
Abstract
This chapter first provides a brief background of how hydrogenation mechanisms have evolved over the years leading to the blossom of catalytic systems with metal-ligand cooperativity. The main body of the chapter focuses specifically on complexes supported by ligands of the type HN(CH2CH2PR2)2. The discussion of hydrogenation systems is organized based on the central metals including Ru, Fe, Os, Rh, Co, Ir, Ni, Pd, Mo, W, Mn, and Re (in that particular order). Substrates involved in these hydrogenation reactions include olefins, aldehydes, ketones, esters, amides, epoxides, nitriles, imines, N-heterocycles, CO2 (to formate or methanol), silyl formates, CO (to ethylene glycol or methanol), and cyclic carbonates. When appropriate, the presence or the lack of metal-ligand cooperativity in these catalytic systems is highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
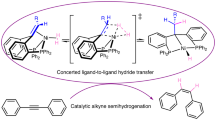
The development of well-defined transition metal-based catalysts for hydrogenation reactions has been an active research area for almost half a century [1,2,3,4]. Early efforts were focused on catalytic hydrogenation of C=C (or C ≡ C) bonds. The generalized and simplified reaction mechanism involves oxidative addition of H2 and coordination of the C=C bond to the metal (Scheme 1, Cycle A). These two steps can occur in either order, as exemplified by Wilkinson’s RhCl(PPh3)3 catalyst for hydrogenating olefins (H2 first) [5] and Halpern’s [(CHIRAPHOS)Rh(solvent)2]+ catalyst for hydrogenating α-aminoacrylic acid derivatives (C=C bond first) [6]. In any case, subsequent C=C insertion into the metal-hydrogen bond followed by reductive elimination of the hydrogenation product completes the catalytic cycle. Hydrogenation reactions can also be catalyzed by a monohydride such as RuHCl(PPh3)3, whose mechanism (Scheme 1, Cycle B) usually features hydrogenolysis of a metal alkyl intermediate generated from C=C insertion [7].

Generalized mechanisms for catalytic hydrogenation of C=C bonds
Catalytic hydrogenation of C=O bonds in aldehydes and ketones, especially those without a neighboring heteroatom to assist carbonyl coordination, was developed much later. Noyori attributed the difficulty to the preferred coordination mode adopted by the carbonyl group [8]. Unlike olefinic substrates, simple aldehydes and ketones often coordinate to metals via the oxygen lone pair instead of the π system [9], which places the carbonyl carbon far away from the hydride to be delivered (Scheme 2, Pathway A). To overcome this issue, Noyori proposed to design catalysts with an acidic hydrogen strategically situated in the ligand scaffold so that it can protonate or form a hydrogen bond with the carbonyl oxygen, forcing an η2-coordination mode for the C=O bond (Scheme 2, Pathway B). Alternatively, in an outer-sphere mechanism, the hydrogen-bonded substrate is brought to the close proximity of the hydride ligand for the desired hydride transfer.

Hydride transfer pathways
The concept of metal-ligand cooperativity described above has significantly advanced the field of homogeneous hydrogenation. In particular, the E–H···O interaction illustrated in Pathway B (Scheme 2) potentially activates the carbonyl group and deemphasizes the role that the metal needs to play. It is therefore not a coincidence that the past decade has witnessed a rapid development of hydrogenation catalysts targeting more challenging substrates such as esters [10, 11] and amides [12, 13] and/or focusing on first-row transition metals including iron [14, 15] and cobalt [16]. Some of these hydrogenation (pre)catalysts as well as the earlier ones developed by Shvo [17] and Noyori [18, 19] are highlighted in Fig. 1.

Representative hydrogenation (pre)catalysts (acidic and hydridic hydrogens are highlighted)
The vast majority of metal-ligand bifunctional catalysts used for hydrogenation reactions contain at least one NH or NH2 donor, which can be preinstalled prior to complexation or formed under hydrogenation conditions (e.g., hydrogenation of ligand C=N bonds) [20]. Although occasionally it is possible to synthesize the H–M–N–H-type complex first [21], most catalytic systems generate this active species in situ from various precatalysts (Scheme 3). Effective catalyst activation strategies include (1) removal of HX (X = Cl, Br, etc.) by a strong base followed by H2 activation [11], (2) hydrogenolysis of a metal alkyl species [16], and (3) unmasking the hydride from the corresponding borohydride complex with heating or in the presence of a BH3 scavenger [22].

Catalyst activation strategies
It had been hypothesized that hydrogenation of C=O bonds catalyzed by H–M–N–H-type complexes would proceed via a concerted H+/H− transfer to the substrate followed by heterolytic cleavage of H2 by the resulting amido species (Scheme 4) [23]. The lost catalytic activity in replacing NH with an NMe donor group is usually an indication of metal-ligand bifunctional catalysis [24]. However, DFT calculations [25] and kinetic studies [26] suggest that the mechanism is more nuanced than initially thought. For example, the delivery of H+/H− to the substrate can be asynchronous, and the alcohol product can serve as a proton shuttle for H2 activation. Furthermore, the metal-bound NH functionality may merely play the role of stabilizing the transition states (through hydrogen bonding interactions) rather than participating in H+ transfer [27]. There are also a number of hydrogenation systems in which alkylation of the NH functionality still results in an active catalyst [28]. Nevertheless, the success of employing H–M–N–H-type complexes as hydrogenation catalysts is evident and likely to provide the momentum to develop new catalysts featuring this particular structural motif.

Simplified catalytic cycle and transition state
This chapter focuses specifically on complexes supported by ligands of the type HN(CH2CH2PR2)2 (RPNHP for short), which are arguably among the most extensively studied hydrogenation catalysts in recent years [29]. Our discussion starts with how these ligands are made and how they are used to prepare the PNP-type complexes. The subsequent overview of hydrogenation catalysis is organized based on the metals, starting from the more popular group 8 elements, transitioning to those in groups 9 and 10, and concluding with mid-transition metals.
2 Ligand Synthesis and Coordination Modes
The more frequently used RPNHP ligands (R = iPr, Cy, Ad or 1-adamantyl, tBu) are commercially available in the neat form or as a THF solution, whereas PhPNHP is typically sold as a hydrochloride salt. If needed, they can be synthesized from [H2N(CH2CH2Cl)2]Cl in one or few steps, depending on the properties of the phosphorus substituents (Scheme 5). Synthesis of PhPNHP or other aryl-substituted ligands is readily accomplished by refluxing [H2N(CH2CH2Cl)2]Cl with the corresponding secondary phosphine in the presence of KOtBu [30,31,32]. Introducing alkyl groups as the phosphorus substituents requires nitrogen protection with a trimethylsilyl group prior to the addition of a lithium dialkylphosphide for the nucleophilic substitution reaction [33,34,35,36]. Hydrolysis of the resulting Me3SiN(CH2CH2PR2)2 restores the NH moiety, which is occasionally performed in the presence of nBu4NF [37] or a 2 M solution of H2SO4 [35] to promote the N–Si bond cleavage. For purification purpose, the crude products are sometimes protonated by a dilute aqueous HCl solution to yield the hydrochloride salts as precipitates [30, 32, 34], and the free RPNHP ligands are released following the treatment with NaOH or KOH.

Synthesis of achiral RPNHP ligands
Chiral RPNHP ligands are also known in the literature (Scheme 6). Chirality has been introduced through the use of a phosphide derived from (2S,5S)-2,5-dimethyl-1-phenylphospholane [38] or an enantiomerically pure secondary phosphine-borane H3B•PH(R)Me (R = tBu, Cy) [39]. In the latter case, lithiation of H3B•PH(R)Me and the subsequent nucleophilic substitution reaction are stereospecific, resulting in stereo-retention at the phosphorus center. In contrast, the in situ generated Li[H3BPPhMe] is configurationally unstable. Synthesis of the corresponding chiral RPNHP ligand thus relies on the use of (SP)-(1-hydroxyethyl)methylphenylphosphine-borane as a masked secondary phosphine-borane and Me3SiN(CH2CH2I)2 as a more reactive electrophile to minimize the chance for racemization [39]. The borane-protected RPNHP ligands can be handled in air, and the removal of BH3 by HBF4•OEt2 is often carried out right before complexation.

Synthesis of chiral RPNHP ligands
The RPNHP ligands or their deprotonated form [N(CH2CH2PR2)2]− (abbreviated here as RPNP) have been employed to make complexes of virtually every metal in groups 4–11 [40]. The coordination chemistry of these ligands is rich, exhibiting a variety of modes including κ1-N [41], κ2-P,N [42], κ2-P,P [43], κ3-P,N,P, and μ2-P,P [44]. As far as hydrogenation catalysts are concerned, the κ3-P,N,P coordination mode is most relevant, because it not only provides an entry to the H–M–N–H species but also stabilizes the metal complexes. As tridentate ligands, RPNHP or RPNP can adopt a meridional or facial configuration, depending on the phosphorus substituents, metals, and ancillary ligands. To illustrate this point, Fig. 2 summarizes the solid-state structures of (RPNHP)FeX2 [45,46,47] and (RPNHP)CoX2 [48,49,50,51,52,53] known to date. The solution structures of (iPrPNHP)FeCl2 probed by Mössbauer and magnetic circular dichroism spectroscopy also suggest that these PNP-type ligands are flexible in binding with metals [45].

Solid-state structures of (RPNHP)MX2 studied by X-ray crystallography
3 Group 8 Metal Systems
3.1 Ruthenium Catalysts
3.1.1 Synthesis of (Pre)catalysts
Synthetic routes to hydrogenation (pre)catalysts involving ruthenium-based PNP-type complexes are summarized in Scheme 7. Complex (PhPNHP)RuHCl(CO) was first developed by Takasago International Corporation with a trademark name of Ru-MACHO [32, 54]. It was originally isolated as a mixture of syn and anti (referring to the relative configuration of NH and RuH) isomers from the reaction of PhPNHP with RuHCl(CO)(PPh3)3 performed in refluxing toluene, although minor modifications to the procedures could lead to the anti isomer only [55, 56]. The presence of two isomers is deemed to be unimportant for the hydrogenation reactions because catalyst activation by a base (Scheme 3) removes the NH hydrogen. The synthetic approach has been successfully extended to other RPNHP ligands [55,56,57] including the one bearing chiral phospholane rings [58]. Substitution of the chloride in Ru-MACHO, iPrRuHCl, and CyRuHCl for BH4− and H− has been accomplished through the addition of NaBH4 [32, 56, 59] and NaBEt3H [56, 60], respectively. The latter reaction with the sterically crowded tBuRuHCl, however, produces a five-coordinate ruthenium hydride, likely due to a facile H2 elimination from the initial product tBuRuH2 [56]. The phenyl analog PhRuH2 synthesized from the NaBEt3H method has a low purity because of rapid decomposition [56]. It can alternatively be synthesized from Ru-MACHO and KOtBu (or KN(SiMe3)2) under H2, though contaminated with ~5% of Ru-MACHO [21].

Synthetic routes to ruthenium-based hydrogenation (pre)catalysts
Other ruthenium precursors have been used to prepare PNP-type hydrogenation (pre)catalysts. The reaction of Ru(COD)(2-methylallyl)2 with tBuPNHP under 5 bar of H2 produces a mixture of tBuRuH2(H2) and tBuRuH(H2) (Scheme 7, Method B) [61]. Pure tBuRuH(H2) can be obtained by stirring the mixture under argon, and its reaction with a primary alcohol also affords tBuRuH as a result of alcohol dehydrogenation and decarbonylation [62]. Using (p-cymene)RuCl2(NHC) (NHC = 1,3-dimethylimidazol-2-ylidene) as the ruthenium source provides an opportunity to incorporate an N-heterocyclic carbene into the catalyst structure. As illustrated in Scheme 7 (Method C), its reaction with a RPNHP ligand can lead to a neutral or cationic pincer complex depending on the solvent used [63].
3.1.2 Hydrogenation of Esters, Ketones, and Their Derivatives
Both Ru-MACHO and Ru-MACHO-BH are commercially available, and among the complexes shown in Scheme 7, they are the most extensively studied ones for catalytic hydrogenation reactions. In 2012, Takasago International Corporation reported that Ru-MACHO mixed with NaOMe was effective for hydrogenation of esters to alcohols (Eq. 1) [54]. The catalytic system is amenable to benzyloxy, piperidinyl, or l-menthoxy group at the α-position but problematic with methoxy or dimethylamino group at the β-position (e.g., MeOCH2CH2CO2Me and Me2NCH2CH2CO2Me). Most remarkably, hydrogenation of methyl (R)-lactate can be performed at room temperature on a multiton scale with minimal erosion to the optical purity (Eq. 2).


In a subsequent report [21], Ikariya demonstrated that Ru-MACHO was efficient in catalyzing hydrogenation of α-difluorinated esters with turnover numbers (TONs) as high as 20,000 (Eq. 3). Functional groups tolerated in this transformation include C=C bonds (terminal or internal), α-pyridyl, and α-thienyl rings. In addition to Ru-MACHO, PhRuH2 and trans-(PhPNHP)RuCl2(CO) are also capable of catalyzing the hydrogenation reactions, although the dichloride complex displays a lower reactivity. For certain substrates (R′ = H, F, Cl, CF3), the hydrogenation process can be stopped at the hemiacetal stage, and in general the selectivity for R′CF2CH(OH)OR″ is improved by lowering the H2 pressure, temperature, and/or the amount of NaOMe. α-Monofluorinated esters can also be hydrogenated under the catalytic conditions; however, the fluorinated primary alcohol products partially undergo cyclization to form epoxides. In a closely related study [64], Lazzari and Cassani showed similar results with RfCO2Me (Rf = C3F7 or C5F11), which led to the isolation of highly fluorinated primary alcohols (Eq. 4).


Another application of the ruthenium-catalyzed ester hydrogenation reactions is in the synthesis of the fragrance hydroxyambran (or 2-cyclododecylpropan-1-ol) [65]. As shown in Scheme 8, hydrogenation of the isomeric mixture of esters with 10% Pd/C provides ethyl 2-cyclododecylpropanoate by saturating all C=C bonds. The second step for ester hydrogenation can be catalyzed at 180°C in toluene by Ru-MACHO (0.2 mol%) in conjunction with NaOMe (2 mol%) or at 150°C in diglyme by Ru-MACHO-BH (1 mol%) alone, both under 50 bar H2. The homogeneous ester hydrogenation can be performed first, although hydrogenation of the resulting mixture of alcohols with 10% Pd/C is plagued by deoxygenation.

Synthesis of hydroxyambran via hydrogenation reactions
In collaboration with Procter & Gamble Company, we studied the hydrogenation of fatty acid methyl esters (FAMEs) under neat conditions [66]. Starting from FAMEs derived from coconut oil, fatty alcohols can be obtained in high yields when the hydrogenation reaction is catalyzed at 135°C by Ru-MACHO (0.07–1.1 mol%, nNaOMe/nRu ~9, 35.5–52.7 bar H2) or Ru-MACHO-BH (0.13–1.0 mol%, 35.5–69.9 bar H2). The catalytic reaction with Ru-MACHO and NaOMe has also been conducted on the kilogram scale with a TON of 1860. Direct hydrogenation of coconut oil to fatty alcohols is feasible under base-free conditions, which involve Ru-MACHO-BH (2.6–2.8 wt%) operating at 135°C under 52.7 bar H2. Hydrogenation of FAMEs containing C=C bonds is more challenging, likely due to the presence of peroxide impurities. Dumeignil and Gauvin recently developed a purification procedure involving 18 h of treatment of FAMEs with basic alumina followed by drying with 3 Å molecular sieves for 48 h [67]. The prepurified FAMEs can undergo smooth hydrogenation to fatty alcohols catalyzed by Ru-MACHO-BH or iPrRuHBH4 (Eq. 5). Depressurizing the system and then reheating the reaction mixture to 130°C under N2 offers a one-pot, two-step synthesis of wax esters. It should be noted that, compared to Ru-MACHO-BH, the isopropyl derivative iPrRuHBH4 shows slightly higher catalytic activity in both hydrogenation and dehydrogenation steps and substantially higher overall selectivity for wax esters.

In 2016, Ogata and Kayaki developed a series of NHC-ligated ruthenium PNP-type catalysts for ester hydrogenation that operate under milder conditions [63]. In particular, complexes PhRuCl2(NHC) and ArRuCl2(NHC) outperform Ru-MACHO in hydrogenating methyl benzoate at 80°C under 10 bar H2. Further optimization of the catalytic conditions showed that in the presence of KOtBu or NaOMe, PhRuCl2(NHC) was active at 50°C even under a balloon pressure of H2, converting various esters to the corresponding alcohols (Eq. 6).

Dialkyl oxalates belong to a special class of esters for which hydrogenation of the first carbonyl group significantly impacts the reactivity of the remaining carbonyl group. A 2013 report by Beller demonstrated that hydrogenation of diethyl oxalate with Ru-MACHO or Ru-MACHO-BH in the presence of NaOEt yielded ethylene glycol exclusively (Scheme 9) [68]. Interestingly, replacing the catalyst with iPrRuHCl or iPrRuH2 under otherwise the same conditions afforded ethyl glycolate only. Further investigation of Ru-MACHO-BH under base-free conditions suggested that the hydrogenation process could stop at the glycolate stage under a lower temperature (60°C) and after a shorter reaction time (1 h). These results imply that the second hydrogenation step is more difficult. As another example of Ru-MACHO-BH differentiating the reactivity of two ester functionalities, MeOCOCH2CO2tBu was subjected to similar hydrogenation conditions (0.54 mol% [Ru], 5.4 mol% NaOEt, 60 bar H2, 100°C, in THF, 3 h), resulting in a partial hydrogenation product with the sterically more hindered carbonyl group left unreacted (i.e., HOCH2CH2CO2tBu as the product) [68]. A closely related substrate is the oxamate illustrated in Eq. 7. The hydrogenation reaction was carried out under more demanding conditions, which unsurprisingly led to complete hydrogenation to ethylene glycol [69].


Complete and partial hydrogenation of diethyl oxalate
Given the higher electrophilicity of the carbonyl carbons, ketones should be more readily hydrogenated than esters. Thus, for molecules containing both ketone and ester functionalities, it is possible to fine-tune the reaction conditions so that one or both carbonyl groups are hydrogenated. This was demonstrated by Tang and Xiao in their study of Ru-MACHO-catalyzed hydrogenation of α-keto esters [70]. Using NaHCO3 as the base additive paired with relatively low H2 pressure (10 bar) and temperature (25°C) leads to α-hydroxy esters almost exclusively (Scheme 10). In contrast, using a stronger base NaOtBu and raising the H2 pressure to 50 bar and temperature to 80°C result in 1,2-diols with high selectivity (86–100%). Selective hydrogenation of γ-keto esters, in principle, could generate γ-hydroxy esters in an analogous way, although the base additive required for catalyst activation also promotes intramolecular transesterification. Very recently, Paixão and Nielsen reported such conversion with TONs of up to 7,400 by employing Ru-MACHO as the precatalyst and NaOEt as the base (Eq. 8) [71]. Under similar conditions, the related ruthenium complexes including Ru-MACHO-BH, PhRuH2, and the commercially available iPrRuHCl also catalyze the hydrogenation of ethyl levulinate to γ-valerolactone, albeit less effectively.


Hydrogenation of α-keto esters
Incorporating a chiral RPNHP ligand into the catalyst structure should allow ketones to be hydrogenated enantioselectively. In a recent report, Junge and Beller tested the catalytic activities of R*RuHCl (syn/anti mixture or pure anti isomer) in hydrogenation of acetophenone and cyclohexyl methyl ketone (Eq. 9) [58]. While the conversion is quantitative, the enantioselectivity is low, suggesting room for improvement in future ligand screening.

Since hemiacetals and aldehydes are intermediates during ester hydrogenation, they can be readily reduced to alcohols under the hydrogenation conditions optimized for esters. Obviously, many other transition metal complexes can also catalyze this process. Employing Ru-MACHO-BH as the hydrogenation precatalyst has some advantage due to the fact that a base additive is not needed, which is ideal for base-sensitive substrates. In exploring precursors to the new antibiotic nemonoxacin, Clarke used this specific ruthenium complex to catalyze the hydrogenation of a hemiacetal and an aldehyde made from asymmetric hydroformylation reactions [72]. Under the conditions outlined in Scheme 11, the alcohol products are obtained with retention of stereochemistry. It is interesting to note that the ester functionality is intact during the hydrogenation process.

Hydrogenation of α-chiral hemiacetals and aldehydes
In addition to esters, ketones, hemiacetals, and aldehydes, amides have also been explored as substrates for the ruthenium-catalyzed hydrogenation reactions, although the conditions are much harsher. In 2018, Tu reported the hydrogenation of lactams to amino alcohols catalyzed by Ru-MACHO-BH (Eq. 10) [73]. The high temperature of 150°C is critical to the success of the hydrogenation process. According to the catalyst activation mechanism (Scheme 3), a base additive is normally not needed for Ru-MACHO-BH to be catalytically active. In fact, Ru-MACHO-BH does show some catalytic activity for hydrogenating N-phenyl-2-pyrrolidone. However, the addition of K3PO4 significantly enhances the catalytic efficiency (96% vs. 64% yield). The NHC-ligated complex PhRuCl2(NHC) displays slightly lower activity (82% yield), whereas the methylated PNP pincer complexes (PhPNMeP)RuHCl(CO) and (PhPNMeP)RuH(BH4)(CO) (PhPNMeP = MeN(CH2CH2PPh2)2) are completely inactive, suggesting the importance of the NH moiety. Under similar conditions, hydrogenation of unprotected lactams (e.g., caprolactam and azocan-2-one) and oxazolidinones (e.g., 3-phenyloxazolidin-2-one) is also possible, providing the corresponding amino alcohols in high yields.

3.1.3 Hydrogenation of Other Bonds
Substrates that can be hydrogenated with the aforementioned ruthenium catalysts go beyond those containing carbonyl groups. Very recently, Gunanathan showed that Ru-MACHO along with KOtBu was effective and selective for the hydrogenation of epoxides to secondary alcohols (Eq. 11) [74]. This transformation proceeds via direct hydrogen transfer from the presumed active species PhRuH2 rather than by a two-step process involving epoxide-to-ketone isomerization followed by ketone hydrogenation. Functional groups compatible with the catalytic conditions are very similar to those observed in ester hydrogenation, except that herein terminal C=C bonds are also hydrogenated. Hydrogenation of chiral epoxide R-glycidol, however, gives a complex mixture, perhaps due to the interference by KOtBu. Another limitation of the catalytic system is that internal epoxides resist hydrogenation.

The strategy of using the ruthenium-based PNP pincer complexes for hydrogenation reactions has been extended to nitrile reduction. Two reports on this topic appeared in 2015, but featured different RPNHP ligands. Both Ru-MACHO and Ru-MACHO-BH were shown by Beller to catalyze the hydrogenation of nitriles to primary amines, although the former required KOtBu to activate the catalyst [75]. One of the challenges for nitrile hydrogenation is selectivity, as the intermediates can be trapped by the initially produced primary amines, which lead to secondary amines, secondary imines, and/or tertiary amines as by-products. Under the conditions summarized in Eq. 12, a variety of aliphatic and aromatic nitriles are converted to primary amines with high selectivity. Lowering the temperature or catalyst loading or hydrogenating long-chain nitriles such as dodecanenitrile, however, erodes selectivity for the primary amines. The catalytic system exhibits high functional group tolerance including the preservation of ester functionalities. Substrates that fail to react include furan-2-carbonitrile, 2-methyl-3-butenenitrile, and 6-bromohexanenitrile. Prechtl focused on the study of ruthenium complexes supported by the more bulky ligand tBuPNHP. Hydrogenation of benzonitrile and p-tolunitrile catalyzed by tBuRuH2(H2)/tBuRuH(H2) or tBuRuH was optimized to favor the secondary imines (Eq. 13), although hydrogenation of p-bromobenzonitrile suffered from moderate yield and low selectivity, and hydrogenation of heptyl cyanide catalyzed by tBuRuH2(H2)/tBuRuH(H2) afforded predominantly octylamine [76]. Under similar catalytic conditions, externally added amines can trap the primary imine intermediates, leading to efficient hydrogenative coupling of nitriles to secondary imines (Eq. 14). Finally, switching the solvent from toluene to iPrOH and raising the temperature from 50°C to 90°C render tBuRuH more selective for the formation of primary amines (Eq. 15). However, varying amounts of R′CH2N=CMe2 (0–29%) were also observed due to dehydrogenation of the solvent iPrOH to acetone.




For the reactions shown in Eqs. 13 and 14, a small amount of secondary amines was often detected, suggesting that it is possible to develop ruthenium-catalyzed hydrogenation of imines. In 2017, Tang reported such process with an objective to develop a diastereoselective route to convert chiral α-ketimino esters to chiral aryl glycine derivatives [77]. As illustrated in Eq. 16, at 25–40°C, Ru-MACHO in combination with NaOEt is effective for the C=N bond hydrogenation, which provides N-tert-butylsulfinyl-protected α-amino esters with high diastereoselectivity.

3.1.4 Hydrogenation Reactions Related to CO2 or CO Reduction
Homogeneous hydrogenation of CO2 or CO to liquid fuels such as methanol has been subject to extensive studies in recent years, which, to some degree, is propelled by the development of PNP-type hydrogenation catalysts. Conversion of CO2 to methanol is formally a six-electron reduction process, and each hydrogenation event reduces formal oxidation state of the carbon by two. Based on this analysis, reduction of CO to methanol is formally a four-electron reduction process. Conversion of CO2 or CO to oxalate or ethylene glycol requires odd number of electrons to be transferred (Fig. 3), which usually involves a radical intermediate or a process separate from hydrogenation. For a more systematic discussion, hydrogenation reactions described in this section are organized based on how formal oxidation state of the carbon changes: (A) +4 to +2, (B) +2 to −2, (C) +2 to +3 to −1, and (D) +4 to −2.

Compounds relevant to CO2 or CO reduction
Hydrogenation of CO2 to formic acid in organic solvents is an endergonic process (ΔG0298 = +32.8 kJ mol−1). The thermodynamics can be improved by performing the reaction in water (ΔG0298 = −4.0 kJ mol−1) and/or adding a base to convert formic acid to a formate salt [78]. Direct hydrogenation of bicarbonate to formate is also thermodynamically favorable. For PNP-type catalytic systems, Beller reported in 2014 that transfer hydrogenation of HCO3− (or CO2) to HCO2− with MeOH was efficiently catalyzed by Ru-MACHO or iPrRuHCl in an alkaline solution [79]. It was noted that hydrogen pressure was built up during the reaction, consistent with catalytic methanol dehydrogenation. To confirm that the in situ generated H2 was responsible for bicarbonate reduction, Ru-MACHO was tested as a hydrogenation catalyst for NaHCO3, which, under the conditions outlined in Eq. 17, afforded HCO2Na in 71% yield. A more recent report by Treigerman showed that this hydrogenation process could be conducted at 70°C in iPrOH-H2O mix solvent and the catalyst could be reused at least three times with an overnight rest of the catalyst between two consecutive runs [80]. Czaun, Prakash, and Olah carried out a more detailed study of Ru-MACHO- and Ru-MACHO-BH-catalyzed hydrogenation of bicarbonate (Eq. 18) as well as hydrogenation of CO2 assisted by a hydroxide (Eq. 19) or a carbonate (Eq. 20) [81]. The reverse reaction, dehydrogenation of formate, was also catalyzed by Ru-MACHO or Ru-MACHO-BH. To demonstrate the reversible hydrogen storage in formate salts, Ru-MACHO-BH was employed to catalyze CO2 hydrogenation (75 bar, pH2: pCO2 = 3: 1) in the presence of NaOH followed by dehydrogenation under an atmospheric pressure, a process that was repeated at 70°C for six times without a significant loss of the catalytic activity. Interestingly, the NH moiety is not needed here; (PhPNMeP)RuHCl(CO) catalyzes the hydrogenation and the dehydrogenation reactions with a comparable or better efficiency than Ru-MACHO and Ru-MACHO-BH.




Hydrogenation of CO2 along with a primary or secondary amine to generate a formamide is also a formally two-electron reduction process (+4 to +2 for the change in formal oxidation state of the carbon). In 2015, Ding reported that in the presence of KOtBu (0.1 mol%), Ru-MACHO, iPrRuHCl, CyRuHCl, AdRuHCl, tBuRuHCl, and the methylated complex (PhPNMeP)RuHCl(CO) were all effective in catalyzing N-formylation of morpholine under H2 and CO2 (35.5 bar each, 0.1 mol% [Ru], 120°C, in THF) [82]. Using Me2NH as the amine (also as a base) and lowering the catalyst loading of Ru-MACHO to 0.000093 mol% produced DMF with TONs of up to 599,000. The catalyst showed remarkably high stability under the catalytic conditions. With a catalyst loading of 0.002 mol%, Ru-MACHO was reused 11 times without the concern for a brief exposure to air between runs. Further hydrogenation of formamides to methanol (formal oxidation state change from +2 to −2) is possible but needs to be performed under a higher temperature and in the presence of KOtBu. As illustrated in Scheme 12, N-formylation of morpholine followed by hydrogenation of the resulting formamide in the same reactor produces methanol in 36% yield along with the unreacted formamide.

One-pot, two-step hydrogenation of CO2 to methanol
Another example of changing formal oxidation state of the carbon from +2 to −2 involves catalytic hydrogenation of silyl formates to methanol. A recent report by Hong showed that silyl formates were first prepared from silanes and CO2 catalyzed by Rh2(OAc)4-K2CO3 or RuCl3•H2O [83]. The subsequent hydrogenation reactions can be catalyzed by Ru-MACHO combined with KOtBu but more efficiently by Ru-MACHO-BH, which does not require a base. Under the optimized conditions (Eq. 21), various silyl formates (R′3Si = trialkyl, aryldialkyl, and alkyldiaryl groups) are converted to methanol and the corresponding silanols. Hydrogenation of silyl formates bearing an electron-donating aryl group (e.g., R′3Si = Me2(p-MeOC6H4)Si or Me2(p-MeC6H4)Si) under 10 bar H2 is complicated by the formation of R′3SiOMe and R′3SiOSiR′3 as the by-products. The selectivity for methanol and silanols can, however, be improved by raising the H2 pressure to 80 bar or by adding 0.1 equiv. of methanol. For the latter strategy, methanol attacks silyl formates to yield silanols and methyl formate, which is in turn readily hydrogenated to 2 equiv. of methanol under the catalytic conditions.

When K3PO4 is used as the catalyst, secondary amines can react with CO (30 bar) at 140°C to give formamides. This reaction coupled with formamide hydrogenation provides an indirect route of hydrogenation of CO to methanol. The challenge lies in the fact that the carbonylation step is favored by an alcoholic solvent, whereas the hydrogenation step is favored by a relatively nonpolar solvent such as toluene. To solve this problem, Prakash designed a one-pot, two-step process in which carbonylation of piperidine or diethylenetriamine (DETA) was carried out in ethanol first [84]. A ruthenium catalyst (Ru-MACHO or Ru-MACHO-BH), toluene, and H2 were then added to the reactor, and following hydrogenation, methanol was produced in 75–80% yield. Direct hydrogenation of CO to methanol was made possible by using DETA as the amine and toluene-EtOH (1:1) as the mix solvent (Eq. 22). The reaction was performed in a closed system, providing methanol in 59% yield (or a TON of 539 based on the amount of Ru-MACHO-BH used) along with formamides in 15% yield.

Similarly, palladium-catalyzed oxidative carbonylation of piperidine provides an oxamide that can be hydrogenated to ethylene glycol, representing an indirect method of hydrogenating CO to ethylene glycol. The overall process involves changes of formal oxidation state of the carbon from +2 to +3 and then to −1. To this end, Li and Beller reported in 2016 that oxidative carbonylation of piperidine was best catalyzed by Pd(acac)2-P(o-tol)3 using compressed air as the source of oxidant [85]. Hydrogenation of the resulting oxamide is affected by Ru-MACHO or Ru-MACHO-BH (0.1–1 mol% loading, in toluene) at 160°C under 60 bar H2 using KOtBu (2–10 mol%) as the additive. Combining these two steps in one reactor is difficult, and the exchange of solvents and a filtration through silica gel are required after the formation of the oxamide (Scheme 13).

Piperidine-mediated conversion of CO to ethylene glycol
For the hydrogenation of CO2 to methanol (with a change of the carbon formal oxidation state from +4 to −2), one strategy is to use cyclic carbonates as surrogates for CO2, which can bypass formic acid (incompatible with metal hydrides) or formate salts (thermodynamic sinks). This was successfully demonstrated in 2012 by Ding who studied ruthenium-catalyzed hydrogenation of ethylene carbonate [55]. Among the precatalysts screened, Ru-MACHO performs significantly better than the analogous complexes bearing alkyl groups as the phosphorus substituents (i.e., iPrRuHCl, CyRuHCl, AdRuHCl, and tBuRuHCl) with TONs as high as 87,000. In this case, the NH moiety is crucial for the catalysis because the methylated complex (PhPNMeP)RuHCl(CO) fails to hydrogenate ethylene carbonate. Under the optimized conditions (Eq. 23), various cyclic carbonates are converted to diols and methanol in almost quantitative yields. The hydrogenation strategy was further applied to poly(propylene carbonate) with an Mw of 1,000,698, giving 1,2-propylene glycol and methanol in high yield (Eq. 24). Hydrogenation of (R)-propylene carbonate under similar conditions generates racemic 1,2-propylene glycol, presumably due to the reversibility of the hydrogenation process.


The seminal work by Sanford in 2015 demonstrated that direct catalytic hydrogenation of CO2 to methanol could be accomplished via tandem catalysis of Me2NH promoted by Ru-MACHO-BH (Scheme 14) [86]. The proposed mechanism involves equilibrium between CO2 and dimethylammonium dimethylcarbamate (DMC), which can be hydrogenated to formic acid (trapped as dimethylammonium formate or DMFA) and DMF, respectively. The most challenging step is the hydrogenation of DMF to methanol, a process requiring temperatures as high as 155°C. Under such conditions, the ruthenium catalyst also starts to decompose. To maximize the yield for methanol, a temperature ramp strategy was developed so that a sufficient amount of DMF and DMFA could be accumulated at 95°C. The subsequent hydrogenation carried out at 155°C provides methanol with TONs of up to 550 and DMF-DMFA with combined TONs of up to 1870.

Ruthenium-catalyzed hydrogenation of CO2 in the presence of Me2NH
In addition to Me2NH, polyamines can also be employed to assist CO2 hydrogenation. Olah and Prakash reported in 2016 that pentaethylenehexamine (PEHA) combined with a catalytic amount of Ru-MACHO or Ru-MACHO-BH promoted the hydrogenation of CO2 to methanol in an etherate solvent (e.g., THF, 1,4-dioxane, diglyme, or triglyme) [87]. After extensive optimization of the reaction, it was determined that with this new catalytic system, the temperature ramp strategy and the addition of K3PO4 were unnecessary. At 155°C under 75 bar H2/CO2 (3: 1 or 9: 1), methanol was obtained with TONs of up to 1,200 and the catalyst was reused five times with 75% of the initial activity retained. CO2 can also be captured from simulated air (400 ppm of CO2 in 80% N2 and 20% O2) by an aqueous solution of PEHA and then subjected to hydrogenation conditions (155°C, 50 bar H2, Ru-MACHO-BH as the catalyst, 55 h), which provides methanol in 79% yield.
Additional improvements to the catalytic system include hydrogenation of the captured CO2 (from pure CO2 or simulated air) using various polyamines in a biphasic mixture of water and 2-methyltetrahydrofuran (2-MeTHF). This allows an easy separation of the catalyst (in 2-MeTHF layer) from the hydrogenation products (in water layer). Depending on the temperature applied, the hydrogenation product can be a formate salt [88] or predominantly methanol [89] (Scheme 15). Both processes have shown excellent recyclability of the catalyst (4–5 runs).

CO2 capture and the subsequent hydrogenation reaction in a biphasic mixture
The polyamines play important roles in determining the yield and selectivity of the hydrogenation process. For hydrogenation of the captured CO2 to formate (in 1,4-dioxane, 50°C, 50 or 80 bar H2), 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,1,3,3-tetramethylguanidine (TMG), and 1,8-diazabicycloundec-7-ene (DBU) outperform PEHA and branched polyethyleneimines (BPEI, Mw = 800) in terms of the formate yield [88]. For hydrogenation of the captured CO2 to methanol (in 2-MeTHF, 145°C, 70 bar H2), PEHA gives a higher methanol yield than BPEI (Mw = 800 or 25,000), linear polyethyleneimines (LPEI, Mw = 2,500 or 100,000), and poly(allylamine) (PAA, Mw = 10,000). The latter three polyamines also produce more formate and formamide as the by-products [89]. In a related study, Kayaki also used BPEI (Mn = 600) and LPEI (Mn = 2,500, 5,000, 25,000, or 250,000) to assist CO2 hydrogenation, although the reactions were carried out in THF only [90]. At 100°C under 100 bar H2 and 100 bar CO2, ruthenium complexes including Ru-MACHO, Ru-MACHO-BH, CyRuHCl, and PhRuCl2(NHC) were shown to be similarly effective in converting CO2 and the polymers to N-formylated PEI with 67–90% CHO content. Further hydrogenation of the N-formylated PEI to methanol or direct hydrogenation of CO2 to methanol assisted by BPEI or LPEI is best catalyzed by Ru-MACHO-BH at 140–160°C under 80 bar H2/CO2 (3:1 or 7:1).
The nature of the phosphorus substituents also plays critical roles in determining the catalytic efficiency. Although Ru-MACHO, iPrRuHCl, and CyRuHCl (in the presence of K3PO4) all prove to be active precatalysts for the hydrogenation of formamides to methanol [91, 92], for polyamine-assisted CO2 hydrogenation, Ru-MACHO (or Ru-MACHO-BH) appears to be the best choice for maximizing methanol yield [89, 92]. A recent mechanistic study by Prakash offered very insightful information about why the phenyl groups are beneficial for the hydrogenation reaction [92]. Evidently, during CO2 to methanol conversion, a small amount of CO (~0.2%) is generated, which poisons those ruthenium catalysts bearing alkyl substituents. In fact, during PEHA-assisted CO2 hydrogenation, the resting state of the catalyst was identified as a cationic bis(carbonyl) hydride complex (Scheme 16). For the phenyl derivative, the CO is more labile due to weaker donation from the phosphorus atoms, allowing [(PhPNHPh)Ru(CO)2H]+ to reenter the catalytic cycle by forming the active species PhRuH2. Such process is less favorable for the alkyl derivatives.

Involvement of the cationic bis(carbonyl) hydride species during the catalytic hydrogenation reaction
3.2 Iron Catalysts
3.2.1 Synthesis of (Pre)catalysts
The recent surge in developing base metal catalysis has prompted many research groups to design iron-based hydrogenation catalysts. A logical extension of the work shown in the previous section would be replacing ruthenium with iron, although the chemistry of the ruthenium PNP-type complexes cannot be simply extrapolated to the iron systems. This is already reflected by how the iron-based (pre)catalysts are made (Scheme 17). First of all, iron analogs of the ruthenium precursors RuHCl(CO)(PPh3)3 and Ru(COD)(2-methylallyl)2 do not exist. While the catalysis community enjoys the use of Ru-MACHO and Ru-MACHO-BH, both of which are commercially available, Fe-MACHO remains elusive, and Fe-MACHO-BH has a very limited lifetime in solution [93]. Nevertheless, in 2013, Beller first reported the synthesis of iPrFeHBH4, which involved the treatment of trans-(iPrPNHP)FeBr2(CO) (made from iPrPNHP and FeBr2(THF)2 under 1 bar CO) with excess NaBH4 in EtOH (Scheme 17, Method A) [94]. Reducing the amount of NaBH4 to 1 equiv. led to the isolation of iPrFeHBr [95], which was alternatively prepared in THF from trans-(iPrPNHP)FeBr2(CO) using NaBEt3H as the hydride source [94]. Depending on the reaction time and work-up procedures, both iPrFeHBH4 and iPrFeHBr can be isolated as a mixture of syn and anti isomers or as a pure anti isomer, although it is expected to have no impact on the catalytic performance. This synthetic strategy has been extended to other ligand systems including EtPNHP [93, 96], CyPNHP [97, 98], the phospholane-based PNP ligand [58], and the P-stereogenic PNP ligands [39]. It is worth pointing out that (S,S)-(tBuMePCH2CH2)2NH adopts the facial coordination mode upon formation of the dibromide complex, whose reaction with NaBEt3H must be carried out in CH2Cl2 instead of THF to avoid degradation. The three P-chiral precatalysts, MePhFeHBr, MeCyFeHBr, and MetBuFeHBr, decompose quickly in solution; therefore, they should be prepared right before use [39]. The more commonly used iron precatalyst iPrFeHBH4 can also be synthesized from the dichloride complex trans-(iPrPNHP)FeCl2(CO) and NaBH4 (10 equiv) in MeCN-EtOH, although applying this protocol to trans-(CyPNHP)FeCl2(CO) fails to generate CyFeHBH4 cleanly [99].

Synthetic routes to iron-based hydrogenation (pre)catalysts
The five-coordinate complex iPrFeH can be obtained from dehydrohalogenation of iPrFeHBr [100] or iPrFeHCl (made from trans-(iPrPNHP)FeCl2(CO) and nBu4NBH4) [101] with KOtBu (Scheme 17, Method B). The cyclohexyl analog CyFeH is also available using this method [101]. Preparing the isocyanide derivatives iPrFeH(CNArMe2) and iPrFeH(CNArOMe) follows similar procedures (Method C) [102]. The key challenge here is in the synthesis of trans-(iPrPNHP)FeCl2(CNR). To avoid the undesired cationic bis(isocyanide) complexes, isocyanides must be diluted and added slowly to (iPrPNHP)FeCl2.
3.2.2 Hydrogenation of Esters, Ketones, and Their Derivatives
In 2014, the Beller group [24] and our group [95] independently reported that iPrFeHBH4 was effective in catalyzing the hydrogenation of esters including lactones to alcohols (Eq. 25). Functional groups tolerated under the catalytic conditions include CF3, MeO, pyridyl, furyl, benzothiazolyl, and isolated C=C bonds. In contrast, nitrile groups and conjugate C=C bonds are hydrogenated along with the carbonyl groups, and phenol-type functionality shuts down the catalysis completely. For further applications (Fig. 4), iPrFeHBH4 has been utilized to catalyze the hydrogenation of a dodecapeptide, which is a precursor to the drug molecule Alisporivir [24], and an industrial sample CE-1270, which is derived from coconut oil and used in surfactant production [95]. As with the ruthenium system, iPrFeHBH4 has also been tested for direct catalytic hydrogenation of coconut oil (2.0 wt% catalyst loading, 135°C, 52.7 bar H2), although the fatty alcohol yield is low (12%) due to the low thermal stability of the catalyst as well as its sensitivity toward impurities [66].


A dodecapeptide and an industrial sample CE-1270 used in iron-catalyzed hydrogenation reactions
The complex iPrFeHBH4 is a precatalyst; under heating, it releases BH3 to generate the active species trans-(iPrPNHP)FeH2(CO) [22, 24]. This process can be facilitated by the addition of Et3N to trap BH3, resulting in a more efficient catalytic system [22]. However, using other bases such as KOtBu and Na2CO3 can reduce the alcohol yield [24]. In the presence of KOtBu (or NaOMe) and under H2, iPrFeHBr is also converted to trans-(iPrPNHP)FeH2(CO), thus catalyzing ester hydrogenation, although it can be complicated by base-promoted transesterification with the alcohol products [95].
To understand the substituent effects, Beller replaced the isopropyl groups in iPrFeHBH4 with ethyl or cyclohexyl groups and studied the catalytic performance of these new borohydride complexes [96]. Consistent with the steric argument, at a relatively low temperature of 60°C, EtFeHBH4 performs better than iPrFeHBH4, which is in turn more reactive than CyFeHBH4 for the hydrogenation of methyl benzoate. Notably, Me2NCH2CH2CO2Me, which is not a viable substrate for the Ru-MACHO system, can be smoothly hydrogenated to Me2N(CH2)3OH at 100°C under 30 bar H2 using EtFeHBH4 as the precatalyst (1 mol%). It should be emphasized here that temperature and H2 pressure play profound roles in controlling the activation, stability, and reactivity of the borohydride complexes and ultimately their catalytic efficiency. A closely related study by Langer showed a decreasing reactivity order of iPrFeHBH4 > CyFeHBH4 > EtFeHBH4 when the hydrogenation of methyl benzoate was conducted as 100°C under 10 bar H2 (Eq. 26) [93].

Catalytic hydrogenation of levulinates, which contain two different types of carbonyl groups, has been explored with the iron PNP-type complexes. Very recently, Paixão and Nielsen showed that at 60°C under 10 bar H2, iPrFeHBH4 combined with KOMe catalyzed the hydrogenation of ethyl levulinate to γ-valerolactone with <10% conversion in 3 h (Scheme 18) [71]. This level of activity is lower than the analogous ruthenium complexes including iPrRuHCl and Ru-MACHO-BH. With a catalytic amount of iPrFeHBr (0.05 mol%) and a stoichiometric amount of KOH, levulinic acid is also hydrogenated at 100°C under 50.7 bar H2, giving γ-valerolactone with a TON of 540 in 5 h [103]. Under base-free conditions with EtFeHBH4 (Scheme 18), methyl levulinate can be fully converted to a diol in high yield [96].

Catalytic hydrogenation of alkyl levulinates
Simple ketones are much more reactive; therefore, their hydrogenation to alcohols can be carried out under milder reaction conditions. For example, hydrogenation of 4-methoxyacetophenone catalyzed by 1 mol% iPrFeHBH4 or iPrFeH alone or by iPrFeHBr with 10 mol% KOtBu takes place at room temperature under 6.5 bar H2, which affords the alcohol product quantitatively in 8 h [104]. Hydrogenation of acetophenone catalyzed by iPrFeH (0.2 mol%) is operative at room temperature under 1 bar H2 [105].
The chiral precatalysts illustrated in Scheme 17 have been designed specifically for asymmetric hydrogenation of ketones. With the exception of (S,S)-MetBuFeHBr, which, after activation by KOtBu, fails to catalyze the hydrogenation of acetophenone [39], all other precatalysts promote ketone hydrogenation at 20–40°C under 5.5–50 bar H2. While the conversions are high, ee’s (ee = enantiomeric excess) for the alcohol products are typically low or moderate (0–64%). The more selective iron-based catalysts for ketone (and imine) hydrogenation are those developed by Morris with chirality built between nitrogen and phosphorus donors. These unsymmetrical P-NH-P′-type ligand systems are beyond the scope of this review. Interested readers are directed to several recent papers for more details [106,107,108,109].
For other carbonyl-based substrates, three research groups have independently investigated iron-catalyzed hydrogenation of amides, which generates amines and alcohols via C–N bond cleavage. In 2016, Langer demonstrated that EtFeHBH4 was effective for the hydrogenation of PhCONHR″ (R″ = Ar, Me), CF3CONHPh, PhCONMe2, and γ-lactams bearing an N-aryl group (Eq. 27) [93]. Under similar conditions, δ-lactams such as N-phenyl-2-piperidone and N-bis(trifluoromethyl)phenyl-2-piperidone failed to be hydrogenated. About the same time, Sanford reported that the addition of a weak base such as K3PO4 and Et3N could enhance the catalytic activity of CyFeHBH4 in amide hydrogenation [97]. Compared to CyFeHBH4, iPrFeHBH4 and EtFeHBH4 are less efficient. Under the optimized conditions (Eq. 27), CH3CONHPh, CH3CONPh2, PhCONHPh, and ArCONPh2 (Ar = Ph or a more electron-withdrawing aryl group) are converted to the corresponding amines and alcohols in high yields. In contrast, hydrogenation of PhCONMe2 and CF3CONHBn is low yielding, and hydrogenation of 4-MeOC6H4CONPh2 and 4-Me2NC6H4CONPh2 is negligible. In a 2017 report, Bernskoetter employed a low catalyst loading of iPrFeH (0.07 mol%) and performed the reaction at 100°C under 30.4 bar H2 (in THF), which resulted in TONs of 50–160 in 4 h for the hydrogenation of R′CONHPh (R′ = Me, Ph, CF3) [110]. The addition of LiOTf and HCONHPh as additives was shown to enhance the catalytic activity of iPrFeH. Under the modified catalytic conditions (Eq. 27), PhCONHMe remains to be unreactive.

3.2.3 Hydrogenation of Other Bonds
The iron-based PNP-type complexes are capable of catalyzing the hydrogenation of polar bonds other than the C=O bonds described above. As demonstrated by Beller in 2014, hydrogenation of aromatic and aliphatic nitriles can be catalyzed by iPrFeHBH4 at 70–130°C under 30 bar H2 (Eq. 28) [111]. The catalytic system shows excellent selectivity for primary amines, which can be conveniently isolated as hydrochloride salts following acidification by HCl. A wide variety of functional groups such as MeO, halogens, NH2, pyridyl, indolyl, and thienyl are amenable to the catalytic conditions. In contrast, NO2 and phenol-type groups shut down the reaction. Most remarkably, hydrogenation of 4-MeOCOC6H4CN at 130°C is selective for the nitrile functionality despite having a reducible ester group. Hydrogenation of cinnamonitrile followed by acidification produces trans-[PhCH=CHCH2NH3]Cl with the C=C bond almost intact, further highlighting the high chemoselectivity (>25: 1). Here, the presence of the NH moiety is critical to the success of the hydrogenation process. A control experiment using (iPrPNMeP)FeH(CO)(BH4) as the catalyst did not yield any hydrogenation product. In a follow-up study, Beller showed that CyFeHBH4 was similarly effective, whereas EtFeHBH4 became inactive when the catalyst loading was reduced from 1 mol% to 0.5 mol% [98]. According to that study, temperature is very critical for the outcome of the hydrogenation. Hydrogenation of PhCN performed below 70°C leads mainly to the secondary imine PhCH=NCH2Ph.

N-heterocycles have been studied as potential organic hydrogen storage materials through reversible acceptorless dehydrogenation and hydrogenation reactions, both of which require a catalyst. In 2014, Jones reported that iPrFeHBr, when activated by KOtBu, was effective for the hydrogenation of quinoline derivatives to 1,2,3,4-tetrahydroquinaldines (Eq. 29) [100]. Related N-heterocycles including 2-methylindole and 2,6-lutidine are also hydrogenated under similar conditions. As expected, iPrFeHBH4 also serves a precatalyst (without a base additive) for this process, although it is less active, resulting in 89% of quinoline being hydrogenated even at a higher temperature of 110°C. According to DFT calculations by Surawatanawong, the first hydrogenation event converts quinoline to 1,4-dihydroquinoline, which undergoes base-assisted isomerization to 3,4-dihydroquinoline [112]. Further hydrogenation of the C=N bond furnishes the 1,2,3,4-tetrahydroquinaldine product.

Typically, olefins are not considered viable substrates for hydrogenation systems that operate via metal-ligand cooperation. However, when the C=C bonds are significantly polarized, they can accept H− and H+ from H–M–N–H-type complexes in a similar way as carbonyl groups. In a recent study, Jones demonstrated this concept in studying iron-catalyzed hydrogenation of styrene and its derivatives [113]. At room temperature under an atmospheric H2 pressure, styrene is converted to ethylbenzene quantitatively in 24 h when iPrFeHBr (5 mol%) mixed with KOtBu (15 mol%) or iPrFeH (5 mol%) is employed as the catalyst. The borohydride complex iPrFeHBH4 is significantly less active due to the need to remove BH3, which is usually favored at elevated temperatures. Under the optimized conditions (Eq. 30), substituted styrenes, especially those containing electron-withdrawing groups, undergo C=C bond hydrogenation smoothly. Because the reaction conditions are very mild, other reducible functional groups such as ester, pyridyl, and CN are tolerated, although hydrogenation of 4-cyanostyrene is sluggish due to catalyst inhibition by substrate coordination. Hydrogenation of trans-PhCH=CHCOCH3 eventually gives the fully saturated product PhCH2CH2CH(OH)CH3. At the early stage of the reaction, C=O hydrogenation is faster than C=C hydrogenation. Consistent with a mechanism featuring metal-ligand cooperativity, weakly polarized C=C bonds such as those in 1-hexene and tert-butylethylene resist hydrogenation, and the methylated complex (iPrPNMeP)FeH(CO)(BH4) shows no catalytic activity even at 100°C.

3.2.4 Hydrogenation Reactions Related to CO2 or CO Reduction
Combining iron catalysis with CO2 reduction addresses many sustainability-related challenges [114]. Like the ruthenium-based systems described earlier, iron-based PNP-type complexes have also been explored in variety of transformations that are associated with CO2 reduction. Once again, the discussion here is organized based on how formal oxidation state of the carbon changes during hydrogenation (Fig. 3).
For an example involving a change of +4 to +2 in carbon oxidation state, Hazari and Schneider showed in 2014 that hydrogenation of CO2 (1:1 mixture with a total pressure of 70 bar) could be catalyzed by CyFeH at 80°C in the presence of 300 equiv. DBU, which yielded formate with a TON of 186 in 12 h [101]. Adding 150 equiv. LiBF4 to the reaction mixture improves the TON to 289 in 4 h. Detailed mechanistic studies by Hazari and Bernskoetter suggest that the Lewis acid disrupts the intramolecular hydrogen bonding interaction between the NH moiety and the formato group and facilitates the release of HCO2− from iron [115]. Further screening of Lewis acids reveals that the hydrogenation reaction is best carried out in the presence of LiOTf with an optimal DBU to LiOTf ratio of 7.5 to 1. Under such conditions, hydrogenation of CO2 catalyzed by iPrFeH and CyFeH gives formate with TONs of 6,030 and 8,910, respectively (Scheme 19). The borohydride complex iPrFeHBH4 displays a lower catalytic activity. Similar to the ruthenium-based catalytic systems, iron-catalyzed hydrogenation of CO2 to the formate stage does not require the presence of the NH moiety. As a matter of fact, (iPrPNMeP)FeH(CO)BH4 and (CyPNMeP)FeH(CO)BH4 are significantly more active with an about 30-fold increase in formate yield. For additional modification to the catalyst structure, Hazari and Bernskoetter incorporated different isocyanide ligands into the PNP pincer system. The five-coordinate complexes iPrFeH(CNArMe2) and iPrFeH(CNArOMe) prove to be less active than the CO analog iPrFeH [102]. The second-generation isocyanide-based catalysts supported by the methylated PNP ligand iPrPNMeP show some improvement over iPrFeH(CNArMe2) and iPrFeH(CNArOMe); however, they are still less effective than the corresponding CO derivatives [116].

Hydrogenation of CO2 to formate catalyzed by various iron-based PNP pincer complexes
Another formally two-electron reduction process with CO2 is N-formylation of amines, as mentioned in the ruthenium systems (Scheme 12). For iron-based catalysts, Bernskoetter compared the activity of iPrFeH, its adduct with HCONHPh, (iPrPNMeP)FeH(CO)BH4, and trans-(iPrPNMeP)FeH2(CO) for the N-formylation of morpholine [117]. Under the conditions outlined in Eq. 31, the reaction catalyzed by iPrFeH generates the formamide with a TON of 1930. The catalytic performance is slightly better than the HCONHPh adduct but worse than the methylated PNP complexes, again illustrating that the NH moiety is not needed for CO2 reduction to the formate stage.

The iron-catalyzed amide hydrogenation has already been described in the previous section (see Eq. 27). Hydrogenation of formamides to methanol is singled out and discussed here due to its relevance to CO2 reduction (which changes the carbon oxidation state from +2 to −2). Under Sanford’s conditions (0.33 mol% CyFeHBH4, 1.66 mol% K3PO4, 20 bar H2, 110°C, 3 h), N-formylmorpholine, HCONHAr, and HCONPh2 are hydrogenated to methanol with TONs of up to 300 [97]. Hydrogenation of HCONHMe and HCONH2 is problematic, providing methanol with only 1–12% yield. Bernskoetter’s system (0.018 or 0.07 mol% iPrFeH, 30.4 bar H2, 100°C, 4 h) hydrogenates N-formylmorpholine, HCONHAr, and HCONPh2 to methanol with TONs typically falling in the range of 1,190–4,430 [110]. Hydrogenation of HCONMePh under the same conditions is low yielding (TON = 60) but can be improved by adding 20 equiv. of HCONHPh (TON = 1,300). The overall hydrogenation process consumes 2 equiv. of H2 (for a formally four-electron reduction process), first converting formamides to hemiaminals and then to methanol. This requires decomposition of hemiaminals to formaldehyde and amines, a process that can be catalyzed by iron or the formamide substrates, depending on the nitrogen substituents [118].
The process of CO to ethylene glycol via oxamide described in Scheme 13 has also been studied with iron-based PNP pincer complexes (i.e., iPrFeHBH4, CyFeHBH4, EtFeHBH4, and trans-(EtPNHP)FeBr2(CO)), although the focus is on the second step that hydrogenates the oxamide to ethylene glycol [85]. With 0.2 mol% an iron catalyst and 1–1.5 mol% KOtBu, after 6 h, only 18–53% of the oxamide is hydrogenated. However, using 2 mol% EtFeHBH4 along with 5 mol% KOH and extending the reaction time to 24 h leads to a full conversion of the oxamide with 77% of the hydrogenation products attributed to ethylene glycol (Eq. 32).

Direct hydrogenation of CO2 to methanol assisted by amines, which changes the carbon oxidation state from +4 to −2, is more challenging with iron catalysts due to their relatively low thermal stability. The ruthenium systems described earlier operate most efficiently at 140–160°C. A thermal stability study of iPrFeHBH4 conducted by Jones showed that at 140°C this compound decomposed completely in 4 h [100]. Nevertheless, some of the iron-based PNP pincer complexes have been tested for this transformation. Olah and Prakash’s strategy of using PEHA to capture CO2 and iPrFeHBr to hydrogenate the captured CO2 (145°C, 70 bar H2, 72 h) failed to produce any methanol. Instead, formate and formamide were detected with NMR yields of 20% and 18%, respectively [89]. On the other hand, hydrogenation of the captured CO2 in a biphasic mixture (as illustrated in Scheme 15) was successful with iPrFeHBr at 55°C under 50 bar H2, which, after 10 h, gave formate in 96% yield. Like the Ru-MACHO-BH system, the iron catalyst can be reused at least four times without losing the catalytic activity [88].
Given the results in Eq. 31 and the fact that CyFeHBH4 and iPrFeH catalyze the hydrogenation of formamides to methanol [97, 110], one might expect some catalytic activity from these iron complexes for CO2 hydrogenation to methanol assisted by amines. A recent study by Bernskoetter suggests that these two steps are incompatible, thus preventing them being carried out in a single reactor [119]. In particular, CO2 poisons the catalyst during formamide hydrogenation. Furthermore, water (generated from CO2 hydrogenation) deactivates the catalytically active species. To solve these issues, N-formylation of morpholine was first catalyzed by iPrFeH in the presence of 3 Å sieves (Scheme 20). The resulting mixture was filtered to remove the sieves as well as ammonium carbamate salt of morpholine. A portion of the filtered mixture was then subjected to the second hydrogenation step catalyzed by iPrFeH. This two-step procedure provides methanol with a net TON of 590.

A two-step hydrogenation of CO2 to methanol catalyzed by an iron complex
3.3 Osmium Catalysts
Osmium complexes have been rarely explored for hydrogenation reactions. DFT calculations on trans-(iPrPNHP)MH2(CO) (M = Fe, Ru, Os) suggest that hydrogenation of MeCN is best catalyzed by iron and ruthenium and hydrogenation of methyl benzoate is best catalyzed by ruthenium [120]. Such predications have not been validated experimentally. The only known osmium system involving the PNP-type ligand is the one developed by Gusev, who treated the iPrPNHP ligand with OsHCl(CO)(PPh3)3 much like for the synthesis of iPrRuHCl (Scheme 21) [57]. The isolated product iPrOsHCl was identified as an isomeric mixture, which underwent dehydrochlorination with KOtBu followed by dehydrogenation of iPrOH to yield the dihydride complex iPrOsH2. What is remarkable about these osmium hydride complexes is that both iPrOsHCl and iPrOsH2 are air and moisture stable in solution.

Synthesis of osmium-based hydrogenation (pre)catalysts
In terms of hydrogenation reactions, iPrOsHCl and iPrOsH2 have been evaluated to catalyze the hydrogenation of hexyl octanoate, cis-3-hexenyl hexanoate, and triglycerides [121]. As for the analogous ruthenium and iron complexes, iPrOsHCl needs to be activated by a strong base such as NaOtBu. The best conditions for hydrogenating hexyl octanoate (in toluene) involve 0.1 mol% iPrOsH2 (loaded in air) at 220°C under 55.2 bar H2 for 24 h, which results in 87% conversion of the ester with high selectivity for the alcohol products. The mixture of iPrOsHCl and NaOtBu shows slightly lower activity. The hydrogenation reaction is operative under neat conditions, and loading the catalysts under an inert atmosphere improves the yield by 6–15%. Hydrogenation of cis-3-hexenyl hexanoate with the osmium catalysts saturates the C=C bond first, during which process the catalysts also degrade, showing no activity toward the ester functionality. To circumvent the issue, hydrogenation of cis-3-hexenyl hexanoate and seed oil (a mixture of canola and soybean oil) is first performed with Pd/C, a heterogeneous catalyst, to saturate the C=C bonds (Scheme 22). After filtration to remove Pd/C, the resulting saturated esters are subjected to hydrogenation catalyzed by iPrOsH2, which reduces the esters with a 60–90% conversion.

A two-step process for the hydrogenation of seed oil
4 Group 9 Metal Systems
4.1 Rhodium Catalysts
As mentioned in Introduction, rhodium holds historical significance in hydrogenation catalysis, particularly for the early efforts to hydrogenate C=C bonds. It is thus somewhat surprising that there is very little development of the PNP-ligated rhodium complexes as catalysts for the modern-day hydrogenation reactions. In 1984, Taqui Khan reported the synthesis of (PhPNHP)RhCl from the reaction of [RhCl(COE)2]2 (COE = cyclooctene) with PhPNHP in benzene [122]. In a series of subsequent reports, this specific PNP complex was shown to catalyze the hydrogenation of cyclohexene [122], 1-heptene [123], and 1-pentene [124] at 10–50°C under 0.4–1 bar H2. The proposed mechanism is analogous to the one for Wilkinson’s RhCl(PPh3)3 catalyst, which involves H2 activation followed by olefin coordination [125]. Based on the NMR analysis, oxidative addition of H2 to (PhPNHP)RhCl produces thee dihydride complexes with the formula (PhPNHP)RhH2Cl. The major product (90%) is consistent with cis-(PhPNHP)RhH2Cl with the PhPNHP ligand adopting the meridional configuration [126]. A more recent study by Jagirdar showed that (PhPNHP)RhH2Cl was unable to catalyze the hydrogenation of aldehydes, ketones, imines, and CO2 at 50°C under 20 bar H2 [127]. These results do not rule out the possibility of using the rhodium-based PNP-type complexes for the hydrogenation of polar bonds, because the hydrogenation reactions were attempted under base-free conditions and the active species could be (PhPNHP)RhH3.
4.2 Cobalt Catalysts
4.2.1 Synthesis of (Pre)catalysts
In contrast to the limited examples for the PNP-type complexes of rhodium, many cobalt derivatives have been studied, including spectroscopic observation and crystallographic characterization of (iPrPNHP)CoH2Cl and (iPrPNHP)CoH3 [128]. While these Co(III) hydrides have yet to be employed for hydrogenation reactions, close to a dozen cobalt complexes supported by the RPNHP ligands have been prepared (Scheme 23) and evaluated as hydrogenation catalysts.

Synthetic routes to cobalt-based hydrogenation (pre)catalysts
As illustrated in Scheme 23, the first class of cobalt PNP-type complexes feature an alkyl or aryl donor, and the synthesis starts with (pyr)2Co(CH2SiMe3)2 (pyr = pyridine) (Method A). Its ligand substitution reaction with CyPNHP gives CyCoCH2TMS, which can be protonated on the nitrogen by Brookhart’s acid, [H(OEt2)2]BArF4, to form a cationic complex [CyCoCH2TMS]+ [16]. Treatment of [CyCoCH2TMS]+ with 1-phenylethanol results in a Co(III) hydride [CyCoHAr]+, which appears to dehydrogenate the alcohol and then activate the C–H bond of the dehydrogenation product, acetophenone [129].
The reaction of a RPNHP ligand with CoCl2 or CoBr2 provides another entry to cobalt-based PNP-type complexes (Method B). Exposure of iPrCoCl2 and iPrCoBr2 to CO produces iPrCoCl2(CO) and iPrCoBr2(CO) [48, 49, 52], and reduction of iPrCoCl2 with 1 equiv. of NaBH4 generates a Co(I) species iPrCoCl [48]. The latter compound can react with CO to yield a cationic bis(carbonyl) complex [iPrCo(CO)2]+ [48], which can alternatively be prepared from the reaction of iPrPNHP with CoCl(PPh3)3 under CO [130].
4.2.2 Applications for Catalytic Hydrogenation Reactions
The first hydrogenation system involving the cobalt-based PNP-type complexes appeared in a 2012 report by Hanson [16]. In that study, 1:1 mixture of CyCoCH2TMS and [H(OEt2)2]BArF4, which essentially generated [CyCoCH2TMS]+ in situ, was shown to catalyze the hydrogenation of terminal and disubstituted (1,1- or 1,2-) olefins at 25°C under an atmospheric H2 pressure (Eq. 33). Without the acid, CyCoCH2TMS alone is almost completely inactive. Aldehydes, ketones, and aldimines are also viable substrates under same conditions or a slightly higher temperature and/or H2 pressure. At 25°C, hydrogenation of C=C bonds is unaffected by the presence of an ester, carboxylic acid, amine, or alcohol group in the olefin substrate and only slightly inhibited by water.

In a follow-up study, Hanson used the methylated PNP complex [(CyPNMeP)CoCH2SiMe3]BArF4 to probe the role that NH moiety could play during the hydrogenation reactions [129]. Evidently, the NH functionality is not needed for olefin hydrogenation but absolutely required for ketone hydrogenation (performed at 25–60°C under 1 bar H2). The lack of metal-ligand cooperation in olefin hydrogenation has been supported by DFT calculations [131]. The proposed mechanism involves C=C bond insertion into the Co–H bond of [(CyPNHP)CoH]BArF4 or [(CyPNMeP)CoH]BArF4 followed by hydrogenolysis of the cobalt alkyl species, in which NH or NMe does not directly participate. The Co(III) hydride [CyCoHAr]+ also shows good activity for hydrogenating styrene under ambient conditions but limited activity for hydrogenating acetophenone even at 60°C under 4.1 bar H2 [129].
Under more forcing conditions, carbonyl groups can be hydrogenated, not only by [CyCoCH2TMS]+ but also by the methylated derivative [(CyPNMeP)CoCH2SiMe3]BArF4. Jones reported in 2017 that both cationic complexes were effective catalysts for ester (or lactone) hydrogenation at 120°C under 55 bar H2 (Eq. 34) [132]. As expected, hydrogenation of α,β-unsaturated esters with [CyCoCH2TMS]+ results in both C=C and C=O bonds being reduced, although C=C bond hydrogenation appears to be faster. In contrast to olefin hydrogenation described earlier, carboxylic acid interferes with ester hydrogenation. No hydrogenation product was observed when adipic acid monoethyl ester was employed as the substrate. The uniqueness about this cobalt-based catalytic system is that methyl esters usually give lower alcohol yields when compared to the corresponding ethyl esters. Mechanistic investigation focusing on methyl benzoate revealed that [CyCoCH2TMS]+ lost its catalytic activity by forming [(CyPNHP)Co(κ1-OCOPh)(κ2-OCOPh)]BArF4, presumably via methane elimination. Similar to the mechanism proposed for olefin hydrogenation, [(CyPNHP)CoH]BArF4 or [(CyPNMeP)CoH]BArF4 is thought to be the active species, although according to DFT calculations, some of intermediates during ester hydrogenation feature a significant distortion of the PNP ligand from the meridional geometry [133].

Under similar conditions (100–140°C, 50 bar H2), the cobalt complexes listed in Scheme 23, Method B, when activated by NaOMe, all display some level of catalytic activity for the hydrogenation of methyl benzoate [48]. The best precatalyst is PhCoCl2, which promotes the hydrogenation of various esters including lactones (Eq. 35). Unlike the catalytic system shown in Eq. 34, here C=C bonds can be tolerated. Substrates that lead to low alcohol yields include PhCO2tBu (due to sterics) and chloro- or bromo-substituted methyl benzoate (due to dehalogenation). This particular catalytic system proves to operate via metal-ligand cooperation; control experiments using the methylated complex (PhPNMeP)CoCl2 did not yield any hydrogenation products.

The cobalt-based PNP-type complexes can also be used to catalyze the hydrogenation of other multiple bonds including those in nitriles and N-heterocycles. In 2018, we reported that catalytic hydrogenation of PhCN could be affected by iPrCoCl2 or iPrCoBr2 in the presence of NaHBEt3, forming PhCH=NCH2Ph exclusively as the hydrogenation product (Scheme 24) [134]. Adding 1 equiv. of CyNH2 to the reaction generated PhCH=NCy selectively, which represents a hydrogenative coupling process. The selectivity of nitrile hydrogenation can be altered to favor primary amines, as demonstrated by Beller in a more recent study [135]. Among the PNP-ligated cobalt dihalide complexes shown in Scheme 23, PhCoCl2 is the most active precatalyst, converting various aromatic and aliphatic nitriles to primary amines (Scheme 24). Functional groups tolerated under the catalytic conditions include F, Cl, NH2, OMe, pyridyl, and pyrrolidyl groups; however, carbonyl groups in esters, ketones, and aldehydes are also hydrogenated along with the nitrile groups. The nature of the catalytically active species is ill-defined here, although all experiments suggest that the hydrogenation process is homogeneous. The lack of reactivity with the methylated complex (PhPNMeP)CoCl2 also supports a metal-ligand cooperative mechanism.

Cobalt-catalyzed hydrogenation of nitriles leading to secondary imines or primary amines
As a further exploration of N-heterocycles as organic hydrogen storage materials, Jones studied the ability of [CyCoCH2TMS]+ to catalyze the hydrogenation of these molecules [136]. Under the conditions shown in Eq. 36, the hydrogenation process takes place very slowly, accepting 2 equiv. of H2 to saturate one nitrogen-containing ring. In contrast to the iron-based catalytic system (Eq. 29), 2,6-lutidine is not a viable substrate for the cobalt catalyst. Analogous to the olefin hydrogenation catalyzed by [CyCoCH2TMS]+, the NH moiety is not needed here.

Catalytic hydrogenation of CO2 has not been explored extensively with the cobalt-based PNP-type complexes described above. The only known example is Bernskoetter’s study of [iPrCo(CO)2]+ as a potential catalyst [130]. Under the conditions outlined in Eq. 37, hydrogenation of CO2 gives the formate with 450 turnovers. Similar to the iron-based system (Scheme 19), the methylated complexes [(iPrPNMeP)Co(CO)2]Cl and [(CyPNMeP)Co(CO)2]Cl are more superior catalysts than [iPrCo(CO)2]+ for CO2 hydrogenation, increasing the TON by 64- or 53-fold [137]. Once again, for CO2 hydrogenation of to the formate stage, the metal-ligand bifunctional catalysts do not appear to have any advantage.

4.3 Iridium Catalysts
4.3.1 Synthesis of (Pre)catalysts
Iridium-based PNP-type complexes are also known in the literature. To develop a hydrogenation catalyst, Taqui Khan treated [Ir(COE)2Cl]2 with the hydrochloride salt of PhPNHP in refluxing benzene, which resulted in a compound with the formula (PhPNHP)IrCl, presumably PhIrCl as shown in Scheme 25 [122]. A more recent synthesis by Jagirdar employed [Ir(COD)Cl]2 and the neutral ligand PhPNHP, which formed [(PhPNHP)Ir(COD)]Cl with the COD ligand still bound to iridium [127]. Subsequent hydrogenation produced an air-stable Ir(III) dihydride PhIrH2Cl, which was further converted to PhIrH3 via dehydrochlorination under H2. It is interesting to note that PhIrH3 exists as a 1:1 isomeric mixture with the PNP ligand adopting either meridional or facial coordination mode. In contrast, the isopropyl analog iPrIrH3 displays the meridional mode only. This trihydride complex can be prepared from dehydrochlorination of the dihydride iPrIrH2Cl followed by dehydrogenation of iPrOH [138]. iPrIrH2Cl is commercially available but can be made from iPrPNHP and [Ir(COE)2Cl]2 in iPrOH at 80°C. It is also worth to point out that in the solid form iPrIrH2Cl is air stable and iPrIrH3 is moderately air stable.

Synthesis of iridium-based hydrogenation (pre)catalysts
4.3.2 Applications for Catalytic Hydrogenation Reactions
The use of iridium-based PNP-type complexes for catalytic hydrogenations reactions can be traced back to 1984, when Taqui Khan studied the hydrogenation of cyclohexene catalyzed by PhIrCl [122]. This reaction operates over the temperature range 20–50°C under 0.4–1 bar H2 and proceeds via an initial H2 activation to form PhIrH2Cl [125]. The catalytic system that really takes advantage of metal-ligand cooperativity is the one developed by Abdur-Rashid in 2009 [139]. It was reported that aldehyde and ketone hydrogenation could be catalyzed by iPrIrH2Cl activated with KOtBu or by iPrIrH3 under base-free conditions. The catalysts are remarkably active at room temperature; the TONs for acetophenone hydrogenation are as high as 30,000 (Eq. 38). Hydrogenation of benzalacetone and β-ionone is chemoselective for the C=O bonds; however, hydrogenation of 2-cyclohexen-1-one produces a 1:1 mixture of allyl alcohol and the fully saturated alcohol. In a related study, Jagirdar examined the catalytic activity of the phenyl derivatives (PhIrH3 and PhIrH2Cl/KOtBu) in hydrogenation reactions, which were carried out at 50°C in methanol under 20 bar H2 with a catalyst loading of 0.1 mol% [127]. In addition to aldehydes and ketones, imines such as PhCH=NPh and PhCH=NBn are hydrogenated, albeit with moderate conversions (31–49% over 6 h). In contrast, methyl benzoate and styrene are completely unreactive.

Abdur-Rashid’s iridium complex iPrIrH2Cl has also been utilized to catalyze the hydrogenation of alkyl levulinates to γ-valerolactone (Eq. 39) [71]. The reaction is enhanced by added ethanol or methanol, and under the optimized conditions, γ-valerolactone was obtained with TONs of up to 9,300. Compared to Ru-MACHO, iPrIrH2Cl is more active, although the ruthenium catalyst can be reused three times without noticeable catalyst decomposition.

Esters can be hydrogenated with the iridium-based PNP-type complexes, although the reaction must be conducted at higher temperatures and under higher H2 pressures. In 2014, Beller showed that in the presence of NaOMe and at 130°C under 50 bar H2, both iPrIrH2Cl and iPrIrH3 were efficient for catalytic hydrogenation of methyl benzoate [140]. Based on the proposed mechanism, iPrIrH3 is the active species transferring H+/H− to the ester substrate, and therefore the base should not be needed for iPrIrH3. However, the addition of NaOMe does improve the conversion and yield, suggesting that the base plays multiple roles during the reaction. The catalytic system (Eq. 40) can tolerate functional groups including halogens, MeO, pyridyl, and furyl groups. Hydrogenation of p-NCC6H4CO2Me and PhCH=CHCO2Me leads to saturation of C≡N, C=O, and C=C bonds. Hydrogenation of phthalic anhydride, on the other hand, can stop at the lactone stage to give phthalide in 71% yield.

Another important type of carbonyl substrates for the iridium-catalyzed hydrogenation reactions is CO2. In 2011, Hazari reported a very facile CO2 insertion process with iPrIrH3, resulting in an iridium formate complex iPrIrH2(OCHO) that is air stable and features a hydrogen bond between the NH group and the formato group (Eq. 41) [141]. iPrIrH2(OCHO) was then employed to catalyze the hydrogenation of CO2 in an aqueous solution of KOH (1 M), providing HCO2K with TONs of up to 348,000 (Scheme 26). The trihydride iPrIrH3 can also be used as the catalyst, although precaution needs to be taken to exclude oxygen from the reactor. Very recently, Jagirdar demonstrated that the phenyl derivative PhIrH3 (a 1:1 isomeric mixture) reacted with CO2 (1 bar at room temperature) to form an insertion product analogous to iPrIrH2(OCHO) [127]. Hydrogenation of CO2 in MOH (M = Li, Na, K) with PhIrH3 or PhIrH2Cl produced HCO2M with much lower TONs of 65–144, although the hydrogenation reactions were tested under relatively low temperatures and pressures. In studying N-formylation of morpholine, Ding also examined the catalytic activity of iPrIrH2Cl (activated by KOtBu), which, under the conditions shown in Scheme 26, generated the formamide with a TON of 720 [82]. An attempt to use ethylene carbonate as CO2 surrogate had limited success with iPrIrH2Cl/KOtBu as the catalyst (0.1 mol%); at 140°C under 50.7 bar H2, ethylene glycol was obtained with only 10% yield [55]. The ruthenium system shown in Eq. 23 is far more reactive.


Iridium-catalyzed hydrogenation of CO2 or N-formylation of morpholine
5 Group 10 Metal Systems
Group 10 metals bearing the PNP-type ligands have been rarely used as hydrogenation catalysts. The only known example of a nickel system is the one developed by Hanson in 2012 [142]. As summarized in Scheme 27, the reaction of Ni(diglyme)Br2 with CyPNHP produces a cationic PNP pincer nickel bromide complex, which can be converted to the hydride [CyNiH]+ using NaBH4 followed by anion exchange with NaBPh4. The neutral hydride CyNiH is available from [CyNiH]+ through deprotonation with KH.

Synthesis of PNP-ligated nickel hydride complexes
Complex [CyNiH]+ proves to be an active catalyst for the hydrogenation of styrene, α-methylstyrene, and tert-butylethylene at 80°C under 4.1 bar H2 (Eq. 42) [142]. Hydrogenation of 1-octene affords n-octane and internal octenes as a result of the competing olefin isomerization process. Under similar conditions, aldehydes are reduced to alcohols but in a non-catalytic manner. The neutral hydride CyNiH is also an active catalyst, although it is less reactive than [CyNiH]+. The methylated complex (CyPNMeP)NiH]BPh4 shows similar activity to [CyNiH]+, suggesting that here a metal-ligand cooperative mechanism is not involved.

The analogous palladium and platinum hydrides have not been reported in the literature. The most relevant study is a 1988 report by Taqui Khan, who used [(PhPNHP)PdCl]Cl (made from Pd(COD)Cl2 and the hydrochloride salt of PhPNHP in benzene) to catalyze hydrogenation of cyclohexene [143]. The reaction was shown to operate at 10–40°C under 0.4–1 bar H2 and proceed via a palladium hydride intermediate.
6 Group 6 Metal Systems
Mid-transition metal complexes supported by the PNP-type ligands have been studied. For group 6 metal systems, chromium complexes have never been utilized to catalyze hydrogenation reactions, although (RPNHP)CrCl2 [144] and (RPNHP)CrCl3 [145] have been known for many years. In contrast, PNP-ligated molybdenum and tungsten complexes have been developed specifically for various hydrogenation processes. They belong to two different types of complexes, each with a d6 electron configuration and isoelectronic to (RPNP)Fe(CO)H, which have already been established as active hydrogenation catalysts.
6.1 Nitrosyl Complexes
To synthesize the desired nitrosyl complexes (RPNP)M(NO)CO (M = Mo, W), Berke used M(NO)(CO)4(AlCl4) as the metal precursors, which were shown to react with the iPrPNHP ligand to form (iPrPNHP)M(NO)(CO)Cl (Scheme 28) [146]. Upon further treatment with NaN(SiMe3)2, the five-coordinate complexes iPrMoNO and iPrWNO were isolated as highly air-sensitive materials.

Synthesis of Mo- and W-based hydrogenation catalysts bearing a nitrosyl ligand
Activation of H2 by iPrMoNO and iPrWNO is feasible but slow at room temperature, forming a mixture of two isomers because of the availability of two sides (NO side vs. CO side) for H2 to approach (Eq. 43). As expected for other H–M–N–H-type complexes, reduction of polar bonds is likely to occur with these hydrides. Indeed, Berke demonstrated that iPrMoNO and iPrWNO were efficient catalysts for the hydrogenation of aldimines bearing various aryl substituents (Scheme 29) [146]. Substrates that are unreactive under the catalytic conditions include p-NO2C6H4CH=NPh, PhCH=NiBu, and surprisingly PhCHO. Acetophenone can be hydrogenated but with a low yield of 32%. Under slightly modified conditions, both aliphatic and aromatic nitriles are hydrogenated with selectivity favoring the secondary imines [147]. This implies that hydrogenation of the intermediate R′CH=NH to R′CH2NH2 is less competitive than the reaction of R′CH=NH with R′CH2NH2 to yield R′CH=NCH2R′. In both catalytic processes, iPrMoNO displays high activity than iPrWNO, which is also confirmed by DFT calculations [148].


Molybdenum- and tungsten-catalyzed hydrogenation of imines and nitriles
Attempts have also been made to use iPrMoNO and iPrWNO to catalyze CO2 hydrogenation [149]. The in situ generated hydrides (see Eq. 43) were shown to undergo CO2 insertion to generate molybdenum and tungsten formate complexes, which could be converted back to iPrMoNO and iPrWNO through the addition of NaN(SiMe3)2. Unfortunately, catalytic hydrogenation of CO2 (pH2 = 70 bar, pCO2 = 10 bar, 140°C) in the presence of a base and with iPrMoNO or iPrWNO (5 mol%) failed to produce HCO2− with a yield greater than 5%. This is likely due to the poisoning of the catalysts by CO2 to form carbamate species (Eq. 44), as separately studied.

6.2 Bis(Carbonyl) Complexes
The bis(carbonyl) system illustrated in Fig. 5 has recently been explored, though only focusing on molybdenum complexes. As shown in Scheme 30, treatment of iPrPNHP with Mo(CO)6 and Mo(PPh3)2(CO)2(MeCN)2 leads to ligand substitution, which generates iPrMo(CO)3 and iPrMoNCMe, respectively [150]. The latter reaction needs to be carried out in THF. Switching the solvent to CH2Cl2 can oxidize Mo(0) to Mo(I), giving the chloride complex iPrMoCl. The analogous bromide complex iPrMoBr is isolated as the minor product from the reaction of iPrPNHP with Mo(η3-allyl)(PPh3)2(CO)2(MeCN)2Br (the major product is iPrMoNCMe).

Molybdenum and tungsten complexes isoelectronic to (RPNP)Fe(CO)H

Synthesis of molybdenum-based precatalysts bearing at least two CO ligands
Both iPrMoCl and iPrMoNCMe, when activated by NaHBEt3, catalyze the hydrogenation of acetophenone, whereas iPrMo(CO)3 shows no activity [150]. Under the optimized conditions for iPrMoCl (Scheme 31), acetophenones substituted by F, MeO, MeS, and CF3 groups are all successfully hydrogenated to the corresponding alcohols in high yields. The reaction of (E)-PhCOCH=CHPh results in both double bonds being reduced. Styrene derivatives are also viable substrates, although a higher temperature of 130°C is required. Hydrogenation reactions performed at 100°C allow the conversion of N-methylformanilides to N-methylanilines with C=C bond and ester functionality intact [151]. Other functional groups including PhCH2O, Me2N, CN, and NO2 are also compatible with the catalytic conditions; however, the product yields are low to moderate (6–52%). Amides of the type R′CONPhR″ (R′ = Me, CF3, Ph) are more challenging substrates, which typically give 11–28% yields for the hydrogenation products. The bromide complex iPrMoBr shows similar activity to iPrMoCl but outperforms iPrMoNCMe. The iPrMo(CO)3 is completely inactive. A detailed mechanistic study [151] focusing on iPrMoCl suggests that NaHBEt3 reduces the Mo(I) complex to several Mo(0) species including Na[(iPrPNHP)Mo(CO)2H] and Na[(iPrPNP)Mo(CO)2]. These two complexes represent the H–M–N–H and M–N molecules characteristic of metal-ligand bifunctional hydrogenation catalysts.

Hydrogenation reactions catalyzed by iPrMoCl-NaHBEt3
7 Group 7 Metal Systems
7.1 Manganese Catalysts
There has been an increasing interest in developing manganese-based hydrogenation catalysts [152]. This is in part motivated by the fact that manganese is the third most abundant transition metal (after iron and titanium) in the Earth’s crust. For PNP-type complexes, manganese species isoelectronic to (RPNP)Fe(CO)H would be (RPNP)Mn(CO)2. To date, strategies of using inexpensive sources of manganese such as MnCl2 to make these Mn(I) complexes have not had much success. For example, (iPrPNHP)MnCl2 prepared from iPrPNHP and MnCl2 does not bind CO [153]. Successful routes to the carbonyl complexes have relied on the use of MnBr(CO)5 as the metal precursor. As illustrated in Scheme 32, the reaction of RPNHP with MnBr(CO)5 can lead to four different structures, which depend on the nature of phosphorus substituents. With medium-sized alkyl groups (R = iPr, Cy, Et), a mixture of neutral dicarbonyl and cationic tricarbonyl complexes (e.g., iPrMnBr and [iPrMn(CO)3]+) is obtained [154]. The RPNHP ligands all adopt the meridional coordination mode, except that in [EtMn(CO)3]+, the EtPNHP ligand occupies three facial coordination sites [37]. The dicarbonyl and tricarbonyl complexes are separable due to solubility difference, although higher temperatures and longer reaction times usually facilitate the conversions to the neutral products. The phospholane-based chiral ligand, (S,S)-(CyMePCH2CH2)2NH, and (S,S)-(tBuMePCH2CH2)2NH can generate the cationic tricarbonyl complexes only [155, 156], whereas the more bulky ligand tBuPNHP leads to further extrusion of CO to yield [tBuMn(CO)2]+ [154]. The reaction of the phenyl-substituted ligand PhPNHP gives the neutral dicarbonyl complex PhMnBr [157]. Synthesis of the five-coordinate complexes iPrMn(CO)2 and EtMn(CO)2 has been accomplished via dehydrobromination of iPrMnBr [154, 158], EtMnBr [37], and [EtMn(CO)3]+ [37] with a strong base. The hydride iPrMnH is available from the reaction of iPrMnBr with NaHBEt3 [153].

Synthesis of manganese-based (pre)catalysts
The seminal work by Beller in 2016 showed that in the presence of NaOtBu, iPrMnBr were efficient in catalyzing the hydrogenation of nitriles, ketones, and aldehydes (Scheme 33) [153]. The relative difficulty for hydrogenating these substrates is reflected by the temperature and H2 pressure employed. As expected, aldehydes are the easiest ones to react. Various functional groups including halogens, CF3, NH2, pyridyl, furyl, and isolated C=C bonds are tolerated under these conditions. The nitrile hydrogenation shows excellent selectivity for primary amines. Hydrogenation of PhCH=CHCN produces the allylic and fully saturated amines. In contrast, hydrogenation of α,β-saturated aldehydes is highly selective for the C=O bonds, forming the allylic alcohols exclusively. Under the conditions for ketone hydrogenation, other reducible functional groups such as ester, lactam, and cyclopropyl ring are unaffected. The proposed mechanism involves an outer-sphere hydrogen transfer from iPrMnH to the substrates followed by regeneration of the hydride through the reaction of iPrMn(CO)2 with H2. Consistent with this mechanism, iPrMnH catalyzes the hydrogenation of benzonitrile under similar but base-free conditions, although for some unknown reason, the addition of NaOtBu improves the catalytic efficiency. The cyclohexyl derivative CyMnBr is slightly less reactive than iPrMnBr in nitrile hydrogenation. Using iPrMnBr and CyMnBr as the precatalysts is more advantageous due to their high stability in air.

Manganese-catalyzed hydrogenation of nitriles, ketones, and aldehydes
Given the availability of chiral RPNHP ligands, it is possible to develop manganese-based PNP-type catalysts for asymmetric hydrogenation of ketones. Since high temperatures often erode enantioselectivity, the reaction conditions need to be further optimized. Using phospholane-based complex [R*Mn(CO)3]+ as the precatalyst and tert-amyl alcohol as the solvent, the Beller group was able to perform ketone hydrogenation at 40°C (Scheme 34) [58, 155]. The remarkable feature about this catalytic system is that aliphatic ketones are hydrogenated to alcohols with ee’s in the 51–83% range, which is typically difficult to achieve with other catalysts. However, bulky ketones including AdCOMe, tBuCOMe, and tBuCOnPr result in low alcohol yields, and ketones of the type PhCOR′ (R′ = Me, Et, nPr, Cy) give low ee’s (11–19%). In a related study, Mezzetti explored P-stereogenic PNP-type manganese complexes as asymmetric hydrogenation catalysts [156]. Under the best conditions for (S,S)-[MetBuMn(CO)3]+ (Scheme 34), acetophenone derivatives are hydrogenated to the corresponding alcohols with ee’s up to 55%. Both experimental and computational studies support the involvement of (RPNP)Mn(CO)2 and (RPNHP)Mn(CO)2H in the catalytic cycle. It is interesting to know that the iron precatalyst (S,S)-MetBuFeHBr (shown in Scheme 17) is about 30 times more active than (S,S)-[MetBuMn(CO)3]+. This is due to the formation of a more stable alkoxide species with manganese, which is generated after hydrogen transfer from (RPNHP)Mn(CO)2H to the ketone substrate.

Manganese-catalyzed asymmetric hydrogenation of ketones
Esters are comparatively more challenging substrates for hydrogenation. In a 2016 report, Beller showed that at 100°C under 30 bar H2, iPrMnBr and CyMnBr (activated with KOtBu) displayed very limited catalytic activity for the hydrogenation of methyl benzoate [37]. Lan and Liu recently also reported that at 120°C under 45 bar H2, iPrMnBr and PhMnBr (activated with KOtBu) catalyzed the hydrogenation of the ketone part of methyl 4-acetylbenzoate with only 3–9% of the ester functionality being reduced [159]. However, Beller demonstrated that both EtMnBr and [EtMn(CO)3]+ were effective hydrogenation catalysts for esters, converting methyl benzoate to benzyl alcohol in 97% yield. Under the conditions outlined in Eq. 45, various aromatic and aliphatic esters including lactones can be hydrogenated to alcohols. The catalytic system shows excellent functional group compatibility, similar to the iron system described earlier (Eq. 25).

Like the ruthenium- and iron-based catalytic systems, the manganese PNP-type complexes have been tested for the hydrogenation of amides and N-heterocycles. Hydrogenation of PhCONHPh proved to be unsuccessful with iPrMnBr (2 mol%, 110°C, 30 bar H2) [159] and [tBuMn(CO)2]+ (4 mol%, 130°C, 50 bar H2) [160] in the presence of KOtBu as the activator. Catalytic hydrogenation of quinoline was shown to be feasible with the neutral dicarbonyl complexes, although the conversions were low (Eq. 46) [159]. DFT calculations suggest that the lack of activity is in part due to the low hydricity of the manganese hydride intermediate. To improve the catalysts, one of the phosphorus donor groups was replaced by a pyridine or imidazole ring, which not only increases the hydricity but also creates a less crowded environment [159, 160].

As suggested by DFT calculations [161], catalytic hydrogenation of CO2 to the formate stage should be possible with the manganese-based PNP-type complexes (e.g., iPrMn(CO)2), although such process has not been validated experimentally. The closest work was done by Prakash, who used iPrMnBr and CyMnBr to catalyze the N-formylation of morpholine, benzylamine, and N-methylbenzylamine (Eq. 47) [162]. Here, the isopropyl derivative iPrMnBr is more efficient than CyMnBr. Other amines including amylamine and N,N′-dimethylethylenediamine can also be converted to the formamides, but the yields are much lower (25–53%). The in situ generated N-formylmorpholine and HCONHBn can be further hydrogenated (at 150°C under 70–80 bar H2) to methanol with TONs of up to 36, again using iPrMnBr as the catalyst (0.5 mol% loading). Hydrogenation of pure N-formylmorpholine under the same conditions gives a substantially higher TON (128) for methanol. Unfortunately, direct hydrogenation of CO2 to methanol assisted by these amines including PEHA [89] has failed to produce any meaningful amount of methanol. This is likely due to the poisoning of the catalyst by CO2 during the formamide hydrogenation, as described in the iron system (Scheme 20).

Two indirect methods of making methanol have been developed with the manganese-based PNP-type catalysts, one involving cyclic carbonates as CO2 surrogate and the other involving CO to methanol. In 2018, Leitner demonstrated the first approach by using iPrMnBr as the precatalyst and NaOtBu as the activator or the in situ generated iPrMn(CO)2 [158]. Under the best conditions for ethylene carbonate (0.1 mol% iPrMn(CO)2, 60 bar H2, 120°C), ethylene glycol and methanol were obtained with TONs of 620 and 400, respectively. As shown in Eq. 48, this process can be extended to other five- and six-membered cyclic carbonates.

Very recently, Checinski and Beller designed a CO-to-methanol process that utilized a nitrogen-containing promoter to capture CO in the form of formamide [163]. The subsequent manganese-catalyzed formamide hydrogenation was expected to produce methanol and regenerate the promoter. After an extensive computational screening and experimental validation, scatole and pyrrole were identified as the best promoters that balance the difficulty of amine carbonylation with that of hydrogenation to methanol. Under the optimized conditions shown in Eq. 49, methanol is obtained with high TONs. Other manganese-based PNP-type complexes have also been tested. Compared to iPrMnBr, CyMnBr, EtMnBr, and [EtMn(CO)3]+ are slightly less active, whereas PhMnBr and [tBuMn(CO)2]+ are almost inactive. The methylated complex (EtPNMeP)Mn(CO)2Br also displays limited reactivity, suggesting that in this case, the presence of the NH moiety is critical to the success of the hydrogenation process.

7.2 Rhenium Catalysts
Rhenium-based PNP-type complexes have been prepared following the procedures established for the manganese system. The reaction of iPrPNHP with ReBr(CO)5 produces [iPrRe(CO)3]+ in which the PNP ligand adopts the facial coordination mode (Eq. 50) [164]. In contrast, the phospholane-based PNP ligand coordinates to the Re(CO)3+ fragment in a meridional fashion [58].

Both [iPrRe(CO)3]+ and [R*Re(CO)3]+ have been employed to catalyze the hydrogenation of ketones. Under the optimized conditions for [iPrRe(CO)3]+ (Scheme 35), most functional groups are tolerated with cyano, phenol, and boric acid being the exceptions [164]. While isolated C=C bonds and internal C ≡ C bonds are intact during the hydrogenation, α,β-unsaturated ketones typically give saturated alcohols as the major or sole products. Hydrogenation of R′COCH2CH2CO2Me provides γ-butyrolactones, although a higher loading of [iPrRe(CO)3]+ (5 mol%) and KOtBu (10 mol%) is needed. Using the chiral precatalyst [R*Re(CO)3]+ results in low ee for hydrogenating acetophenone and moderate ee for hydrogenating cyclohexyl methyl ketone [58]. This level of enantioselectivity is lower than that achieved with the manganese analog but comparable to the ruthenium- and iron-based catalytic systems.

Rhenium-catalyzed hydrogenation of ketones
8 Summary and Outlook
The use of the RPNHP ligands to design hydrogenation catalysts has been a fruitful patch in the field of homogeneous catalysis. As shown in this chapter, transition metal complexes supported by these ligands along with strong-field ligands such as CO, NO, and isocyanides have been so extensively studied that most of the mid- and late-transition metals have been involved. Some of the hydrogenation processes do not require the NH moiety. Examples include hydrogenation of weakly polarized C=C bonds and hydrogenation of CO2 to formate. However, metal-ligand cooperativity enabled by the NH functionality does have advantage for the more challenging hydrogenation processes such as CO2 hydrogenation to methanol and amide hydrogenation.
We envision that interests in using RPNHP ligated complexes for catalytic hydrogenation reactions will continue to grow in the future. In particular, group 5 and group 11 metals have not been explored to build PNP-type complexes specifically for hydrogenation reactions. A recent computation study focusing on (iPrPNHP)M(NO)2H (M = V, Nb, Ta; see Fig. 6) suggests that they are promising catalysts [165]. Inspired by the structure of the active site of [Fe]-hydrogenase, Yang has computationally designed PNP-type complexes of iron [166] and cobalt [167] that contain acylmethylpyridinol as the ancillary ligand. These molecules present significant synthetic challenges but may provide a path for synthetic chemists to identify more robust and active hydrogenation catalysts.

Computationally designed hydrogenation catalysts
References
de Vries JG, Elsevier CJ (eds) (2007) The handbook of homogeneous hydrogenation. Wiley-VCH, Weinheim
Clarke ML (2012) Cat Sci Technol 2:2418
Werkmeister S, Junge K, Beller M (2014) Org Process Res Dev 18:289
Alig L, Fritz M, Schneider S (2019) Chem Rev 119:2681
Osborn JA, Jardine FH, Young JF, Wilkinson G (1966) J Chem Soc A:1711
Halpern J (1982) Science 217:401
Hallman PS, McGarvey BR, Wilkinson G (1968) J Chem Soc A:3143
Noyori R, Ohkuma T (2001) Angew Chem Int Ed 40:40
Shambayati S, Crowe WE, Schreiber SL (1990) Angew Chem Int Ed Engl 29:256
Zhang J, Leitus G, Ben-David Y, Milstein D (2006) Angew Chem Int Ed 45:1113
Saudan LA, Saudan CM, Debieux C, Wyss P (2007) Angew Chem Int Ed 46:7473
Ito M, Koo LW, Himizu A, Kobayashi C, Sakaguchi A, Ikariya T (2009) Angew Chem Int Ed 48:1324
Balaraman E, Gnanaprakasam B, Shimon LJW, Milstein D (2010) J Am Chem Soc 132:16756
Casey CP, Guan H (2007) J Am Chem Soc 129:5816
Sui-Seng C, Freutel F, Lough AJ, Morris RH (2008) Angew Chem Int Ed 47:940
Zhang G, Scott BL, Hanson SK (2012) Angew Chem Int Ed 51:12102
Shvo Y, Czarkie D, Rahamim Y, Chodosh DF (1986) J Am Chem Soc 108:7400
Ohkuma T, Ooka H, Hashiguchi S, Ikariya T, Noyori R (1995) J Am Chem Soc 117:2675
Doucet H, Ohkuma T, Murata K, Yokozawa T, Kozawa M, Katayama E, England AF, Ikariya T, Noyori R (1998) Angew Chem Int Ed 37:1703
Sui-Seng C, Haque FN, Hadzovic A, Pütz A-M, Reuss V, Meyer N, Lough AJ, Zimmer-De Iuliis M, Morris RH (2009) Inorg Chem 48:735
Otsuka T, Ishii A, Dub PA, Ikariya T (2013) J Am Chem Soc 135:9600
Qu S, Dai H, Dang Y, Song C, Wang Z-X, Guan H (2014) ACS Catal 4:4377
Sandoval CA, Ohkuma T, Muñiz K, Noyori R (2003) J Am Chem Soc 125:13490
Werkmeister S, Junge K, Wendt B, Alberico E, Jiao H, Baumann W, Junge H, Gallou F, Beller M (2014) Angew Chem Int Ed 53:8722
Dub PA, Gordon JC (2017) ACS Catal 7:6635
Smith NE, Bernskoetter WH, Hazari N (2019) J Am Chem Soc 141:17350
Dub PA, Gordon JC (2018) Nat Rev Chem 2:396
Dub PA, Scott BL, Gordon JC (2017) J Am Chem Soc 139:1245
Werkmeister S, Neumann J, Junge K, Beller M (2015) Chem Eur J 21:12226
Wilson ME, Nuzzo RG, Whitesides GM (1978) J Am Chem Soc 100:2269
Hermosilla P, López P, García-Orduña P, Lahoz FJ, Polo V, Casado MA (2018) Organometallics 37:2618
Kuriyama W, Matsumoto T, Ino Y, Ogata O (2011) PCT Int Appl WO/2011/048727 A1
Danopoulos AA, Wills AR, Edwards PG (1990) Polyhedron 9:2413
Ma W, Cui S, Sun H, Tang W, Xue D, Li C, Fan J, Xiao J, Wang C (2018) Chem Eur J 14:13118
Abdur-Rashid K, Graham T, Tsang C-W, Chen X, Guo R, Jia W, Amoroso D, Sui-Seng C (2008) PCT Int Appl WO/2008/141439 A1
Meiners J, Friedrich A, Herdtweck E, Schneider S (2009) Organometallics 28:6331
Elangovan S, Garbe M, Jiao H, Spannenberg A, Junge K, Beller M (2016) Angew Chem Int Ed 55:15364
Burk MJ, Feaster JE, Harlow RL (1991) Tetrahedron Asymmetry 2:569
Huber R, Passera A, Gubler E, Mezzetti A (2018) Adv Synth Catal 360:2900
Schneider S, Meiners J, Askevold B (2012) Eur J Inorg Chem:412
Coles SJ, Edwards PG, Hursthouse MB, Read PW (1994) J Chem Soc Chem Commun:1967
Coles SJ, Danopoulos AA, Edwards PG, Hursthouse MB, Read PW (1995) J Chem Soc Dalton Trans:3401
Sherwood R, Gonzàlez de Rivera F, Wan JH, Zhang Q, White AJP, Rossell O, Hogarth G, Wilton-Ely JDET (2015) Inorg Chem 54:4222
García-Seijo MI, Habtemariam A, Parsons S, Gould RO, García-Fernández ME (2002) New J Chem 26:636
Fillman KL, Bielinski EA, Schmeier TJ, Nesvet JC, Woodruff TM, Pan CJ, Takase MK, Hazari N, Neidig ML (2014) Inorg Chem 53:6066
Ma W, Cui S, Sun H, Tang W, Xue D, Li C, Fan J, Xiao J, Wang C (2018) Chem Eur J 24:13118
Nguyen DH, Morin Y, Zhang L, Trivelli X, Capet F, Paul S, Desset S, Dumeignil F, Gauvin RM (2017) ChemCatChem 9:2652
Junge K, Wendt B, Cingolani A, Spannenberg A, Wei Z, Jiao H, Beller M (2018) Chem Eur J 24:1046
Rozenel SS, Kerr JB, Arnold J (2011) Dalton Trans 40:10397
Shao Z, Fu S, Wei M, Zhou S, Liu Q (2016) Angew Chem Int Ed 55:14653
Lagaditis PO, Schluschaß B, Demeshko S, Würtele C, Schneider S (2016) Inorg Chem 55:4529
Fu S, Chen N-Y, Liu X, Shao Z, Luo S-P, Liu Q (2016) J Am Chem Soc 138:8588
Zhou W, Wei Z, Spannenberg A, Jiao H, Junge K, Jung H, Beller M (2019) Chem Eur J 25:8459
Kuriyama W, Matsumoto T, Ogata O, Ino Y, Aoki K, Tanaka S, Ishida K, Kobayashi T, Sayo N, Saito T (2012) Org Process Res Dev 16:166
Han Z, Rong L, Wu J, Zhang L, Wang Z, Ding K (2012) Angew Chem Int Ed 51:13041
Zhang L, Raffa G, Nguyen DH, Swesi Y, Corbel-Demailly L, Capet F, Trivelli X, Desset S, Paul S, Paul J-F, Fongarland P, Dumeignil F, Gauvin RM (2016) J Catal 340:331
Bertoli M, Choualeb A, Lough AJ, Moore B, Spasyuk D, Gusev DG (2011) Organometallics 30:3479
Garbe M, Wei Z, Tannert B, Spannenberg A, Jiao H, Bachmann S, Scalone M, Junge K, Beller M (2019) Adv Synth Catal 361:1913
Touge T, Aoki K, Nara H, Kuriyama W (2012) PCT Int Appl WO/2012/144650 A1
Nielsen M, Alberico E, Baumann W, Drexler H-J, Junge H, Gladiali S, Beller M (2013) Nature 495:85
Choi J-H, Schloerer NE, Berger J, Prechtl MHG (2014) Dalton Trans 43:290
Choi J-H, Heim LE, Ahrens M, Prechtl MHG (2014) Dalton Trans 43:17248
Ogata O, Nakayama Y, Nara H, Fujiwhara M, Kayaki Y (2016) Org Lett 18:3894
Lazzari D, Cassani MC, Bertola M, Casado Moreno F, Torrente D (2013) RSC Adv 3:15582
Deutsch J, Jiménez Pinto J, Doerfelt S, Martin A, Köckritz A (2015) Flavour Fragr J 30:101
Fairweather NT, Gibson MS, Guan H (2015) Organometallics 34:335
Nguyen DH, Raffa G, Morin Y, Desset S, Capet F, Nardello-Rataj V, Dumeignil F, Gauvin RM (2017) Green Chem 19:5665
Ziebart C, Jackstell R, Beller M (2013) ChemCatChem 5:3228
Satapathy A, Gadge ST, Bhanage BM (2017) ChemSusChem 10:1356
Gao S, Tang W, Zhang M, Wang C, Xiao J (2016) Synlett:1748
Padilla R, JØrgensen MSB, Paixão MW, Nielsen M (2019) Green Chem 21:5195
Pittaway R, Fuentes JA, Clarke ML (2017) Org Lett 19:2845
Chen J, Wang J, Tu T (2018) Chem Asian J 13:2559
Thiyagarajan S, Gunanathan C (2019) Org Lett 21:9774
Neumann J, Bornschein C, Jiao H, Junge K, Beller M (2015) Eur J Org Chem:5944
Choi J-H, Prechtl MHG (2015) ChemCatChem 7:1023
Wei Q, Zhang F, Zhao X, Wang C, Xiao J, Tang W (2017) Org Biomol Chem 15:5468
Wang W-H, Himeda Y, Muckerman JT, Manbeck GF, Fujita E (2015) Chem Rev 115:12936
Liu Q, Wu L, Gülak S, Rockstroh N, Jackstell R, Beller M (2014) Angew Chem Int Ed 53:7085
Treigerman Z, Sasson Y (2018) ACS Omega 3:12797
Kothandaraman J, Czaun M, Goeppert A, Haiges R, Jones J-P, May RB, Prakash GKS, Olah GA (2015) ChemSusChem 8:1442
Zhang L, Han Z, Zhao X, Wang Z, Ding K (2015) Angew Chem Int Ed 54:6186
Koo J, Kim SH, Hong SH (2018) Chem Commun 54:4995
Kar S, Goeppert A, Prakash GKS (2019) J Am Chem Soc 141:12518
Dong K, Elangovan S, Sang R, Spannenberg A, Jackstell R, Junge K, Li Y, Beller M (2016) Nat Commun 7:12075
Rezayee NM, Huff CA, Sanford MS (2015) J Am Chem Soc 137:1028
Kothandaraman J, Goeppert A, Czaun M, Olah GA, Prakash GKS (2016) J Am Chem Soc 138:778
Kothandaraman J, Goeppert A, Czaun M, Olah GA, Prakash GKS (2016) Green Chem 18:5831
Kar S, Sen R, Goeppert A, Prakash GKS (2018) J Am Chem Soc 140:1580
Yoshimura A, Watari R, Kuwata S, Kayaki Y (2019) Eur J Inorg Chem:2375
Kothandaraman J, Kar S, Sen R, Goeppert A, Olah GA, Prakash GKS (2017) J Am Chem Soc 139:2549
Kar S, Sen R, Kothandaraman J, Goeppert A, Chowdhury R, Munoz SB, Haiges R, Prakash GKS (2019) J Am Chem Soc 141:3160
Schneck F, Assmann M, Balmer M, Harms K, Langer R (2016) Organometallics 35:1931
Alberico E, Sponholz P, Cordes C, Nielsen M, Drexler H-J, Baumann W, Junge H, Beller M (2013) Angew Chem Int Ed 52:14162
Chakraborty S, Dai H, Bhattacharya P, Fairweather NT, Gibson MS, Krause JA, Guan H (2014) J Am Chem Soc 136:7869
Elangovan S, Wendt B, Topf C, Bachmann S, Scalone M, Spannenberg A, Jiao H, Baumann W, Junge K, Beller M (2016) Adv Synth Catal 358:820
Rezayee NM, Samblanet DC, Sanford MS (2016) ACS Catal 6:6377
Lange S, Elangovan S, Cordes C, Spannenberg A, Jiao H, Junge H, Bachmann S, Scalone M, Topf C, Junge K, Beller M (2016) Cat Sci Technol 6:4768
Koehne I, Schmeier TJ, Bielinski EA, Pan CJ, Lagaditis PO, Bernskoetter WH, Takase MK, Würtele C, Hazari N, Schneider S (2014) Inorg Chem 53:2133
Chakraborty S, Brennessel WW, Jones WD (2014) J Am Chem Soc 136:8564
Bielinski EA, Lagaditis PO, Zhang Y, Mercado BQ, Würtele C, Bernskoetter WH, Hazari N, Schneider S (2014) J Am Chem Soc 136:10234
Smith NE, Bernskoetter WH, Hazari N, Mercado BQ (2017) Organometallics 36:3995
Yi Y, Liu H, Xiao L-P, Wang B, Song G (2018) ChemSusChem 11:1474
Bonitatibus Jr PJ, Chakraborty S, Doherty MD, Siclovan O, Jones WD, Soloveichik GL (2015) Proc Natl Acad Sci U S A 112:1687
Chakraborty S, Lagaditis PO, Förster M, Bielinski EA, Hazari N, Holthausen MC, Jones WD, Schneider S (2014) ACS Catal 4:3994
Lagaditis PO, Sues PE, Sonnenberg JF, Wan KY, Lough AJ, Morris RH (2014) J Am Chem Soc 136:1367
Sonnenberg JF, Lagaditis PO, Lough AJ, Morris RH (2014) Organometallics 33:6452
Smith SAM, Lagaditis PO, Lüpke A, Lough AJ, Morris RH (2017) Chem Eur J 23:7212
Seo CSG, Tannoux T, Smith SAM, Lough AJ, Morris RH (2019) J Org Chem 84:12040
Jayarathne U, Zhang Y, Hazari N, Bernskoetter WH (2017) Organometallics 36:409
Bornschein C, Werkmeister S, Wendt B, Jiao H, Alberico E, Baumann W, Junge H, Junge K, Beller M (2014) Nat Commun 5:4111
Sawatlon B, Surawatanawong P (2016) Dalton Trans 45:14965
Xu R, Chakraborty S, Bellows SM, Yuan H, Cundari TR, Jones WD (2016) ACS Catal 6:2127
Bernskoetter WH, Hazari N (2017) Acc Chem Res 50:1049
Zhang Y, MacIntosh AD, Wong JL, Bielinski EA, Williard PG, Mercado BQ, Hazari N, Bernskoetter WH (2015) Chem Sci 6:4291
Curley JB, Smith NE, Bernskoetter WH, Hazari N, Mercado BQ (2018) Organometallics 37:3846
Jayarathne U, Hazari N, Bernskoetter WH (2018) ACS Catal 8:1338
Suàrez LA, Culakova Z, Balcells D, Bernskoetter WH, Eisenstein O, Goldberg KI, Hazari N, Tilset M, Nova A (2018) ACS Catal 8:8751
Lane EM, Zhang Y, Hazari N, Bernskoetter WH (2019) Organometallics 38:3084
Jiao H, Junge K, Alberico E, Beller M (2016) J Comput Chem 37:168
Acosta-Ramirez A, Bertoli M, Gusev DG, Schlaf M (2012) Green Chem 14:1178
Taqui Khan MM, Khan BT, Begum S, Nazeeruddin K (1984) J Mol Catal 26:207
Taqui Khan MM, Khan BT, Begum S, Ali SM (1986) J Mol Catal 34:283
Taqui Khan MM, Khan BT, Begum S (1987) React Kinet Catal Lett 34:105
Taqui Khan MM, Khan BT, Begum S (1986) J Mol Catal 34:9
Taqui Khan MM, Rao ER, Siddiqui MRH, Khan BT, Begum S, Ali SM, Reddy J (1988) J Mol Catal 45:35
Ramaraj A, Nethaji M, Jagirdar BR (2019) J Organomet Chem 883:25
Rozenel SS, Padilla R, Camp C, Arnold J (2014) Chem Commun 50:2612
Zhang G, Vasudevan KV, Scott BL, Hanson SK (2013) J Am Chem Soc 135:8668
Mills MR, Barnes CL, Bernskoetter WH (2018) Inorg Chem 57:1590
Jing Y, Chen X, Yang X (2015) Organometallics 34:5716
Yuwen J, Chakraborty S, Brennessel WW, Jones WD (2017) ACS Catal 7:3735
Chowdhury A, Biswas S, Pramanik A, Sakar P (2019) Dalton Trans 48:16083
Dai H, Guan H (2018) ACS Catal 8:9125
Schneekönig J, Tannert B, Hornke H, Beller M, Junge K (2019) Cat Sci Technol 9:1779
Xu S, Chakraborty S, Yuan H, Jones WD (2015) ACS Catal 5:6350
Spentzos AZ, Barnes CL, Bernskoetter WH (2016) Inorg Chem 55:8225
Clarke ZE, Maragh PT, Dasgupta TP, Gusev DG, Lough AJ, Abdur-Rashid K (2006) Organometallics 25:4113
Chen X, Jia W, Guo R, Graham TW, Gullons MA, Abdur-Rashid K (2009) Dalton Trans:1407
Junge K, Wendt B, Jiao H, Beller M (2014) ChemCatChem 6:2810
Schmeier TJ, Dobereiner GE, Crabtree RH, Hazari N (2011) J Am Chem Soc 133:9274
Vasudevan KV, Scott BL, Hanson SK (2012) Eur J Inorg Chem:4898
Taqui Khan MM, Khan BT, Begum S (1988) J Mol Catal 45:305
McGuinness DS, Brown DB, Tooze RP, Hess FM, Dixon JT, Slawin AMZ (2006) Organometallics 25:3605
McGuinness DS, Wassercheid P, Keim W, Hu C, Englert U, Dixon JT, Grove C (2003) Chem Commun:334
Chakraborty S, Blacque O, Fox T, Berke H (2014) Chem Asian J 9:328
Chakraborty S, Berke H (2014) ACS Catal 4:2191
Wei Z, Junge K, Beller M, Jiao H (2017) Cat Sci Technol 7:2298
Chakraborty S, Blacque O, Berke H (2015) Dalton Trans 44:6560
Leischner T, Spannenberg A, Junge K, Beller M (2018) Organometallics 37:4402
Leischner T, Suarez LA, Spannenberg A, Junge K, Nova A, Beller M (2019) Chem Sci 10:10566
Zell T, Langer R (2018) ChemCatChem 10:1930
Elangovan S, Topf C, Fischer S, Jiao H, Spannenberg A, Baumann W, Ludwig R, Junge K, Beller M (2016) J Am Chem Soc 138:8809
Fu S, Shao Z, Wang Y, Liu Q (2017) J Am Chem Soc 139:11941
Garbe M, Junge K, Walker S, Wei Z, Jiao H, Spannenberg A, Bachmann S, Scalone M, Beller M (2017) Angew Chem Int Ed 56:11237
Passera A, Mezzetti A (2019) Adv Synth Catal 361:4691
Jang YK, Krückel T, Rueping M, El-Sepelgy O (2018) Org Lett 20:7779
Kaithal A, Hölscher M, Leitner W (2018) Angew Chem Int Ed 57:13449
Wang Y, Zhu L, Shao Z, Li G, Lan Y, Liu Q (2019) J Am Chem Soc 141:17337
Papa V, Carbero-Antonino JR, Alberico E, Spannenberg A, Junge K, Junge H, Beller M (2017) Chem Sci 8:3576
Rawat KS, Pathak B (2017) Cat Sci Technol 7:3234
Kar S, Goeppert A, Kothandaraman J, Prakash GKS (2017) ACS Catal 7:6347
Ryabchuk P, Stier K, Junge K, Checinski MP, Beller M (2019) J Am Chem Soc 141:16923
Wei D, Roisnel T, Darcel C, Clot E, Sortais J-B (2017) ChemCatChem 9:80
Wei Z, Junge K, Beller M, Jiao H (2018) CR Chim 21:303
Yang X (2015) Chem Commun 51:13098
Ge H, Jing Y, Yang X (2016) Inorg Chem 55:12179
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Ekanayake, D.A., Guan, H. (2020). Hydrogenation Reactions Catalyzed by PNP-Type Complexes Featuring a HN(CH2CH2PR2)2 Ligand. In: van Koten, G., Kirchner, K., Moret, ME. (eds) Metal-Ligand Co-operativity. Topics in Organometallic Chemistry, vol 68. Springer, Cham. https://doi.org/10.1007/3418_2020_63
Download citation
DOI: https://doi.org/10.1007/3418_2020_63
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-68915-5
Online ISBN: 978-3-030-68916-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)