
Abstract
One of the goals of personalized medicine is to utilize biomarkers to sub-classify patients into risk groups that can be used to guide recommendations for therapy. In addition to classical risk factors, gene signatures and genomics are being developed as a means to biologically characterize tumors and to stratify patients according to the risk associated with the specific molecular aberrations present in their disease. Gene signatures and genomics are currently being investigated as a personalized medicine strategy for many cancers, but have been studied most extensively in breast cancer. In this disease, the results of genetic signatures have come to influence current recommendations for adjuvant chemotherapy for appropriately selected patient populations. In this chapter, we will review the use of gene signatures and genomics in the development of personalized oncology, with an emphasis on applications for breast cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Breast Cancer
- Triple Negative Breast Cancer
- Recurrence Score
- Breast Conservation Therapy
- Massively Parallel Signature Sequencing
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Personalized Medicine and Biomarkers
Cancer often exhibits both inter-patient and intra-tumoral genetic heterogeneity, even among patients with the same primary malignancy (Schilsky 2010; De Palma and Hanahan 2012). In order to assess an individual patient’s prognosis, often clinical or pathologic features are utilized, such as age, tumor size, lymph node status, histology, grade, and margin status, among other features depending on the specific malignancy. With a steadily growing understanding of the biologic basis for the heterogeneity of cancer, there is considerable interest in biomarker development and the implementation of personalized medicine (Ely 2009; Ginsburg and Willard 2009). A biomarker is defined by the NCI as a biological molecule that is found in the blood, other body fluids, or tissue that is a sign of a condition or disease. Biomarkers have many uses in oncology, including risk assessment (BRCA mutation analysis), screening (PSA), prognosis (Oncotype DX), and prediction of response to therapy (HER2 amplification) (Henry and Hayes 2012). In addition, several biomarkers, such as CEA, PSA, and CA 15-3, can be used to monitor response to therapy and to detect recurrent disease. An important distinction should be made regarding the difference between prognostic and predictive biomarkers. Prognostic biomarkers provide information on patient outcome, independent of treatment. Predictive biomarkers, however, can be used to predict the likelihood of a response to a given therapy. While prognostic biomarkers may be useful to determine the natural history of disease, predictive biomarkers may be better suited to provide a means to personalize therapy (La Thangue and Kerr 2011).
As many key regulators of cellular processes have been identified, including oncogenes and tumor suppressor genes, it was anticipated that normal cellular function might be restored by counteracting a specific genetic abnormality. However, the complexity of the dysregulation that develops during carcinogenesis is perhaps greater than was originally anticipated. In addition, the frequent crosstalk between pathways has sometimes hindered development of novel cancer therapies that have meaningful clinical efficacy (De Palma and Hanahan 2012; Mendelsohn et al. 2012).
The concept of personalized medicine, at first glance, might seem to imply that each individual patient would receive a unique combination of therapies specific to the genetic aberrations present in his/her malignancy. However, as a practical matter, personalized medicine in oncology often uses predictive biomarkers to identify subgroups of patients who are likely to benefit from a specific targeted therapy. With many of the recently developed targeted therapies, often only a small proportion of patients, on the order of 10−20 %, will harbor the specific molecular aberration and be expected to respond to the targeted therapy. Instead of exposing a large population of patients to additional systemic therapy, treatment efficacy could be maximized by administering this therapy only to those most likely to derive a benefit. In this way, patients who are unlikely to benefit from the therapy can avoid the additional side effects and cost. The overall goal of this strategy of personalized medicine is to define specific subgroups of patients and to tailor therapy accordingly (La Thangue and Kerr 2011). It should be emphasized that risk stratification in personalized medicine using biomarkers is currently only additive in the context of clinical/anatomic staging. Gene expression profiling is one method that has been used clinically to define patient subgroups and to administer personalized medicine. We will next review the methods and statistical issues related to gene expression profiling.
2 Introduction to Gene Expression Profiling and Associated Technologies
2.1 Methods of Gene Expression Profiling
Gene expression is the formation of a functional gene product that is created from the information contained within a gene. Gene expression profiling involves assessing the relative amounts of mRNA produced from various genes. The first step of gene expression profiling involves isolation and purification of the mRNA from the test sample. In the case of tumor samples, this is often obtained from formalin-fixed paraffin-embedded (FFPE) tissue, such as biopsy or surgical resection specimens. Although this method is feasible, formalin-fixation and processing of a tissue specimens results in chemical modification and degradation of RNA (Medeiros et al. 2007). Therefore, when possible, frozen specimens or fresh specimens stored in special RNA-preserving solution are desired, as they yield more intact RNA. Regardless of the method of preservation, delays between tissue harvesting and specimen fixation should be minimized, in order to reduce RNA degradation. Once the mRNA is isolated and purified, it is converted to complementary DNA (cDNA) by reverse transcription.
The microarray technique utilizes an array of gene-specific DNA probes or oligos, spotted onto a slide (Quackenbush 2006; Tefferi et al. 2002; Hamilton 2012). Each spot contains a specific DNA sequence corresponding to a gene. The relative level of gene expression for each gene can be determined for a sample by fluorescently-labeling the cDNA and then hybridizing the labeled cDNA with the DNA microarray. The relative fluorescence intensity, which corresponds to the level of gene expression, can then be determined for each gene (Fig. 1). The Mammaprint is an example of a microarray-based test designed to assess a woman’s risk of metastasis from breast cancer, and will be discussed in more detail later in this chapter (Kim et al. 2009).

Overview of microarray analysis. Reprinted with permission from Quackenbush (2006)
Alternatively, gene expression can be assessed with a quantitative PCR (qPCR) based approach, in which the cDNA obtained from a sample is amplified using gene-specific primers and labeled probes. This method requires creation of specific primers and probes for each gene of interest, and logistically this limits the number of genes that can be analyzed. Oncotype DX is an example of a qPCR-based test which can be used to assess recurrence risk for women with ER-positive, lymph node-negative breast cancer. The Oncotype DX assay will be discussed further later in this chapter (Kim et al. 2009).
2.2 Validation of Gene Expression Profiling
Prior to clinical application, the use of a specific biomarker or a panel of biomarkers must be thoroughly studied and validated (Simon et al. 2009). Teutsch et al. outline three key components to evaluation of a genetic test: analytic validity, clinical validity, and (Henry and Hayes 2012; Teutsch et al. 2009). The first component, analytic validity, is defined as the ability of a test to accurately and reliably measure the genotype of interest. The analytic validity includes assessment of the sensitivity, specificity, precision (reproducibility), and assay robustness (resistance to small changes in assay parameters). The second component, clinical validity, is defined as the ability of a test to accurately and reliably predict the clinical event. This includes determination of the positive and negative predictive values of the test. Clinical validity requires analysis of an independent cohort to validate the original findings. This is often performed with an initial analysis of a “test set” and confirmation with an independent “validation set”, which may be an independent cohort of patients at the same institution or, alternatively, patients at another institution. The third component in evaluating a new biomarker is clinical utility, which is defined as evidence of improved measurable clinical outcome, and added value to patient management decision-making compared with current management. In other words, does the use of the new biomarker provide additional information that changes patient management? Also, the extent of the effect of the biomarker is considered. For example, does the biomarker status correspond to a small percentage change in outcome or does it confer a large, several-fold difference? Is this difference enough for clinicians to change management?
Microarray data analysis enables us to monitor the expression level of genes and changes in the expression patterns with respect to pathologic conditions at a genome scale. There are two approaches to analyze the data—supervised or unsupervised analysis. In supervised analysis, distinct groups of genes or samples (i.e. patient vs. normal) are identified and differences in expression profiles between the groups are evaluated. On the other hand, in unsupervised analysis, sets of genes or samples with similar expression profiles are grouped, and their common clinical and physiological and/or biological features are identified. If there are pre-existing clusters (patients and normal or different known tumor types), supervised analysis is more appropriate. However, unsupervised analysis is a powerful method for identifying new clusters (uncharacterized pathways of dysregulated gene expression, new tumor subtypes etc.) Clustering analysis generates distinct groups of genes or samples based on their similarity of expression profiles and may be hierarchical or non-hierarchical. In hierarchical clustering, the relationships among objects within and between groups are specified and represented as dendrograms (Hastie et al. 2009). In this way, samples with similar expression patterns are grouped together within branches of a sample dendrogram, and in like manner, genes with consistent expression patterns within sample groups cluster in gene dendrogram branches. These results indicate cellular and molecular features differentiating groups of samples, which may be important for diagnosis or prognosis, and furthermore identify key transcriptional and signaling pathways that may be targeted by new or existing therapies. One pitfall with microarray data analysis is the issue of multiple hypothesis testing. By definition, microarray experiments simultaneously test the expression of thousands of genes, often assessing gene expression using a much lower number of individual patient samples. This creates a statistical problem known as multiple hypothesis testing. Data analysis solutions to this problem exist, but extreme caution should be used in interpreting the results of a microarray study with relatively low numbers of patient samples (<25), thousands of gene probes and a single patient/tumor data set. The false discovery rate (FDR), which is a modified p value used to adjust for multiple comparisons, is often reported when groups of patient samples are compared for gene expression using microarrays. In general, FDR values <0.05 are acceptable for statistical significance in microarray studies, and most current studies employ additional methods to reduce the false positive rate (Benjamini and Hochberg 1995; Storey and Tibshirani 2003). In addition, a number of more advanced statistical methods are currently available for data analysis, many of which evaluate biologically meaningful gene sets or pathways (Hastie et al. 2009; Tseng et al. 2012).
In summary, prior to implementation of a new biomarker or gene signature into clinical practice, it must be evaluated for analytic validity, clinical validity, and clinical utility. Large validation studies are required. The highest level of evidence for a new genetic signature would be a prospective clinical study that is designed with assessment of the biomarker as the primary objective of the trial (Henry and Hayes 2012). This would minimize bias in subject selection and standardize sample handling and assay conditions. Gene expression profiling has been perhaps most extensively studied in breast cancer. We will next review the development and implementation of personalized medicine in breast cancer on the basis of gene expression.
3 Defining Patient Subgroups in Breast Cancer on the Basis of Gene Expression
3.1 Classical Studies of Global Gene Expression in Breast Cancer
In 2000, a seminal paper by Perou et al. characterized the gene expression patterns for 42 women with breast cancer (Perou et al. 2000). Most of the specimens analyzed were breast cancer, but a few samples of normal breast tissue were also examined. Some patients had multiple specimens studied, such as the primary tumor and a lymph node metastasis, and in some cases samples were obtained before and after chemotherapy administration. Microarrays were performed, with over 8,000 genes analyzed. Using a hierarchical clustering method, genes were grouped based on the similarity of their patterns of expression. The authors found that there was significant variation in gene expression patterns among the tumor specimens. Interestingly, samples from the same patient, such as from a primary tumor and a lymph node, or before and after chemotherapy, were more similar to each other than to any other sample, in terms of their gene expression pattern. The “intrinsic” gene subset is a subset of 496 genes which showed greater variation between unrelated samples than was seen between samples from the same patient. This subset of genes includes specific clusters of genes, such as the luminal cluster, HER2 (Erb-B2) cluster, proliferation cluster, and basal cluster. The “molecular portraits” of gene expression examined in this study led to identification of the intrinsic subtypes of breast cancer. In the first publication, the subtypes identified were ER+/luminal-like, basal-like, Erb-B2+, and normal breast (Perou et al. 2000). Further investigation led to the finding that the luminal subtype could potentially be divided into two or three subgroups, termed luminal A, luminal B, and luminal C, each with a unique gene expression pattern (Sorlie et al. 2001). Alternatively, two luminal subgroups, luminal A and luminal B, could be described. Importantly, the clinical outcome of patients was evaluated for each of the subtypes, including luminal A, luminal B, luminal C, normal breast-like, Erb-B2+, and basal-like. Significant differences were seen in both relapse-free survival and overall survival, with basal-like and Erb-B2+ subtypes having the worst outcome and luminal A subtype showing the best outcome (Fig. 2). In a subsequent analysis of 115 breast cancers, the subtypes were further refined (Sorlie et al. 2003), to include the luminal A, luminal B, basal, Erb-B2+, and normal breast-like subtypes. The cluster dendrogram for tumors, when divided into these five subtypes (luminal A, luminal B, normal breast-like, Erb-B2+, and basal-like), can be seen in Fig. 3. Also, the full cluster diagram and gene expression patterns can be seen in Fig. 4. Subtype characterization was confirmed by cluster analyses performed on independent data sets of cohorts of patients from other institutions. The clinical outcomes of different subtypes were assessed and revealed differences in overall survival and time to distant metastases. One other interesting finding from this work was that BRCA1 mutation was strongly associated with predisposition to the basal tumor phenotype.

Clinical outcome for patients based on tumor subtype. Kaplan-Meier survival curves demonstrate the relapse-free survival (RFS) for breast cancer patients, as classified by tumor subtype. Reprinted with permission from Hu et al. (2006)

Cluster dendrogram demonstrating tumor subtypes. This dendogram shows the clustering of the tumors into five tumor subgroups. Branches for tumors with low correlation to any tumor subtype are shown in gray. Reprinted with permission from Sorlie et al. (2003)

Hierarchical clustering of breast tumors using the intrinsic gene set, with full cluster (left) and specific gene clusters shown in more detail (right), including the Erb-B2+ (C), luminal B (D), basal (E), normal breast-like (F), and luminal A (G) clusters. Reprinted with permission from Sorlie et al. (2003)
More recently, a new breast cancer intrinsic gene list containing 1300 genes was evaluated and validated in independent data sets (Hu et al. 2006). This analysis stratified patients into five subtypes, luminal A, luminal B, basal-like, HER2+, and normal breast-like, using a 105 tumor training set and validation set of 311 tumor samples (compiled from three independent studies). Clinical outcomes for the subtypes were significantly different in terms of relapse-free and overall survival. Multivariate analysis demonstrated that tumor subtype was prognostic of relapse-free, disease-specific, and overall survival, independent of standard clinical factors such as tumor size, lymph node status, and tumor grade.
3.2 Implications of Breast Cancer Subtype for Systemic Therapy Selection
Determination of breast tumor subtype is clinically valuable, as the subtype (often clinically defined by immunohistochemical profile) frequently guides decisions for systemic therapy. For example, women with luminal A or luminal B (ER positive) breast cancer, are known to derive benefit from adjuvant hormone therapy (Davies et al. 2011), and therefore most will receive anti-estrogen therapy in the form of tamoxifen or an aromatase inhibitor. Patients with the Erb-B2+(HER2+) subtype will often receive Herceptin (trastuzumab), which is a monoclonal antibody against the HER2/neu receptor. Women with HER2-positive breast cancer who are either lymph node-positive or high-risk node negative have been found to benefit from Herceptin therapy (Smith et al. 2007; Perez et al. 2011) and many current studies are exploring combinations of anti-HER2 targeted therapies for these patients (Gianni et al. 2012). The basal subtype or the overlapping subtype known as “triple negative breast cancer” continues to receive significant research attention due to its relative poor prognosis and lack of targeted therapeutic strategies. Information provided by the intrinsic subtypes (using an immunohistochemical approach) has been adopted by 2011 St. Gallen Consensus Conference (Goldhirsch et al. 2011) (Table 1).
4 Gene Signatures and Clinical Decision Making in Breast Cancer
Fortunately, the majority of women diagnosed with breast cancer do not have metastatic disease at the time of diagnosis, and are in a clinically curable situation. However, after surgery, all women have some degree of risk of relapse, at local, regional, and distant sites. The magnitude of the risk of relapse can be quite different depending on the individual woman, and can be assessed using clinical, pathologic, and treatment-related variables. Over the past decade, gene signatures for women with breast cancer have provided significant additional prognostic information. The greatest clinical impact of these assays has been risk stratification in relatively low risk patient populations (ER positive and node negative). Specifically, Mammaprint is a commercially available gene expression assay that has been used to predict recurrence in patients with node negative cancers. Oncotype DX is a commercially available, clinically validated gene expression assay that is used to guide recommendations for the use of systemic chemotherapy in addition to anti-estrogen therapy for patients with ER positive, node negative disease. It is important to note that clinical use of any gene signature should be considered only for independently validated assays performed on large datasets, and care should be taken to apply use of the signature only to patients for whom the assay has been validated. Currently, clinical use of Oncotype DX in node positive and/or ER negative disease is considered experimental.
4.1 Mammaprint
One of the earliest gene signatures for breast cancer was the Mammaprint, or the Amsterdam 70-gene prognostic signature. This signature was initially characterized in a cohort of 98 primary breast cancer patients, which included 34 women that developed distant metastatic disease within 5 years and 44 women who did not (van’t Veer et al. 2002). There were also 20 patients with BRCA1 or BRCA2 germline mutations included in the analysis. For the sporadic cases, all women were <55 years old, had a tumor size <5 cm, and were lymph node negative. RNA was isolated from frozen tumor samples and supervised analysis of the microarray data identified a set of 70 genes that allowed discrimination between patients with good and poor prognosis, with an accuracy of 83 %. The Mammaprint allows a binary classification, either a good prognosis or poor prognosis signature (Fig. 5). Women in the poor prognostic group based on this signature have a significantly increased risk of developing distant metastatic disease within 5 years (odds ratio, OR = 28). On multivariate analysis including classical prognostic factors, the Mammaprint signature was an independent predictor of outcome.

Patient outcome based on Mammaprint signature. The metastasis-free survival (left panel) and overall survival (right panel) of breast cancer patients with good prognosis signature and poor prognosis signature are shown. Reprinted with permission from van de Vijver et al. (2002)
Further validation of the Mammaprint gene signature was performed in a cohort of 295 patients (van de Vijver et al. 2002). All patients were less than 53 years old, had a primary tumor size of less than 5 cm, and in this cohort 151 were lymph node negative, while 144 were lymph node positive. Overall survival at 10 years was 95 % for those with good prognosis signature and was 55 % for those with poor prognosis signature. Probability of remaining free of distant metastases was 85 % for those with good prognosis signature and 51 % for those with poor prognosis signature. However, it should be noted that 61 patients in this study were also members of the original cohort used to develop the signature. Independent validation studies were subsequently conducted, including an analysis of 302 patients from five European centers (Buyse et al. 2006). Patients included in this analysis were <61 years old, lymph node negative, tumor size <5 cm, and did not receive adjuvant systemic therapy. Median follow-up was 13.6 years. The 70-gene prognostic signature remained an independent prognostic factor for development of distant metastases and overall survival, with unadjusted hazard ratios of 2.32 and 2.79, respectively.
There is also interest in assessing risk of distant metastatic disease in older women, who may not tolerate chemotherapy well. Identification of older women that have a low risk of distant metastatic disease might allow avoidance of the considerable toxicity associated with chemotherapy in this group of women. The Mammaprint gene signature has also been evaluated for women between the ages of 55 and 70 (Mook et al. 2010). In this analysis, frozen tumor specimens from 148 women, aged 55−70 years old with tumor size <5 cm and negative lymph nodes, were analyzed and assigned either good or poor prognosis based on their 70-gene signature. The 70-gene prognosis signature was prognostic of breast cancer-specific survival (P = 0.036). Distant metastasis-free survival at 5 years was 93 % for patients with a good prognosis signature and 72 % for those with poor prognosis signature, but this difference was not statistically different in this cohort (P = 0.07).
Currently, there are several ongoing clinical trials designed to further characterize the utility of Mammaprint. MINDACT (Microarray In Node-negative and 1−3 positive lymph node Disease may Avoid ChemoTherapy; EORTC 10041), is a Phase III prospective randomized study comparing Mammaprint with clinical-pathological assessment (Adjuvant! Online) in selecting patients with 0−3 positive lymph nodes for adjuvant chemotherapy. In the trial, women with discordant prognostic assessment on Mammaprint and Adjuvant! Online will be randomized for the decision of adjuvant chemotherapy based on either Mammaprint or Adjuvant! Online risk status. All women with high risk scores on both Mammaprint and Adjuvant! Online will receive chemotherapy and all women with low risk scores on both will not receive chemotherapy. Accrual of 6,600 patients has been achieved, with results currently pending.
Other ongoing trials include PROMIS, which is a prospective registry study to assess the impact of Mammaprint on systemic therapy decision making for patients with an intermediate Oncotype DX score. NBRST, is a prospective registry study designed to measure outcomes based on molecular subgroups, determined by Mammaprint and other profiles, for patients undergoing neoadjuvant chemotherapy or endocrine therapy. Similarly, MINT I, is a study designed to test the ability of Mammaprint (in combination with other factors) to predict response to neoadjuvant chemotherapy. Of note, initial studies using Mammaprint were conducted on frozen tissue, but currently the assay can be performed on fresh, frozen, or formalin-fixed paraffin-embedded specimens.
4.2 Oncotype DX
The Oncotype DX is a real-time quantitative reverse-transcriptase-polymerase-chain-reaction (RT-PCR) assay of 21 prospectively selected genes designed for use in fixed, paraffin-embedded tumor specimens. Paik and colleagues first developed a real-time RT-PCR assay to quantify gene expression of 250 candidate genes and subsequently analyzed the relation between breast cancer recurrence and gene expression in a preliminary inquiry of 447 patients (Paik et al. 2004). From this analysis, they selected a panel of 16 cancer-related genes and five reference genes. The cancer-related genes included those involved in proliferation and invasion, among others. An algorithm was designed to calculate a Recurrence Score (RS) based on the levels of expression of these genes. The RS ranged from 0 to 100, with higher scores reflecting greater likelihood of distant recurrence. Patients were divided into three risk categories based on their RS: low-risk (RS < 18), intermediate-risk (RS 18−30), and high-risk (RS > 30). Paik et al. demonstrated the ability of the Oncotype DX assay to predict the likelihood of distant recurrence for women with ER-positive, lymph node-negative breast cancer (Paik et al. 2004). In this analysis, tumor samples from 668 women treated with tamoxifen on NSABP B-14 were evaluated. NSABP B-14 was a clinical trial of ER-positive, node-negative breast cancer, in which women were randomized to tamoxifen versus placebo. The rate of distant recurrence at 10 years was 7 % for those with low-risk RS, 14 % for intermediate-risk RS, and 31 % for high-risk RS (Fig. 6). Recurrence score was found to be an independent prognostic factor on multivariate analysis. RS was also predictive of overall survival. The risk of distant recurrence can be predicted using the RS as a continuous function (Fig. 7).

Stratification of patient outcome based on Oncotype DX risk group. The freedom from distant recurrence is shown for patients in the low, intermediate, and high risk groups. Reprinted with permission from Paik et al. (2004)

Rate of distant metastases as a function of Recurrence Score, determined by Oncotype DX analysis. The risk of distant metastasis at 10 years can be estimated for any given recurrence score, using the continuous function shown here. Reprinted with permission from Paik et al. (2004)
Determination of the RS from the Oncotype DX assay was also found to predict the magnitude of benefit from chemotherapy. An analysis was performed of tumor samples from the NSABP B-20 trial, in which women with ER-positive, node negative breast cancer were randomized to tamoxifen with or without chemotherapy. RT-PCR was successful in the majority (97 %) of blocks that had sufficient remaining specimen and were therefore included in this analysis (n = 651). In NSABP B-20, there was a benefit with the addition of chemotherapy, in terms of local, regional, and distant recurrence. In the current study, the magnitude of benefit from chemotherapy was greatest among patients with high risk RS, and patients in intermediate or low risk groups demonstrated no significant benefit from chemotherapy (Paik et al. 2006). In another study, the Oncotype DX-derived RS was found to be predictive of pathologic complete response (pCR) in patients receiving neoadjuvant chemotherapy (Gianni et al. 2005). Patients with higher RS had a greater probability of having a pCR after completion of neoadjuvant chemotherapy. Based on the findings above, the randomized Phase III TAILORx trial for women with ER-positive, node-negative breast cancer was designed. Women enrolled on this trial with an Oncotype DX RS <11 receive hormone therapy only, and those with a RS >25 receive chemotherapy followed by hormone therapy. Women with RS of 11−25 are randomized to either hormone therapy alone or in combination with chemotherapy. Enrollment has been completed and results are currently pending.
Two preliminary studies have reported on the utility of the Oncotype DX assay in lymph-node positive patients (Dowsett et al. 2010; Albain et al. 2010). Dowsett et al. evaluated the Oncotype DX assay on specimens from the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial (Dowsett et al. 2010). The ATAC trial compared the efficacy of arimidex, tamoxifen, or both for post-menopausal women. In this analysis, Oncotype DX assay was performed for samples of patients on the single agent arms (arimidex only or tamoxifen only; n = 1,372). Prognostic value of Oncotype-based RS was seen both in patients treated with tamoxifen and confirmed for patients treated with arimidex. In addition, RS was predictive of distant recurrence for lymph node negative (n = 872) and also for lymph node positive patients (n = 306). Similarly, Albain et al. investigated the utility of the Oncotype DX assay for post-menopausal women with node-positive, ER-positive breast cancer (Albain et al. 2010). In this retrospective analysis, 367 samples from the Phase III SWOG-8814 trial, in which women were randomized to tamoxifen with or without the addition of chemotherapy, were analyzed. Patients with high-risk RS showed a significant benefit in disease-free survival with the addition of chemotherapy, whereas those with intermediate- or low-risk RS did not. An ongoing Phase III trial, SWOG RxPONDER (S1007), has been initiated to prospectively evaluate the benefit of chemotherapy for women with 1−3 positive lymph nodes, ER-positive, HER2-negative breast cancer with RS of 25 or less. Women meeting eligibility criteria will be randomized to receive endocrine therapy with or without the addition of chemotherapy. The primary endpoint is disease-free survival and enrollment is ongoing.
Oncotype DX assessment is incorporated in the ASCO recommendations for use of tumor markers in breast cancer and in NCCN guidelines to predict the risk of recurrence for women with ER-positive, HER2-negative, node-negative breast cancer treated with tamoxifen. The NCCN panel considers Oncotype DX an option, to be taken into consideration only in the context of other elements of patient risk stratification. Other applications at this time are investigational and require further validation prior to inclusion in clinical decision-making.
Recently there has been interest in molecular characterization of ductal carcinoma in situ, DCIS. DCIS is a non-invasive form of breast cancer that has the potential to develop into invasive disease over time. Adjuvant radiation therapy has been shown to decrease the risk of both non-invasive and invasive recurrence after partial mastectomy for DCIS. There is ongoing interest in identifying women with DCIS with a low risk of recurrence after surgery who would likely derive a small absolute benefit from adjuvant therapy. However, the use of clinical and pathologic factors to identify women with low risk DCIS is not entirely straightforward. Even patients with low or intermediate grade DCIS and negative margins have a measurable risk of recurrence. Solin et al. reported DCIS risk stratification utilizing 12 of the 21 genes in the Oncotype DX assay (Solin et al. 2012). In data presented at the San Antonio Breast Cancer Symposium in 2011, the Oncotype DX assay has been proposed as a means to stratify risk of recurrence in patients with DCIS who have undergone partial mastectomy alone. For this analysis, the Oncotype DX was performed for 327 women (approximately 50 % of patients) enrolled on the ECOG E-5194 study. Women in this study had relatively low risk DCIS, with either low or intermediate grade DCIS ≤ 2.5 cm or high grade DCIS ≤ 1 cm. The “DCIS Score” was determined and women were grouped into three categories: high, intermediate, and low. They found that the DCIS Score obtained from the Oncotype DX assay was a significant independent predictor of ipsilateral breast events (either invasive or DCIS) on multivariate analysis. It was proposed that the DCIS Score could be used to estimate a woman’s risk of recurrence after partial mastectomy for DCIS and potentially guide individual recommendations for adjuvant therapy. However, even the low risk group had a 12 % risk of any breast event (either invasive or non-invasive recurrence). Also, further validation is necessary before this is applied to routine clinical practice.
4.3 Other Novel Gene Signature Assays
Other gene signatures are being developed, including the Breast Cancer Index (BCI), MapQuant DX Genomic Grade Index (GGI), among others (Prat et al. 2011). The Breast Cancer Index (BCI) is an assay for women with ER-positive, node-negative breast cancer, which may assist in prediction of the risk of distant recurrence. Two prognostic quantitative real-time PCR assays, the HOXB13:IL17BR two-gene ratio and the molecular grade index (MGI), are incorporated in the BCI. HOXB13 is an antiapoptotic gene associated with increased risk of recurrence and the IL-17 receptor B is associated with decreased risk of recurrence. The combination of the HOXB13:IL17BR and MGI were found to be prognostic of outcome (Ma et al. 2008). MapQuant DX, also known as Genomic Grade Index or GGI, is a microarray based assay that defines two molecular subgroups that have distinct clinical outcomes (Loi et al. 2007). Early comparisons of the various gene signatures show high rates of concordance in predicting outcome (Fan et al. 2006).
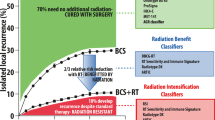
4.4 Gene Expression and Radiation Therapy
The primary purpose and utility of the previously discussed gene signatures was to stratify patients based on risk of distant recurrence. There are several questions regarding gene expression in breast cancer that are more relevant to radiation therapy, and we will review the existing data for each of these questions here. Haffty and Buchholz (2010) have recently written an excellent editorial on recent publications concerning gene expression and local recurrence in breast cancer, and interested readers are encouraged to review their findings (Haffty and Buchholz 2010). In brief, the authors define the following questions regarding gene expression and radiation therapy: (1) Is there a group of women at sufficiently low risk of local recurrence after lumpectomy alone that can be spared radiation therapy? (2) Alternatively, can gene expression signatures be used to identify a group of women at sufficiently high risk of local or regional recurrence after breast conservation (BCT)? Would these patients be better served with dose escalation or the use of radiosensitizers? (3) As breast conservation treatments evolve to include accelerated regimens, hypofractionation and partial breast volumes, can the results of gene expression assist in selection of patients for altered radiotherapy regimens? (4) With respect to postmastectomy irradiation, are our current guidelines oversimplified with respect to tumor biology? Can gene expression profiling be used to identify patients at risk for local regional recurrence after mastectomy?
Over the past few years, several groups have assessed the Oncotype DX and other gene expression signatures for their association with risk of locoregional recurrence. In one study the Oncotype DX RS was shown to predict risk of locoregional recurrence in a cohort of women with node-negative ER-positive breast cancer (Mamounas et al. 2010). In contrast, a similar study evaluated the RS for patients treated with breast conserving surgery, chemotherapy, and radiation therapy on ECOG E2197 (Solin et al. 2012). In this analysis, RS was determined for patients with 1−3 positive lymph nodes or negative lymph nodes with tumor size greater than 1.0 cm. Neither the intrinsic biologic subtype nor the RS was predictive of local or regional recurrence in this cohort, suggesting inability to define a subset of patients that may not require adjuvant radiation as part of breast conservation. Voduc et al. (2010) used an immunohistochemical panel (ER, PR, HER2, EGFR, CK5/6 and Ki-67) and a tissue microarray to classify 2,985 tumors into intrinsic subtypes (luminal A, luminal B, luminal-HER2, Her2 enriched, basal-like or triple negative phenotype non basal) (Voduc et al. 2010) (Fig. 8). A multivariate analysis was then performed to determine the risk of local or regional relapse associated with the intrinsic subtypes after adjusting for standardized risk factors. Luminal A tumors were found to have the lowest risk of locoregional recurrence, and 10-year local relapse-free survival after breast conserving surgery was 92 % (95 % confidence interval (CI) 90−95 %) for this patient population. For patients with HER2-enriched and basal subtypes, the 10-year local relapse-free survival after breast conserving surgery was reduced to 79 % (95 % CI 69−89 %) and 86 % (95 % CI 80−93 %), respectively. Haffty and Buchholz noted that these data were generated prior to the trastuzumab era, and caution that the HER2-enriched data may not be as clinically relevant today as recent data have shown that the addition of trastuzumab is associated with a significant improvement in local control (Haffty and Buchholz 2010). Perhaps the most useful piece of data from this analysis is the fact that luminal A tumors have an excellent prognosis and very low rates of local recurrence after breast conservation treatment.

Local (a) and regional (b) relapse rates, by intrinsic subtype. Violet line, luminal A; light blue, luminal human epidermal growth factor receptor 2 (HER2); dark blue, luminal B; yellow, five-marker negative phenotype; red, basal; beige, HER2 enriched. Reprinted with permission from Voduc et al. (2010)
More recently, Abdulkarim et al. (2011) reported that patients with T1-T2 triple negative breast cancers treated with breast conserving surgery followed by irradiation had a 5-year actuarial locoregional recurrence rate of 4 % compared to 10 % after mastectomy alone (P = 0.027) (Abdulkarim et al. 2011). These results suggest that in field effects of local irradiation are important for control of triple negative breast cancer and provide clinical evidence that tumor biology may influence response to local radiation (Pignol et al. 2011). Most importantly, these results call into question our previous assumption that breast conservation and mastectomy are equivalent treatments for biologically aggressive breast cancer. In this patient population, it appears that minimal surgery followed by local radiotherapy is more effective than a more radical surgery. Finally, we must also revisit our recommendations regarding postmastectomy radiation. These results suggest that our guidelines should be modified to include intrinsic subtype. Additional study will be needed to determine whether postmastectomy radiation can improve local control for triple negative breast cancers (Pignol et al. 2011).
Do we need to develop new gene expression signatures to directly address response to radiation in breast cancer? Initial studies showed relatively little difference in global gene expression profiles from primary breast tumors that recurred locally after breast conservation therapy versus tumors that did not (Kreike et al. 2006). Based on the hypothesis that gene expression patterns related to wound healing would be important for cancer invasion and metastasis, Chang et al. developed the “core serum response” (CSR) gene signature in vitro and then tested the ability of this gene signature to predict outcome in 295 patients treated for early breast cancer (Chang et al. 2004, 2005). In this patient population, the increased expression of the CSR genes (also known as the “wound response signature”) was associated with decreased overall and distant metastasis-free survival. Nuyten et al. then trained (n = 81) and validated (n = 80) a classifier for local recurrence after breast conservation therapy (BCT) using the wound response signature (Nuyten 2006). Most recently, Kreike et al. have compared gene expression profiles from 56 primary breast cancers that recurred after BCT versus 109 primary breast cancers that did not recur after BCT (Kreike et al. 2009). Both supervised and unsupervised methods of classification were used to separate patients based on local recurrence after treatment. In addition, the authors tested many other published gene signatures for the ability to predict local recurrence, including their previously developed wound response signature. In this analysis, the five molecular subtypes [as most recently defined by Hu et al. (2006)] were associated with local recurrence after BCT. Luminal B type and HER2-like tumors had significantly increased local recurrence after BCT versus the other subtypes including the basal/triple negative subtype. Repeat testing of the wound response signature in this data set did not accurately predict local recurrence, emphasizing the importance of multiple validation studies prior to clinical implementation. In a supervised analysis, the authors developed a new 111 gene signature for the prediction of local recurrence after BCT and subsequently validated this signature using separate dataset of 161 patients. The results of these studies are compelling and could potentially allow for treatment intensification for patients at a high risk of local recurrence after BCT. Although these results are promising, rigorous clinical validation studies will be needed before we can consider incorporating these results into clinical practice.
It has been hypothesized that gene expression profiling could be used to develop a signature predictive of response to radiotherapy, although the genetic diversity observed in solid tumors may obscure these effects. Numerous studies have analyzed gene expression patterns before and after radiotherapy, and most of these studies have used in vitro or in vivo model systems (Ogawa et al. 2007). Not surprisingly, many categories of genes are upregulated in response to radiation treatment, and many of these genes regulate cellular responses to stress, cell cycle progression and DNA repair. Torres-Roca et al. (2005) developed a gene signature for radiosensitivity using a panel of 35 cancer cell lines and the results of clonogenic survival assays after 2 Gy (SF2) (Torres-Roca et al. 2005). Gene selection was based upon an fit to a linear regression model of gene expression versus cellular radiosensitivity. Genes selected were then used to build a multivariate model to predict SF2. The initial study identified novel genes implicated in the radiation response (RBAP48 and RGS19). The same group then integrated gene expression and cellular radiosensitivity data from 48 cell lines and used a systems-biology based approach to develop a 10 gene network (AR, cJun, STAT1, PKC, RelA, cABL, SUMO1, CDK1, HDAC1, and IRF1) associated with cellular radiosensitivity (Eschrich et al. 2009a, b). The radiosensitivity index (RSI) is a linear function of expression of the ten genes. The RSI is inversely proportional to the radiosensitivity of the tumor (i.e. a low RSI indicates a more radiosensitive tumor). The RSI has been clinically tested in three datasets (rectal, esophageal, head and neck) for total of 118 patients (Eschrich et al. 2009a, b). Eschrich et al. (2012) recently published the results of RSI testing in two breast cancer datasets that included patients treated with breast conservation and mastectomy (Eschrich et al. 2012). In the first dataset, patients treated with radiotherapy and predicted to be radiosensitive (RS) on the basis of RSI were found to have improved 5-year relapse-free survival versus patients predicted to be radioresistant (RR) (95 vs. 75 %, n = 77). In the second data set, patients treated with radiotherapy and RS on the basis of RSI were found to have improved 5-year distant metastasis free survival versus RR patients (77 vs. 64 %), and RSI was found to be an independent predictor of outcome in ER positive patients treated with radiotherapy. Piening et al. developed a gene signature for radiation induced (RI) and radiation repressed (RR) genes using 12 human lymphoblast cell lines exposed to 5 Gy (Piening et al. 2009). The RI and RR gene sets were then compared to published gene signatures and used to predict outcome after treatment in two published breast cancer patient data sets. The authors note that while many RR genes overlap with the previously well characterized proliferation signature, the RI genes add prognostic information, and the combination of RR and RI genes was able to predict outcome in the published data sets tested. Additional study, including clinical validation and clinical utility studies, will be needed to determine whether RR and RI genes can be used independently to predict outcome to radiotherapy in breast cancer.
Very few studies have explored gene expression differences before and after irradiation in breast cancer using clinical samples. Helland et al. analyzed gene expression from tumor samples from 19 stage III/IV breast cancer patients before and after radiotherapy with 20 Gy (Helland et al. 2006). In that study, several genes were upregulated in irradiated tumors including GPX1, DDB2, GDF15 and CDKN1A. The authors noted that the tumor suppressor gene TP53 was mutated in 39 % of their samples, and gene expression profiles were, not surprisingly, influenced by TP53 mutational status. It should also be noted that this relatively small patient dataset was quite hetereogeneous for biomarker expression (ER/PR/HER2) and instrinsic subtype analysis was not performed.
In summary, recent studies have shown that intrinsic breast cancer subtypes do influence local control after radiation treatment in breast cancer. In general, the luminal A subtype has the lowest risk of local recurrence after radiotherapy. Patients with early stage triple negative breast cancers have improved local control when radiation is used as part of breast conserving therapy compared to similar patients treated with mastectomy. In the research setting, several groups have developed gene signatures associated with radiation response in breast cancer. Additional validation and utility studies will be needed before we can use these signatures in the clinic.
5 Genetic Variation in Cancer and Targeted Therapy
5.1 Nature of Genetic Variation in Cancer
Cancer is classically considered to be a genetic disorder which develops as an evolutionary process, consisting of serial acquisition of somatic mutations and subsequent natural selection. Clones of abnormal cells arise from this process and continue to evolve during oncogenesis. With successive cell divisions, subclones with varying capabilities of proliferation, survival, invasion, and metastasis develop. Some subclones will emerge as dominant, while others will acquire deleterious mutations and are outcompeted by more dominant clones. Depending on the severity of the deleterious mutations, these clones may die or continue to exist as small remnants of the evolutionary history of the malignant process. The somatic mutations may include insertions, deletions, base substitutions, rearrangements, copy number alterations, or epigenetic changes.
Somatic mutations are acquired over one’s lifetime and are randomly distributed in the genome. In addition, germline mutations can also affect one’s susceptibility to cancer. There is some variation in genome sequence among humans, representing approximately 0.1 % of the genome. This genetic variation may range from single nucleotide changes to gross karyotype alterations. Single nucleotide polymorphisms (SNPs) are the most common type of variation representing approximately 90 % of human genome variation, but there are also structural variants (insertions, deletions, inversions, copy number variations), rare variants, and epigenetic differences. One’s risk of cancer may be influenced both by the inherent genetic variation and germline mutations as well as the somatic mutations that occur over one’s lifetime, which may be modulated by lifestyle and environmental factors (Stratton 2011; Stratton et al. 2009).
Although mutations occur throughout the genome, those that by chance occur in certain regions of the genome may be more likely to promote oncogenesis. “Driver mutations” are those that tend to occur in a subset of genes known as the “cancer genes”. Driver mutations confer a growth advantage to the cell and directly contribute to cancer development. Passenger mutations, on the other hand, are those that happen along the way but do not give the cell a growth advantage. Passenger mutations may be detected in a cancer genome, but have not contributed to oncogenesis (Stricker et al. 2011). Some estimates suggest that several, perhaps approximately five, key mutations are required to generate cancer (Stratton 2011; Stratton et al. 2009).
Cancer genes may be functionally classified as dominant or recessive, in terms of their behavior at the cellular level. Dominant cancer genes, or oncogenes, require only one allele to be mutated and often result in constitutive activation. Oncogenes promote cell survival and proliferation. The majority (>80 %) of known of cancer genes are dominantly acting. Recessive cancer genes, or tumor suppressor genes, require both alleles to be altered in order for an effect to be seen. Tumor suppressor genes often play a role in cell cycle regulation, DNA repair, and apoptosis. If only one copy of the tumor suppressor gene is mutated or lost, the other copy can function normally. Examples of tumor suppressor genes include retinoblastoma protein RB1, TP53, BRCA1, and BRCA2. Mutations that affect the DNA repair process may result in an increase in the rate of somatic mutations in the cancer cell lineage (Stratton 2011; Stratton et al. 2009).
5.2 Methods Used to Study Genetic Variation
Initial studies of the genetics of cancer involved cytogenetic studies of chromosomes, with characterization of chromosomal translocations and abnormalities of chromosome copy number. The development of recombinant DNA technology later provided the ability to isolate and sequence portions of the genome associated with frequent rearrangements. In 2000, a draft sequence of the human genome was completed (Lander et al. 2001; Venter et al. 2001). This was a monumental step, which has facilitated further sequencing of cancer genomes, including whole gene families and most protein-coding exons. Nearly one decade after the human genome sequence was announced, the first completely sequenced cancer genomes were published in January 2010 (Pleasance et al. 2010a, b). Many more cancer genomes are being sequenced, and it is estimated that tens of thousands of cancer genomes will be sequenced over the next several years (Fig. 9). Efforts are being led by the International Cancer Genome Consortium and the Cancer Genome Atlas project in the United States.

Timeline of key events in cancer genomics. Reprinted with permission from Stratton (2011)
The primary techniques used to study cancer genomics include: whole genome sequencing, targeted genome sequencing, cancer genotyping, and genome-wide association studies. Whole genome sequencing, as the name implies, determines the entire DNA sequence of a genome. First generation DNA sequencing techniques included Maxam–Gilbert chemical sequencing (Maxam and Gilbert 1977) and Sanger (chain-termination) sequencing (Sanger et al. 1977). Sanger sequencing is very accurate, but is limited by its high cost and low throughput. Newer sequencing methods, termed next-generation sequencing (NGS), have since been developed that have higher throughput and are more economical. Examples of next-generation sequencing techniques include massively parallel signature sequencing (MPSS), pyrosequencing, Illumina sequencing (sequencing by synthesis), SOLiD sequencing, ion semiconductor sequencing, and single molecule real-time (SMRT) sequencing (Tran et al. 2012). With these and other novel sequencing techniques, whole genome sequencing is becoming more affordable and feasible to perform. We have come a long way from the sequencing of the first human genome which cost nearly $3 billion and took a decade to complete, with current cost of approximately $10,000 per genome (Fig. 10).

Cost of genome sequencing over time. Reprinted with permission from Wetterstrand, http://www.genome.gov/sequencingcosts
Targeted genomic sequencing uses a similar approach, but limits sequencing efforts to specific regions or genes of interest. By sequencing only specific portions of the genome, targeted genomic sequencing is both efficient and cost-effective. The regions of interest may be a limited number of genes, the whole exome (portion of the genome formed by exons), or the cancer genome (portion of the genome containing the cancer genes). Data analysis is also simplified, as only segments of the genome are sequenced and subsequently analyzed (Tran et al. 2012). Our institution is among the institutions that now offer targeted genomic sequencing of multiple cancer genes available for clinical use.
Cancer genotyping refers to a method of determining whether a specific known cancer gene mutation is present in a tumor. As common mutations of cancer genes are continually being identified, it can be relatively straightforward to assess whether these specific mutations are present in a patient’s tumor. Genotyping of clinical specimens is performed with high-throughput genotyping platforms, such as Taqman OpenArray Genotyping, Affymetrix genotyping arrays, and MassARRAY (Tran et al. 2012).
Another method of studying genetic variation is through genome-wide association study (GWAS). In this type of analysis, typically two cohorts are studied, those with the disease of interest (cases) and those without the disease (controls). GWAS looks for associations between single-nucleotide polymorphisms (SNPs) and a disease (Manolio 2010). A SNP is considered to be associated with a disease if it is more common in those with the disease than in the control population. GWAS data is often displayed in a Manhattan plot, which shows the relative association for various SNPs across the genome (Fig. 11). GWAS analysis has some limitations, including the issue of multiple hypothesis testing and the fact that most SNPs identified by GWAS thus far have been typically associated with only a small increase in the risk of disease, with a median odds ratio of 1.33 (Manolio 2010).

Genome-wide association studies (GWAS). A sample Manhattan plot is shown, which displays the P values for all genotyped single-nucleotide polymorphisms (SNPs). Reprinted with permission from Manolio (2010)
5.3 Clinical Cancer Genomics
A human breast cancer genome was sequenced and published in April 2010, only a few months after the first cancer genomes were published (Ding et al. 2010). In this publication the investigators sequenced four samples from a patient with metastatic basal-like breast cancer, including the primary tumor, peripheral blood, a brain metastasis and a xenograft derived from the primary tumor. Several interesting findings were reported, including a number of mutations contained in the primary tumor that were found to be enriched in the metastatic tumor.
Several publications in Nature in 2012 highlighted breast cancer genomics studies, demonstrating considerable progress in our understanding (Comprehensive Molecular Portraits of Human Breast Tumours 2012; Ellis et al. 2012; Curtis et al. 2012; Stephens et al. 2012; Shah et al. 2012; Banerji et al. 2012). Stephens and colleagues examined somatic mutations in 100 breast cancer genomes. They found driver mutations in several known cancer genes and also identified several new cancer genes based on non-random clustering of mutations (Stephens et al. 2012). Shah and colleagues studied mutations in 104 triple negative breast cancers (Shah et al. 2012). They found that the most frequently mutated gene was TP53, which had mutations in 62 % of patients with basal triple negative breast cancer, and 43 % of non-basal triple negative disease. Other genes with frequent mutations included PIK3CA, USH2A, PTEN, and RB1. Interestingly, they discovered that for most tumors, mutations in tumor suppressor genes such as TP53 tended to occur in the highest clonal frequency, suggestive of an early event in the clonal evolution of the tumor. By comparing RNA sequencing with the genome data, they also found that only 36 % of single nucleotide variants were expressed. Curtis et al. (2012) examined the genomes and transcriptomes of nearly 2,000 breast cancers and based on joint clustering of copy number and gene expression data, identified 10 patient subgroups with a range of breast cancer outcomes. Banerji and colleagues determined the sequence of whole exomes for 103 breast cancer patients in Mexico and Vietnam, as well as whole genome sequences for 22 breast cancer/normal pairs. They found frequent mutations in many known breast cancer genes, as well as identified mutations in the CBFB transcription factor gene and a MAGI3-AKT3 fusion which results in constitutive AKT activation (Banerji et al. 2012).
Koboldt et al. from The Cancer Genome Atlas Network studied several hundred patients, 463 of whom were evaluated on five different platforms, including mRNA expression microarrays, DNA methylation chips, SNP arrays, miRNA sequencing, and whole-exome sequencing (Comprehensive Molecular Portraits of Human Breast Tumours 2012). Somatic mutations in only three genes (TP53, PIK3CA and GATA3) occurred at >10 % incidence across breast cancers. Characteristic mutations were found within breast cancer subtypes, including common mutations in GATA3, PIK3CA and MAP3K1 in the luminal A subtype. TP53 was mutated in 84 % of basal-like breast cancers, and copy number analysis demonstrated many similarities between basal-like breast cancers and serous ovarian cancers, including widespread genomic instability, common gains of 1q, 3q, 8q and 12p, and common losses of 4q, 5q and 8p. Integrated analysis of protein phosphorylation and mRNA data identified two subgroups within the HER2+ group. Only 50 % of HER2+ cancers were categorized as HER2 overexpressing by mRNA analysis; the remaining 50 % of HER2+ cancers were within the luminal subtypes. When both HER2 protein and mRNA were overexpressed, increased expression of EGFR, pEGFR, HER2 and pHER2 was observed. Ellis and colleagues conducted either whole exome or whole genome sequencing for 77 patients with ER positive breast cancer from two trials of neoadjuvant aromatase inhibition (Ellis et al. 2012). In patients with aromatase inhibitor resistance, some pathways including TP53, DNA replication, and mismatch repair, were found to be enriched relative to patients sensitive to aromatase inhibition.
In summary, genomics has provided additional insight into the molecular mechanisms that drive breast cancer development. Only a very few genes are mutated at a high frequency across all breast cancers. Characteristic mutations are common within breast cancer subtypes and may help to guide targeted therapy in the future. Many of these mutations are within pathways that regulate the radiation response. Additional study will be needed to determine if individual gene mutations can serve as biomarkers for breast cancer response to radiation.
6 Conclusion
Through remarkable scientific innovation, we have witnessed elaborate gene expression profiling studies as well as the sequencing of entire cancer genomes over the past decade, which provides intricate knowledge about oncogenesis and the drivers of this process. As we gain further understanding of the molecular processes involved, novel therapeutics may be developed and subsequently utilized for the patients most likely to derive a benefit. Considerable progress has been made in this regard in the fields of breast cancer, non-small cell lung cancer, and melanoma, among others, and will likely play an increasing role in the treatment of these and other malignancies in the future.
References
Abdulkarim BS, Cuartero J, Hanson J, Deschenes J, Lesniak D, Sabri S (2011) Increased risk of locoregional recurrence for women with T1–2N0 triple-negative breast cancer treated with modified radical mastectomy without adjuvant radiation therapy compared with breast-conserving therapy. J Clin Oncol 29(21):2852–2858
Albain KS, Barlow WE, Shak S et al (2010) Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 11(1):55–65
Banerji S, Cibulskis K, Rangel-Escareno C et al (2012) Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486(7403):405–409
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57(1):289−300
Buyse M, Loi S, van’t Veer LJ et al (2006) Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst 98(17):1183–1192
Chang HY, Sneddon JB, Alizadeh AA et al (2004) Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol 2(2):E7
Chang HY, Nuyten DS, Sneddon JB et al (2005) Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA 102(10):3738–3743
Comprehensive Molecular Portraits of Human Breast Tumours (2012) Nature 490(7418): 61−70
Curtis C, Shah SP, Chin SF et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486(7403):346–352
Davies C, Godwin J, Gray R et al (2011) Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 378(9793):771–784
De Palma M, Hanahan D (2012) The biology of personalized cancer medicine: facing individual complexities underlying hallmark capabilities. Mol Oncol 6(2):111–127
Ding L, Ellis MJ, Li S et al (2010) Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464(7291):999–1005
Dowsett M, Cuzick J, Wale C et al (2010) Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: a TransATAC study. J Clin Oncol 28(11):1829–1834
Ellis MJ, Ding L, Shen D et al (2012) Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486(7403):353–360
Ely S (2009) Personalized medicine: individualized care of cancer patients. Transl Res 154(6):303–308
Eschrich S, Zhang H, Zhao H et al (2009a) Systems biology modeling of the radiation sensitivity network: a biomarker discovery platform. Int J Radiat Oncol Biol Phys 75(2):497–505
Eschrich SA, Pramana J, Zhang H et al (2009b) A gene expression model of intrinsic tumor radiosensitivity: prediction of response and prognosis after chemoradiation. Int J Radiat Oncol Biol Phys 75(2):489–496
Eschrich SA, Fulp WJ, Pawitan Y et al (2012) Validation of a radiosensitivity molecular signature in breast cancer. Clin Cancer Res 18(18):5134–5143
Fan C, Oh DS, Wessels L et al (2006) Concordance among gene-expression-based predictors for breast cancer. N Engl J Med 355(6):560–569
Gianni L, Zambetti M, Clark K et al (2005) Gene expression profiles in paraffin-embedded core biopsy tissue predict response to chemotherapy in women with locally advanced breast cancer. J Clin Oncol 23(29):7265–7277
Gianni L, Pienkowski T, Im YH et al (2012) Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol 13(1):25–32
Ginsburg GS, Willard HF (2009) Genomic and personalized medicine: foundations and applications. Transl Res 154(6):277–287
Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ (2011) Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St. Gallen international expert consensus on the primary therapy of early breast cancer 2011. Ann Oncol 22(8):1736–1747
Haffty BG, Buchholz TA (2010) Molecular predictors of locoregional recurrence in breast cancer: ready for prime time? J Clin Oncol 28(10):1627–1629
Hamilton SR (2012) Molecular pathology. Mol Oncol 6(2):177–181
Hastie T, Tibshirani R, Friedman J (2009) The elements of statistical learning, 2nd ed. Springer Series
Helland A, Johnsen H, Froyland C et al (2006) Radiation-induced effects on gene expression: an in vivo study on breast cancer. Radiother Oncol 80(2):230–235
Henry NL, Hayes DF (2012) Cancer biomarkers. Mol Oncol 6(2):140–146
Hu Z, Fan C, Oh DS et al (2006) The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics 7:96
Kim C, Taniyama Y, Paik S (2009) Gene expression-based prognostic and predictive markers for breast cancer: a primer for practicing pathologists. Arch Pathol Lab Med 133(6):855–859
Kreike B, Halfwerk H, Kristel P et al (2006) Gene expression profiles of primary breast carcinomas from patients at high risk for local recurrence after breast-conserving therapy. Clin Cancer Res 12(19):5705–5712
Kreike B, Halfwerk H, Armstrong N et al (2009) Local recurrence after breast-conserving therapy in relation to gene expression patterns in a large series of patients. Clin Cancer Res 15(12):4181–4190
La Thangue NB, Kerr DJ (2011) Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nat Rev Clin Oncol 8(10):587–596
Lander ES, Linton LM, Birren B et al (2001) Initial sequencing and analysis of the human genome. Nature 409(6822):860–921
Loi S, Haibe-Kains B, Desmedt C et al (2007) Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol 25(10):1239–1246
Ma XJ, Salunga R, Dahiya S et al (2008) A five-gene molecular grade index and HOXB13:IL17BR are complementary prognostic factors in early stage breast cancer. Clin Cancer Res 14(9):2601–2608
Mamounas EP, Tang G, Fisher B et al (2010) Association between the 21-gene recurrence score assay and risk of locoregional recurrence in node-negative, estrogen receptor-positive breast cancer: results from NSABP B-14 and NSABP B-20. J Clin Oncol 28(10):1677–1683
Manolio TA (2010) Genomewide association studies and assessment of the risk of disease. N Engl J Med 363(2):166–176
Maxam AM, Gilbert W (1977) A new method for sequencing DNA. Proc Natl Acad Sci USA 74(2):560–564
Medeiros F, Rigl CT, Anderson GG, Becker SH, Halling KC (2007) Tissue handling for genome-wide expression analysis: a review of the issues, evidence, and opportunities. Arch Pathol Lab Med 131(12):1805–1816
Mendelsohn J, Ringborg U, Schilsky RL (2012) Personalized cancer—a strategy to counteract an increasing cancer challenge. Mol Oncol 6(2):109–110
Mook S, Schmidt MK, Weigelt B et al (2010) The 70-gene prognosis signature predicts early metastasis in breast cancer patients between 55 and 70 years of age. Ann Oncol 21(4):717–722
Nuyten D, Kreike B, Hart A et al (2006) Predicting a local recurrence after breast-conserving therapy by gene expression profiling. Breast Cancer Res 8(5):R62
Ogawa K, Murayama S, Mori M (2007) Predicting the tumor response to radiotherapy using microarray analysis (Review). Oncol Rep 18(5):1243–1248
Paik S, Shak S, Tang G et al (2004) A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 351(27):2817–2826
Paik S, Tang G, Shak S et al (2006) Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol 24:3726–3734
Perez EA, Romond EH, Suman VJ et al (2011) Four-year follow-up of trastuzumab plus adjuvant chemotherapy for operable human epidermal growth factor receptor 2-positive breast cancer: joint analysis of data from NCCTG N9831 and NSABP B-31. J Clin Oncol 29(25):3366–3373
Perou CM, Sorlie T, Eisen MB et al (2000) Molecular portraits of human breast tumours. Nature 406(6797):747–752
Piening BD, Wang P, Subramanian A, Paulovich AG (2009) A radiation-derived gene expression signature predicts clinical outcome for breast cancer patients. Radiat Res 171(2):141–154
Pignol JP, Rakovitch E, Olivotto IA (2011) Is breast conservation therapy superior to mastectomy for women with triple-negative breast cancers? J Clin Oncol 29(21):2841–2843
Pleasance ED, Stephens PJ, O’Meara S et al (2010a) A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463(7278):184–190
Pleasance ED, Cheetham RK, Stephens PJ et al (2010b) A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463(7278):191–196
Prat A, Ellis MJ, Perou CM (2011) Practical implications of gene-expression-based assays for breast oncologists. Nat Rev Clin Oncol 9(1):48−57
Quackenbush J (2006) Microarray analysis and tumor classification. N Engl J Med 354(23):2463–2472
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74(12):5463–5467
Schilsky RL (2010) Personalized medicine in oncology: the future is now. Nat Rev Drug Discov 9(5):363–366
Shah SP, Roth A, Goya R et al (2012) The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486(7403):395–399
Simon RM, Paik S, Hayes DF (2009) Use of archived specimens in evaluation of prognostic and predictive biomarkers. J Natl Cancer Inst 101(21):1446–1452
Smith I, Procter M, Gelber RD et al (2007) 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet 369(9555):29–36
Solin LJ, Gray R, Goldstein LJ et al (2012) Prognostic value of biologic subtype and the 21-gene recurrence score relative to local recurrence after breast conservation treatment with radiation for early stage breast carcinoma: results from the eastern cooperative oncology group E2197 study. Breast Cancer Res Treat 134(2):683–692
Sorlie T, Perou CM, Tibshirani R et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 98(19):10869–10874
Sorlie T, Tibshirani R, Parker J et al (2003) Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 100(14):8418–8423
Stephens PJ, Tarpey PS, Davies H et al (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486(7403):400–404
Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100(16):9440–9445
Stratton MR (2011) Exploring the genomes of cancer cells: progress and promise. Science 331(6024):1553–1558
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458(7239):719–724
Stricker T, Catenacci DV, Seiwert TY (2011) Molecular profiling of cancer—the future of personalized cancer medicine: a primer on cancer biology and the tools necessary to bring molecular testing to the clinic. Semin Oncol 38(2):173–185
Tefferi A, Bolander ME, Ansell SM, Wieben ED, Spelsberg TC (2002) Primer on medical genomics. Part III: microarray experiments and data analysis. Mayo Clin Proc 77(9):927–940
Teutsch SM, Bradley LA, Palomaki GE et al (2009) The evaluation of genomic applications in practice and prevention (EGAPP) initiative: methods of the EGAPP working group. Genet Med 11(1):3–14
Torres-Roca JF, Eschrich S, Zhao H et al (2005) Prediction of radiation sensitivity using a gene expression classifier. Cancer Res 65(16):7169–7176
Tran B, Dancey JE, Kamel-Reid S et al (2012) Cancer genomics: technology, discovery, and translation. J Clin Oncol 30(6):647–660
Tseng GC, Ghosh D, Feingold E (2012) Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res 40(9):3785–3799
van de Vijver MJ, He YD, van’t Veer LJ et al (2002) A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347(25):1999–2009
van’t Veer LJ, Dai H, van de Vijver MJ et al (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415(6871): 530−536
Venter JC, Adams MD, Myers EW et al (2001) The sequence of the human genome. Science 291(5507):1304–1351
Voduc KD, Cheang MC, Tyldesley S, Gelmon K, Nielsen TO, Kennecke H (2010) Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol 28(10):1684–1691
Wetterstrand KA, DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). http://www.genome.gov/sequencingcosts. Accessed March 29 2013
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Thomas, M.A., Rashmi, R., Payton, J., Zoberi, I., Schwarz, J.K. (2013). Integration of Gene Signatures and Genomic Data into Radiation Oncology Practice. In: Nieder, C., Gaspar, L. (eds) Decision Tools for Radiation Oncology. Medical Radiology(). Springer, Berlin, Heidelberg. https://doi.org/10.1007/174_2013_858
Download citation
DOI: https://doi.org/10.1007/174_2013_858
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-37101-1
Online ISBN: 978-3-642-37102-8
eBook Packages: MedicineMedicine (R0)