107]. Because of its higher stability, it has been characterized in more detail than the previously discussed [Fe4S4(CN)4]4− clusters, and the nature of the [4Fe–4S]0 core is better understood. The average Fe–S bond distance and cluster volume in the NHC-terminated cluster are close to those of the cyanide-terminated cluster, and each is somewhat more symmetric than the reduced Fe protein (Table 5). An absorption band is again observed around 510 nm, which is very close to those seen in [Fe4S4(CN)4]4− and the all-ferrous Fe protein of nitrogenase. The position of this band shows little dependence on the identity of the terminal ligands, indicating that this electronic transition is an intrinsic property of the [4Fe–4S]0 core.
In order to obtain insight into the electronic structure of the [4Fe–4S]0 core and to compare to the biological all-ferrous [4Fe–4S] clusters, detailed EPR and Mössbauer analysis have been carried out on 15 [108]. The parallel-mode X-band EPR spectrum of cluster 15 at 2 K exhibits a resonance with g eff = 16.1 that originates from the M S = ±4 doublet of an S = 4 ground state. The zero-field Mossbauer spectrum of cluster 15 at 100 K exhibits two doublets with a 3:1 intensity ratio, which is different from the cyanide-terminated cluster but in agreement with the data on the all-ferrous Fe protein of nitrogenase [50]. The average isomer shift of the synthetic compound is slightly lower than in the protein (Table 4), which may indicate more covalency at iron. Fitting variable-field Mössbauer spectra illuminated the spin coupling behavior: out of the four high-spin ferrous sites, three are aligned parallel to the cluster spin and the fourth antiparallel, giving a total spin of 4. This conclusion is consistent with computations predicting that a T 2 distortion is favorable. This T 2 distortion (along one Fe-centroid axis) also explains the 3:1 ratio of Mössbauer signals, and is fully consistent with all of the data. The observation of a similar distortion in the synthetic cluster and the protein-bound cluster implies that the 3:1 distortion is not imposed on the cluster by the protein/solvent environment [23, 61]. However, the extent of the distortion can be modulated by changes in terminal ligation or protein environment. Recently, these conclusions were strengthened by a DFT study that also shows a 3:1 pattern in both optimized geometry and calculated 57Fe Mossbauer spectra and supports the idea that the exchange interaction are highly dependent on the core geometry [109].
In summary, systematic variations of the supporting ligands have recently enabled the isolation and detailed study of a synthetic all-ferrous [4Fe–4S] cluster for the first time. These studies have greatly enhanced our understanding of the protein-bound all-ferrous clusters because they show that the S = 4 ground state, the 3:1 ratio of iron environments, and the 510–520 nm electronic absorption band are intrinsic features of the all-ferrous cubane core. The structural parameters for the synthetic compound are known in much greater precision than the protein-bound structure, which facilitates comparison to computations. Challenges for the future include ligand variation to enforce an E-symmetry distortion (2:2 ratio of iron environments), which could give an S = 0 ground state [108]. Isolation of such a cluster could help to understand the feasibility of the proposed “alternative” form of the all-ferrous nitrogenase Fe protein discussed in section 2.3. It will also be interesting to learn whether the NHC ligands can be removed to give reactivity at the highly reduced iron core. Finally, one hopes that the ability to make a large amount of synthetic all-ferrous cubane clusters will allow chemists to test the idea that such clusters can do two-electron transfer as proposed in nitrogenase.
5 Synthetic All-Ferrous Iron–Sulfur Clusters of Nuclearity Eight and Higher
5.1 Eight-Iron Clusters
Eight-iron clusters relevant to all-ferrous iron–sulfur chemistry have three different core structures (Fig. 14). As shown on the left of Fig. 14, [8Fe–6S] cores contain eight irons in a cube, capped on each face by a sulfur, and each iron is coordinated by a terminal ligand to complete its tetrahedral coordination. In the center of Fig. 14, [8Fe–7S] cores have two [4Fe–4S] cubanes that share a μ6-sulfide. In contrast, the [8Fe–8S]0 clusters consist of two [4Fe–4S] cubanes linked via two Fe–S bonds, as illustrated on the right of Fig. 14.

Shapes of [8Fe–6S], [8Fe–7S], and [8Fe–8S] cores. The black circles represent iron atoms, and the white circles represent sulfur atoms. All iron atoms are four-coordinate with a pseudotetrahedral geometry, and their coordination spheres are completed by terminal ligands (not shown)
5.1.1 [8Fe–6S]4+
The [8Fe–6S]4+ core was identified in the compound (PhCH2NEt3)4[Fe8S6I8] (9), which was the first all-ferrous iron–sulfur cluster to be isolated [110]. It was synthesized from a [4Fe–4S]2+ precursor (Eq. 3), and the phosphine is present only in an basket-type intermediate [Fe6S6I2(PR3)4], but not in the final product. An excess of Fe2+ is required for this conversion, or else only amorphous iron sulfides are obtained. Alternatively, the [Fe6S6I2]0 intermediate can be avoided using bidentate phosphine ligated [Fe2I2(dppe)2], which does not form a basket intermediate. Cluster 9 is stable in air for a short time, but only X-ray characterization was given, and the electronic structure and coupling have not been reported.
Building from the observation that [Fe6S6I2(PR3)4] can react with [FeI2(PR3)2] to form a [Fe8S6I8]4− core, a similar reaction was carried out using Ru2+ instead of Fe3+ (Eq. 4) [111]. This reaction also results in the formation of a [8Fe–6S]4+ core, and in this case the core is attached to two octahedral Ru fragments via opposing sulfur atoms, in the compound [(MeCN)4(Ph2MePS)Ru–Fe8S6I8–Ru(Ph2MePS)(MeCN)4]. The structure of this cluster was determined by X-ray diffraction, which evidenced a topologically identical [Fe8S6I8]4− core with an elongation of the [8Fe–6S] core along the Ru–Ru axis. Similar reactions with Ni2+ have also been explored, and they incorporate various numbers of Ni atoms into the cubane core [112, 113].
5.1.2 [8Fe–7S]2+
These clusters are of interest because of their analogy to the P cluster of nitrogenase (Sect. 2.3.2). The development of these clusters has benefitted from the use of organic solvents and very bulky thiolates that protect the cluster. The first example, cluster 18, was isolated from the reaction of Fe[N(SiMe3)2]2, TipSH (Tip = 2,4,6-iPr3C6H2), thiourea SC(NMe2)2, and elemental sulfur (S8) in toluene [114]. Cluster 18 accurately reproduces the [8Fe–7S] core geometry of the reduced PN cluster, even though the Mössbauer spectrum of 18 shows that it has a 6Fe2+2Fe3+ oxidation state corresponding to POX [114]. In an effort to reduce the synthetic cluster to the all-ferrous level, 18 and some of its terminal ligand substituted analogues 19–21 were reduced electrochemically. Cyclic voltammetry showed two quasi-reversible one-electron reductions, which formally lead to the all-ferrous oxidation state (Fig. 15, Table 6) [115]. However, the all-ferrous product has not been isolated. Although mixed-metal clusters are formally outside the scope of this review, it should be mentioned that Mo2Fe6 clusters of the same shape are known that have all of their iron atoms in the ferrous level [116–118].

Structures of synthetic [8Fe–7S] clusters
Bulk preparation of all ferrous [8Fe–7S] clusters has been attempted using chemical reduction of 18 by decamethylcobaltocene (which has a potential of −1.7 V) [119]. However, the product did not correspond to simple reduction: instead, there is activation of a C–H bond of Cp*2Co, affording cluster 22 (Fig. 15). One possible mechanism for this transformation is that a highly reduced cluster intermediate deprotonates Cp*2Co+; if so, then the reduced cluster intermediate (of unknown oxidation level) must be a formidable base. Since the oxidation state of cobalt in the decamethylcobaltocenyl group in the product is unknown, the oxidation state of the iron atoms in the clusters remains unclear.
The related cluster 23 with all-sulfur ligation is in the Fe 3+3 Fe 2+5 oxidation level [120]. It has three quasireversible reductions evident by CV, with the third one (presumably leading to the all-ferrous form) at −1.4 V. This transiently generated species has not been examined further, but considering the apparent reversibility of the redox processes in the CV, it is a promising lead for continued study.
5.1.3 [8Fe–8S]0
5.1.3.1 Synthesis
These clusters are conceptually derived from two [4Fe–4S] clusters through bonds between the iron and sulfur along a common edge of the cube, and thus are termed “edge-bridged.” The neutral all-ferrous clusters [Fe8S8(PR3)6] were first isolated during attempts to generate all-ferrous [4Fe–4S]0 clusters with phosphine as terminal ligands [103, 105, 121]; as noted above, phosphines bind relatively weakly to [4Fe–4S] clusters. Since the edge-bridged clusters have poor solubility, the reaction may also be driven to the eight-iron cluster by precipitation of the product (LeChâtelier’s principle). Similar [8Fe–8S]0 clusters were generated with terminal NHC ligands [107]. On the other hand, cyanide-terminated clusters [Fe4S4(CN)4]4– did not aggregate into edge-bridged dimers, perhaps due to the stronger binding of cyanide or the negative charge on the cluster [106].
Another route to prepare [8Fe–8S] clusters is by direct self-assembly from FeCl2(PR3)2, a sulfur source and terminal ligands. This method (which was mentioned above in the context of Fig. 13) has been demonstrated to lead to [Fe8S8(PCy3)6] (Eq. 5) [105], [Fe8S8(PiPr3)6] (Eq. 6) [122] and [Fe8S8(NHC)6] (17 in Fig. 13) [107].
A third synthetic method proceeds through the intermediacy of the all-ferrous heptanuclear compound [Fe7S6(PEt3)5Cl2] by the addition of NHC (Eq. 7) [107]. However, one pitfall of this method is that the seven-iron cluster is not very stable and decomposes over several hours at room temperature. Finally, since the N-heterocyclic carbenes are stronger donors than phosphines, [Fe8S8(NHC)6] can also be generated by terminal ligand substitution from [Fe8S8(PiPr3)6] or [Fe16S16(PiPr3)8] (Eq. 8; see also Fig. 13) [107].
5.1.3.2 Properties
X-ray diffraction has been used to determine the structures of a number of [8Fe–8S] edge-bridged dicubanes [103, 105, 107]. All three [8Fe–8S] clusters, [Fe8S8(PCy3)6], [Fe8S8(PiPr3)6] and [Fe8S8(NHC)6] have idealized C 2h symmetry, with similar dimensions. The bridging Fe2S2 rhomb is rigorously planar, with the intracubane Fe–S bonds consistently longer than the intercubane bonds. The dicubane structure is retained in benzene solution, as shown by 1H NMR spectroscopy.
In keeping with the symmetry of the core, the Mössbauer spectra of [8Fe–8S]0 clusters share a pattern of one quadrupole doublet (25% in intensity) with larger quadrupole splitting and one or two doublets (75% in intensity) with smaller quadrupole splitting (Table 7). The average isomer shifts of phosphine-ligated clusters (δ av = 0.60 mm/s) and NHC-ligated clusters (δ av = 0.62 mm/s) are similar and consistent with the all-ferrous oxidation level. The similarities in geometric and electronic structure of [4Fe–4S]0 and [8Fe–8S]0 clusters indicate that the spin ordering for the edge-bridged dicubanes is the same as that within the [4Fe–4S]0 fragments (Fig. 3) [123]. The diamagnetic ground states of [8Fe–8S]0 clusters are proposed to come from antiferromagnetic coupling of two [4Fe–4S]0 fragments (Fig. 16).

Proposed spin ordering model for [8Fe–8S]0 edge-bridged dicubanes
5.1.3.3 Reactivity
The reactivity of [Fe8S8(PCy3)6] has not been studied extensively, because it is insoluble in all common solvents. However, it reacts with chloroform or SCl2 to give a mixture containing the all-ferrous cluster, [Fe8S6(PCy3)4Cl4] [105]. The [8Fe–6S]4+ core of this molecule has the same structural motif described above in Sect. 5.1.1.
The analogous [Fe8S8(PiPr3)6] has been studied more thoroughly, as it is soluble in solvents such as benzene, toluene and THF [103, 122]. Treatment of this dicubane cluster with the oxidant [Cp2Fe]+ results in cleavage of edge-bridged Fe–S bonds and yields monocubane species with [4Fe–4S]+ cores. When oxidation occurs in the presence of a bulky siloxide, it is possible to isolate neutral clusters with a single siloxide ligand (Fig. 17) [122].

Oxidation of [Fe8S8(PiPr3)6] with formation of [4Fe–4S]+ clusters
Further study on [8Fe–8S]0 clusters, particularly the new stabilized NHC-terminated clusters, is needed in order to understand the reaction chemistry of this all-ferrous core. Since edge-bridged dicubane [2Mo–6Fe–8S] clusters have been demonstrated to be precursors to clusters with the topology of the PN cluster of nitrogenase [118, 124], it will be interesting to see if similar core conversion can be realized in [8Fe–8S]0 clusters to form new all-iron analogues of the P cluster.
5.2 Sixteen-Iron Clusters
5.2.1 Synthesis
The first [16Fe–16S]0 clusters were discovered as a byproduct in efforts to isolate neutral [Fe4S4(PR3)4] clusters; the same edge-bridging phenomena that give the [8Fe–8S]0 clusters can also bridge additional edges to form [16Fe–16S]0 species [103, 105]. The number of fused cubes seems to be dependent on the crystallization conditions, which control the opportunity to lose phosphine and aggregate [103, 105]. So far, [16Fe–16S]0 cores are limited to phosphine-based clusters, and tetracubanes have not yet been reported with other terminal ligands, such as cyanide and NHC.
5.2.2 Properties
The structures of [16Fe–16S]0 clusters have been determined by X-ray diffraction analysis. In these compounds, the four cubanes each bridge edges in a cyclic fashion so that all four cubanes are equivalent with overall D 4 symmetry (see compound 16 in Fig. 15). The pattern of distances and angles in each cubane and in the bridging rhomb resembles those seen in the dicubane [8Fe–8S]0 cores discussed in Sect. 5.1.3. The solubility of [Fe16S16(PiPr3)8] and [Fe16S16(PtBu3)8] is appreciable in THF, benzene and toluene, while [Fe16S16(PCy3)8] is only slightly soluble in all common solvents. Retention of the core structure is evident from the number of peaks in the 1H NMR spectra in benzene [103, 105].
The [16Fe–16S]0 clusters have been studied by CV and Mössbauer (Table 8) [103]. In THF, [Fe16S16(PtBu3)8] is oxidized in three steps at E 1/2 = −0.33 V, +0.15 V, and +0.46 V, defining a one-electron-transfer series [16Fe–16S]0/1+/2+/3+. Since the monocubane cluster with the same terminal ligand [Fe4S4(PtBu3)4] exhibits a [4Fe–4S]+/0 redox potential of E 1/2 = −0.51 V, it is apparent that the fusion of the four individual cubanes makes the reduction to the all-ferrous state slightly easier, presumably because the edge-bridging cube is less electron-donating than a phosphine. Since a number of closely spaced one-electron oxidations are observed, it suggests that there is electronic interaction among the clusters, where the oxidation of one influences the potential of the others.
The zero field 57Fe Mössbauer spectra of [Fe16S16(PtBu3)8] and [Fe16S16(PtPr3)8] verify the all-ferrous oxidation level with average isomer shifts of δ 0.61–0.63 mm/s. In analogy to [8Fe–8S]0 clusters, the spectra of [16Fe–16S]0 clusters share the pattern of two quadrupole doublets in an intensity ratio of 3:1, indicating that fusion does not significantly influence the spin coupling within each [4Fe–4S] cubane.
5.2.3 Reactivity
The reductive cleavage of ligand substrates by [16Fe–16S]0 clusters has been reported [122]. Upon addition of the oxidants I2, RSSR, or RSeSeR in the presence of phosphine, tetracubane [Fe16S16(PiPr3)8] dissociates into cubane fragments, each of which captures one phosphine ligand and one monoanion generated from reductive cleavage of the oxidant (Fig. 18). Since [Fe16S16(PiPr3)8] is in equilibrium with [Fe8S8(PiPr3)6] in the presence of phosphine, it seems likely that the reaction is stepwise, with dissociation to [Fe8S8(PiPr3)6] preceding an oxidation reaction like that shown in Fig. 17.

Reduction of substrates by [16Fe–16S] clusters
6 Conclusion
The study of highly reduced iron–sulfur clusters is both long standing and undergoing current development. Research studies on these iron–sulfur clusters represent an interesting confluence of insights from metalloprotein isolation, spectroscopy, and synthetic work, and show the valuable interplay between these areas of bioinorganic research. Several themes have emerged. First, the coupling of protonation and reduction is evident in a number of the highly reduced biological clusters, and the influence of protons (whether as hydrogen bonds or as discrete proton transfers) plays a critical role in modulating their behavior. This fits into an increasing realization in bioinorganic chemistry that the “second coordination sphere” tunes redox potentials over a wide range [125, 126]. It is possible that the biological all-ferrous clusters described here (particularly the Fe protein of nitrogenase) use these tuning influences to enable multielectron transfer, which is of general interest in the context of solar energy research [127].
The synthesis of iron–sulfur clusters in highly reduced states has been a challenge for the synthetic chemistry community because of the sensitivity of the complexes and their tendency to decompose through a range of pathways. Key advances described above are the observation and isolation of all-ferrous clusters, particularly the N-heterocyclic carbene based [4Fe–4S]0 clusters that could be directly compared to spectroscopy on biological all-ferrous clusters. This recent work has confirmed the oxidation state of the spectroscopically observed biological clusters, and given more detail into the geometric and electronic structure. It is clear that many more advances remain for future investigations.
References
Beinert H, Holm RH, Münck E (1997) Science 277:653
Beinert H (2000) J Biol Inorg Chem 5:2
Johnson DC, Dean DR, Smith AD, Johnson MK (2005) Annu Rev Biochem 74:247
Fontecave M (2006) Nat Chem Biol 2:171
Que L Jr, Holm RH, Mortenson LE (1975) J Am Chem Soc 97:463
Johnson MK, Duderstadt RE, Duin EC (1999) Adv Inorg Chem 47:1
Seino H, Hidai M (2011) Chem Sci 2:847
Crack JC, Green J, Thomson AJ, Le Brun NE (2012) Curr Opin Chem Biol 16:35
White MF, Dillingham MS (2012) Curr Opin Struct Biol 22:94
Brandt U (2006) Annu Rev Biochem 75:69
Beinert H, Kennedy MC, Stout CD (1996) Chem Rev 96:2335
Canfield DE (2005) Annu Rev Earth Planet Sci 33:1
Hagen KS, Reynolds JG, Holm RH (1981) J Am Chem Soc 103:4054
Blöchl E, Keller M, Wächtershäuser G, Stetter KO (1992) Proc Natl Acad Sci USA 89:8117
Huber C, Wächtershäuser G (1997) Science 276:245
Heinen W, Lauwers AM (1996) Orig Life Evol Biosph 22:131
Dörr M, Käßbohrer J, Grunert R, Kreisel G, Brand WA, Werner RA, Geilmann H, Apfel C, Robl C, Weigand W (2003) Angew Chem Int Ed 42:1540
Bill E (2012) Hyperfine Interact 205:139
Ogino H, Inomata S, Tobita H (1998) Chem Rev 98:2093
Lee SC, Holm RH (2004) Chem Rev 104:1135
Lee SC, Holm RH (2003) Proc Natl Acad Sci USA 100:3595
Rao PV, Holm RH (2004) Chem Rev 104:527
Torres RA, Lovell T, Noodleman L, Case DA (2003) J Am Chem Soc 125:1923
Dey A, Francis EJ, Adams MWW, Babini E, Takahashi Y, Fukuyama K, Hodgson KO, Hedman B, Solomon EI (2007) Science 318:1464
Im SC, Lam KY, Lim MC, Ooi BL, Sykes AG (1995) J Am Chem Soc 117:3635
Im SC, Kohzuma T, McFarlane W, Gaillard J, Sykes AG (1997) Inorg Chem 36:1388
Im SC, Worrall JAR, Liu G, Aliverti A, Zanetti G, Luchinat C, Bertini I, Sykes AG (2000) Inorg Chem 39:1755
Yoo SJ, Meyer J, Münck E (1999) J Am Chem Soc 121:10450
Verhagen M, Link TA, Hagen WR (1995) FEBS Lett 361:75
Leggate EJ, Bill E, Essigke T, Ullmann GM, Hirst J (2004) Proc Natl Acad Sci USA 101:10913
Duff JLC, Breton JLJ, Butt JN, Armstrong FA, Thomson AJ (1996) J Am Chem Soc 118:8593
Moreno C, Macedo AL, Moura I, Moura JJG, LeGall J (1994) J Inorg Biochem 53:219
Hirst J, Jameson GNL, Allen JWA, Armstrong FA (1998) J Am Chem Soc 120:11994
Mayer SM, Lawson DM, Gormal CA, Roe SM, Smith BE (1999) J Mol Biol 292:871
Peters JW, Stowell MHB, Soltis SM, Finnegan MG, Johnson MK, Rees DC (1997) Biochemistry 36:1181
Zimmermann R, Orme-Johnson WH, Münck E, Shah VK, Brill WJ, Henzl MT, Rawlings J (1978) Biochim Biophys Acta 537:185
Pierik AJ, Wassink H, Haaker H, Hagen WR (1993) Eur J Biochem 212:51
McLean PA, Papaefthymiou V, Ormejohnson WH, Münck E (1987) J Biol Chem 262:12900
Lanzilotta WN, Christiansen J, Dean DR, Seefeldt LC (1998) Biochemistry 37:11376
Danyal K, Mayweather D, Dean DR, Seefeldt LC, Hoffman BM (2010) J Am Chem Soc 132:6894
Lowe DJ, Fisher K, Thorneley RNF (1993) Biochem J 292:93
Chan JM, Christiansen J, Dean DR, Seefeldt LC (1999) Biochemistry 38:5779
Danyal K, Dean DR, Hoffman BM, Seefeldt LC (2011) Biochemistry 50:9255
Huyett JE, Chan JM, Christiansen J, Dean DR, Seefeldt LC (1999) J Inorg Biochem 74:173
Fisher K, Lowe DJ, Tavares P, Pereira AS, Huynh BH, Edmondson D, Newton WE (2007) J Inorg Biochem 101:1649
Tezcan FA, Kaiser JT, Mustafi D, Walton MY, Howard JB, Rees DC (2005) Science 309:1377
Thorneley RNF, Lowe DJ (1996) J Biol Inorg Chem 1:576
Watt GD, Reddy KRN (1994) J Inorg Biochem 53:281
Vincent KA, Tilley GJ, Quammie NC, Streeter I, Burgess BK, Cheesman MR, Armstrong FA (2003) Chem Commun 2590
Yoo SJ, Angove HC, Burgess BK, Hendrich MP, Münck E (1999) J Am Chem Soc 121:2534
Angove HC, Yoo SJ, Burgess BK, Münck E (1997) J Am Chem Soc 119:8730
Musgrave KB, Angove HC, Burgess BK, Hedman B, Hodgson KO (1998) J Am Chem Soc 120:5325
Angove HC, Yoo SJ, Münck E, Burgess BK (1998) J Biol Chem 273:26330
Strop P, Takahara PM, Chiu HJ, Angove HC, Burgess BK, Rees DC (2001) Biochemistry 40:651
Guo ML, Sulc F, Ribbe MW, Farmer PJ, Burgess BK (2002) J Am Chem Soc 124:12100
Lowery TJ, Wilson PE, Zhang B, Bunker J, Harrison RG, Nyborg AC, Thiriot D, Watt GD (2006) Proc Natl Acad Sci USA 103:17131
McKenna CE, Gutheil WG, Wei S (1991) Biochim Biophys Acta 1075:109
Collins JM, Uppal R, Incarvito CD, Valentine AM (2005) Inorg Chem 44:3431
Nyborg AC, Johnson JL, Gunn A, Watt GD (2000) J Biol Chem 275:39307
Erickson JA, Nyborg AC, Johnson JL, Truscott SM, Gunn A, Nordmeyer FR, Watt GD (1999) Biochemistry 38:14279
Hans M, Buckel W, Bill E (2008) J Biol Inorg Chem 13:563
Mayerle JJ, Frankel RB, Holm RH, Ibers JA, Phillips WD, Weiher JF (1973) Proc Natl Acad Sci USA 70:2429
Mascharak PK, Papaefthymiou GC, Frankel RB, Holm RH (1981) J Am Chem Soc 103:6110
Mayerle JJ, Denmark SE, Depamphilis BV, Ibers JA, Holm RH (1975) J Am Chem Soc 97:1032
Wong GB, Kurtz DM, Holm RH, Mortenson LE, Upchurch RG (1979) J Am Chem Soc 101:3078
Ballmann J, Dechert S, Bill E, Ryde U, Meyer F (2008) Inorg Chem 47:1586
Ballmann J, Sun XR, Dechert S, Schneider B, Meyer F (2009) Dalton 4908
Ballmann J, Albers A, Demeshko S, Dechert S, Bill E, Bothe E, Ryde U, Meyer F (2008) Angew Chem Int Ed 47:9537
Beardwood P, Gibson JF (1992) J Chem Soc Dalton 2457
Beardwood P, Gibson JF (1985) J Chem Soc Chem Commun 1345
Beardwood P, Gibson JF (1985) J Chem Soc Chem Commun 102
Beardwood P, Gibson JF (1988) Polyhedron 7:1911
Beardwood P, Gibson JF (1986) J Chem Soc Chem Commun 490
Ballmann J, Sun X, Dechert S, Bill E, Meyer F (2007) J Inorg Biochem 101:305
Albers A, Demeshko S, Dechert S, Bill E, Bothe E, Meyer F (2011) Angew Chem Int Ed 50:9191
Saouma CT, Kaminsky W, Mayer JM (2012) J Am Chem Soc 134:7293
Ballmann J, Dechert S, Demeshko S, Meyer F (2009) Eur J Inorg Chem 3219
DePamphilis BV, Averill BA, Herskovitz T, Que L, Holm RH (1974) J Am Chem Soc 96:4159
Wong GB, Bobrik MA, Holm RH (1978) Inorg Chem 17:578
Corazza F, Floriani C, Zehnder M (1987) Dalton 709
Dorfman JR, Girerd JJ, Simhon ED, Stack TDP, Holm RH (1984) Inorg Chem 23:4407
Mukherjee RN, Stack TDP, Holm RH (1988) J Am Chem Soc 110:1850
Berno P, Floriani C, Chiesivilla A, Guastini C (1989) Dalton 551
Vela J, Stoian S, Flaschenriem CJ, Münck E, Holland PL (2004) J Am Chem Soc 126:4522
Rodriguez MM, Stubbert BD, Scarborough CC, Brennessel WW, Bill E, Holland PL (2012) Angew Chem Int Ed 51: 8247
Henkel G, Tremel W, Krebs B (1981) Angew Chem Int Ed 20:1033
Hagen KS, Christou G, Holm RH (1983) Inorg Chem 22:309
Cambray J, Lane RW, Wedd AG, Johnson RW, Holm RH (1977) Inorg Chem 16:2565
Zhou CY, Raebiger JW, Segal BM, Holm RH (2000) Inorg Chim Acta 300:892
Balch AL (1969) J Am Chem Soc 91:6962
Tezuka M, Yajima T, Tsuchiya A, Matsumoto Y, Uchida Y, Hidai M (1982) J Am Chem Soc 104:6834
Nakazawa M, Mizobe Y, Matsumoto Y, Uchida Y, Tezuka M, Hidai M (1986) Bull Chem Soc Jpn 59:809
Tomohiro T, Uoto K, Okuno H (1990) Chem Commun 194
Tanaka M, Tanaka K, Tanaka T (1982) Chem Lett 767
Tanaka K, Tanaka M, Tanaka T (1981) Chem Lett 895
DuBois MR (1989) Chem Rev 89:1
Schwartz A, Van Tamelen EE (1977) J Am Chem Soc 99:3189
Van Tamelen EE, Gladysz JA, Miller JS (1973) J Am Chem Soc 95:1347
Schrauzer GN, Kiefer GW, Tano K, Doemeny PA (1974) J Am Chem Soc 96:641
Cleland WE, Holtman DA, Sabat M, Ibers JA, DeFotis GC, Averill BA (1983) J Am Chem Soc 105:6021
Snyder BS, Holm RH (1988) Inorg Chem 27:2339
Reynolds MS, Holm RH (1988) Inorg Chem 27:4494
Zhou HC, Holm RH (2003) Inorg Chem 42:11
Tyson MA, Demadis KD, Coucouvanis D (1995) Inorg Chem 34:4519
Goh C, Segal BM, Huang JS, Long JR, Holm RH (1996) J Am Chem Soc 118:11844
Scott TA, Berlinguette CP, Holm RH, Zhou HC (2005) Proc Natl Acad Sci USA 102:9741
Deng L, Holm RH (2008) J Am Chem Soc 130:9878
Chakrabarti M, Deng L, Holm RH, Münck E, Bominaar EL (2009) Inorg Chem 48:2735
Chakrabarti M, Münck E, Bominaar EL (2011) Inorg Chem 50:4322
Pohl S, Opitz U (1993) Angew Chem Int Ed 32:863
Pohl S, Barklage W, Saak W, Opitz U (1993) Chem Commun 1251
Saak W, Pohl S (1991) Angew Chem Int Ed 30:881
Junghans C, Saak W, Pohl S (1994) Chem Commun 2327
Ohki Y, Sunada Y, Honda M, Katada M, Tatsumi K (2003) J Am Chem Soc 125:4052
Ohki Y, Imada M, Murata A, Sunada Y, Ohta S, Honda M, Sasamori T, Tokitoh N, Katada M, Tatsumi K (2009) J Am Chem Soc 131:13168
Zhang YG, Holm RH (2003) J Am Chem Soc 125:3910
Zhang Y, Holm RH (2004) Inorg Chem 43:674
Berlinguette CP, Miyaji T, Zhang YG, Holm RH (2006) Inorg Chem 45:1997
Ohki Y, Murata A, Imada M, Tatsumi K (2009) Inorg Chem 48:4271
Ohki Y, Ikagawa Y, Tatsumi K (2007) J Am Chem Soc 129:10457
Cai LS, Segal BM, Long JR, Scott MJ, Holm RH (1995) J Am Chem Soc 117:8863
Deng LA, Majumdar A, Lo WN, Holm RH (2010) Inorg Chem 49:11118
Chakrabarti M, Deng L, Holm RH, Münck E, Bominaar EL (2010) Inorg Chem 49:1647
Berlinguette CP, Holm RH (2006) J Am Chem Soc 128:11993
Grove LE, Xie J, Yikilmaz E, Karapetyan A, Miller A-F, Brunold TC (2008) Inorg Chem 47:3993
Marshall NM, Garner DK, Wilson TD, Gao Y-G, Robinson H, Nilges MJ, Lu Y (2009) Nature 462:113
Karlsson S, Boixel J, Pellegrin Y, Blart E, Becker H-C, Odobel F, Hammarström L (2010) J Am Chem Soc 132:17977
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Yao, W., Gurubasavaraj, P.M., Holland, P.L. (2012). All-Ferrous Iron–Sulfur Clusters. In: Rabinovich, D. (eds) Molecular Design in Inorganic Biochemistry. Structure and Bonding, vol 160. Springer, Berlin, Heidelberg. https://doi.org/10.1007/430_2012_81
Download citation
DOI: https://doi.org/10.1007/430_2012_81
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-43598-4
Online ISBN: 978-3-662-43599-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)