
Abstract
Drug addiction is a chronic brain disease characterized by compulsive drug-seeking and drug-taking behaviors despite the major negative consequences. Current well-established neuronal underpinnings of drug addiction have promoted the substantial progress in understanding this disorder. However, non-neuronal mechanisms of drug addiction have long been underestimated. Fortunately, increased evidence indicates that neuroimmune system, especially Toll-like receptor 4 (TLR4) signaling, plays an important role in the different stages of drug addiction. Drugs like opioids, psychostimulants, and alcohol activate TLR4 signaling and enhance the proinflammatory response, which is associated with drug reward-related behaviors. While extensive studies have shown that inhibition of TLR4 attenuated drug-related responses, there are conflicting findings implicating that TLR4 signaling may not be essential to drug addiction. In this chapter, preclinical and clinical studies will be discussed to further evaluate whether TLR4-based neuroimmune pharmacotherapy can be used to treat drug addiction. Furthermore, the possible mechanisms underlying the effects of TLR4 inhibition in modulating drug-related behaviors will also be discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Drug addiction is a chronic brain disease characterized by compulsive drug-seeking and drug-taking behaviors despite the major negative consequences (Cheron and Kerchove d'Exaerde 2021). It is one of the leading causes of disability and fatality worldwide today, with a huge annual cost related to crime, reduced work productivity and health care (Nestler and Luscher 2019). Current studies focusing on neuronal adaptations have yielded much progress in the research of drug addiction. For example, it is suggested that molecular, synaptic, and neurocircuitry neuroadaptations combine to promote the transition to drug addiction, which is comprised of increased incentive salience, decreased reward, increased stress, and decreased executive function (Wise and Koob 2014). However, non-neuronal underpinnings of drug addiction have long been underestimated (Kashima and Grueter 2017). Fortunately, a growing body of studies indicate that neuroimmune system plays an important role in the different stages of drug addiction, including binge/intoxication, withdrawal, and relapse (Hutchinson et al. 2012; June et al. 2015; Northcutt et al. 2015).
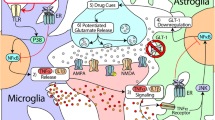
Toll-like receptors (TLRs) are a family of pattern recognition receptors (PPRs) in the innate immune system which detect and respond both to exogenous pathogen associated molecular patterns (PAMPs) and endogenous danger associated molecular patterns (DAMPs) (Koropatnick et al. 2004; Hennessy et al. 2010; Connolly and O'Neill 2012). Toll-like receptor 4 (TLR4) is one of the TLRs and its activation leads to enhanced production of proinflammatory cytokines and chemokines. In the brain, TLR4 is mainly expressed in glial cells like microglia and astrocytes (Vaure and Liu 2014). Upon recognition of PAMPs or DAMPs, TLR4 signals through two distinct pathways, the myeloid differentiation primary response protein 88 (MyD88)-dependent and MyD88-independent pathway (Kawai and Akira 2007a). In the MyD88-dependent pathway, the signal transduces through activation of Interleukin-1 receptor associated kinases (IRAKs, like IRAK1 and IRAK4) and TNF receptor associated factor 6 (TRAF6), which subsequently promotes the phosphorylation of inhibitors of nuclear factor κB kinases (IKK). The activation in turn leads to the NFκB activation and the production of proinflammatory cytokines and chemokines (Kawai and Akira 2007b). Alternatively, in MyD88-independent pathway, the adaptor protein TRIF, TRAF3 and interferon regulatory factor 3 (IRF3) are involved (Takeda and Akira 2005) (Fig. 1).

Schematic representation of the role of toll-like receptor 4 (TLR4) signaling in drug addiction. It should be noted that the non-neuronal mechanism underlying drug addiction is not clear. Drugs of abuse bind to the accessory receptor of TLR4, MD-2, and activate the downstream signaling which comprised of two distinct pathways (MyD88-dependent and MyD88-independent pathways). This activation leads to the transcription of proinflammatory regulators like TNF-α and IL-β and enhances non-neuronal alterations, which subsequently act in concert with neuronal adaptations and contribute to drug reward-related behaviors, withdrawal and relapse
TLR4 signaling is suggested to be involved in several neuropsychiatric disorders, including major depressive disorders, neurodegenerative disorders, and impulsive control (Nie et al. 2018; Landreth and Reed-Geaghan 2009; Aurelian et al. 2016; Garcia Bueno et al. 2016; Liu et al. 2019). As drugs of abuse can be considered as “exogenous,” it is recognized that drugs of different class activate TLR4 signaling and induce proinflammatory responses. Emerging evidence has suggested the important role of TLR4 signaling in regulating drug addiction (Crews et al. 2017). In this chapter, we will discuss the preclinical and clinical evidence of TLR4 signaling modulation in drug addiction (i.e., opioid, psychostimulants, and alcohol addiction), in order to evaluate whether TLR4-based neuroimmune pharmacotherapy can be used as novel treatment for this disorder. Furthermore, we will also discuss the possible mechanisms underlying the effects of TLR4 antagonism in regulating drug-related behaviors.
2 Role of TLR4 Signaling in Drug Addiction
2.1 Opioid
Although the major targets of opioids in the brain are opioid receptors, which probably mediate most of the effects of opioids within the CNS, growing evidence has demonstrated that opioids can also interact with TLRs, among which the TLR4 is best studied in opioid addiction. In vitro evidence suggests that the molecular interaction between the opioid system and TLR4 is complex. The opioid antagonist naloxone inhibited the classic TLR4 agonist LPS-induced secretion of IL-β and morphological changes of microglia in mixed brain cell cultures (Das et al. 1995). In contrast, both morphine and fentanyl could activate TLR4 in unstimulated cells, even though the activation level was much lower than that was stimulated by LPS (Hutchinson et al. 2010). Morphine exposure could elevate TLR4 protein and mRNA expression as well as activate TLR4-related signaling pathways in the Nucleus Accumbens (NAc) (Schwarz and Bilbo 2013). Interestingly, morphine and fentanyl could attenuate LPS-induced activation of TLR4 in a non-competitive manner (Hutchinson et al. 2010). These findings suggest that opioids might interact with TLR4 and act as its partial agonists. Besides in vitro reports, many behavioral studies have explored the role of TLR4 in mediating the effects of opioids, including addictive properties (Gabr et al. 2021).
Many pharmacological studies using the TLR4 antagonists such as (+)-naloxone and lipopolysaccharide from Rhodobacter sphaeroides (LPS-RS) have implicated that TLR4 participates in the development of opioid addiction and relapse. (+)-Naloxone blocked morphine-induced conditioned place preference (CPP), remifentanil self-administration, drug-induced reinstatement of heroin-seeking behavior, and dopamine release in the NAc (Hutchinson et al. 2012; Yue et al. 2020). Another study found that microinjection of TLR4 antagonist LPS-RS into the ventral tegmental area (VTA) prevented the conditioning and maintenance, but not expression, of morphine-induced CPP (Chen et al. 2017). In the same study, it was suggested that the STAT3 might mediate the function of TLR4 since LPS-RS prevented morphine-induced activation of STAT3 in the VTA (Chen et al. 2017). Interestingly, microinjection of LPS-RS into the NAc did not affect drug-induced reinstatement of heroin-seeking, suggesting that the NAc might not be the critical brain site where TLR4 regulates opioid addiction (Yue et al. 2020). Consistent with pharmacological findings, global deletion of tlr4 or myd88 gene prevented oxycodone-induced CPP in mice (Hutchinson et al. 2012). Studies that evaluated the effects of ibudilast also provided some implications on the role of TLR4 in opioid addiction. Ibudilast is principally a Phosphodiesterase 4 (PDE4) inhibitor but also exerts antagonist property at TLR4. Moreover, ibudilast could decrease morphine-induced dopamine release in the NAc in rodents (Bland et al. 2009).
Opioid withdrawal has been demonstrated to participate in the development of opioid addiction via a negative reinforcing mechanism (Koob and Volkow 2010). Several pharmacological studies have indicated that TLR4 also regulates opioid withdrawal. The TLR4 antagonist (+)-naloxone could significantly attenuate the μ opioid receptor antagonist (−)-naloxone-precipitated withdrawal behavior in morphine-dependent rats (Hutchinson et al. 2010). The TLR4 antagonist ibudilast reduced spontaneous withdrawal-induced hyperactivity in rats (Hutchinson et al. 2009). In contrast, the genetic deletion of TLR4 genes did not affect opioid withdrawal. Compared to wildtype Balb/c mice, both TLR4-KO and MyD88-KO mice (Balb/c background) showed similar degrees of naloxone-precipitated jumping behavior, an animal model of opioid withdrawal (Liu et al. 2011). A more recent study also reported similar findings that both TLR4 mutant and null mice showed normal morphine withdrawal behaviors (Mattioli et al. 2014). These findings suggest that the tlr4 gene might not be critical for opioid withdrawal. However, it should be noted that global deletion of TLR4 or MyD88 genes may result in changes in many other genes that could compensate for the loss in the function of TLR4 signaling. Therefore, future studies using conditional deletion of TLR4 are required to address the role of the tlr4 gene in the development of opioid addiction.
Nevertheless, not all literature supports the view that TLR4 mediates opioid addiction. Acute injection of (+)-naltrexone did not affect incubated cue-induced heroin-seeking or extended access heroin self-administration. Whereas chronic administration of (+)-naltrexone reduced incubated cue-induced heroin-seeking but did not affect ongoing extended access heroin self-administration (Theberge et al. 2013). One explanation is that TLR4 signaling might only participate in some particular opioid addiction-related behaviors. Furthermore, many factors such as opioid dose, history of drug use, and treatment strategy (i.e., acute or chronic treatment) are essential factors that might dramatically influence the pharmacological effects of TLR4 antagonists on opioid addiction.
In clinical settings, TLR4 antagonist ibudilast was tested for its efficacy in attenuating opioid-related effects. Ibudilast was shown to reduce ratings of drug liking following 15 mg of oxycodone and heroin craving (Metz et al. 2017). Meanwhile, ibudilast also decreased drug breakpoint under the 15 mg but not 30 mg oxycodone condition in a progressive-ratio oxycodone self-administration task, suggesting that ibudilast attenuated, at least to some extent, the reinforcing effects of oxycodone (Metz et al. 2017). On the contrary, ibudilast was unable to consistently affect subjective effect ratings of oxycodone in opioid-dependent volunteers in another study (Cooper et al. 2017). Nevertheless, it decreased ratings of withdrawal symptoms on some SOWS items during detoxification (Cooper et al. 2016).
2.2 Psychostimulants
2.2.1 Cocaine
Cocaine activates innate immune system within the brain through its interaction with TLR4 (Cearley et al. 2011; Coller and Hutchinson 2012), possibly in a region-specific manner (Burkovetskaya et al. 2020). Cocaine docked to the same binding site of MD-2 as the classical TLR4 agonist LPS and increased the proinflammatory responses. This effect is associated with cocaine-induced dopamine release and cocaine reward, an effect that can be blocked by TLR4 antagonist (+)-naloxone (Northcutt et al. 2015). Pretreatment of (+)-naloxone or LPS-RS attenuated cocaine-induced elevation of extracellular dopamine in the NAc, while they alone did not affect the dopamine signaling. Meanwhile, non-TLR4 modulator, neurotensin, did not affect cocaine-induced dopamine elevation, suggesting the specificity to TLR4 receptor. Moreover, pretreatment of TLR4 antagonists blocked the development of cocaine CPP and self-administration, while sparing food-maintained responses (Northcutt et al. 2015). Consistently, TLR4 mutant mice showed less responses to cocaine self-administration and cocaine reward learning, suggesting the importance of TLR4 in cocaine reinforcement (Kashima and Grueter 2017; Northcutt et al. 2015).
However, inconsistent findings suggest that TLR4 may not be crucial for cocaine-related behavioral and neurochemical alterations. Tanda and colleagues found that (+)-naloxone or (+)-naltrexone did not decrease cocaine or heroin-induced dopamine levels in the NAc shell (Tanda et al. 2016). Both antagonists attenuated cocaine or remifentanil self-administration at a higher dose that decreased food-maintained responding as well, suggesting a lack of selectivity on reward behaviors (Tanda et al. 2016). In addition, (+)-naloxone did not interact with cocaine subjective effects in the drug-discrimination studies (Tanda et al. 2016). It is further shown that a TLR4 agonist reactivated microglia, suppressed striatal synaptic strength, and finally decreased cocaine-induced sensitization (Lewitus et al. 2016). These results challenge the current knowledge of TLR4 in cocaine addiction, yet call for further examination and clarification of the role of TLR4 in cocaine-related responses.
A recent clinical study showed that cocaine users had a significant increase in IL-6 compared with control group, demonstrating an activation of the immune system (Moreira et al. 2016). Nonetheless, there are few clinical studies examining the effect of neuroimmune modulators in regulating cocaine addiction. More clinical investigations focusing on the possibility of neuroimmune signaling as novel therapeutic target for cocaine addiction are needed.
2.2.2 Methamphetamine
Methamphetamine (METH) exposure activates glia cells and enhances proinflammatory cytokines release (Goncalves et al. 2008; Loftis et al. 2011; Nakajima et al. 2004). Indeed, METH was shown to bind to MD-2, the key receptor of TLR4 and enhanced CD11b and IL-6 in mRNAs in the VTA (Wang et al. 2019). Increased evidence suggests that modulation of TLR4 can reduce METH-related behavioral and neurochemical effects (Fujita et al. 2012; Narita et al. 2006; Zhang et al. 2006). TLR4 antagonists (+)-naloxone and LPS-RS reduced METH-induced elevation of dopamine in the NAc (Wang et al. 2019). Ibudilast, AV1013, and minocycline decreased METH-induced behavioral sensitization, drug-primed and cue-induced METH-seeking (Snider et al. 2012; Beardsley et al. 2010), METH-induced conditioned place preference (CPP) (Fujita et al. 2012; Chen et al. 2009) and METH self-administration (Snider et al. 2013). These findings indicate an essential role of glia activation underlying the rewarding effects of METH. Interestingly, it is also implicated that cannabinoids Δ9-tetrahydrocannabinol and cannabidiol might be effective for protection of METH-induced inflammation through modulation of TLR4 and NF-κB signaling (Majdi et al. 2019).
Clinical studies also yielded inspiring results that neuroimmune modulators could be effective against METH-related symptoms. Initially, a case study reported that minocycline significantly improved the psychotic symptoms in METH use disorders (Tanibuchi et al. 2010). Later, in an early-stage study, ibudilast reduced several METH-related subjective effects including High, Good, Stimulated and Like, suggesting its effect in attenuating the reward-associated subjective effects of METH (Worley et al. 2016). Moreover, ibudilast is also shown to improve the attention performance during the early abstinence from METH dependence (Birath et al. 2017). All these results implicated that neuroimmune modulators may have protective effects on METH-related disorders. However, a most recent clinical trial showed that ibudilast did not affect METH abstinence (Heinzerling et al. 2020). This randomized, placebo-controlled trial included 64 patients with METH use disorders for the 12-week ibudilast treatment and urine specimen was collected for drug screen and study assessments (Heinzerling et al. 2020). Ibudilast was well tolerated yet did not alter METH abstinence (Heinzerling et al. 2020). No significant correlation between serum ibudilast levels and METH use during treatment for patients was observed (Heinzerling et al. 2020). These results seem discouraging, yet it is still early to conclude that ibudilast has no effect on METH-related actions. No further assessment on the effect of ibudilast on METH intake or craving was provided. Indeed, a pilot clinical study showed that ibudilast could reduce METH-induced elevation of peripheral markers of inflammation, which may underlie the mechanisms of METH addiction. As such, more research investigating the effects of TLR4 modulation in METH-taking or relapse could add valuable information to the field.
2.2.3 Nicotine
Currently, there are no studies examining the role of TLR4 in nicotine addiction. Although it is suggested that nicotine increased the expression of TLR4 and also upregulated TLR4-related proinflammatory responses in vitro (Yin et al. 2014; Hu et al. 2012; Xu et al. 2014), less is known about whether TLR4 is involved in nicotine reward or withdrawal. Interestingly, a recent clinical study showed a potential association between TLR4 polymorphism and lifetime smoking (Zerdazi et al. 2017). Based on the study from 514 bipolar disorder patients, El-Hadi and colleagues found that rs10759932 was significantly associated with tobacco smoking (Zerdazi et al. 2017). This finding suggests the involvement of TLR4 in smoking, or further, nicotine addiction. However, studies also suggest that nicotine attenuates neuroinflammation induced by microglia activation in the brain (Park et al. 2007; Lutz et al. 2014), possibly through TLR4 signaling (Li et al. 2021). Nicotine and its metabolite cotinine targeted TLR4 co-receptor, MD-2, and inhibited LPS-induced production of TNF-α and nitric oxide, and further blocked microglia activation (Li et al. 2021). Moreover, this effect cannot be abolished by nicotinic acetylcholine receptor (nAChR) inhibitor or nAChRs siRNA (Li et al. 2021). These results seem inconsistent and add more complexity to the role of TLR4 in nicotine response.
2.3 Ethanol
Neuroinflammation contributes to the establishment of addiction of several substances, including alcohol. In vitro and in vivo studies have shown that ethanol produces neuroinflammation at least partially through TLR4 signaling pathway and leads to the activation of NFκB (Blanco et al. 2005; Fernandez-Lizarbe et al. 2009). For example, adolescent binge drinking increases the TLR4 expression in the adult prefrontal cortex, which is correlated with deficits in reversal learning and increased preservative behaviors (Vetreno and Crews 2012). Bing drinking also promoted the IL-1β mRNA expression in the basolateral amygdala (BLA). Consistently, intra-BLA infusions of IL-1 receptor antagonist (IL-1Ra) decreased the alcohol consumption without altering sucrose drinking and locomotion in mice (Marshall et al. 2016). Furthermore, studies utilized TLR4 transgenic animal models showed that TLR4 deficiency prevented ethanol-induced neuroinflammation along with synaptic changes and long-term behavioral and cognitive alterations (Fernandez-Lizarbe et al. 2009; Pascual et al. 2017; Montesinos et al. 2015; Montesinos et al. 2016; Montesinos et al. 2018; Shukla et al. 2018). Consistently, TLR4 antagonists like (+)-Naltrexone and Nalmefene prevented TLR4 activation and inhibited alcohol-induced upregulations of proinflammatory responses as well as alcohol intake and reward (Jacobsen et al. 2018a, b; Montesinos et al. 2017). However, a recent comprehensive study showed that TLR4 may not be essential to excessive alcohol drinking (Harris et al. 2017). Using different species, different tests of alcohol consumption, and different methods to inhibit TLR4 signaling, they found that TLR4 inhibition did not affect the drinking-in-the-dark or two-bottle choice chronic ethanol intake or ethanol self-administration (Harris et al. 2017). This study questioned the essentiality of TLR4 in alcohol reward. Nevertheless, they did agree on the effect of TLR4 modulation in alcohol-induced sedation and GABA receptor function (Harris et al. 2017).
Despite the complex results from preclinical studies, much efforts have been put on whether TLR4-related neuroimmune responses regulate alcohol intake in patients with alcohol use disorders (AUD). Studies showed that AUD patients had altered TLR4 methylation, which is correlated with alcohol consumption patterns (Karoly et al. 2017, 2018). Post-mortem human also showed upregulated TLR4-related immunoreactivity cells that correlated with lifetime alcohol consumption (Crews et al. 2013), although alcohol withdrawal may have differentiated effects (Donnadieu-Rigole et al. 2016). In a randomized, placebo-controlled clinical study, however, ibudilast did not affect the subjective responses to alcohol. Meanwhile, it attenuated alcohol-induced stimulant and mood-altering effects in patients with more depressive symptoms (Ray et al. 2017), while other appetitive responses, like craving for high-fat/high-sugar diet, were not altered (Cummings et al. 2018). These results raised a question whether improvement of depressive symptomatology should be considered as a measurement for potential pharmacotherapies. Nevertheless, we are still at the very beginning to examine TLR4 as promising therapeutic target for the treatment of alcohol addiction, more comprehensive studies with larger sample size are warranted.
3 Possible Mechanisms Underlying the Role of TLR4 Signaling in Drug Addiction
Apart from the traditional neuronal mechanisms which involves dopaminergic, glutamatergic, and GABAergic system, drugs of abuse-induced glia activation are believed to contribute to the development of drug addiction. Opioid, psychostimulants, and alcohol all bind to the accessory receptor Myeloid Differentiation factor 2 (MD-2) and activate TLR4. This activation promotes the release of proinflammatory cytokines and chemokines, which subsequently alters the neuroadaptations and synaptic plasticity that is related to drug-induced aberrant behaviors. TLR4 is showed to play a role in NAc synaptic physiology and drug reward behavior (Kashima and Grueter 2017). TLR4-KO animals demonstrated a significantly decreased α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor/N-methyl-D-aspartate (NMDA) receptor ratio (A/N ratio) in the NAc core, suggesting a decrease in postsynaptic strength caused by a reduced AMPAR transmission or increased NMDAR transmission (Kashima and Grueter 2017). Meanwhile, TLR4-KO D1(−) MSNs showed significant slower NMDAR decay kinetics compared with WT, suggesting an altered NMDAR stoichiometry (Kashima and Grueter 2017). Because altered NMDARs in the NAc MSNs are related to behavioral adaptations affecting motivation and reward-associated learning, it is further shown that TLR4-KO mice exhibit deficits in long-term depression in the NAc core, paralleled with deficits in drug reward learning (Kashima and Grueter 2017). These results showed a direct association between TLR4 and drug-induced neuroadaptations.
The downstream effectors of TLR4 may also play a part in regulating drug addiction. Our recent study examined the role of IRAK4, a downstream molecule of TLR4 signaling, in opioid addiction. We found that IRAK4 antagonist PF06650833 reduced cue-induced reinstatement of morphine-seeking and fentanyl-seeking (Wu et al. 2021). Morphine self-administration induced activation of IRAK4 in the NAc, which was accompanied by increases in IKKα/β activity and expression level of soluble TNF-α (Wu et al. 2021). Furthermore, microinjection of RF06650833 into the NAc reduced cue-induced reinstatement of morphine-seeking (Wu et al. 2021). As IRAK4 is one of the keynotes of the TLR4 signaling cascade, our results might suggest that TLR4 might participate in the cue-induced reinstatement of morphine-seeking via the IRAK4 signaling pathway.
Immune factors like TNF-α and IL-β that are involved in the modulation of synaptic functions probably participate in drug reward as well. TNF-α is a key effector in the TLR4 signaling, and inhibition of TNF-α abolishes TLR4-mediated responses (Kawai and Akira 2010; Eidson et al. 2017). It is reported that TNF-α is involved in cocaine-induced plasticity (Lewitus et al. 2016). Drugs of abuse activate the glia cells in the NAc, which subsequently enhance the production of TNF-α. TNF-α is known to regulate the internalization of synaptic AMPA receptors (Lewitus et al. 2014). A recent study showed that cocaine activates striatal microglia and promotes TNF-α production, which suppresses the glutamatergic synaptic strength in the NAc core (Lewitus et al. 2016). Besides the AMPARs, TNF-α is also suggested to regulate the activity of presynaptic metabotropic glutamate receptors and GABAA receptors (Bezzi et al. 2001; Stellwagen et al. 2005; Pascual et al. 2012; Domercq et al. 2006). Like TNF-α, IL-β is also activated by TLR4 (Latz et al. 2013). IL-β is associated with long-term potentiation which underlies learning and memory, thus is implicated with drug-related aberrant memory (Rizzo et al. 2018). IL-β decreases glutamate supply through the inhibition of glial glutamate transporter activity, resulting in the attenuation of glutamate-glutamine cycle-dependent GABA synthesis. Moreover, IL-β also participates in the regulation of postsynaptic GABA receptor activity. These modulations are widely associated with synaptic plasticity which may contribute to TLR4 signaling-related neuroadaptations (Wang et al. 2000).
The activation of TLR4 by drugs of abuse produces neuroinflammation as well as neurodegeneration within key brain regions that are involved in drug addiction (Alfonso-Loeches et al. 2010; Pascual et al. 2011; Alfonso-Loeches et al. 2012). Conversely, inhibition of TLR4 abolishes the proinflammatory responses and blocks cell damage (Blanco et al. 2005). For example, neurodegenerations in the prefrontal cortex are associated with the loss of executive functions over behavioral inhibition or a lack of inhibitory control over mesolimbic areas, which may consequently promote the drug-taking behaviors (Crews et al. 2011, 2015). Generally, the loss of control over progression from initial recreational drug use to compulsive drug-taking may promote the development of drug addiction (Wu and Li 2020). Although much evidence has implicated the role of TLR4 and its signaling in drug addiction, the exact mechanisms and process remain unknown. Nevertheless, it should be kept in mind that drugs of abuse activation of TLR4 signaling may work in conjunction with the traditional well-established neuronal mechanisms, as the modulation of central immune system alone did not alter drug-related behaviors (Coller and Hutchinson 2012).
4 Future Directions
While extensive studies have suggested that TLR4 and its signaling play an important role in drug addiction, many questions remain to be answered before TLR4 modulators could be used as potential treatments for alleviating drug abuse-related symptoms. Firstly, conflicting results from preclinical studies suggest the complex effects of TLR4 in regulating drug addiction. Future comprehensive studies that examine the effect of TLR4 modulation in different drug class from different drug addiction stages (i.e. binge/intoxication, withdrawal and relapse) will help establish whether TLR4 is a promising and novel therapeutic target to treat drug addiction. Secondly, the mechanism underlying the effect of TLR4 modulations in drug addiction is no clear. More studies carefully investigate how TLR4-related activation contribute to the progression of drug addiction are urgently needed. More importantly, to answer how TLR4-related non-neuronal system communicate and synergize with the well-known neuronal system will help tremendously in understanding the mechanisms underlying drug addiction. Last but not least, increased recognition of TLR4 in regulating drug addiction leads to a growing interest in clinical investigations. However, we are still far away from reaching a solid conclusion from clinical settings that TLR4 modulators could be potential pharmacotherapies for drug addiction. Future randomized and placebo-controlled clinical studies with large sample size, which examine the long-term safety, tolerability, and efficacy of TLR4-based neuroimmune pharmacotherapies are warranted.
5 Conclusion
Drugs of abuse activate TLR4 and its signaling and enhance the production of proinflammatory cytokines and chemokines. Modulations of TLR4 and its signaling are shown to be involved in addiction to drugs from different class, including psychostimulants, opioids, and alcohol. Furthermore, increased evidence has suggested that TLR4 and related glial cell modulators could be potential treatments for addiction-related behaviors. This is a thriving topic that requires more comprehensive studies for both target validation and clinical efficacy verification to reshape the treatment for drug addiction.
References
Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, Guerri C (2010) Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci 30(24):8285–8295
Alfonso-Loeches S, Pascual M, Gomez-Pinedo U, Pascual-Lucas M, Renau-Piqueras J, Guerri C (2012) Toll-like receptor 4 participates in the myelin disruptions associated with chronic alcohol abuse. Glia 60(6):948–964
Aurelian L, Warnock KT, Balan I, Puche A, June H (2016) TLR4 signaling in VTA dopaminergic neurons regulates impulsivity through tyrosine hydroxylase modulation. Transl Psychiatry 6:e815
Beardsley PM, Shelton KL, Hendrick E, Johnson KW (2010) The glial cell modulator and phosphodiesterase inhibitor, AV411 (ibudilast), attenuates prime- and stress-induced methamphetamine relapse. Eur J Pharmacol 637(1–3):102–108
Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E et al (2001) CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 4(7):702–710
Birath JB, Briones M, Amaya S, Shoptaw S, Swanson AN, Tsuang J et al (2017) Ibudilast may improve attention during early abstinence from methamphetamine. Drug Alcohol Depend 178:386–390
Blanco AM, Valles SL, Pascual M, Guerri C (2005) Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J Immunol 175(10):6893–6899
Bland ST, Hutchinson MR, Maier SF, Watkins LR, Johnson KW (2009) The glial activation inhibitor AV411 reduces morphine-induced nucleus accumbens dopamine release. Brain Behav Immun 23(4):492–497
Burkovetskaya ME, Small R, Guo L, Buch S, Guo ML (2020) Cocaine self-administration differentially activates microglia in the mouse brain. Neurosci Lett 728:134951
Cearley CN, Blindheim K, Sorg BA, Krueger JM, Churchill L (2011) Acute cocaine increases interleukin-1beta mRNA and immunoreactive cells in the cortex and nucleus accumbens. Neurochem Res 36(4):686–692
Chen H, Uz T, Manev H (2009) Minocycline affects cocaine sensitization in mice. Neurosci Lett 452(3):258–261
Chen JX, Huang KM, Liu M, Jiang JX, Liu JP, Zhang YX et al (2017) Activation of TLR4/STAT3 signaling in VTA contributes to the acquisition and maintenance of morphine-induced conditioned place preference. Behav Brain Res 335:151–157
Cheron J, Kerchove d'Exaerde A (2021) Drug addiction: from bench to bedside. Transl Psychiatry 11(1):424
Coller JK, Hutchinson MR (2012) Implications of central immune signaling caused by drugs of abuse: mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacol Ther 134(2):219–245
Connolly DJ, O'Neill LA (2012) New developments in toll-like receptor targeted therapeutics. Curr Opin Pharmacol 12(4):510–518
Cooper ZD, Johnson KW, Pavlicova M, Glass A, Vosburg SK, Sullivan MA et al (2016) The effects of ibudilast, a glial activation inhibitor, on opioid withdrawal symptoms in opioid-dependent volunteers. Addict Biol 21(4):895–903
Cooper ZD, Johnson KW, Vosburg SK, Sullivan MA, Manubay J, Martinez D et al (2017) Effects of ibudilast on oxycodone-induced analgesia and subjective effects in opioid-dependent volunteers. Drug Alcohol Depend 178:340–347
Crews FT, Zou J, Qin L (2011) Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun 25(Suppl 1):S4–S12
Crews FT, Qin L, Sheedy D, Vetreno RP, Zou J (2013) High mobility group box 1/toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry 73(7):602–612
Crews FT, Sarkar DK, Qin L, Zou J, Boyadjieva N, Vetreno RP (2015) Neuroimmune function and the consequences of alcohol exposure. Alcohol Res 37(2):331–41, 44–51
Crews FT, Walter TJ, Coleman LG Jr, Vetreno RP (2017) Toll-like receptor signaling and stages of addiction. Psychopharmacology 234(9–10):1483–1498
Cummings JR, Tomiyama AJ, Ray LA (2018) Does the neuroimmune modulator ibudilast alter food craving? Results in a sample with alcohol use disorder. J Addict Med 12(5):410–417
Das KP, McMillian MK, Bing G, Hong JS (1995) Modulatory effects of [Met5]-enkephalin on interleukin-1 beta secretion from microglia in mixed brain cell cultures. J Neuroimmunol 62(1):9–17
Domercq M, Brambilla L, Pilati E, Marchaland J, Volterra A, Bezzi P (2006) P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J Biol Chem 281(41):30684–30696
Donnadieu-Rigole H, Mura T, Portales P, Duroux-Richard I, Bouthier M, Eliaou JF et al (2016) Effects of alcohol withdrawal on monocyte subset defects in chronic alcohol users. J Leukoc Biol 100(5):1191–1199
Eidson LN, Inoue K, Young LJ, Tansey MG, Murphy AZ (2017) Toll-like receptor 4 mediates morphine-induced neuroinflammation and tolerance via soluble tumor necrosis factor signaling. Neuropsychopharmacology 42(3):661–670
Fernandez-Lizarbe S, Pascual M, Guerri C (2009) Critical role of TLR4 response in the activation of microglia induced by ethanol. J Immunol 183(7):4733–4744
Fujita Y, Kunitachi S, Iyo M, Hashimoto K (2012) The antibiotic minocycline prevents methamphetamine-induced rewarding effects in mice. Pharmacol Biochem Behav 101(2):303–306
Gabr MM, Saeed I, Miles JA, Ross BP, Shaw PN, Hollmann MW et al (2021) Interaction of opioids with TLR4-mechanisms and ramifications. Cancers (Basel) 13(21):5274
Garcia Bueno B, Caso JR, Madrigal JL, Leza JC (2016) Innate immune receptor toll-like receptor 4 signalling in neuropsychiatric diseases. Neurosci Biobehav Rev 64:134–147
Goncalves J, Martins T, Ferreira R, Milhazes N, Borges F, Ribeiro CF et al (2008) Methamphetamine-induced early increase of IL-6 and TNF-alpha mRNA expression in the mouse brain. Ann N Y Acad Sci 1139:103–111
Harris RA, Bajo M, Bell RL, Blednov YA, Varodayan FP, Truitt JM et al (2017) Genetic and pharmacologic manipulation of TLR4 has minimal impact on ethanol consumption in rodents. J Neurosci 37(5):1139–1155
Heinzerling KG, Briones M, Thames AD, Hinkin CH, Zhu T, Wu YN et al (2020) Randomized, placebo-controlled trial of targeting neuroinflammation with ibudilast to treat methamphetamine use disorder. J Neuroimmune Pharmacol 15(2):238–248
Hennessy EJ, Parker AE, O'Neill LA (2010) Targeting toll-like receptors: emerging therapeutics? Nat Rev Drug Discov 9(4):293–307
Hu SX, Sui HX, Jin HJ, Ni XY, Liu XX, Xue MQ et al (2012) Lipopolysaccharide and dose of nicotine determine the effects of nicotine on murine bone marrow-derived dendritic cells. Mol Med Rep 5(4):1005–1010
Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL et al (2009) Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast). Brain Behav Immun 23(2):240–250
Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX et al (2010) Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun 24(1):83–95
Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J et al (2012) Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J Neurosci 32(33):11187–11200
Jacobsen JHW, Buisman-Pijlman FTA, Mustafa S, Rice KC, Hutchinson MR (2018a) The efficacy of (+)-Naltrexone on alcohol preference and seeking behaviour is dependent on light-cycle. Brain Behav Immun 67:181–193
Jacobsen JHW, Buisman-Pijlman FT, Mustafa S, Rice KC, Hutchinson MR (2018b) Antagonising TLR4-TRIF signalling before or after a low-dose alcohol binge during adolescence prevents alcohol drinking but not seeking behaviour in adulthood. Neuropharmacology 128:460–473
June HL, Liu J, Warnock KT, Bell KA, Balan I, Bollino D et al (2015) CRF-amplified neuronal TLR4/MCP-1 signaling regulates alcohol self-administration. Neuropsychopharmacology 40(6):1549–1559
Karoly HC, Thayer RE, Hagerty SL, Hutchison KE (2017) TLR4 methylation moderates the relationship between alcohol use severity and gray matter loss. J Stud Alcohol Drugs 78(5):696–705
Karoly HC, Ellingson JM, Hutchison KE (2018) Interactions between TLR4 methylation and alcohol consumption on subjective responses to an alcohol infusion. Alcohol Alcohol 53(6):650–658
Kashima DT, Grueter BA (2017) Toll-like receptor 4 deficiency alters nucleus accumbens synaptic physiology and drug reward behavior. Proc Natl Acad Sci U S A 114(33):8865–8870
Kawai T, Akira S (2007a) TLR signaling. Semin Immunol 19(1):24–32
Kawai T, Akira S (2007b) Signaling to NF-kappaB by toll-like receptors. Trends Mol Med 13(11):460–469
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol 11(5):373–384
Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35(1):217–238
Koropatnick TA, Engle JT, Apicella MA, Stabb EV, Goldman WE, McFall-Ngai MJ (2004) Microbial factor-mediated development in a host-bacterial mutualism. Science 306(5699):1186–1188
Landreth GE, Reed-Geaghan EG (2009) Toll-like receptors in Alzheimer’s disease. Curr Top Microbiol Immunol 336:137–153
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13(6):397–411
Lewitus GM, Pribiag H, Duseja R, St-Hilaire M, Stellwagen D (2014) An adaptive role of TNFalpha in the regulation of striatal synapses. J Neurosci 34(18):6146–6155
Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D (2016) Microglial TNF-alpha suppresses cocaine-induced plasticity and behavioral sensitization. Neuron 90(3):483–491
Li H, Peng Y, Lin C, Zhang X, Zhang T, Wang Y et al (2021) Nicotine and its metabolite cotinine target MD2 and inhibit TLR4 signaling. Innovation (N Y) 2(2):100111
Liu L, Coller JK, Watkins LR, Somogyi AA, Hutchinson MR (2011) Naloxone-precipitated morphine withdrawal behavior and brain IL-1beta expression: comparison of different mouse strains. Brain Behav Immun 25(6):1223–1232
Liu JF, Wu R, Li JX (2019) Toll of mental disorders: TLR-mediated function of the innate immune system. Neurosci Bull 35(4):771–774
Loftis JM, Choi D, Hoffman W, Huckans MS (2011) Methamphetamine causes persistent immune dysregulation: a cross-species, translational report. Neurotox Res 20(1):59–68
Lutz JA, Kulshrestha M, Rogers DT, Littleton JM (2014) A nicotinic receptor-mediated anti-inflammatory effect of the flavonoid rhamnetin in BV2 microglia. Fitoterapia 98:11–21
Majdi F, Taheri F, Salehi P, Motaghinejad M, Safari S (2019) Cannabinoids Delta(9)-tetrahydrocannabinol and cannabidiol may be effective against methamphetamine induced mitochondrial dysfunction and inflammation by modulation of toll-like type-4(toll-like 4) receptors and NF-kappaB signaling. Med Hypotheses 133:109371
Marshall SA, Casachahua JD, Rinker JA, Blose AK, Lysle DT, Thiele TE (2016) IL-1 receptor signaling in the basolateral amygdala modulates binge-like ethanol consumption in male C57BL/6J mice. Brain Behav Immun 51:258–267
Mattioli TA, Leduc-Pessah H, Skelhorne-Gross G, Nicol CJ, Milne B, Trang T et al (2014) Toll-like receptor 4 mutant and null mice retain morphine-induced tolerance, hyperalgesia, and physical dependence. PLoS One 9(5):e97361
Metz VE, Jones JD, Manubay J, Sullivan MA, Mogali S, Segoshi A et al (2017) Effects of ibudilast on the subjective, reinforcing, and analgesic effects of oxycodone in recently detoxified adults with opioid dependence. Neuropsychopharmacology 42(9):1825–1832
Montesinos J, Pascual M, Pla A, Maldonado C, Rodriguez-Arias M, Minarro J et al (2015) TLR4 elimination prevents synaptic and myelin alterations and long-term cognitive dysfunctions in adolescent mice with intermittent ethanol treatment. Brain Behav Immun 45:233–244
Montesinos J, Pascual M, Rodriguez-Arias M, Minarro J, Guerri C (2016) Involvement of TLR4 in the long-term epigenetic changes, rewarding and anxiety effects induced by intermittent ethanol treatment in adolescence. Brain Behav Immun 53:159–171
Montesinos J, Gil A, Guerri C (2017) Nalmefene prevents alcohol-induced neuroinflammation and alcohol drinking preference in adolescent female mice: role of TLR4. Alcohol Clin Exp Res 41(7):1257–1270
Montesinos J, Pascual M, Millan-Esteban D, Guerri C (2018) Binge-like ethanol treatment in adolescence impairs autophagy and hinders synaptic maturation: role of TLR4. Neurosci Lett 682:85–91
Moreira FP, Medeiros JR, Lhullier AC, Souza LD, Jansen K, Portela LV et al (2016) Cocaine abuse and effects in the serum levels of cytokines IL-6 and IL-10. Drug Alcohol Depend 158:181–185
Nakajima A, Yamada K, Nagai T, Uchiyama T, Miyamoto Y, Mamiya T et al (2004) Role of tumor necrosis factor-alpha in methamphetamine-induced drug dependence and neurotoxicity. J Neurosci 24(9):2212–2225
Narita M, Miyatake M, Narita M, Shibasaki M, Shindo K, Nakamura A et al (2006) Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology 31(11):2476–2488
Nestler EJ, Luscher C (2019) The molecular basis of drug addiction: linking epigenetic to synaptic and circuit mechanisms. Neuron 102(1):48–59
Nie X, Kitaoka S, Tanaka K, Segi-Nishida E, Imoto Y, Ogawa A et al (2018) The innate immune receptors TLR2/4 mediate repeated social defeat stress-induced social avoidance through prefrontal microglial activation. Neuron 99(3):464–79 e7
Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA et al (2015) DAT isn’t all that: cocaine reward and reinforcement require toll-like receptor 4 signaling. Mol Psychiatry 20(12):1525–1537
Park HJ, Lee PH, Ahn YW, Choi YJ, Lee G, Lee DY et al (2007) Neuroprotective effect of nicotine on dopaminergic neurons by anti-inflammatory action. Eur J Neurosci 26(1):79–89
Pascual M, Balino P, Alfonso-Loeches S, Aragon CM, Guerri C (2011) Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav Immun 25(Suppl 1):S80–S91
Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A (2012) Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A 109(4):E197–E205
Pascual M, Montesinos J, Montagud-Romero S, Forteza J, Rodriguez-Arias M, Minarro J et al (2017) TLR4 response mediates ethanol-induced neurodevelopment alterations in a model of fetal alcohol spectrum disorders. J Neuroinflammation 14(1):145
Ray LA, Bujarski S, Shoptaw S, Roche DJ, Heinzerling K, Miotto K (2017) Development of the neuroimmune modulator ibudilast for the treatment of alcoholism: a randomized, placebo-controlled, human laboratory trial. Neuropsychopharmacology 42(9):1776–1788
Rizzo FR, Musella A, De Vito F, Fresegna D, Bullitta S, Vanni V et al (2018) Tumor necrosis factor and interleukin-1beta modulate synaptic plasticity during neuroinflammation. Neural Plast 2018:8430123
Schwarz JM, Bilbo SD (2013) Adolescent morphine exposure affects long-term microglial function and later-life relapse liability in a model of addiction. J Neurosci 33(3):961–971
Shukla PK, Meena AS, Rao R, Rao R (2018) Deletion of TLR-4 attenuates fetal alcohol exposure-induced gene expression and social interaction deficits. Alcohol 73:73–78
Snider SE, Vunck SA, van den Oord EJ, Adkins DE, McClay JL, Beardsley PM (2012) The glial cell modulators, ibudilast and its amino analog, AV1013, attenuate methamphetamine locomotor activity and its sensitization in mice. Eur J Pharmacol 679(1–3):75–80
Snider SE, Hendrick ES, Beardsley PM (2013) Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur J Pharmacol 701(1–3):124–130
Stellwagen D, Beattie EC, Seo JY, Malenka RC (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 25(12):3219–3228
Takeda K, Akira S (2005) Toll-like receptors in innate immunity. Int Immunol 17(1):1–14
Tanda G, Mereu M, Hiranita T, Quarterman JC, Coggiano M, Katz JL (2016) Lack of specific involvement of (+)-naloxone and (+)-naltrexone on the reinforcing and neurochemical effects of cocaine and opioids. Neuropsychopharmacology 41(11):2772–2781
Tanibuchi Y, Shimagami M, Fukami G, Sekine Y, Iyo M, Hashimoto K (2010) A case of methamphetamine use disorder treated with the antibiotic drug minocycline. Gen Hosp Psychiatry 32(5):559.e1–3
Theberge FR, Li X, Kambhampati S, Pickens CL, St Laurent R, Bossert JM et al (2013) Effect of chronic delivery of the toll-like receptor 4 antagonist (+)-naltrexone on incubation of heroin craving. Biol Psychiatry 73(8):729–737
Vaure C, Liu Y (2014) A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol 5:316
Vetreno RP, Crews FT (2012) Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and toll-like receptors in the adult prefrontal cortex. Neuroscience 226:475–488
Wang S, Cheng Q, Malik S, Yang J (2000) Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J Pharmacol Exp Ther 292(2):497–504
Wang X, Northcutt AL, Cochran TA, Zhang X, Fabisiak TJ, Haas ME et al (2019) Methamphetamine activates toll-like receptor 4 to induce central immune signaling within the ventral tegmental area and contributes to extracellular dopamine increase in the nucleus accumbens shell. ACS Chem Neurosci 10(8):3622–3634
Wise RA, Koob GF (2014) The development and maintenance of drug addiction. Neuropsychopharmacology 39(2):254–262
Worley MJ, Swanson AN, Heinzerling KG, Roche DJ, Shoptaw S (2016) Ibudilast attenuates subjective effects of methamphetamine in a placebo-controlled inpatient study. Drug Alcohol Depend 162:245–250
Wu R, Li JX (2020) Toll-like receptor 4 signaling and drug addiction. Front Pharmacol 11:603445
Wu R, Liu J, Vu J, Huang Y, Dietz DM, Li JX (2021) Interleukin-1 receptor-associated kinase 4 (IRAK4) in the nucleus accumbens regulates opioid-seeking behavior in male rats. Brain Behav Immun 101:37–48
Xu Y, Zhang Y, Cardell LO (2014) Nicotine exaggerates LPS-induced airway hyperreactivity via JNK-mediated up-regulation of toll-like receptor 4. Am J Respir Cell Mol Biol 51(3):370–379
Yin Y, Hou G, Li E, Wang Q, Kang J (2014) PPARgamma agonists regulate tobacco smoke-induced toll like receptor 4 expression in alveolar macrophages. Respir Res 15:28
Yue K, Tanda G, Katz JL, Zanettini C (2020) A further assessment of a role for toll-like receptor 4 in the reinforcing and reinstating effects of opioids. Behav Pharmacol 31(2&3):186–195
Zerdazi EH, Oliveira J, Vorspan F, Bennabi M, Jamain S, Etain B et al (2017) TLR4 gene polymorphism associated with lifetime cigarette smoking in bipolar disorder. J Neuroimmunol 305:96–101
Zhang L, Kitaichi K, Fujimoto Y, Nakayama H, Shimizu E, Iyo M et al (2006) Protective effects of minocycline on behavioral changes and neurotoxicity in mice after administration of methamphetamine. Prog Neuro-Psychopharmacol Biol Psychiatry 30(8):1381–1393
Acknowledgements
RW was partially supported by the National Natural Science Foundation of China (Grant 81701340) and Natural Science Foundation of Jiangsu Province (Grant BK 20170517). JXL was supported by National Institutes of Health National Institute on Drug Abuse (Grant R01DA047967).
Conflict of Interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Liu, J., Li, JX., Wu, R. (2022). Toll-Like Receptor 4: A Novel Target to Tackle Drug Addiction?. In: Kumar, V. (eds) Toll-like Receptors in Health and Disease. Handbook of Experimental Pharmacology, vol 276. Springer, Cham. https://doi.org/10.1007/164_2022_586
Download citation
DOI: https://doi.org/10.1007/164_2022_586
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-06511-8
Online ISBN: 978-3-031-06512-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)